

Graphical abstract
Keywords: COVID-19, SARS-CoV-2, Parsimony principle, Phylogenetic relationship, Basal clade, RNA proofreading capability
Abstract
The COVID-19 pandemic is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) whose origin is still shed in mystery. In this study, we developed a method to search the basal SARS-CoV-2 clade among collected SARS-CoV-2 genome sequences. We first identified the mutation sites in the SARS-CoV-2 whole genome sequence alignment. Then by the pairwise comparison of the numbers of mutation sites among all SARS-CoV-2s, the least mutated clade was identified, which is the basal clade under parsimony principle. In our first analysis, we used 168 SARS-CoV-2 sequences (GISAID dataset till 2020/03/04) to identify the basal clade which contains 33 identical viral sequences from seven countries. To our surprise, in our second analysis with 367 SARS-CoV-2 sequences (GISAID dataset till 2020/03/17), the basal clade has 51 viral sequences, 18 more sequences added. The much larger NCBI dataset shows that this clade has expanded with 85 unique sequences by 2020/04/04. The expanding basal clade tells a chilling fact that the least mutated SARS-CoV-2 sequence was replicating and spreading for at least four months. It is known that coronaviruses have the RNA proofreading capability to ensure their genome replication fidelity. Interestingly, we found that the SARS-CoV-2 without its nonstructural proteins 13 to 16 (Nsp13-Nsp16) exhibits an unusually high mutation rate. Our result suggests that SARS-CoV-2 has an unprecedented RNA proofreading capability which can intactly preserve its genome even after a long period of transmission. Our selection analyses also indicate that the positive selection event enabling SARS-CoV-2 to cross species and adapt to human hosts might have been achieved before its outbreak.
1. Introduction
The coronavirus disease 2019 (COVID-19) has been reported in over 200 countries (WHO, 2020). It is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is clustered with the SARS-CoVs of bat in a clade and regarded as a SARS-like virus (Xu et al., 2020). Although it has a lower mortality rate than SARS-CoV, SARS-CoV-2 exhibits a high contagious transmission pattern with low detectability, making this virus more threatening than any coronavirus before. Now COVID-19 becomes a global hazard instead of a regional crisis.
The COVID-19 epidemic first broke out in early December 2019, in Wuhan City, China. The news reported that SARS-CoV-2 was associated with a sea food market in Wuhan, but its origin is still shed in mystery. Even the evolutionary relationship within SARS-CoV-2s is very obscure. The bat RaTG13 coronavirus is usually used as the outgroup to root the SARS-CoV-2′s phylogenetic tree in a lot of practices (Forster et al., 2020). However, the bat RaTG13 coronavirus was discovered in 2013 and the great divergence between the bat virus and SARS-CoV-2s created a problem for phylogenetic inference, called long-branch attraction (Gribaldo and Philippe, 2002). That is the fast evolving SARS-CoV-2 branches which would be wrongfully placed close to the bat RaTG13 coronavirus as if they were the ancestors to the other SARS-CoV-2s.
In this work, we used a parsimony method to determine which SARS-CoV-2 clade is the most basal clade to the others. Our method does not need any outgroup and thus circumvents the long-branch attraction trap. To ours surprise, we identified an expanding basal clade in this work, which suggests that SARS-CoV-2 has an extraordinary RNA proofreading capability for protecting its genome from mutation. Additionally, we investigated the selection process for all SARS-CoV-2 coding sequences in this work and did not observe any significant positive selection event, which proposes that the positive selection enabling SARS-CoV-2 to cross species and adapt to human hosts has already achieved before its outbreak in Wuhan in early December 2019. By comparing SARS-CoV-2′s nonstructural proteins with their SARS-CoV’s counterparts, we found the fast evolving SARS-CoV virus (406592_Shenzhen_2020) harbors an premature termination codon at nonstructural protein 12 (Nsp12). Truncated nonstructural proteins of SARS-CoV-2 might affect its replication fidelity and susceptibility to antiviral drugs, such as Remdesivir (Agostini et al., 2018). Finally, the much larger NCBI dataset shows that the least mutated SARS-CoV-2 sequence was replicating and spreading for at least four months.
2. Data and method
2.1. SARS-CoV-2 genome data
430 SARS-CoV-2 whole genome sequences were downloaded from GISAID. Due to the continuous incoming SARS-CoV-2 genome data on GISAID, we downloaded the dataset in two batches. The first batch of 184 SARS-CoV-2 genome data was obtained till 2020/03/04 and the second batch of 246 SARS-CoV-2 genome data was obtained till 2020/03/17. After removing the low-coverage (shorter than 29,000 base pairs) and low-quality sequences (with more than 100 ambiguous nucleotides), 367 SARS-CoV-2 sequences were kept for the next step analysis. For a much large dataset, we retrieved the SARS-CoV-2 genome data from Coronavirus genomes of NCBI Datasets (https://www.ncbi.nlm.nih.gov/datasets/coronavirus/genomes/). We only downloaded the sequence of human host. The downloaded dataset has 7007 complete SARS-CoV-2 genome sequences. After removing the low-coverage (shorter than 29,000 base pairs) and low-quality sequences (with more than 100 ambiguous nucleotides), 4678 SARS-CoV-2 genome sequences were kept for the further analysis. The collection dates for GISAID data were retrieved from GISAID cov2020 acknowledgement table (from 2019/12/30 to 2020/03/13) and the collection dates for NCBI data were from GBFF file (2019/12/30 to 2020/07/07).
2.2. Genome sequence alignment
We used MUSCLE 3.8.31 to perform the alignment for all SARS-CoV-2 sequences with default parameters (Edgar, 2004). The alignment results were trimmed with an in-house Perl script to remove any gap or ambiguous site. In our first analysis (GISAID dataset till 2020/03/04), the final alignment of SARS-CoV-2s has 168 sequences and 29,347 nucleotide sites. In our second analysis (GISAID dataset till 2020/03/17), the final alignment of SARS-CoV-2s has 367 sequences and 29,188 nucleotide sites. For the NCBI SARS-CoV-2 dataset, the final alignment of 4678 SARS-CoV-2 genome sequences has 28,683 nucleotide sites.
2.3. Pair-wise point mutation calculation among all SARS-CoV-2 sequences
In our first analysis, there are 211 mutation sites found in the final SARS-CoV-2 alignment. In our second analysis, there are 390 mutation sites in the final SARS-CoV-2 alignment. Each SARS-CoV-2 sequence was compared to the other SARS-CoV-2 sequences from the final alignment in pair-wise fashion. By doing so, the number of point mutations was calculated for each SARS-CoV-2 sequence. Under parsimony principle, the SARS-CoV-2 sequence that has the least number of point mutations are regarded as the ancestors (Fig. 1 ).
Fig. 1.

The illustration of our parsimony method for determining the most basal SARS-CoV-2s.
2.4. Substitution, selection and phylogenetic analyses
Substitution matrices for SARS-CoV-2s were computed with MEGA X (Kumar et al., 2018). MEGA X (Hyphy model) and Datamonkey (branch-site model) were used to detect the positive selection sites among SARS-CoV-2s (Weaver et al., 2018). The SARS-CoV-2′s phylogenetic tree was constructed with FastTree 2.1.7 using the GTR model and gamma distribution (Price et al., 2010). RAxML v8.2.12 and MrBayes 3.2.6 were also used to construct the phylogenetic trees of SARS-CoV-2s (Ronquist et al., 2012, Stamatakis, 2014).
3. Result
3.1. Transition-transversion biases and mutation rate in SARS-CoV-2s
In our first analysis (GISAID dataset till 2020/03/04), using the genomic DNA alignments of 168 SARS-CoV-2s, we calculated the transition-transversion biases for SARS-CoV-2s (Table 1 ). Our first analysis shows that the transition-transversion bias is 3.058 in SARS-CoV-2s. More C to T and T to C substitutions are observed in SARS-CoV-2s. In our second analysis (GISAID dataset till 2020/03/17), the genomic DNA alignment of 367 SARS-CoV-2s was used to calculated the transition-transversion bias. There are more A to G or G to A transition in the larger dataset and the transition-transversion bias increases to 3.955 (Table 2 ). Using the number of point mutations in the least mutated clade, we calculate the mutation rate for SARS-CoV-2 as follows. We assume that there is no mutation in the least mutated clade and 365 total point mutations are shared by the rest 97 SARS-CoV-2 clades (Supplemental data 2). There are average 3.76 point mutations in each SARS-CoV-2. Our final alignment has 29,188 nucleotide sites. So the mutation rate is 1.29 × 10−4 per nucleotide. The SARS-CoV-2 epidemic in Wuhan started on the early December of 2019 and the final collection date of our second dataset is 2020/03/13 (almost four months) (Wu et al., 2020). Thus, the mutation rate 3.87 × 10−4 per nucleotide per year for the alignment length of 29,188 nucleotide sites. For a complete SARS-CoV-2 genome with 29,903 nucleotides (Wu et al., 2020), the mutation rate is 3.95 × 10−4 per nucleotide per year, which is almost 8 times lower than the mutation rate of SARS-CoV and 3 times lower than that of MERS-CoV (Table 3 ). The mutation rates and the transition-transversion bias for SARS-CoV and MERS-CoV are collected from literatures (2004, xxxx, Cotten et al., 2014, Pavlovic-Lazetic et al., 2005, Zhang et al., 2016). The transition-transversion bias also shows that SARS-CoV-2 has a much stable genome than SARS-CoV and MERS-CoV (Table 3).
Table 1.
Maximum composite likelihood estimate of the pattern of nucleotide substitution in SARS-CoV-2s with 168 viral sequences.
| A | T | C | G | |
|---|---|---|---|---|
| A | – | 3.78 | 2.15 | 7.57 |
| T | 3.51 | – | 20.84 | 2.29 |
| C | 3.51 | 36.55 | – | 2.29 |
| G | 11.58 | 3.78 | 2.15 | – |
The nucleotide frequencies are 29.92% (A), 32.18% (T/U), 18.34% (C), and 19.56% (G). The transition/transversion rate ratios are k1 = 3.297 (purines) and k2 = 9.681 (pyrimidines).
Table 2.
Maximum composite likelihood estimate of the pattern of nucleotide substitution in SARS-CoV-2s with 367 viral sequences.
| A | T | C | G | |
|---|---|---|---|---|
| A | – | 3.08 | 1.75 | 9.27 |
| T | 2.87 | – | 20.79 | 1.87 |
| C | 2.87 | 36.58 | – | 1.87 |
| G | 14.2 | 3.08 | 1.75 | – |
The nucleotide frequencies are 29.96% (A), 32.19% (T/U), 18.30% (C), and 19.55% (G). The transition/transversion rate ratios are k1 = 4.95 (purines) and k2 = 11.863 (pyrimidines).
Table 3.
Mutation rates and transition vs. transversion (Ts/Tv) biases across three human infected coronaviruses.
| Species | Mutation rate (mutation per nt per year) | Ts/Tv bias |
|---|---|---|
| SARS-CoV | 3.01 × 10−3 | 2.61 |
| MERS-CoV | 1.12 × 10−3 | 1.87 |
| SARS-CoV-2 | 3.95 × 10−4 | 3.95 |
3.2. Point mutation in SARS-CoV-2 and selection analyses
In our first analysis (GISAID dataset till 2020/03/04), the final alignment of 168 SARS-CoV-2 sequences has 29,347 nucleotides and only contains 211 non-conserved sites (Supplemental data 1). There are four recurrent point mutations which appear in more than 10 sequences. Using the virus sequence from a seafood market’s worker as the reference (GISAID ID: EPI_ISL_402125), we annotated these four recurrent point mutations. They are C to T mutation at reference position 8782 (alignment position 8466, AGC to AGT, synonymous substitution, serine, ORFlab protein), G to T mutation at reference position 26,144 (alignment position 25818, GGT to GTT, non-synonymous substitution, glycine to valine, ORF3a protein), T to C mutation at reference position 28,144 (alignment position 27817, TTA to TCA, non-synonymous substitution, leucine to serine, ORF8 protein), and C to T mutation at reference position 29,095 (alignment position 28768, TTC to TTT, synonymous substitution, phenylalanine, N protein).
The similarity among SARS-CoV-2 sequences is quite high. 168 SARS-CoV-2 sequences can be further divided into 98 clades according to their sequence identities (Supplemental data 2). The sequences with 100% alignment identity are classified as one clade. The largest clade has 33 SARS-CoV-2 sequences while the smallest one has only one. The numbers of point mutations were calculated among 98 clades in a pair-wise fashion. Our result shows that the largest clade has the least number of point mutations among 98 clades, which was identified as basal clade. Its 33 sequences come from the seven countries of China, Japan, Singapore, Thailand, United States, South Korea, and Nepal. In China, the clade covers at least eight provinces and regions including, Zhejiang (Hangzhou), Hubei (Wuhan), Jiangsu, Chongqing, Anhui (Hefei), Taiwan, Guangdong, and Hong Kong.
In our second analysis (GISAID dataset till 2020/03/17), the final alignment of 367 SARS-CoV-2 sequences has 29,188 nucleotides and only contains 390 non-conserved sites (Supplemental data 3). 367 SARS-CoV-2 sequences can be divided into 215 clades according to their sequence identities (Supplemental data 4). The sequences with 100% alignment identity are classified as one clade. The basal clade now contains 51 sequences. 18 SARS-CoV-2 sequences and three more countries, England, Ireland and Netherlands, are added to the basal clade. There are twenty point mutations recurrent in more than 10 sequences. With more observed mutations, we investigated the possible positive selection events for all SARS-CoV-2 coding sequences with the Hyphy model (MEGA X) and the branch-site model (Datamonkey), but we did not detect any significant positive selection signal. This indicates that the observed mutations in 367 SARS-CoV-2 sequences are most likely to be neutral. Notably, 406592_Shenzhen_2020 has the largest number of mutations in both our analyses.
3.3. Phylogenetic analysis and classification of SARS-CoV-2s
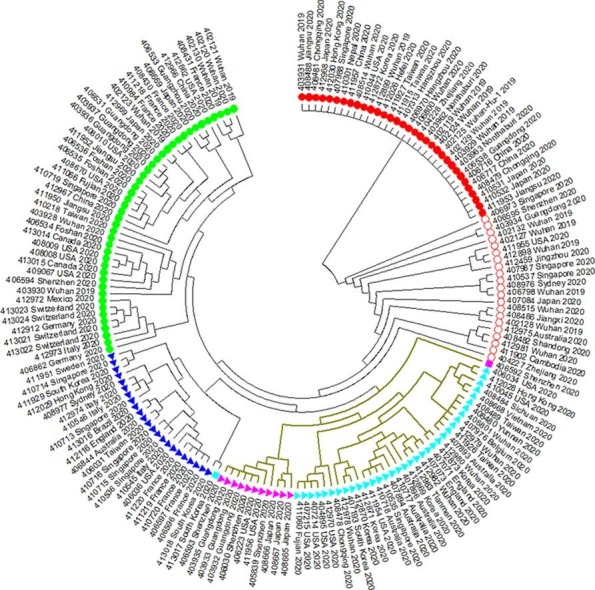
In our first analysis (GISAID dataset till 2020/03/04), we constructed a maximum likelihood tree with 168 SARS-CoV-2 sequences and rooted it with the least mutated clade (Fig. 2 and Supplemental Fig. 1). The tree shows that SARS-CoV-2s can be divided into five major clades. Three of them have at least one dominant mutation which is recurrent in more than 10 SARS-CoV-2s. We classified 21 SARS-CoV-2s as the intermediate type between the basal clade and the other four major clades. They do not form a single clade in the maximum likelihood tree.
Fig. 2.

The maximum likelihood tree of 168 SARS-CoV-2s. Red filled circle  indicates clade 1 (basal clade). Red hollow circle
indicates clade 1 (basal clade). Red hollow circle  indicates the intermediate type between the basal clade and the other four clades. Light-blue triangle
indicates the intermediate type between the basal clade and the other four clades. Light-blue triangle  indicates clade 2 (C to T at 8782). Magenta triangle
indicates clade 2 (C to T at 8782). Magenta triangle  indicates clade 3 (C to T at 8782 and C to T at 29095). Dark-blue triangle
indicates clade 3 (C to T at 8782 and C to T at 29095). Dark-blue triangle  indicates the basal clade 4 (G to T at 26144). Green filled circle
indicates the basal clade 4 (G to T at 26144). Green filled circle  indicates clade 4 (no dominant mutation). Colored squares
indicates clade 4 (no dominant mutation). Colored squares  and
and  indicate two specific sequences. Khaki branch indicates T to C substitution at 27,817 (leucine to serine change). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
indicate two specific sequences. Khaki branch indicates T to C substitution at 27,817 (leucine to serine change). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In Fig. 2, clade 1 is the basal clade which contains 33 SARS-CoV-2 sequences. These sequences are identical. Clade 2 and 3 share the C to T mutation at reference position 8782 while Clade 3 has the additional C to T mutation at reference position 29095. Clade 2 has 35 sequences and Clad 3 has 11 ones. Clade 4 contains 23 SARS-CoV-2 sequences with the G to T mutation at reference position 26144, which causes a glycine to valine change in ORF3a protein. Clade 5 contains 43 SARS-CoV-2 sequences with no dominant mutation (a mutation recurrent in more than 10 sequences).
The T to C mutation at reference position 28,144 that causes a leucine to serine change in ORF8 protein can be found in all SARS-CoV-2s in Clade 2, 3 and two special sequences marked with squares in Fig. 2 (413017_Sourth_Korea_2020 and 406592_Shenzhen_2020). Since the basal clade has T at reference position 28144, leucine is the ancestral state, not serine. 413017_South_Korea_2020 also has the G to T mutation at reference position 26144. 406592_Shenzhen_2020 also has the C to T mutation at reference position 29095.
In our second analysis (GISAID dataset till 2020/03/17), we constructed a maximum likelihood tree with 367 SARS-CoV-2 sequences and rooted it with the expanded least mutated clade (Fig. 3 ). Due to no positive selection detected among SARS-CoV-2s, we did not annotate or classify any mutation in our second maximum likelihood tree. The added 199 sequences in our second analysis are mostly incorporated in our second tree’s clades, instead of forming a single clade of their own. Because SARS-CoV-2s have a very slow evolutionary rate, only a few informative sites so far can be used for their phylogenetic analysis, which render the trees only suitable for the probable estimation of SARS-CoV-2s’ evolutionary relationship and classification. Thus, both RAxML and MrBayes trees show a very poor performance in resolving SARS-CoV-2′s phylogeny (Supplemental Figs. 3 and 4). These two methods require a substantial amount of informative sites to reconstruct a reasonably accurate phylogenetic tree. Such difficulty has also been stated by RAxML’s authors (Benoit Morel et al., 2020).
Fig. 3.

The maximum likelihood tree of 367 SARS-CoV-2s. Red filled circle indicates clade 1 (basal clade). Red hollow circle indicates the intermediate type between the basal clade and the other four clades. Light-blue triangle indicates clade 2 (C to T at 8782). Magenta triangle indicates clade 3 (C to T at 8782 and C to T at 29095). Dark-blue triangle indicates the basal clade 4 (G to T at 26144). Green filled circle indicates clade 4 (no dominant mutation). Colored squares and indicate two specific sequences. Yellow hollow circle  indicates 199 added SARS-CoV-2 sequences in our second analysis. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
indicates 199 added SARS-CoV-2 sequences in our second analysis. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We further constructed a maximum likelihood tree for the much larger NCBI SARS-CoV-2 dataset. There are total 4678 SARS-CoV-2 sequences in the phylogenetic tree of the NCBI dataset (Fig. 4 ). For such a large dataset, we identified the basal clade using the seafood market worker’s SARS-CoV-2 (EPI_ISL_402125) whose NCBI accession ID is NC_045512. It is the first SARS-CoV-2 genome sequence obtained in the Wuhan COVID-19 epidemic (Wu et al., 2020). We found that the basal clade had expanded with 85 sequences (Supplemental data 5). Two more countries, France and Pakistan, were added to the basal clade. The SARS-CoV-2 sequence of this clade was lastly found in Milwaukee county of Wisconsin, USA by 2020/04/04 (NCBI accession ID: MT706448). Using the seafood market worker’s SARS-CoV-2 sequence (EPI_ISL_402125 and NC_045512) as the reference, we found nine major recurrent mutations in the NCBI dataset (Table 4 ). These mutations can classify most SARS-CoV-2s in NCBI dataset into six groups (Fig. 4). There are 470 SARS-CoV-2s in group one, 213 SARS-CoV-2s in group two, 783 SARS-CoV-2s in group three, 952 SARS-CoV-2s in group four, 660 SARS-CoV-2s in group five, and 1504 SARS-CoV-2s in group six, respectively. Frustratingly, multiple geographic locations could be found in each group due to SARS-CoV-2′s low mutability. It looks like that the SARS-CoV-2s have already been everywhere.
Fig. 4.

The maximum likelihood tree of 4678 SARS-CoV-2s. Red filled circle indicates the basal clade. Yellow hollow circle indicates group 1 with no major recurrent mutation. Magenta hollow circle  indicates group 2 with C to T mutation at reference position 8782. Dark-olive-green hollow circle
indicates group 2 with C to T mutation at reference position 8782. Dark-olive-green hollow circle  indicates group 3 with C to T mutation at reference position 8782, C to T mutation at reference position 17747, A to G mutation at reference position 17858, and C to T mutation at reference position 18060. Dark-blue hollow circle
indicates group 3 with C to T mutation at reference position 8782, C to T mutation at reference position 17747, A to G mutation at reference position 17858, and C to T mutation at reference position 18060. Dark-blue hollow circle  indicates group 4 with C to T mutation at reference position 3037, C to T mutation at reference position 14408, and A to G mutation at reference position 23403. Light-blue hollow circle
indicates group 4 with C to T mutation at reference position 3037, C to T mutation at reference position 14408, and A to G mutation at reference position 23403. Light-blue hollow circle  indicates group 5 with C to T mutation at reference position 3037, C to T mutation at reference position 14408, A to G mutation at reference position 23403, and G to T mutation at reference position 25563. Green hollow circle
indicates group 5 with C to T mutation at reference position 3037, C to T mutation at reference position 14408, A to G mutation at reference position 23403, and G to T mutation at reference position 25563. Green hollow circle  indicates group 6 with C to T mutation at reference position 1059, C to T mutation at reference position 3037, C to T mutation at reference position 14408, A to G mutation at reference position 23403, and G to T mutation at reference position 25563. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
indicates group 6 with C to T mutation at reference position 1059, C to T mutation at reference position 3037, C to T mutation at reference position 14408, A to G mutation at reference position 23403, and G to T mutation at reference position 25563. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 4.
Nine major recurrent mutations found in the NCBI dataset.
| Reference Position | Reference nucleotide | Mutation | Gene | Reference codon | Alternative Codon | Amino Acid Change |
|---|---|---|---|---|---|---|
| 1059 | C | C > T | Orf1ab | ACC | ATC | T to I |
| 3037 | C | C > T | Orf1ab | TTC | TTT | F to F |
| 8782 | C | C > T | Orf1ab | AGC | AGT | S to S |
| 14,408 | C | C > T | Orf1ab | CCT | CTT | P to L |
| 17,747 | C | C > T | Orf1ab | CCT | CTT | P to L |
| 17,858 | A | A > G | Orf1ab | TAT | TGT | Y to C |
| 18,060 | C | C > T | Orf1ab | CTC | CTT | L to L |
| 23,403 | A | A > G | S | GAT | GGT | D to G |
| 25,563 | G | G > T | Orf3a | CAG | CAT | Q to H |
The first identified SARS-CoV-2 genome sequence (EPI_ISL_402125 and NC_045512) is used as the reference.
3.4. Fast evolving SARS-CoV-2 does not have nonstructural proteins 13–16
One SARS-CoV-2 sequence (406592_Shenzhen_2020) is especially notable in our point mutation comparison. It has the fastest evolutionary rate among all SARS-CoV-2s and a transition/transversion rate of 0.93. We compared it with the seafood market worker’s SARS-CoV-2 (EPI_ISL_402125) and found that it at least accumulated 27 point mutations. 20 out of 27 mutations are non-synonymous ones which result in amino acid changes (Table 5 ). 13 out of 20 non-synonymous mutations occurred in the orf1ab replicase gene which is responsible for RNA proofreading during virus replication in coronaviruses (Denison et al., 2011). After being processed by viral proteinases, orf1ab replicase gene produces 16 mature nonstructural proteins (Nsp1 to Nsp16) (Denison et al., 2011). Each nsp has its own unique function. Using SARS-CoV sequence with NCBI accession number NC_004718 as a template, we annotated the non-synonymous mutations in orf1ab replicase gene according to their positions in nsp genes (Table 6 ). The nsp2 gene is not required for viral replication (Graham et al., 2006). The nsp8, nsp12, and nsp15 genes encode for a hexadecamer with putative processivity activities, RNA-dependent RNA polymerase (RdRp), and endoribonuclease (EndoU), respectively (Denison et al., 2011). Interestingly, due to a premature stop codon mutation in its nsp12 gene, this fast evolving SARS-CoV-2 lacks nsp13 (RNA helicase-ATPase), nsp14 (exoribonuclease and methyltransferase), nsp15 (endoribonuclease), and nsp16 (RNA 2′-O-methyltransferase). Especially, nsp14 is essential for replication fidelity in coronavirus (Eckerle et al., 2010, Eckerle et al., 2007). This result partly explains why 406592_Shenzhen_2020 accumulates a large number of mutations in its genome.
Table 5.
The non-synonymous mutations in EPI_ISL_406592.
| Gene | Gene length | Nucleotide position from exon start | AA position from exon start | Reference (EPI_ISL_402125) | Mutation (EPI_ISL_406592) | Amino Acid Change |
|---|---|---|---|---|---|---|
| Orf1ab1 | 13,203 | 1904 | 635 | TTT | TCT | F to S |
| 3536 | 1179 | GAT | GCT | D to A | ||
| 4378 | 1460 | GAA | GGA | E to G | ||
| 4391 | 1464 | CGG | CCG | R to P | ||
| 4463,4464 | 1488 | GGT | GTA | G to V | ||
| 4474 | 1492 | TCT | CCT | S to P | ||
| 6043 | 2015 | AGC | GGC | S to G | ||
| 6521 | 2174 | ACT | AGT | T to S | ||
| 6569 | 2190 | ATT | AGT | I to S | ||
| 7826 | 2609 | CTC | CAC | L to H | ||
| Orf1ab2 | 8091 | 2169 | 723 | TAT | TAA | Y to * |
| 6848 | 2283 | TTC | TAC | F to Y | ||
| S | 3822 | 3385 | 1129 | GTA | CTA | V to L |
| 3822 | 1262 | GAG | GGG | E to G | ||
| Orf3a | 828 | 716 | 239 | GAG | GTG | E to V |
| 749 | 250 | GAC | GTC | D to V | ||
| M | 669 | 233 | 78 | GGT | GCT | G to A |
| Orf8 | 366 | 251 | 84 | TTA | TCA | L to S |
Note: * Asterisk indicates a stop codon. Orf1ab1 and orf1ab2 are two exons of one gene.
Table 6.
The non-synonymous mutations in EPI_ISL_406592′s nonstructural proteins.
| Gene | Gene length | Nucleotide position from exon start | AA position from exon start | SARS-Cov | Reference (EPI_ISL_402125) | Mutation (EPI_ISL_406592) | Amino Acid Change |
|---|---|---|---|---|---|---|---|
| nsp2 | 3942 | 1364 | 455 | CTT | TTT | TCT | F to S |
| nsp8 | 1302 | 506 | 169 | CTT | CTT | CAT | L to H |
| nsp12 | 5676 | 2169 | 723 | TAT | TAT | TAA | Y to * |
| nsp15 | 2226 | 695 | 232 | TTC | TTC | TAC | F to Y |
Note: * Asterisk indicates a stop codon.
4. Discussion
This work is designed to resolve the evolutionary relationship within SARS-CoV-2s after its outbreak. In our first analysis (GISAID dataset till 2020/03/04), we found a basal clade with 33 sequences which are from seven countries. In our second analysis (GISAID dataset till 2020/03/17), the basal clade expanded into ten countries with 51 sequences. The much larger NCBI dataset shows that this clade has expanded with 85 sequences. For the basal SARS-CoV-2s, the earliest collection date is 2019/12/26 and the latest collection date is 2020/04/04, so they were immune to mutations for at least three months. Due to their average four-day incubation time, SARS-CoV-2s could spread before we could detect it (Linton et al., 2020). Their extraordinary RNA proofreading capability further blurred the evolutionary relationship among SARS-CoV-2s. Thus, the origin of SARS-CoV-2 remains unknown. The best way to answer this question is to find the SARS-CoV-2′s natural host.
SARS-CoV-2 is a perfect virus in terms of transmission. It has an incubation period of about 4 days (Linton et al., 2020). In an extreme case, an incubation period of 27 days has been reported. The asymptomatic SARS-CoV-2 carriers could transmit this virus to others (Bai et al., 2020). That’s why it is really difficult to detect SARS-CoV-2 in the initial stage of its transmission. Our first analysis (GISAID dataset till 2020/03/04) shows that the basal clade has 33 identical SARS-CoV-2 sequences, about one fifth of total sequences used in our first analysis, gathered from all over the Pan-Pacific region. Our second analysis (GISAID dataset till 2020/03/17) shows that the basal clade expands to a total of 51 identical SARS-CoV-2s and covers ten countries. The very SARS-CoV-2 found in Wuhan’s COVID-19 outbreak appeared in Europe afterwards. The basal SARS-CoV-2 must have infected tens of thousands of people, if not hundreds of thousands. For the number of people it infected and the geographic region it covered, the basal SARS-CoV-2 is a super virus for its high contagiousness and low detectability.
We used three different methods to construct SARS-CoV-2′s phylogenetic trees based on three batches of data. None of them yields a satisfactory result. Two taxa with faraway geographic locations are often clustered together. Thus, the phylogenetic analysis for SARS-CoV-2 is more suitable for its classification rather than its transmission route. So far, the origin of SARS-CoV-2 is still unknown. The phylogenetic tree of the NCBI dataset shows that at least its classification is reliable and the SARS-CoV-2 with the same major mutations are usually clustered together. The problem is that the SARS-CoV-2s from the same group always cover several continents (Supplemental data 3). It sends the alarming message from SARS-CoV-2: when you found one of them, they have been already everywhere.
The transition-transversion bias for SARS-CoV-2 is 3.058 in our first analysis and 3.955 in our second analysis. The increasing transition-transversion bias suggests that SARS-CoV-2′s genome becomes more stable during its global transmission. We did not detect any significant positive selection signal in its genome, which further proposes that the handful mutations we observed in SARS-CoV-2s are mostly neutral. For the SARS-CoV-2′s evolution, we have to ask such a question: why does this virus evolve so slowly? The answer might lie in its orf1ab protein which encodes replicase polyproteins responsible for SARS-CoV-2′s RNA proofreading capability and replication fidelity. The SARS-CoV-2 lost its nsp13 to nsp16 and has a very fast evolutionary rate. It has been proved that nsp14 is required for replication fidelity and mediates the antiviral effect of Remdesivir in coronaviruses (Agostini et al., 2018, Denison et al., 2011). So far, we still do not understand the biological properties of SARS-CoV-2′s nsp genes. They may hold the key to combat this virus.
In conclusion, SARS-CoV-2 is a highly stable and contagious virus with low detectability. If COVID-19′s patients does not get a proper medical care, the virus will unleash its decimating power, a mortality rate much higher than influenza. Its debut was an ultra-spreading event and COVID-19 has already become a global crisis now. Every harsh measure should be taken in order to contain it.
5. Compliance and ethics
All authors declare no potential competing interest. This work used the online data and ethical approval is not applicable.
6. Authors’ contributions
LBS, ZZ and FNH collected and primarily processed data. LBS, ZZ and FNH analyzed the data and produced the figures. LBS and FNH wrote the first draft of this manuscript. LBS, ZZ and FNH revised the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgement
We thank scientific communities all over the world for their selfless effort in this pandemic. Special gratitude to GISAID and NCBI for the SARS-CoV-2 data they provide.
Consent for publication
All authors consented the right for publication.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ympev.2020.107017.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
Supplementary figure 1.
Supplementary figure 2.

Supplementary figure 3.
Supplementary figure 4.

References
- 2004. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303, 1666-1669. [DOI] [PubMed]
- Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X., Smith E.C., Case J.B., Feng J.Y., Jordan R., Ray A.S., Cihlar T., Siegel D., Mackman R.L., Clarke M.O., Baric R.S., Denison M.R. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio. 2018;9 doi: 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Yao L., Wei T., Tian F., Jin D.Y., Chen L., Wang M. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA. 2020 doi: 10.1001/jama.2020.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit Morel, P.B., Lucas Czech, Ben Bettisworth, Lukas Hübner, Sarah Lutteropp, Dora Serdari, Evangelia-Georgia Kostaki, Ioannis Mamais, Alexey M Kozlov, Pavlos Pavlidis, Dimitrios Paraskevis, Alexandros Stamatakis, 2020. Phylogenetic analysis of SARS-CoV-2 data is difficult. Preprint at https://www.biorxiv.org/content/10.1101/2020.08.05.239046v1. [DOI] [PMC free article] [PubMed]
- Cotten M., Watson S.J., Zumla A.I., Makhdoom H.Q., Palser A.L., Ong S.H., Al Rabeeah A.A., Alhakeem R.F., Assiri A., Al-Tawfiq J.A., Albarrak A., Barry M., Shibl A., Alrabiah F.A., Hajjar S., Balkhy H.H., Flemban H., Rambaut A., Kellam P., Memish Z.A. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio. 2014;5 doi: 10.1128/mBio.01062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M.R., Graham R.L., Donaldson E.F., Eckerle L.D., Baric R.S. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNABiol. 2011;8:270–279. doi: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle L.D., Becker M.M., Halpin R.A., Li K., Venter E., Lu X., Scherbakova S., Graham R.L., Baric R.S., Stockwell T.B., Spiro D.J., Denison M.R. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle L.D., Lu X., Sperry S.M., Choi L., Denison M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007;81:12135–12144. doi: 10.1128/JVI.01296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster P., Forster L., Renfrew C., Forster M. Phylogenetic network analysis of SARS-CoV-2 genomes. PNAS. 2020;117:9241–9243. doi: 10.1073/pnas.2004999117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Sims A.C., Baric R.S., Denison M.R. The nsp2 proteins of mouse hepatitis virus and SARS coronavirus are dispensable for viral replication. Adv. Exp. Med. Biol. 2006;581:67–72. doi: 10.1007/978-0-387-33012-9_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaldo S., Philippe H. Ancient phylogenetic relationships. Theor. Popul. Biol. 2002;61:391–408. doi: 10.1006/tpbi.2002.1593. [DOI] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton N.M., Kobayashi T., Yang Y., Hayashi K., Akhmetzhanov A.R., Jung S.M., Yuan B., Kinoshita R., Nishiura H. Incubation period and other epidemiological characteristics of 2019 novel coronavirus infections with right truncation: a statistical analysis of publicly available case data. J. Clin. Med. 2020;9 doi: 10.3390/jcm9020538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic-Lazetic G.M., Mitic N.S., Tomovic A.M., Pavlovic M.D., Beljanski M.V. SARS-CoV genome polymorphism: a bioinformatics study. Genomics, Proteom. Bioinf. 2005;3:18–35. doi: 10.1016/S1672-0229(05)03004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Hohna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S., Shank S.D., Spielman S.J., Li M., Muse S.V., Kosakovsky Pond S.L. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018;35:773–777. doi: 10.1093/molbev/msx335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, 2020. WHO Coronavirus Disease (COVID-19) Dashboard.
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., Yuan M.L., Zhang Y.L., Dai F.H., Liu Y., Wang Q.M., Zheng J.J., Xu L., Holmes E.C., Zhang Y.Z. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., Zhong W., Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020 doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Shen L., Gu X. Evolutionary dynamics of MERS-CoV: potential recombination, positive selection and transmission. Sci. Rep. 2016;6:25049. doi: 10.1038/srep25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.