

Abstract
Rising numbers of cases of multidrug- and extensively drug-resistant Pseudomonas aeruginosa over recent years have created an urgent need for novel therapeutic approaches to cure potentially fatal infections. One such approach is virulence attenuation where anti-virulence compounds, designed to reduce pathogenicity without affording bactericidal effects, are employed to treat infections. P. aeruginosa uses the pqs quorum sensing (QS) system, to coordinate the expression of a large number of virulence determinants as well as bacterial-host interactions and hence represents an excellent anti-virulence target.
We report the synthesis and identification of a new series of thiazole-containing quinazolinones capable of inhibiting PqsR, the transcriptional regulator of the pqs QS system. The compounds demonstrated high potency (IC50 < 300 nM) in a whole-cell assay, using a mCTX:PpqsA-lux-based bioreporter for the P. aeruginosa PAO1-L and PA14 strains. Structural evaluation defined the binding modes of four analogues in the ligand-binding domain of PqsR through X-ray crystallography. Further work showed the ability of 6-chloro-3((2-pentylthiazol-4-yl)methyl)quinazolin-4(3H)-one (18) and 6-chloro-3((2-hexylthiazol-4-yl)methyl)quinazolin-4(3H)-one (19) to attenuate production of the PqsR-regulated virulence factor pyocyanin. Compounds 18 and 19 showed a low cytotoxic profile in the A549 human epithelial lung cell line making them suitable candidates for further pre-clinical evaluation.
Keywords: Pseudomonas aeruginosa, Quorum sensing, Inhibitors, X-ray crystal structure, PqsR
Graphical abstract

Highlights
-
•
Pseudomonas aeruginosa – a Gram-negative pathogenic bacterium.
-
•
Quorum sensing – a cell-to-cell communication strategy used by microbes to coordinate the production of various virulence factors.
-
•
Inhibitors of PqsR – small molecules that inhibit the signal of Pseudomonas aeruginosa Quinolone Signal Receptor.
-
•
X-ray crystal structure – used to demonstrate the 3-D structural orientation of the ligand in the protein structure.
1. Introduction
Pseudomonas aeruginosa (PA), a Gram-negative pathogenic bacterium found widely in nature, is a common cause of infection in immunocompromised patients. Chronic and recurring infections are widespread, and PA infection is frequently associated with respiratory failure, reduced pulmonary function and mortality within cystic fibrosis patients [[1], [2], [3]]. A rise in multidrug resistant cases of PA has raised this organism to a priority class pathogen of critical importance by the World Health Organisation [4].
In order to combat the threat of antimicrobial resistance, novel strategies are required to provide long-lasting solutions, which drastically reduce the rate of emergence of resistance due to the high selective pressure posed by current antibiotic treatments. One such approach is the use of alternative treatments which can attenuate virulence within bacterial populations without directly killing the infectious organisms and/or sensitise these populations to the action of existing antibiotics [5,6]. The net result is a non-pathogenic population which can be cleared by the immune system or which may become sensitive to existing antimicrobials [[6], [7], [8], [9]].
Extensive work associated with this approach has investigated inhibition of quorum sensing systems within bacterial populations. Quorum sensing (QS) is a cell-to-cell communication strategy used by microbes to coordinate the production of numerous traits including virulence factors in a population-dependent manner. QS relies on the production and sensing of small diffusible signal molecules known as autoinducers (AIs) or QS signal molecules (QSSMs) [[10], [11], [12]]. Although most commonly observed as an intraspecies event, QS is also seen at the interspecies and inter-kingdom level through bacteria-bacteria communication systems as well as in bacteria-fungi mixed populations [[13], [14], [15]].
During infection QS in PA controls the production of virulence traits such as the phenazine, pyocyanin and the siderophore pyoverdine, which are capable of cytotoxic effects against mammalian cells and iron scavenging respectively, as well as those responsible for bacterial motility and biofilm formation [[16], [17], [18], [19], [20], [21], [22]].
PA utilises three closely interlinked QS systems to fully elicit its pathogenicity; the las and rhl systems which operate via N-acylhomoserine lactones as their QSSMs (1, 2), whereas the pqs system uses alkylquinolones (AQ), namely 2-heptylquinolin-4(1H)-one (HHQ, 3) and the Pseudomonas Quinolone Signal (2-heptyl-3-hydroxy-4(1H)-quinolone, PQS, 4) as their cognate signals (Fig. 1) [7,11,[23], [24], [25]].
Fig. 1.

Known QSSMs in P. aeruginosa include N-acylhomoserine lactones which regulate 1 the rhl and 2las systems, and 3, 4 HHQ and PQS which regulate the pqs system.
The pqs QS system of PA requires the pqsABCDE operon for the biosynthesis of HHQ 3, the precursor of PQS 4, though the pqsE gene product is also known to play both a biosynthetic and a key regulatory role in the regulation of multiple virulence factors [[26], [27], [28], [29], [30], [31]]. HHQ is converted to PQS via the monooxygenase PqsH. The expression of the pqsABCDE operon is controlled by the LysR-type transcriptional regulator PqsR upon binding its cognate ligands, PQS and HHQ [32,33].
Inhibition of the pqs system can result in a reduction in the production of pyocyanin and alkylquinolones as well as many other virulence traits [32,[34], [35], [36], [37]]. As such, PqsR has been validated as a target for the inhibition of the pqs system and hence virulence using both in vitro assays and in vivo in mouse infection models [37].
Herein we report the discovery of a new series of compounds with high potency against PqsR in two different PA strains (PAO1-L and PA14) that represent the two major genomic groups. Binding of compounds 6, 12, 18 and 19 to this regulatory protein is shown through co-crystallisation into the ligand-binding domain (LBD) of PqsR [32,36]. The lead compounds (18, 19) were shown to attenuate pyocyanin production and demonstrated very low cytotoxicity against mammalian cells, supporting their suitability for further studies.
2. Results and discussion
2.1. SAR-based design and synthesis of PqsR inhibitors
Previous research has shown that quinazolinone scaffolds can provide the basis for PqsR antagonists, in part due to the similarity this core shares with the endogenous ligands PQS and HHQ [32]. Therefore, it was unsurprising that an in silico screen of the University of Nottingham Managed Chemical Compound Collection (MCCC, a collection of 85,000 diverse compounds) gave several hit compounds bearing this structural motif. One such compound, 5 (Fig. 2), containing a thiazole group proved interesting for further structure activity relationship (SAR) exploration.
Fig. 2.

Hit compound 5 found through in silico and subsequent in vitro testing of the MCCC compound library, was optimised to yield 6. Replacing the thiazole moiety with an oxadiazole, as in 7, yielded an inactive compound when screened at 10 μM ligand concentration.
Initial testing of 5 showed an activity for PAO1-L of 13.2 ± 2.73 μM in a mCTX-PpqsA-lux luminescence based bioreporter assay. Further optimisation yielded 6, with an improved activity of 1.0 ± 0.42 μM. Furthermore, homologous oxadiazole 7 was found to be inactive at 10 μM, providing evidence for the importance of the thiazole ring for inhibiting PqsR. As such, a SAR study was conducted around 6, primarily focussing on 2-substitution of the thiazole.
We envisaged that variation to the isopropyl group of compound 6 was the most promising area to explore with regards to improving potency. A range of alternate functionalities varying from alkyl chains to substituted amines and small aromatic groups provided a diverse set of compounds to explore the space available within the LBD of the PqsR protein where these ligands were believed to bind (Fig. 3).
Fig. 3.

Structures of all synthesised compounds carried through to in vitro testing in PAO1-L.
A four-step synthetic procedure (Scheme 1) provided a robust route to the desired 2,4-disubstituted thiazoles bearing alkyl, amino and aryl functionalities. Initially, 2-amino-5-chlorobenzoic acid was condensed in formamide to give the corresponding 6-chloroquinazolin-4(3H)-one 36. Reaction with chloroacetone gave the intermediate 37, which was brominated through refluxing in acetic acid with bromine to afford the α-bromoketone 38. Thiazole formation in ethanol with a range of thioamides gave the 2,4-disubstituted thiazoles 6, 8–26.
Scheme 1.

Synthetic route to compounds 6, 8–26: (i) formamide, 150 °C, 16 h; (ii) NaH, NMP, 0 °C, 30 min, then chloroacetone, 0 °C, 1 h; (iii) bromine, acetic acid, 65 °C, 16 h; (iv) thioamide or thiourea, ethanol, 80 °C, 16 h; for complete structure of the compounds, see Fig. 3.
In addition to this series of 2,4-substituted thiazoles (6, 8–26), a 2,5-substituted thiazole 35 analogous to 6 was synthesised to observe whether altering the orientation of the thiazole ring could improve efficacy, as was purported through a ligand docking screen in silico. Furthermore, in a bid to decrease lipophilicity, a range of 2-amino-5-substituted thiazoles were synthesised in a two-step route from 6-chloroquinazolin-4(3H)-one (36).
Synthesis of the 5-substituted thiazole 35 required an alternate four-step synthetic route (Scheme 2). Initially, bromomalonaldehyde was condensed with 2-methylpropanethioamide, to give 2-isopropylthiazole-5-carbaldehyde, which was immediately treated with hydroxylamine hydrochloride affording 2-isopropylthiazole-5-carbaldehyde oxime (39). Reduction of the oxime using zinc powder and hydrochloric acid gave the primary amine (40).
Scheme 2.

Synthetic route to 35: (i) ethanol, rt, 4 h, then NH2OH·HCl, TEA, rt, 2 h; (ii) HCl, zinc powder, 60 °C, 2 h; (iii) DMFDMA, 100 °C, 2 h; (iv) acetic acid, ethanol, 100 °C, 16 h.
Separately, 2-amino-5-chlorobenzoic acid was treated with DMFDMA to give methyl 5-chloro-2-(((dimethylamino)methylene)amino)benzoate (41) [38]. Cyclisation under acidic conditions with methylamino thiazole (40) gave the desired product 6-chloro-3-((2-isopropylthiazol-5-yl)methyl)quinazolin-4(3H)-one (35).
The 2-amino-5-substituted thiazoles (27–31) were prepared from reacting 6-chloroquinazolin-4(3H)-one (36) with 2-chloro-5-(chloromethyl)thiazole, to give 6-chloro-3-((2-chlorothiazol-5-yl)methyl)quinazolin-4(3H)-one (34). Displacement of the chlorine at the 2-position of the thiazole with five different amines under neutral conditions gave products 27–31 in yields of 21–84% (Scheme 3).
Scheme 3.

Synthetic route to 27–31: (i) KOH, TBAI, 2-chloro-5-(chloromethyl)thiazole, toluene, 70 °C, 1 h; (ii) amine, DMSO, 100–130 °C, 16 h; for complete structure of the compounds, see Fig. 3.
In addition to variations about the thiazole, changes to the halogen decorating the quinazolinone core could alter binding to PqsR significantly. It was believed that the 6-chloro functionality in 6 could bind into a deep pocket within the LBD of PqsR, and possibly form a hydrogen bond with Thr265, as reported before. However, compounds reported by Ilangovan et al. contained a chlorine at the 7-position of the bicyclic core [32]. Therefore, in order to fully investigate the binding mode of this series, a 7-chloroquinazolin-4(3H)-one subset was synthesised.
Two compounds were synthesised bearing a 7-chloro substitution on the bicyclic ring, with variations on the thiazole: 32 contained a tert-butyl group at the thiazole 2-position, and 33 a pentyl chain. The synthetic route was similar to that by which the 6-chloro series was synthesised (Scheme 4), though chlorination of quinazolinone 43 yielded the desired α-haloketone 44. Subsequent thiazole formation led to the synthesis of 32 and 33.
Scheme 4.

Synthetic route to 32–34: (i) formamide, 150 °C, 16 h; (i) NaH, NMP, 0 °C, 30 min, then chloroacetone, 0 °C, 1 h; (iii) NCS, sulfuric acid, DCM, 40 °C, 6 h; (iv) thioamide, acetic acid, ethanol, 80 °C, 16 h; for complete structure of the compounds, see Fig. 3.
2.2. Inhibition of the pqs system by potential PqsR antagonists
A PqsR-dependent bioreporter assay was used to assess the degree to which the compounds synthesised in the SAR study could inhibit the pqs QS system. Introduction of a mCTX:PpqsA-lux transcriptional into the chromosomal CTX sites of PA strains PAO1-L and PA14 results in the production of bioluminescence at high bacterial cell densities in cultures where PQS and HHQ have reached their threshold activation concentrations for PqsR activation and in turn the PpqsA promoter [39]. Consequently, inhibitors of PqsR reduce bioluminescence levels in these strains. Initially 96-well plates containing the PAO1-L mCTX:PpqsA-lux bioreporter strain grown in lysogeny broth (LB) were treated with a single concentration of 10 μM of each compound to determine which compounds were active. As a threshold level for compound selection, a 50% reduction of pqs activity compared to the DMSO control was set, where pqs activity was a ratio of luminescence over OD600. Only compounds above the 50% threshold progressed for IC50 determination.
Of the 29 compounds tested, 14 compounds showed >50% inhibition at a 10 μM concentration and their concentration-dose response curves were generated. Importantly, the active compounds were found to not inhibit growth compared to the DMSO control (Fig. S2).
From the SAR study, it was apparent that short, unbranched alkyl chains led to a loss of activity, (see compounds 8–11). Furthermore, the addition of heteroatoms into the alkyl chain at the 2-position of the thiazole appeared to reduce activity. Of the seven amino alkyl chain containing compounds, only 25 (IC50 = 3.5 μM in PAO1-L) had inhibitory activity at a ligand concentration of 10 μM. The presence of a tertiary amine did not improve activity, and the inclusion of an additional heteroatom led to no inhibition (compound 24).
Compounds 23 and 26, both containing three heteroatoms in their side chains, were found to be inactive, further suggesting that increase in polarity in the side chain leads to inactivity. In the case of 23 this may be due to the polarity of the nitro group; its charged state may result in the loss of activity in the whole cell-based assay as a result of poor cell penetration. Compound 26 had the lowest ClogP value of all compounds in the series (1.31). Notably, no compound with a ClogP value under 3.00 showed any activity, which also suggests that difficulty in penetrating the lipid bilayer contributes significantly to the inactivity of these compounds.
For the given series, 6-chloro substitution on the quinazolinone ring demonstrated improved activity over a 7-chloro substituted scaffold. Compound 32 (IC50 = 2.9 μM) was only marginally more active than the lead compound 45 (IC50 = 5.0 μM, Fig. 4) of a previously described 7-chloro substituted scaffold [32]. Furthermore, compound 33 was inactive when tested at a ligand concentration of 10 μM. Comparable 6-chloro substituted compounds 12 (IC50 = 397 nM) and 18 (IC50 = 313 nM) both showed sub-micromolar activity, with 12 displaying seven-fold stronger activity in strain PAO1-L compared with the weakly active 32.
Fig. 4.

A previously described PqsR antagonist (45) featured a quinazolinone scaffold with a 7-chloro substituted ring. Removal of the chlorine atom led to a 10-fold drop in activity, whilst a 6-chlorine substituted ring was inactive. By contrast, the compound series described in this article found 6-chlorine substitution to be optimal. Both 45 and an endogenous ligand, NHQ (46) were successfully crystallised in the PqsR ligand-binding domain [32].
Moreover, inverting the heteroatoms within the thiazole ring reduced activity, as indicated by the difference in activities of 6 (IC50 = 1.0 μM) and 35; the 2,5- disubstituted thiazole 35 was inactive at a concentration of 10 μM, whereas the 2,4-analogue 6 was previously shown to have an activity of 1.0 μM. This inversion of the thiazole ring may have further contributed to the inactivity of compounds 27–31 in addition to the increased polarity in the side chain.
One argument rationalising the intolerance of heteroatoms may be poor bacterial uptake, or high rates of transporter-based efflux [40]. Although a whole cell approach removes the ability to define a binding event in the context of the assay itself, in combination with X-ray crystallography this methodology allows for definitive selection of compounds which both bind to the desired molecular target, and also have a proven efficacy.
Dose response curves were obtained for all 14 compounds exhibiting >50% inhibition in the 10 μM spot test, with activities ranging from 244 nM to 3.55 μM in the bioreporter strain PAO1-L. Active compounds were then assayed against the clinically-relevant strain PA14 (Table 1). In general, the compounds displayed similar activities in both strains PAO1-L and PA14.
Table 1.
In vitro data for all compounds defined as active (inhibits pqs system to below 50% at 10 μM). NA denotes inactive compounds which showed a remaining activity above 50% at 10 μM (remaining activity for not active compounds is shown in brackets). Reported values are mean ± SD of n = 2 replicates. cLogP values were calculated using MarvinSketch based on the algorithm outlined by Viswanadhan et al. [41].
| Compound | IC50 value in PAO1-L/nM | IC50 value in PA14/nM | Predicted cLogP values |
|---|---|---|---|
| 6 | 1046 ± 419 | 1578 ± 358.0 | 3.38 |
| 8 | NA (83%) | NA | 2.14 |
| 9 | NA (52%) | NA | 3.03 |
| 10 | NA (79%) | NA | 2.84 |
| 11 | NA (104%) | NA | 2.09 |
| 12 | 397 ± 141.5 | 650 ± 35.1 | 3.94 |
| 13 | 1216 ± 44.0 | 1333 ± 192.5 | 3.57 |
| 14 | 1563 ± 674.0 | 1720 ± 882.0 | 3.73 |
| 15 | 1112 ± 879.5 | 646 ± 90.2 | 4.27 |
| 16 | 1572 ± 1119 | 639 ± 73.5 | 4.02 |
| 17 | 1360 ± 501.0 | 683 ± 156.9 | 4.02 |
| 18 | 313 ± 156.2 | 342 ± 39.4 | 4.17 |
| 19 | 298 ± 182.0 | 265 ± 3.4 | 4.62 |
| 20 | 1327 ± 189.5 | 1308 ± 272.0 | 3.10 |
| 21 | 2048 ± 1241.0 | 2916 ± 1005.5 | 3.64 |
| 22 | 244 ± 49.6 | 123 ± 20.9 | 4.04 |
| 23 | NA (89%) | NA | 3.98 |
| 24 | NA (85%) | NA | 2.13 |
| 25 | 3545 ± 2934.0 | NA | 3.50 |
| 26 | NA (108%) | NA | 1.31 |
| 27 | NA (72%) | NA | 4.00 |
| 28 | NA (83%) | NA | 4.02 |
| 29 | NA (101%) | NA | 2.17 |
| 30 | NA (67%) | NA | 3.55 |
| 31 | NA (85%) | NA | 2.18 |
| 32 | 2942 ± 808.0 | NA | 3.94 |
| 33 | NA (106%) | NA | 4.17 |
| 34 | NA (95%) | NA | 2.93 |
| 35 | NA (65%) | NA | 3.43 |
The pharmacological evaluation shed further light on the importance of the 2-substitution of the thiazole. Regarding the length and branching of the alkyl chain, two clear trends became evident. It was apparent that an increased chain length improved potency, with the two most potent inhibitors bearing a hexyl (19, IC50 = 298 nM) and pentyl chain (18, IC50 = 313 nM) respectively. Moreover, increased branching at the 1-position of the alkyl chain greatly improved potency: compound 12 (IC50 = 397 nM) containing a tert-butyl side chain demonstrated potent activity in strain PAO1-L.
Strain PAO1-L is a moderately virulent laboratory adapted strain routinely used to screen for PqsR inhibitors [34,42]. However, given the genomic diversity of PA strains, it is essential to test isolates belonging to the two major genomic groups. Hence, we also screened the inhibitors against the highly virulent PA14 strain [43]. It was therefore encouraging to see that activities were comparable between these strains. Moreover, for a few select compounds (15–17, 19, 22) potencies were shown to be greater in PA14, suggesting broad activity across the two main PA clades.
Three compounds (18, 19 and 22, IC50 = 244 nM), were selected for X-ray crystallography based on the high potency across both strains. Compound 12 was also chosen for further study, due to its excellent potency in PAO1-L. Furthermore, compound 6 was selected for X-ray crystallography studies to provide a comparison between the first optimised compound in the series, and the most potent compounds.
2.3. Structure of PqsR ligand-binding domain complexed with compounds 6, 12, 18 and 19
Compounds 6, 12, 18 and 19 were successfully soaked into a truncated form of PqsR (PqsR94-309) containing the ligand-binding domain of the protein (see S3-6 for additional data including electron density plots and crystallographic table of data collection and refinement). All four ligands occupied a similar space to that exhibited by a previously described quinazolinone-based inhibitor 45 and two endogenous ligands (HHQ, 3, and NHQ, 46) bearing quinolone scaffolds [32,44]. In each case, the quinazolinone core occupies the previously defined B pocket of the LBD [32], and the alkyl chain extends into the open A pocket (Fig. 5). In addition, no major conformational changes occurred to the protein backbone in each of the crystal soaking experiments, as well as the previously reported PqsR crystal structures.
Fig. 5.

(a) Overlaying the crystal structures of 12 (green), 18 (cyan) and HHQ (3, orange, PDB entry 6Q7U) shows that the core quinazolinone structures occupy almost identical spaces within the LBD, with the alkyl chains extending out to the LBD entrance. Importantly, the carbonyl groups of 12 and 18 face towards Thr265, enabling a H-bond to form, whereas the carbonyl of HHQ faces the opposite wall of the LBD in the direction of the B-sub pocket; (b) The same trends are observed between 6 (gold), 19 (magenta) and 45 (yellow, PDB entry 4JVI). The chlorine atoms of 6, 19 and 45 directly overlap, demonstrating its importance in locking the structure’s conformation NHQ (46, PDB entry 4JVD) followed the above trends, but was omitted for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
As with 45, the electron density contours around the halogen atom in each crystal, suggesting that the chlorine has locked the ligand into its defined orientation with the quinazolinone core occupying the B pocket of the PqsR LBD. However, whilst in 45 the carbonyl faces towards the B sub-pocket, the electron density maps suggest that the carbonyl groups of 6, 12, 18 and 19 all face towards the opposite face of the LBD in the direction of Thr265. The key result of this is that it enables a hydrogen bond to form between the carbonyl of each of the four compounds with the hydroxyl group of Thr265 (Fig. 6). This is further supported by the observation that the hydroxyl component of Thr265 faces towards the carbonyl in all four structures, in contrast to 45 where it faces towards the chlorine atom and elicits a polar interaction with the halogen.
Fig. 6.

The binding of 6 into the PqsR LBD as a surface representation (a), and the key binding interactions observed between the carbonyl of 6 and Thr265, and the sulfur atom of 6 and Tyr258 (b). Below these are analogous representations for 12 (c–d), 18 (e–f) and 19 (g–h). In each surface representation the chlorine atom is buried deep into the B pocket, anchoring the ligand in place. Binding modes are seen to be similar in each example, with the hydroxyl of Thr265 turned to face the carbonyl of the quinazolinone, thereby eliciting a H-bond. Furthermore, the sulfur is angled towards the phenol of Tyr258 enabling a polar interaction.
The importance of this hydrogen bond was further emphasised by the lower activities of compounds 32 and 33, both featuring a 7-chloro substitution. Repositioning of the chlorine atom removes the possibility of forming a hydrogen bond between the carbonyl and Thr265, hence the significant observed loss of potency.
The thiazole moieties were shown to further contribute to the activity of the series through interactions with Tyr258. In all four crystal structures, the sulfur atom was shown to orient itself facing the tyrosine, likely due to the formation of a polar interaction between the sulfur atom and the phenol group. The inactivity of oxadiazole 7 suggests that the presence of the sulfur aids in binding to the LBD, further implicating the thiazole as an integral component of the series. The role of the sulfur is further highlighted by the inactivity of 35, whereby the 2,5-thiazole regioisomer is unable to form a polar interaction with Tyr258, leading to a lower potency when compared to the 2,4-thiazole analogue 6.
The majority of residues in the LBD of PqsR bear aliphatic side chains. As such, hydrophobic interactions are seen to drive activity in both endogenous ligands and synthetic inhibitors. The SAR study indicated that increasing the length and branching of the aliphatic substitution at the 2-position of the thiazole dramatically improved potency. This is observed in the crystal structures, whereby the aliphatic chain forms hydrophobic interactions with the side chains of residues in the A pocket of the LBD. Compounds 6 (IC50 = 1.0 μM) and 12 (IC50 = 397 nM) are weaker inhibitors than 18 (IC50 = 313 nM) and 19 (IC50 = 298 nM), as they have shorter alkyl chains unable to form as many hydrophobic interactions with the protein. In particular, 18 and 19 are able to reach Ile186 and Leu189 at the far edge of the A pocket, creating a larger network of hydrophobic interactions and improving the activity over shorter chain analogues such as 6 and 12 (Fig. S4).
2.4. Reduction of pyocyanin production by the PqsR inhibitors
To demonstrate a link between the inhibitory effects of 18 and 19 on PqsR and hence virulence gene expression, pyocyanin production in the PAO1-L was quantified after overnight incubation with either 18 or 19 at a concentration three times higher than their IC50, or a vehicle (DMSO) control. Both compounds reduced pyocyanin production to 23% and 36% respectively (Fig. 7), compared with the control. These results indicate that these compounds can attenuate the production of this virulence factor through inhibition of PqsR similar to that shown for other PqsR inhibitors [34,45,46].
Fig. 7.
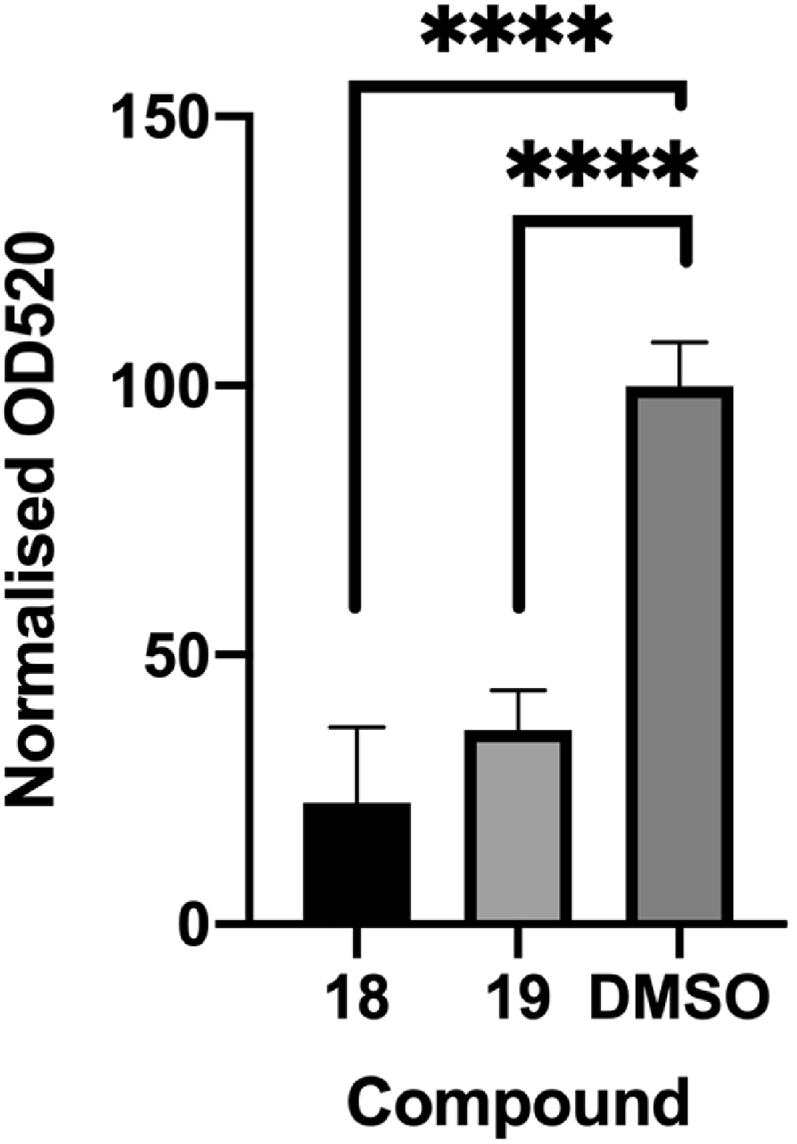
Compounds 18 and 19 were shown to significantly reduce pyocyanin production to 23% and 36% respectively against a control of 0.1% DMSO at 3 × the IC50 value in P. aeruginosa strain PAO1-L.
2.5. Effect of PqsR inhibitors on cytotoxicity of lung epithelial cells
A cytotoxicity study using A549 lung epithelial cells was conducted with 18 and 19, to establish the suitability of these compounds for further studies. Compounds 18 and 19 tested at increasing concentrations ranging from 0.1 to 100 μM showed no significant toxicity (Fig. 8). Testing at higher concentrations was not possible as neither compound was fully soluble above this concentration. However, the data showed that both 18 and 19 are not cytotoxic up to 100 μM. Therefore, the therapeutic index is > 292-fold in 18 and > 377-fold in 19 against the A549 cell line relative to the activities calculated for PA14.
Fig. 8.

Cytotoxicity data for (left) 18 and (right) 19 indicated that neither shows significant toxicity up to 100 μM in A549 lung epithelial cells. The lethal dose (LD50) value could not be calculated, as cell viability was not reduced to < 50% (indicated by the red dashed line). It was not possible to test at higher compound concentrations due to insolubility in the test buffer. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3. Conclusion
In summary, a new series of chloro-3-((2-substituted-thiazole)quinazolin-4(3H)-one compounds was synthesised and tested in a whole cell bioreporter assay to determine their ability to inhibit PqsR, and subsequently reduce P. aeruginosa pyocyanin production. A SAR study showed key contributing functionalities, particularly a long or branched alkyl chain at the 2-position of the thiazole. These studies revealed a preference for chlorine substitution on the 6-position of the quinazolinone ring.
X-ray crystallography into the LBD of PqsR confirmed a binding mode similar to that seen in previously described quinazolinone- and quinolone-based inhibitor-bound crystal structures. Further assays confirmed that the most active compounds 18 and 19 were capable of significantly reducing production of the toxin pyocyanin, whilst remaining non-toxic to eukaryotic cells, providing a basis for further pre-clinical studies.
4. Experimental
4.1. Chemistry – general methods
Chemicals and solvents were provided by Fisher Scientific UK, Acros Organics, Sigma-Aldrich, Merck Millipore or Fluorochem. All reactions were monitored by TLC using Merck Silica Gel 60 Å F254 TLC plates or by LC-MS. Unless otherwise stated, all compounds were dried under high vacuum either at rt or within an oven at 40 °C. LC-MS data was collected on a Shimadzu UFLCXR HPLC system coupled to an Applied Biosystems API 2000 LC/MS/MS electrospray ionization (ESI). The column used was a Phenomenex Gemini-NX 3 μm-110Â C18, 50 × 2mm at 40 °C. The flow rate was 0.5 mL/min, the UV detection was at 220 nm and 254 nm. The LC-MS ran for 1 min at 5% B; 5–98% B over 2 min, 98% B for 2 min, 98 to 5% B over 0.5 min and then 5% for 1 min where solvent A: 0.1% formic acid in water; solvent B: acetonitrile. Unless otherwise stated compounds reported had a purity >95%. NMR spectroscopy was performed using a Bruker AV(III) HD 400 NMR spectrometer equipped with a 5 mm BBFO+ probe, recording 1H and 13C NMR at 400.25 MHz and 100.66 MHz respectively; or a Bruker AV(III) 500 NMR spectrometer equipped with a 5 mm dual 1H/13C helium-cooled cryoprobe, recording 1H and 13C NMR at 500.13 MHz and 125.77 MHz respectively. NMR data was processed using iNMR (version 5.5.7) referencing spectra to residual solvents. Chemical shifts are quoted as δ: values in ppm; coupling constants J are given in Hz and multiplicities are described as follows: s - singlet, d - doublet, t - triplet, q - quartet, qi - quintet, sep – septet, m – multiplet, app – apparent, br - broad.
4.1.1. General procedure for the preparation of thiazoles 6, 8–23, 32, 33
A solution of the appropriate thioamide or thiourea (0.13 mmol) and α-haloketone 38 or 44 (0.06 mmol) in EtOH (4 mL) was refluxed at 80 °C for 16 h. a After cooling to room temperature, the reaction mixture was concentrated in vacuo and purified through flash column chromatography. Chromatography was run with a gradient of ethyl acetate/petroleum ether (1:2) to pure ethyl acetate. b
-
a
Where compound 44 was used as the α-haloketone, 600 μL of acetic acid was added to aid in solubilizing the starting material.
-
b
Excepting compounds 11 and 26 which were run in DCM/methanol (19:1).
4.1.1.1. 6-Chloro-3-((2-isopropylthiazol-4-yl)methyl)quinazolin-4(3H)-one (6)
Obtained 6 (18 mg, 90%) as a white crystal: 1H NMR (DMSO‑d6, 400 MHz) δ 8.54 (s, 1H), 8.05 (d, J = 1.68 Hz, 1H), 7.83 (dd, J = 8.61, 1.72 Hz, 1H), 7.70 (d, J = 8.69 Hz, 1H), 7.39 (s, 1H), 5.25 (s, 2H), 3.24–3.17 (m, 1H), 1.26 (d, J = 6.84 Hz, 6H) ppm; 13C NMR (DMSO‑d6 101 MHz) δ 177.7, 158.9, 150.0, 148.5, 146.6, 134.5, 131.4, 129.5, 125.1, 122.9, 115.7, 45.4, 32.4, 22.8 ppm; LC-MS (+ESI) calculated for C15H14ClN3OS m/z 320.1 (M + H), found m/z 320.1 (M + H).
4.1.1.2. 6-Chloro-3-((2-methylthiazol-4-yl)methyl)quinazolin-4(3H)-one (8)
Obtained 8 (17 mg, 90%) as an orange crystal: 1H NMR (CDCl3, 400 MHz) δ 8.34 (s, 1H), 8.25 (d, J = 1.24 Hz, 1H), 7.69–7.63 (m, 2H), 7.20 (s, 1H), 5.22 (s, 2H), 2.66 (s, 3H) ppm; 13C NMR (CDCl3 101 MHz) δ 159.9, 149.4, 146.9, 146.6, 134.7, 133.1, 129.2, 126.2, 123.2, 117.7, 45.4, 29.7, 19.1 ppm; LC-MS (+ESI) calculated for C13H10ClN3OS m/z 292.0 (M + H), found m/z 292.1 (M + H).
4.1.1.3. 6-Chloro-3-((2-(trifluoromethyl)thiazol-4-yl)methyl)quinazolin-4(3H)-one (9)
Obtained 9 (3.5 mg, 16%) as a yellow solid: 1H NMR (CDCl3, 400 MHz) δ 8.35 (s, 1H), 8.24 (d, J = 1.64 Hz, 1H), 7.71 (s, 1H), 7.69–7.66 (m, 2H), 5.32 (s, 2H) ppm; LC-MS (+ESI) calculated for C13H7ClF3N3OS m/z 346.0 (M + H), found m/z 346.1 (M + H).
4.1.1.4. 6-Chloro-3-((2-ethylthiazol-4-yl)methyl)quinazolin-4(3H)-one (10)
Obtained 10 (14 mg, 70%) as a white solid: 1H NMR (CDCl3, 400 MHz) δ 8.28 (s, 1H), 8.15 (s, 1H), 7.61–7.55 (m, 2H), 7.14 (s, 1H), 5.16 (s 2H), 2.90 (q, J = 7.52 Hz, 2H), 1.27 (t, J = 7.56 Hz, 3H) ppm; 13C NMR (CDCl3 101 MHz) δ 173.9, 159.7, 149.1, 147.0, 146.5, 134.6, 132.9, 129.1, 126.0, 123.1, 117.1, 45.3, 26.8, 14.0 ppm; LC-MS (+ESI) calculated for C14H12ClN3OS m/z 306.0 (M + H), found m/z 306.1 (M + H).
4.1.1.5. 2-(4-((6-Chloro-4-oxoquinazolin-3(4H)-yl)methyl)thiazol-2-yl)acetonitrile (11)
Obtained 11 (18 mg, 90%) as a red-orange solid: 1H NMR (CDCl3, 400 MHz) δ 8.32 (s, 1H), 8.23 (d, J = 1.80 Hz, 1H), 7.70–7.64 (m, 2H), 7.43 (s, 1H), 5.24 (s, 2H), 4.06 (s, 2H) ppm; 13C NMR (CDCl3 101 MHz) δ 159.8, 158.4, 150.6, 146.6, 146.5, 134.8, 133.3, 129.2, 126.1, 123.1, 119.8, 115.0, 45.3, 22.2 ppm; LC-MS (+ESI) calculated for C14H9ClN4OS m/z 317.0 (M + H), found m/z 317.2 (M + H).
4.1.1.6. 3-((2-(Tert-butyl)thiazol-4-yl)methyl)-6-chloroquinazolin-4(3H)-one (12)
Obtained 12 (9.0 mg, 43%) as a white crystal: 1H NMR (CDCl3, 400 MHz) δ 8.43 (s, 1H), 8.27 (d, J = 1.80 Hz, 1H), 7.70–7.65 (m, 2H), 7.19 (s, 1H), 5.26 (s, 2H), 1.40 (s, 9H) ppm; 13C NMR (CDCl3 101 MHz) δ182.4, 159.9, 148.9, 147.1, 146.6, 134.7, 133.1, 129.2, 126.2, 123.2, 116.7, 45.3, 37.8, 30.9 ppm; LC-MS (+ESI) calculated for C16H16ClN3OS m/z 334.1 (M + H), found m/z 334.2 (M + H).
4.1.1.7. 6-Chloro-3-((2-isobutylthiazol-4-yl)methyl)quinazolin-4(3H)-one (13)
Obtained 13 (11 mg, 53%) as a silver crystal: 1H NMR (DMSO‑d6, 400 MHz) δ 8.54 (s, 1H), 8.07 (d, J = 2.36 Hz, 1H), 7.86 (dd, J = 8.69, 2.40 Hz, 1H), 7.72 (d, J = 8.69 Hz, 1H), 7.42 (s, 1H), 5.25 (s, 2H), 2.77 (d, J = 7.08 Hz, 2H), 1.95 (qt, J = 6.72, 6.72 Hz, 1H), 0.88 (d, J = 6.65 Hz, 6H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 170.1, 158.9, 150.1, 148.6, 146.6, 134.5, 131.4, 129.5, 125.1, 122.9, 116.4, 45.3, 41.2, 29.1, 21.9 ppm; LC-MS (+ESI) calculated for C16H16ClN3OS m/z 334.1 (M + H), found m/z 334.1 (M + H).
4.1.1.8. 3-((2-Butylthiazol-4-yl)methyl)-6-chloroquinazolin-4(3H)-one (14)
Obtained 14 (29 mg, 88%) as a yellow crystal: 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 8.23 (dd, J = 2.00, 0.87 Hz, 1H), 7.67–7.60 (m, 2H), 7.19 (s, 1H), 5.22 (s, 2H), 2.92 (t, J = 7.74 Hz 2H), 1.71 (tt, J = 7.70, 7.70 Hz, 2H), 1.38 (tq, J = 7.45, 7.45 Hz, 3H), 0.90 (t, J = 7.38 Hz, 5H) ppm; 13C NMR (101 MHz, DMSO) δ 171.4, 158.9, 150.0, 148.6, 146.7, 134.5, 131.4, 129.6, 125.1, 122.9, 116.2, 45.4, 32.2, 31.4, 21.5, 13.5 ppm; LC-MS (+ESI) calculated for C16H16ClN3OS m/z 334.1 (M + H), found m/z 334.1 (M + H).
4.1.1.9. 6-Chloro-3-((2-(pentan-2-yl)thiazol-4-yl)methyl)quinazolin-4(3H)-one (15)
Obtained 15 (11 mg, 45%) as a yellow oily solid: 1H NMR (CDCl3, 400 MHz) δ 8.37 (s, 1H), 8.25 (d, J = 1.52 Hz, 1H), 7.69–7.63 (m, 2H), 7.20 (s, 1H), 5.24 (s, 2H), 3.14 (qt, J = 6.96, 6.96 Hz, 1H), 1.77–1.53 (m, 2H), 1.33–1.25 (m, 5H), 0.88 (t, J = 7.32 Hz, 3H) ppm; 13C NMR (CDCl3 101 MHz) δ 178.6, 159.8, 148.8, 147.0, 146.5, 134.7, 133.1, 129.1, 126.2, 123.2, 116.7, 45.3, 39.9, 38.3, 21.4, 20.3, 13.9 ppm; LC-MS (+ESI) calculated for C17H18ClN3OS m/z 348.1 (M + H), found m/z 348.2 (M + H).
4.1.1.10. 6-Chloro-3-((2-(2-methylbutyl)thiazol-4-yl)methyl)quinazolin-4(3H)-one (16)
Obtained 16 (20 mg, 91%) as a colourless oil: 1H NMR (CDCl3, 400 MHz) δ 8.35 (s, 1H), 8.25 (d, J = 1.64 Hz, 1H), 7.69–7.63 (m, 2H), 7.21 (s, 1H), 5.24 (s, 2H), 2.94 (dd, J = 14.61, 6.08 Hz, 1H), 2.75 (dd, J = 14.61, 8.09, 1H), 1.89–1.80 (m, 1H), 1.46–1.14 (m, 2H), 0.92–0.87 (m, 6H) ppm; 13C NMR (CDCl3 101 MHz) δ 171.6, 159.7, 149.2, 146.9, 146.6, 134.7, 129.2, 126.2, 123.0, 117.3, 45.4, 40.3, 36.0, 29.7, 29.1, 19.0, 11.3 ppm; LC-MS (+ESI) calculated for C17H18ClN3OS m/z 348.1 (M + H), found m/z 348.2 (M + H).
4.1.1.11. 6-Chloro-3-((2-isopentylthiazol-4-yl)methyl)quinazolin-4(3H)-one (17)
Obtained 17 (13 mg, 71%) as a white crystal: 1H NMR (CDCl3, 400 MHz) δ 8.36 (s, 1H), 8.25 (d, J = 1.48 Hz, 1H), 7.69–7.63 (m, 2H), 7.20 (s, 1H), 5.23 (s, 2H), 2.95 (t, J = 7.61 Hz, 2H), 1.65–1.62 (m, 3H), 0.93 (d, J = 6.04 Hz, 6H); 13C NMR (CDCl3 101 MHz) δ 172.9, 159.8, 149.2, 146.9, 134.7, 133.0, 129.2, 126.2, 123.3, 117.2, 45.4, 38.9, 31.5, 29.7, 27.7, 23.3 ppm; LC-MS (+ESI) calculated for C17H18ClN3OS m/z 348.1 (M + H), found m/z 348.2 (M + H).
4.1.1.12. 6-Chloro-3-((2-pentylthiazol-4-yl)methyl)quinazolin-4(3H)-one (18)
Obtained 18 (20 mg, 90%) as a white crystal: 1H NMR (CDCl3, 400 MHz) δ 8.35 (s, 1H), 8.26 (d, J = 1.92 Hz, 1H), 7.69–7.63 (m, 2H), 7.20 (s, 1H), 5.23 (s, 2H), 2.93 (t, J = 7.68 Hz, 2H), 1.77–1.69 (m, 2H), 1.37–1.33 (m, 4H), 0.90 (t, J = 6.93 Hz, 3H) ppm; 13C NMR (CDCl3 101 MHz) δ 172.8, 159.9, 149.1, 147.0, 146.6, 134.7, 133.1, 129.2, 126.2, 123.2, 117.2, 45.4, 33.4, 31.3, 29.6, 22.3, 13.9 ppm; LC-MS (+ESI) calculated for C17H18ClN3OS m/z 348.1 (M + H), found m/z 348.2 (M + H).
4.1.1.13. 6-Chloro-3-((2-hexylthiazol-4-yl)methyl)quinazolin-4(3H)-one (19)
Obtained 19 (15 mg, 66%) as a white crystal: 1H NMR (CDCl3, 400 MHz) δ 8.36 (s, 1H), 8.25 (d, J = 1.72 Hz, 1H), 7.69–7.64 (m, 2H), 7.21 (s, 1H), 5.23 (s, 2H), 2.94 (t, J = 7.89 Hz, 2H), 1.74 (tt, J = 7.64, 7.64 Hz, 2H), 1.40–1.33 (m, 2H), 1.30–1.27 (m, 4H), 0.86 (t, J = 6.93 Hz, 3H) ppm; 13C NMR (CDCl3 101 MHz) δ 172.8, 159.8, 149.1, 146.9, 146.5, 134.7, 133.1, 129.1, 126.1, 123.2, 117.2, 45.4, 33.4, 31.4, 29.9, 28.7, 22.4, 14.0 ppm; LC-MS (+ESI) calculated for C18H20ClN3OS m/z 362.1 (M + H), found m/z 362.3 (M + H).
4.1.1.14. 6-Chloro-3-((2-(furan-2-yl)thiazol-4-yl)methyl)quinazolin-4(3H)-one (20)
Obtained 20 (17 mg, 80%) as a white crystal: 1H NMR (CDCl3, 400 MHz) δ 8.40 (s, 1H), 8.26 (d, J = 1.88 Hz, 1H), 7.67 (d, J = 2.20 Hz, 1H), 7.66 (s, 1H), 7.50 (d, J = 1.09 Hz, 1H), 7.32 (s, 1H), 6.97 (d, J = 3.64 Hz, 1H), 6.52 (dd, J = 3.36, 1.80 Hz, 1H), 5.29 (s, 2H) ppm; 13C NMR (CDCl3 101 MHz) δ 159.9, 158.9, 150.6, 148.5, 146.9, 146.6, 143.9, 134.8, 133.2, 129.2, 126.2, 123.2, 117.3, 112.3, 109.5, 45.4 ppm; LC-MS (+ESI) calculated for C16H10ClN3O2S m/z 344.0 (M + H), found m/z 344.1 (M + H).
4.1.1.15. 6-Chloro-3-((2-(2-(furan-2-yl)vinyl)thiazol-4-yl)methyl)quinazolin-4(3H)-one (21)
Obtained 21 (12 mg, 51%) as a cream solid. A minor impurity remained, giving a final purity of 91.7% as determined by 1H NMR: 1H NMR (DMSO‑d6, 400 MHz) δ 8.60 (s, 1H), 8.09 (d, J = 2.32 Hz, 1H), 7.88 (dd, J = 8.73, 2.36 Hz, 1H), 7.77–7.73 (m, 2H), 7.54 (s, 1H), 7.29 (d, J = 16.09 Hz, 1H), 7.08 (d, J = 16.09 Hz, 1H), 6.79 (d, J = 3.24 Hz, 1H), 6.59 (dd, J = 2.88, 1.64 Hz, 1H), 5.30 (s, 2H) ppm; 13C NMR (101 MHz, DMSO) δ 166.0, 159.0, 151.7, 151.1, 148.6, 146.7, 144.5, 134.6, 131.5, 129.6, 125.1, 122.9, 121.5, 118.3, 117.0, 112.7, 112.5, 45.5 ppm; LC-MS (+ESI) calculated for C18H12ClN3O2S m/z 370.0 (M + H), found m/z 370.2 (M + H).
4.1.1.16. 6-Chloro-3-((2-phenylthiazol-4-yl)methyl)quinazolin-4(3H)-one (22)
Obtained 22 (20 mg, 92%) as a white solid: 1H NMR (CDCl3, 400 MHz) δ 8.47 (s, 1H), 8.27 (d, J = 1.72 Hz, 1H), 7.92–7.89 (m, 2H), 7.70–7.64 (m, 2H), 7.43–7.41 (m, 3H), 7.36 (s, 1H), 5.32 (s, 2H) ppm; 13C NMR (CDCl3 101 MHz) δ 169.1, 159.9, 150.7, 147.1, 146.6, 134.7, 133.1, 133.1, 130.4, 129.2, 129.0, 126.5, 126.2, 123.2, 117.9, 45.5 ppm; LC-MS (+ESI) calculated for C18H12ClN3OS m/z 354.0 (M + H), found m/z 354.2 (M + H).
4.1.1.17. 6-Chloro-3-((2-(4-nitrophenyl)thiazol-4-yl)methyl)quinazolin-4(3H)-one (23)
Obtained 23 (15 mg, 48%) as a white crystal: 1H NMR (400 MHz, DMSO‑d6) δ 8.66 (s, 1H), 8.36–8.28 (m, 2H), 8.15 (d, J = 2.08 Hz, 1H), 8.14–8.06 (m, 2H), 7.91–7.83 (m, 2H), 7.75 (d, J = 8.74 Hz, 1H), 5.39 (s, 2H) ppm; LC-MS (+ESI) calculated for C18H11ClN4O3S m/z 399.0 (M + H), found m/z 399.1 (M + H).
4.1.1.18. 6-Chloro-3-((2-((2-methoxyethyl)amino)thiazol-4-yl)methyl)quinazolin-4(3H)-one (24)
Obtained 24 (34 mg, 51%) as a cream solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.47 (s, 1H), 8.08 (d, J = 2.67 Hz, 1H), 7.85 (dd, J = 9.06, 2.70 Hz, 1H), 7.72–7.67 (m, 2H), 6.42 (s, 1H), 5.00 (s, 2H), 3.41 (t, J = 5.39 Hz, 2H), 3.32 (t, J = 4.91 Hz, 2H), 3.21 (s, 3H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 169.1, 158.9, 148.6, 146.6, 146.3, 134.5, 131.3, 129.5, 125.1, 122.9, 102.8, 70.1, 57.9, 45.5, 43.9 ppm; LC-MS (+ESI) calculated for C15H15ClN4O2S m/z 351.1 (M + H), found m/z 350.6 (M + H).
4.1.1.19. 3-((2-(Butylamino)thiazol-4-yl)methyl)-6-chloroquinazolin-4(3H)-one (25)
Obtained 25 (16 mg, 24%) as a white crystal: 1H NMR (DMSO‑d6, 400 MHz) δ 8.46 (s, 1H), 8.08 (d, J = 2.65 Hz, 1H), 7.86 (s, dd, J = 8.68, 2.65 Hz, 1H), 7.72 (d, J = 8.68 Hz, 1H), 7.61 (t, J = 5.67 Hz, 1H), 6.42 (s, 1H), 5.00 (s, 2H), 3.11 (dt, J = 6.87, 5.20 Hz, 2H), 1.46 (tt, J1 = J2 = 7.07 Hz), 1.28 (tq, J = 7.33, 7.33 Hz, 2H), 0.83 (t, J = 7.32 Hz, 3H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 169.4, 158.9, 148.6, 146.7, 146.5, 134.5, 131.3, 129.5, 125.1, 122.9, 102.4, 45.5, 44.2, 30.7, 19.5, 13.6 ppm; LC-MS (+ESI) calculated for C16H17ClN4OS m/z 349.1 (M + H), found m/z 348.9 (M + H).
4.1.1.20. 2-(4-((6-Chloro-4-oxoquinazolin-3(4H)-yl)methyl)thiazol-2-yl)guanidine (26)
Obtained 26 (13 mg, 62%) as a cream solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.55 (s, 1H), 8.08 (d, J = 2.40 Hz, 1H), 7.85–7.83 (m, 1H), 7.70 (dd, J = 8.77, 2.36 Hz, 1H), 7.04 (br s, 4H), 5.06 (s, 2H) ppm; LC-MS (+ESI) calculated for C13H11ClN6OS m/z 335.0 (M + H), found m/z 335.2 (M + H).
4.1.2. General procedure for the preparation of 3-((2-amino)thiazol-5-yl)methyl)quinazolin-4(3H)-one compounds 27–31 from 34
A solution of 34 (0.32 mmol) and appropriate amine (0.96 mmol) in DMSO (0.5 mL) was refluxed at 100–130 °C (dependent on the boiling point of the amine) for 16 h. The reaction mixture was cooled to room temperature, diluted with water and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried over brine and concentrated in vacuo. Purification through flash column chromatography in ethyl acetate/petroleum ether (4:1) to ethyl acetate to methanol/ethyl acetate (19:1) yielded products 27–31 in yields of 21–84%.
4.1.2.1. 6-Chloro-3-((2-(4-methylpiperidin-1-yl)thiazol-5-yl)methyl)quinazolin-4-(3H)-one (27)
Obtained 27 (86 mg, 72%) as a yellow solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.57 (s, 1H), 8.12 (d, J = 2.28 Hz, 1H), 7.88 (dd, J = 8.82, 2.67 Hz, 1H), 7.72 (d, J = 8.70 Hz, 1H), 7.26 (s, 1H), 5.20 (s, 2H), 3.80–3.77 (m, 2H), 2.92 (ddd, J = 3.02, 3.02, 12.44 Hz, 2H), 1.65–1.61 (m, 2H), 1.58–1.51 (m, 1H), 1.10 (dddd, J = 4.14, 4.14, 4.14, 11.93 Hz, 2H), 0.89 (d, J = 6.16 Hz, 3H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 171.7, 159.0, 147.8, 146.6, 140.2, 134.6, 131.6, 129.6, 125.0, 122.6, 119.8, 48.2, 42.2, 32.7, 30.0, 21.5 ppm; LC-MS (+ESI) calculated for C18H19ClN4OS m/z 375.1 (M + H), found m/z 374.6 (M + H).
4.1.2.2. 6-Chloro-3-((2-(cyclohexylamino)thiazol-5-yl)methyl)quinazolin-4-(3H)-one (28)
Obtained 28 (25 mg, 21%) as a brown solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.55 (s, 1H), 8.11 (d, J = 2.52 Hz, 1H), 8.67 (dd, J = 8.55, 2.21 Hz, 1H), 7.72 (d, J = 8.78 Hz, 1H), 7.50 (d, J = 7.48 Hz, 1H), 7.11 (s, 1H), 5.15 (s, 2H), 3.39–3.34 (m, 1H), 1.89–1.84 (m, 2H), 1.69–1.64 (m, 3H), 1.56–1.51 (m, 1H), 1.31–1.11 (m, 4H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 169.0, 159.0, 147.9, 146.6, 139.5, 134.6, 131.6, 129.6, 125.0, 122.7, 117.9, 53.0, 42.3, 32.3, 25.3, 24.4 ppm; LC-MS (+ESI) calculated for C18H19ClN4OS m/z 375.1 (M + H), found m/z 374.6 (M + H).
4.1.2.3. 6-Chloro-3-((2-(2-hydroxyethyl(methyl)amino)thiazol-5-yl)methyl)quinazolin-4-(3H)-one (29)
Obtained 29 (94 mg, 84%) as a yellow solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.57 (s, 1H), 8.12 (d, J = 2.52 Hz, 1H), 7.88 (dd, J = 8.3, 2.51 Hz, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.24 (s, 1H), 5.20 (s, 2H), 4.75 (t, J = 5.74, 1H), 3.55 (dt, J = 6.31, 6.31 Hz, 2H), 3.43 (t, J = 5.75, 2H), 2.99 (s, 3H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 171.1, 159.0, 147.8, 146.6, 140.3, 134.6, 131.6, 129.5, 125.0, 122.6, 119.0, 58.1, 54.7, 42.3, 40.4 ppm; LC-MS (+ESI) calculated for C15H15ClN4O2S m/z 351.1 (M + H), found m/z 350.6 (M + H).
4.1.2.4. 3-((2-(Butylamino)thiazol-5-yl)methyl)-6-chloroquinazolin-4(3H)-one (30)
Obtained 30 (46 mg, 41%) as a yellow solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.54 (s, 1H), 8.11 (d, J = 2.22 Hz, 1H), 7.85 (dd, J = 8.03, 2.22 Hz, 1H), 7.70 (d, J = 7.75 Hz, 1H), 7.56 (t, J = 5.54 Hz, 1H), 7.13 (s, 1H), 5.15 (s, 2H), 3.13 (dt, J = 6.96, 6.96 Hz, 2H), 1.46 (tt, J = 7.01, 7.01 Hz, 2H), 1.29 (tq, J = 7.36, 7.36 Hz, 2H), 0.85 (t, J = 7.41 Hz, 3H) ppm; 13C NMR (CDCl3, 101 MHz) δ 172.0, 160.1, 146.7, 145.9, 139.8, 135.0, 133.5, 129.4, 126.3, 123.2, 118.8, 46.0, 43.2, 31.4, 20.1, 13.8 ppm; LC-MS (+ESI) calculated for C16H17ClN4OS m/z 349.1 (M + H), found m/z 348.7 (M + H).
4.1.2.5. 6-Chloro-3-((2-((2-methoxyethyl)amino)thiazol-5-yl)methyl)quinazolin-4-(3H)-one (31)
Obtained 31 (24 mg, 22%) as a yellow solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.55 (s, 1H), 8.11 (d, J = 2.04 Hz, 1H), 8.08 (s, br, 1H), 7.86 (dd, J = 8.85, 2.85 Hz, 1H), 7.71 (d, J = 8.85 Hz, 1H), 7.21 (s, 1H), 5.16 (s, 2H), 3.43 (t, J = 5.37 Hz, 2H), 3.38–3.34 (m, 2H), 3.23 (s, 3H) ppm; 13C NMR (CDCl3, 101 MHz) δ 171.7, 160.2, 146.9, 145.7, 136.6, 135.1, 133.6, 129.4, 126.3, 123.0, 118.0, 70.5, 59.0, 45.9, 43.2 ppm; LC-MS (+ESI) calculated for C15H15ClN4O2S m/z 351.1 (M + H), found m/z 350.7 (M + H).
4.1.2.6. 3-((2-Tert-butyl)thiazol-4-yl)methyl)-7-chloroquinazolin-4(3H)-one (32)
Obtained 32 (2.0 mg, 11%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H), 8.25 (d, J = 8.56 Hz, 1H), 7.74 (d, J = 1.99 Hz, 1H), 7.47 (dd, J = 8.55, 2.01 Hz, 1H), 7.22 (s, 1H), 5.28 (s, 2H), 1.43 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 182.9, 160.3, 148.7, 148.7, 148.3, 140.8, 128.4, 128.1, 127.0, 120.6, 117.1, 45.2, 38.0, 31.0 ppm; LC-MS (+ESI) calculated for C16H16ClN3OS m/z 334.1 (M + H), found m/z 334.2 (M + H).
4.1.2.7. 7-Chloro-3-((2-pentylthiazol-4-yl)methyl)quinazolin-4(3H)-one (33)
Obtained 33 (2.0 mg, 11%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.57 Hz, 1H), 7.99 (s, 1H), 7.76 (d, J = 2.00 Hz, 1H), 7.47 (dd, J = 8.57, 2.00 Hz, 1H), 4.98 (s, 1H), 3.83 (s, 2H), 2.64 (t, J = 7.55 Hz, 2H), 1.73–1.67 (m 2H), 1.36–1.28 (m, 4H), 0.94–0.86 (m, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 199.0, 197.2, 160.1, 148.3, 147.9, 141.3, 128.5, 128.5, 126.9, 120.2, 53.8, 43.6, 36.5, 31.2, 25.3, 22.4, 14.0 ppm; LC-MS (+ESI) calculated for C17H18ClN3OS m/z 348.1 (M + H), found m/z 348.0 (M + H).
4.1.2.8. 6-Chloro-3-((2-chlorothiazol-5-yl)methyl)quinazolin-4(3H)-one (34)
A solution of 36 (1.00 g, 5.50 mmol) in toluene (15 mL) was treated with KOH (620 mg, 11.10 mmol) and TBAI (193 mg, 0.55 mmol) and heated at 70 °C for 10 min. To this was added 2-chloro-5-(chloromethyl)thiazole (740 μL, 6.60 mmol) dropwise and stirred for 1 h at 70 °C forming a dark brown solution. The reaction mixture was cooled to room temperature, diluted with water and extracted with ethyl acetate (4 × 20 mL). The combined organic layers were dried over Na2SO4 and filtered. The organic layer was concentrated in vacuo and triturated with diethyl ether (3 × 20 mL), yielding 34 (1.53 g, 89%) as a yellow solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.62 (s, 1H), 8.13 (d, J = 2.57 Hz, 1H), 7.89 (dd, J = 8.89, 2.57 Hz, 1H), 7.81 (s, 1H), 7.73 (d, J = 8.61 Hz, 1H), 5.36 (s, 2H) ppm; 13C NMR (CDCl3, 101 MHz) δ 160.1, 154.2, 146.6, 145.3, 141.3, 135.4, 134.2, 134.0, 129.6, 126.3, 123.0, 43.1 ppm; LC-MS (+ESI) calculated for C12H7Cl2N3OS m/z 312.0 (M + H), found m/z 311.8 (M + H).
4.1.2.9. 6-Chloro-3-((2-isopropylthiazol-5-yl)methyl)quinazolin-4(3H)-one (35)
To a solution of intermediate 41 (140 mg, 0.58 mmol) in acetonitrile (5 mL) was added acetic acid (500 μL) and 40 (55 mg, 0.29 mmol) and stirred at 100 °C for 2 h. The reaction mixture was cooled to room temperature, neutralised with saturated NaHCO3, and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried over brine, concentrated in vacuo and purified through HPLC, yielding 35 (2 mg, 2%) as a white solid: 1H NMR (400 MHz, DMSO‑d6) δ 8.63 (s, 1H), 8.12 (d, J = 2.51 Hz, 1H), 7.88 (dd, J = 8.71, 2.53 Hz, 1H), 7.77 (s, 1H), 7.72 (d, J = 8.69 Hz, 1H), 5.37 (s, 2H), 3.20 (sept, J = 6.87 Hz, 1H), 1.27 (d, J = 6.85 Hz, 6H) ppm; 13C NMR (126 MHz, DMSO) δ 178.2, 165.8, 147.9, 146.6, 142.2, 134.8, 131.7, 131.6, 129.7, 125.1, 122.7, 41.8, 32.6, 22.7 ppm; LC-MS (+ESI) calculated for C15H14ClN3OS m/z 320.1 (M + H), found m/z 320.2 (M + H).
4.1.2.10. 6-Chloroquinazolin-4(3H)-one (36)
A solution of 2-amino-5-chlorobenzoic acid (4.99 g, 29.00 mmol) and formamide (20 mL) was refluxed at 150 °C for 16 h, forming a brown precipitate. The reaction mixture was cooled to room temperature and 20 g ice was added. The mixture was left to stand for 1 h, then the precipitate filtered and concentrated in vacuo, yielding 36 (5.13 g, 97%) as brown microcrystals: 1H NMR (DMSO‑d6, 400 MHz) δ 12.44 (br s, 1H), 8.12 (s, 1H), 8.06 (d, J = 2.40 Hz, 1H), 7.85 (dd, J = 8.65, 2.52 Hz, 1H), 7.70 (d, J = 8.77 Hz, 1H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 160.3, 152.6, 146.5, 134.9, 131.5, 130.0, 125.3, 124.4 ppm; LC-MS (+ESI) calculated for C8H5ClN2O m/z 181.0 (M + H), found m/z 181.2 (M + H).
4.1.2.11. 6-Chloro-3-(2-oxopropyl)quinazolin-4(3H)-one (37)
To a solution of 36 (1.60 g, 6.76 mmol) in anhydrous NMP was added NaH (650 mg, 27.08 mmol) in small portions, forming a white precipitate. After stirring for 30 min the solution returned to a clear brown state and chloroacetone (2.4 mL, 29.28 mmol) was added dropwise forming a deep red solution. The solution was concentrated in vacuo and recrystallised in 3:1 hexane/ethyl acetate, yielding 37 (1.80 g, 86%) as white needle crystals: 1H NMR (DMSO‑d6, 400 MHz) δ 8.25 (s, 1H), 8.07 (d, J = 2.40 Hz, 1H), 7.89 (dd, J = 8.73, 2.48 Hz, 1H), 7.74 (d, J = 8.73 Hz, 1H), 4.98 (s, 2H), 2.25 (s, 3H) ppm; 13C NMR (CDCl3, 101 MHz) δ 199.7, 159.87, 146.7, 146.4, 135.0, 133.3, 129.3, 126.1, 122.9, 54.7, 27.5 ppm; LC-MS (+ESI) calculated for C11H9ClN2O2 m/z 237.0 (M + H), found m/z 237.1 (M + H).
4.1.2.12. 3-(3-Bromo-2-oxopropyl)-6-chloroquinazolin-4(3H)-one (38)
To a solution of 37 (460 mg, 1.94 mmol) in acetic acid (10 mL) was added bromine (125 μL, 62.42 mmol) dropwise, forming a deep red solution. The reaction mixture was refluxed at 65 °C for 16 h, forming a red precipitate. The reaction mixture was concentrated in vacuo, diluted with water and neutralised with 2 M NaOH. The organic layer was extracted with ethyl acetate (3 × 20 mL) and washed with water (2 × 20 mL) and brine (1 × 20 mL). The organic layers were combined and concentrated in vacuo and purified through flash column chromatography (1:1 ethyl acetate/petroleum ether), yielding 38 (790 mg, 66%) as a white solid: 1H NMR (DMSO‑d6, 400 MHz) δ 8.27 (s, 1H), 8.08 (d, J = 2.36 Hz, 1H), 7.90 (dd, J = 8.69, 2.40 Hz, 1H), 7.75 (d, J = 8.73 Hz, 1H), 5.13 (s, 2H), 4.57 (s, 2H) ppm; 13C NMR (CDCl3, 101 MHz) δ 194.6, 160.0, 146,7, 146.1, 135.2, 133.6, 129.5, 126.1, 122.8, 52.4, 31.4 ppm; LC-MS (+ESI) calculated for C11H8BrClN2O2 m/z 314.9 (M + H), found m/z 315.1 (M + H).
4.1.2.13. 2-Isopropylthiazole-5-carbaldehyde oxime (39)
Bromomalonaldehyde (150 mg, 1.00 mmol) and 2-methylpropanethioamide (103 mg, 1.00 mmol) were dissolved in EtOH (5 mL) and stirred at room temperature for 4 h. Hydroxylamine hydrochloride (90 mg, 1.30 mmol) and TEA (1 mL) was added and stirred for a further 2 h at room temperature. The reaction mixture was concentrated in vacuo and purified through flash column chromatography in petroleum ether/ethyl acetate (4:1 to 3:2) yielding 39 (70 mg, 34%) as a yellow solid: 1H NMR (DMSO‑d6, 400 MHz) δ 12.29 (s, 1H), 8.28 (s, 1H), 8.02–8.00 (m, 3H), 7.54–7.50 (m, 3H) ppm;
4.1.2.14. (2-Isopropylthiazol-5-yl)methanamine (40)
To a solution of 39 (59 mg, 0.29 mmol) in EtOH (4 mL) was added zinc powder (47 mg, 0.72 mmol) and hydrochloric acid (130 μL, 13 N, 1.72 mmol). The reaction mixture was stirred at 60 °C for 2 h, then cooled to room temperature. The mixture was diluted with water and neutralised with saturated NaHCO3, then extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over brine and concentrated in vacuo yielding 40 (55 mg, 100%) as a yellow solid: 1H NMR (DMSO‑d6, 400 MHz) δ 7.92–7.90 (m, 2H), 7.79 (1H, s), 7.53–7.47 (m, 3H), 5.02 (s, br, 2H), 4.10 (s, 2H) ppm;
4.1.2.15. Methyl 5-chloro-2-(((dimethylamino)methylene)amino)benzoate (41)
A solution of 2-amino-5-chlorobenzoic acid (50 mg, 0.29 mmol) in acetonitrile (5 mL) was treated with 1,1-dimethoxy-N,N- dimethylmethanamine and stirred at 100 °C for 2 h. The intermediate 41 was confirmed through LC-MS and carried through without purification: LC-MS (+ESI) calculated for C11H13ClN2O2 m/z 241.1 (M + H), found m/z 240.7 (M + H).
4.1.2.16. 7-Chloroquinazolin-4(3H)-one (42)
4-chloroanthranilic acid (1.00 g, 5.82 mmol) was refluxed in formamide (10 mL) at 150 °C for 16 h, forming a brown precipitate. The reaction mixture was cooled to room temperature and 20 g ice was added. The mixture was left to stand for 1 h, then the precipitate filtered and concentrated in vacuo, yielding 42 (850 mg, 81%) as a brown solid: 1H NMR (DMSO‑d6, 400 MHz) δ 12.39 (s, 1H), 8.14 (s, 1H), 8.11 (d, J = 8.56 Hz, 1H), 7.72 (d, J = 2.09 Hz, 1H), 7.55 (dd, J = 8.52, 2.11 Hz, 1H) ppm; 13C NMR (DMSO‑d6, 101 MHz) δ 160.1, 149.9, 146.9, 138.9, 128.0, 127.0, 126.4, 121.5 ppm; LC-MS (+ESI) calculated for C8H5ClN2O m/z 181.0 (M + H), found m/z 181.2 (M + H).
4.1.2.17. 7-Chloro-3-(2-oxopropyl)quinazolin-4(3H)-one (43)
Compound 42 (850 mg, 4.70 mmol) was dissolved in NMP (5 mL) and treated with NaH (380 mg, 9.40 mmol). The reaction mixture was stirred at room temperature for 30 min, then chloroacetone (1.1 mL, 14.10 mmol) was added dropwise. The reaction was stirred for 30 min at room temperature, the diluted with water (20 mL), and extracted with ethyl acetate (4 × 15 mL). The combined organic layers were dried over brine, concentrated in vacuo and recrystallised in 2:1 hexane/ethyl acetate, yielding compound 43 (827 mg, 74%) as a silver crystal: 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.52 Hz, 1H), 7.88 (s, 1H), 7.73 (d, J = 2.02 Hz, 1H), 7.47 (dd, J = 8.57, 1.98 Hz, 1H), 4.79 (s, 2H), 2.35 (s, 3H) ppm; 13C NMR (CDCl3, 101 MHz) δ 199.8, 160.4, 149.3, 147.5, 141.0, 128.4, 127.4, 120.5, 77.2, 76.8, 54.8, 27.7 ppm; LC-MS (+ESI) calculated for C11H9ClN2O2 m/z 237.0 (M + H), found m/z 237.1 (M + H).
4.1.2.18. 7-Chloro-3-(3-chloro-2-oxopropyl)quinazolin-4(3H)-one (44)
Compound 43 (235 mg, 1.00 mmol) and NCS (133 mg, 1.00 mmol) were stirred in DCM (5 mL) and H2SO4 (1 mL) at 40 °C for 6 h. The reaction mixture was cooled to room temperature, neutralised with NaOH and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried over brine, concentrated in vacuo and purified through flash column chromatography in petroleum ether/ethyl acetate (7:3 to 3:7) yielding 44 as a white solid (30 mg, 11%): 1H NMR (400 MHz, Acetic Acid-d4) δ 8.43 (s, 1H), 8.26 (d, J = 8.59 Hz, 1H), 7.81 (d, J = 2.05 Hz, 1H), 7.59 (dd, J = 8.63, 2.01 Hz, 1H), 5.23 (s, 2H), 4.52 (s, 2H) ppm; 13C NMR (101 MHz, Acetic Acid-d4) δ 195.7, 160.4, 149.8, 147.7, 141.2, 128.5, 128.4, 125.5, 119.8, 52.7, 46.4 ppm; LC-MS (+ESI) calculated for C11H8Cl2N2O2 m/z 271.0 (M + H), found m/z 270.6 (M + H).
4.2. PqsR bioreporter assay
4.2.1. Bacterial growth conditions
P. aeruginosa strains PAO1-L and PA14 were grown in lysogeny broth (LB). All strains were incubated at 37 °C for 16 h, prior to use when OD600 was approximately 2.5. Strains PAO1-L and PA14 contained a chromosomal mCTX:PpqsA-lux fusion such that light is emitted following activation of the pqsA promoter. Consequently, a reduction in bioluminescence is indicative of pqs system inhibition. Strain BL21 (DE3) contained a pET28:PqsR plasmid, allowing for overexpression of the protein PqsR, and containing a kanamycin resistance cassette. To these cultures was added 100 μg/mL kanamycin for selection.
4.2.2. Bioluminescence reporter gene IC50 and 10 μM spot test assays
Two 1.5 mL Eppendorf tubes were filled with 500 μL of LB broth. To one was added 3.16 μL of a 10 mM stock of a given inhibitor, and to the other 1 μL, resulting in overall concentrations of 63.2 μM and 20 μM respectively. A one in 10 serial dilution of both stock solutions into four further Eppendorf tubes each containing 450 μL of LB broth gave 10 concentrations of the compound from 63.2 μM to 2 nM. Using only the central 60 wells of a Grenier 96 well flat black plate, 100 μL of each concentration was added to three wells for results in triplicate.
Concurrently, an overnight culture of either PAO1-L or PA14 with the relevant antibiotic, grown to an OD600 of between 2.0 and 3.0, was diluted in LB broth to OD600 = 0.02. To each of the wells containing a given concentration of compound in LB, was added 100 μL of the diluted P. aeruginosa, giving overall concentrations of 31.6 μM to 1 nM. The outer wells were filled with 200 μL of LB broth, as well as any unfilled inner wells, and the plate placed into a luminometer-spectrometer (Tecan GENios Pro), running a script at 37 °C over 24 h, with a kinetic cycle measuring OD600 and luminescence every 30 min.
A peak in luminescence was observed always between 8 and 9 h, after which deterioration of the growth curve led to a drop in bioluminescence. As such, readouts of the OD600 and bioluminescence were taken from the highpoint of the luminescence readout, and the results plotted in Graphpad Prism 8, measuring log [concentration] against luminescence (Relative Light Units)/OD600.
A similar methodology was applied for the 10 μM spot test assays, but all compounds were diluted to 10 μM from the 10 mM stock by adding 1 μL of the stock to 500 μL of LB broth, yielding a 20 μM solution. As with the IC50 assays, 100 μL was then added to at least three wells and diluted twofold by the addition of 100 μL of P. aeruginosa at OD600 = 0.02. As all compounds were tested at 10 μM, no serial dilutions of the stock solutions were performed. Data was displayed as a column graph, with all results normalised against the negative control containing 0.1% DMSO (100%).
4.2.3. Crystallography and protein determination
Protein was prepared as previously described by Ilangovan et al. [32] Crystals were grown in 24 well sitting and hanging drop Cryschem plates. Crystallisation reservoir consisted of 100 mM trisodium citrate, 200 mM ammonium acetate and 2-methyl-2,4-pentanediol (MPD). Citrate pH (5.5–6.5) and MPD concentration (3%–10%) were varied along the plate’s X and Y axes respectively, and optimal conditions for crystal soaking of 6, 12, 18 and 19 were found to be a pH of 6.25 containing 6% MPD. Ligands were dissolved in DMSO or a multi-component solvent mixture and allowed to incubate in the crystallisation drop for 24 h.
Diffraction images were integrated with DIALS [47] (via Xia2 or DUI) and scaled in the CCP4 suite. Structures were solved by Molecular Replacement with PHASER [48] using 6Q7W as a search model. Model was refined with REFAC5 [49] with Jellybody restrains and ligand fitted into the Fo-Fc density map using COOT [50]. Ligand restrains were generated in AceDrg [51]. Omit and POLDER maps were generated using Phenix [52]. Finished structures have been deposited in the PDB as 6Z17 (6), 6Z07 (12), 6Z5K (18) and 6YZ3 (19). Ligand description (CIF), is available in supplementary information.
The construct pET28a:pqsR is available upon request.
4.2.4. Pyocyanin quantification
A 5 mL liquid culture of PAO1-L wildtype in LB medium was grown overnight for over 16 h with shaking at 200 rpm. Flasks containing 15 mL overnight PAO1-L culture diluted in LB to an OD600 of 0.05, and either an inhibitor compound at 3 × IC50 value, or an equivalent volume of DMSO control was added and incubated for a further 16 h with shaking at 200 rpm.
An OD600 measurement was taken for each culture after 16 h for normalisation, before each sample was centrifuged for 10 min at 10,000×g and 24 °C. The supernatants were filtered through a 0.22 μm filter, and 7.5 mL collected. To each 7.5 mL filtered supernatant was added 4.5 mL chloroform, and each sample vortexed at 3000 rpm for 10 s. The samples were centrifuged at 10,000 rcf for 10 min at 4 °C, and the aqueous layer discarded. To 3 mL of each sample was added to 1.5 mL 0.2 M HCl and each vortexed for 10 s, followed by centrifugation at 10,000×g for 2 min at 4 °C. Measurement of the OD520 of the aqueous layer of each sample against a 0.2 M HCl control provided the raw data which could subsequently be normalised against the OD600 readings taken after incubation. Adapted from Essar et al. [53].
4.2.5. Cytotoxicity
A549 lung epithelial cells were cultured in Gibco’s DMEM media containing 10% FBS and 1% penicillin-streptomycin at 37 °C with 5% CO2. When 80% confluent, the medium was removed and the cells washed with 2 × 10 mL PBS and 1 × 7 mL trypsin-EDTA. To the flask was added 7 mL trypsin-EDTA and incubated for 5 min at 37 °C, 5% CO2. 7 mL FBS was added to quench trypsinization, and the cells centrifuged at 300 g for 5 min.
The supernatant was removed and the cell pellet washed with DMEM-F12 medium and centrifuged again at 300 g for 5 min. The pellet was then resuspended in 3 mL DMEM-F12 media and diluted further to allow loading of 100 μL of suspension containing 10,000 cells per well into 96 well plates. Cells were incubated for 16 h, at which point 100 μL of each compound was added to the relevant wells to give a total volume of 200 μL. Cells were incubated for a further 16 h, and 20 μL of Alamar blue added to each well. After 5 h incubation, fluorescence was measured with excitation at 510 nm and emission at 590 nm. Values were then normalised against the untreated cell control. Adapted from O’Brien et al. [54].
Author contributions
SG wrote the manuscript with supervision from MS, MC and PW with input from all co-authors. The junior authors performed the experiments and analysed the data with supervision from MS, MC and PW. FS, MS, MC, PW and JE contributed to the study and experimental design. (Synthesis: SG, FS; Biological Assays: SG and FS; X-ray crystallography WR with supervision from JE.)
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Wellcome Trust doctoral training programme in antimicrobials and antimicrobial resistance (SG, WR ref: 108876/B/15/Z) and the JPI-AMR/MRC funded SENBIOTAR program (Ref. MR/N501852/1). FS, JE, PW, MC and MS are funded by the National Biofilms Innovation Centre (NBIC) which is an Innovation and Knowledge Centre funded by the Biotechnology and Biological Sciences Research Council, InnovateUK and Hartree Centre [Award Number BB/R012415/1. We would like to thank our colleagues at Diamond Light Source for the provision of Synchrotron radiation at Beamline I04 under session MX19880-21.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2020.112778.
Abbreviations
- AI
auto inducer
- AQ
alkyl quinolone
- DCM
dichloromethane
- DMEM
Dulbecco’s modified eagle medium
- DMFDMA
N,N-dimethylformamide dimethyl acetate
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- EtOH
ethanol
- FBS
foetal bovine serum
- HHQ
2-heptyl-4-hydroxyquinoline
- HPLC
high performance liquid chromatography
- IC50
half maximal inhibitory concentration
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- LB
lysogeny broth
- LBD
ligand-binding domain
- LC-MS
liquid chromatography-mass spectrometry
- LD50
half maximal lethal dose
- MCCC
managed chemical compound collection
- MPD
2-methyl-2,4-pentanediol
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- NHQ
2-nonyl-4-hydroxyquinoline
- NMP
N-methyl pyrrolidinone
- NMR
nuclear magnetic resonance
- OD600
optical density at 600 nm
- PA
Pseudomonas aeruginosa
- PQS
2-heptyl-3-hydroxy-4(1H)-quinolone
- QS
quorum sensing
- QSSM
quorum sensing signal molecule
- rcf
relative centrifugal force
- SAR
structure-activity relationship
- SD
standard deviation
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TBAI
tetrabutylammonium iodide
- TEA
triethylamine
- THF
tetrahydrofuran
- tris
tris(hydroxymethyl)aminomethane
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Henry R.L., Mellis C.M., Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr. Pulmonol. 1992;12:158–161. doi: 10.1002/ppul.1950120306. [DOI] [PubMed] [Google Scholar]
- 2.Coburn B., Wang P.W., Diaz Caballero J., Clark S.T., Brahma V., Donaldson S., Zhang Y., Surendra A., Gong Y., Elizabeth Tullis D., Yau Y.C.W., Waters V.J., Hwang D.M., Guttman D.S. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015;5:10241. doi: 10.1038/srep10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emerson J., Rosenfeld M., McNamara S., Ramsey B., Gibson R.L. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 2002;34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 4.Tacconelli E., Carrara E., Savoldi A., Harbarth S., Mendelson M., Monnet D.L., Pulcini C., Kahlmeter G., Kluytmans J., Carmeli Y., Ouellette M., Outterson K., Patel J., Cavaleri M., Cox E.M., Houchens C.R., Grayson M.L., Hansen P., Singh N., Theuretzbacher U., Magrini N. WHO pathogens priority list working group. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018;18:318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- 5.Clatworthy A.E., Pierson E., Hung D.T. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 6.Wagner S., Sommer R., Hinsberger S., Lu C., Hartmann R.W., Empting M., Titz A. Novel strategies for the treatment of Pseudomonas aeruginosa infections. J. Med. Chem. 2016;59:5929–5969. doi: 10.1021/acs.jmedchem.5b01698. [DOI] [PubMed] [Google Scholar]
- 7.Soukarieh F., Williams P., Stocks M.J., Cámara M. Pseudomonas aeruginosa quorum sensing systems as drug discovery targets: current position and future perspectives. J. Med. Chem. 2018;61:10385–10402. doi: 10.1021/acs.jmedchem.8b00540. [DOI] [PubMed] [Google Scholar]
- 8.Williams P. Strategies for inhibiting quorum sensing. Emerg. Top. Life Sci. 2017;1:23–30. doi: 10.1042/ETLS20160021. [DOI] [PubMed] [Google Scholar]
- 9.Hurley M.N., Cámara M., Smyth A.R. Novel approaches to the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. Eur. Respir. J. 2012;40:1014–1023. doi: 10.1183/09031936.00042012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams P., Cámara M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009;12:182–191. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Diggle S.P., Winzer K., Chhabra S.R., Worrall K.E., Cámara M., Williams P. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can Be produced in the absence of LasR. Mol. Microbiol. 2003;50:29–43. doi: 10.1046/j.1365-2958.2003.03672.x. [DOI] [PubMed] [Google Scholar]
- 12.Grandclément C., Tannières M., Moréra S., Dessaux Y., Faure D. Quorum quenching: role in nature and applied developments. FEMS Microbiol. Rev. 2016;40:86–116. doi: 10.1093/femsre/fuv038. [DOI] [PubMed] [Google Scholar]
- 13.Ryan R.P., Dow J.M. Diffusible signals and interspecies communication in bacteria. Microbiology (Read.) 2008;154:1845–1858. doi: 10.1099/mic.0.2008/017871-0. [DOI] [PubMed] [Google Scholar]
- 14.Albuquerque P., Casadevall A. Quorum sensing in fungi--a review. Med. Mycol. 2012;50:337–345. doi: 10.3109/13693786.2011.652201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nazik H., Sass G., Ansari S.R., Ertekin R., Haas H., Déziel E., Stevens D.A. 2019. Novel Intermicrobial Molecular Interaction: Pseudomonas Aeruginosa Quinolone Signal (PQS) Modulates Aspergillus Fumigatus Response to Iron. Microbiology (Reading, Engl.) [DOI] [PubMed] [Google Scholar]
- 16.Lau G.W., Hassett D.J., Ran H., Kong F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol. Med. 2004;10:599–606. doi: 10.1016/j.molmed.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Lau G.W., Ran H., Kong F., Hassett D.J., Mavrodi D. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect. Immun. 2004;72:4275–4278. doi: 10.1128/IAI.72.7.4275-4278.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Britigan B.E., Roeder T.L., Rasmussen G.T., Shasby D.M., McCormick M.L., Cox C.D. Interaction of the Pseudomonas aeruginosa secretory products pyocyanin and pyochelin generates hydroxyl radical and causes synergistic damage to endothelial cells. Implications for Pseudomonas-associated tissue injury. J. Clin. Invest. 1992;90:2187–2196. doi: 10.1172/JCI116104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stintzi A., Evans K., Meyer J.-M., Poole K. Quorum sensing and siderophore biosynthesis in Pseudomonas aeruginosa: LasR/LasI mutants exhibit reduced pyoverdine biosynthesis. FEMS Microbiol. Lett. 1998;166:341–345. doi: 10.1111/j.1574-6968.1998.tb13910.x. [DOI] [PubMed] [Google Scholar]
- 20.Glessner A., Smith R.S., Iglewski B.H., Robinson J.B. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of twitching motility. J. Bacteriol. 1999;181:1623–1629. doi: 10.1128/jb.181.5.1623-1629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reimmann C., Ginet N., Michel L., Keel C., Michaux P., Krishnapillai V., Zala M., Heurlier K., Triandafillu K., Harms H., Défago G., Haas D. Genetically programmed autoinducer destruction reduces virulence gene expression and swarming motility in Pseudomonas aeruginosa PAO1. Microbiology (Read.) 2002;148:923–932. doi: 10.1099/00221287-148-4-923. [DOI] [PubMed] [Google Scholar]
- 22.O’Loughlin C.T., Miller L.C., Siryaporn A., Drescher K., Semmelhack M.F., Bassler B.L. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 2013;110:17981–17986. doi: 10.1073/pnas.1316981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J., Zhang L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell. 2015;6:26–41. doi: 10.1007/s13238-014-0100-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rampioni G., Leoni L., Williams P. The art of antibacterial warfare: deception through interference with quorum sensing-mediated communication. Bioorg. Chem. 2014;55:60–68. doi: 10.1016/j.bioorg.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Heeb S., Fletcher M.P., Chhabra S.R., Diggle S.P., Williams P., Cámara M. Quinolones: from antibiotics to autoinducers. FEMS Microbiol. Rev. 2011;35:247–274. doi: 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drees S.L., Fetzner S. PqsE of Pseudomonas aeruginosa acts as pathway-specific thioesterase in the biosynthesis of alkylquinolone signaling molecules. Chem. Biol. 2015;22:611–618. doi: 10.1016/j.chembiol.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 27.Coleman J.P., Hudson L.L., McKnight S.L., Farrow J.M., Calfee M.W., Lindsey C.A., Pesci E.C. Pseudomonas aeruginosa PqsA is an anthranilate-coenzyme A ligase. J. Bacteriol. 2008;190:1247–1255. doi: 10.1128/JB.01140-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drees S.L., Li C., Prasetya F., Saleem M., Dreveny I., Williams P., Hennecke U., Emsley J., Fetzner S. PqsBC, a condensing enzyme in the biosynthesis of the Pseudomonas aeruginosa quinolone signal: crystal structure, inhibition, and reaction mechanism. J. Biol. Chem. 2016;291:6610–6624. doi: 10.1074/jbc.M115.708453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahner J.H., Empting M., Kamal A., Weidel E., Groh M., Börger C., Hartmann R.W. Exploring the chemical space of ureidothiophene-2-carboxylic acids as inhibitors of the quorum sensing enzyme PqsD from Pseudomonas aeruginosa. Eur. J. Med. Chem. 2015;96:14–21. doi: 10.1016/j.ejmech.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Pistorius D., Ullrich A., Lucas S., Hartmann R.W., Kazmaier U., Müller R. Biosynthesis of 2-alkyl-4(1H)-Quinolones in Pseudomonas aeruginosa: potential for therapeutic interference with pathogenicity. Chembiochem. 2011;12:850–853. doi: 10.1002/cbic.201100014. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y.-C., Hussain F., Negm O., Pavia A., Halliday N., Dubern J.-F., Singh S., Muntaka S., Wheldon L., Luckett J., Tighe P., Bosquillon C., Williams P., Cámara M., Martínez-Pomares L. Contribution of the alkylquinolone quorum-sensing system to the interaction of Pseudomonas aeruginosa with bronchial epithelial cells. Front. Microbiol. 2018;9:3018. doi: 10.3389/fmicb.2018.03018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ilangovan A., Fletcher M., Rampioni G., Pustelny C., Rumbaugh K., Heeb S., Cámara M., Truman A., Chhabra S.R., Emsley J., Williams P. Structural basis for native agonist and synthetic inhibitor recognition by the Pseudomonas aeruginosa quorum sensing regulator PqsR (MvfR) PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu C., Kirsch B., Zimmer C., de Jong J.C., Henn C., Maurer C.K., Müsken M., Häussler S., Steinbach A., Hartmann R.W. Discovery of antagonists of PqsR, a key player in 2-alkyl-4-quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem. Biol. 2012;19:381–390. doi: 10.1016/j.chembiol.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 34.Soukarieh F., Vico Oton E., Dubern J.F., Gomes J., Halliday N., de Pilar Crespo M., Ramírez-Prada J., Insuasty B., Abonia R., Quiroga J., Heeb S., Williams P., Stocks M.J., Cámara M. In silico and in vitro-guided identification of inhibitors of alkylquinolone-dependent quorum sensing in Pseudomonas aeruginosa. Molecules. 2018;23:257. doi: 10.3390/molecules23020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatterjee M., D’Morris S., Paul V., Warrier S., Vasudevan A.K., Vanuopadath M., Nair S.S., Paul-Prasanth B., Mohan C.G., Biswas R. Mechanistic understanding of phenyllactic acid mediated inhibition of quorum sensing and biofilm development in Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 2017;101:8223–8236. doi: 10.1007/s00253-017-8546-4. [DOI] [PubMed] [Google Scholar]
- 36.Kitao T., Lepine F., Babloudi S., Walte F., Steinbacher S., Maskos K., Blaesse M., Negri M., Pucci M., Zahler B., Felici A., Rahme L.G. Molecular insights into function and competitive inhibition of Pseudomonas aeruginosa multiple virulence factor regulator. mBio. 2018;9 doi: 10.1128/mBio.02158-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starkey M., Lepine F., Maura D., Bandyopadhaya A., Lesic B., He J., Kitao T., Righi V., Milot S., Tzika A., Rahme L. Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupton J.T., Miller J.F., Bryant R.D., Maloney P.R., Foster B.S. The preparation of aromatic amidino esters and their reaction with primary amines. Tetrahedron. 1987;43:1747–1752. [Google Scholar]
- 39.Fletcher M.P., Diggle S.P., Crusz S.A., Chhabra S.R., Cámara M., Williams P. A dual biosensor for 2-alkyl-4-quinolone quorum-sensing signal molecules. Environ. Microbiol. 2007;9:2683–2693. doi: 10.1111/j.1462-2920.2007.01380.x. [DOI] [PubMed] [Google Scholar]
- 40.Köhler T., Michea-Hamzehpour M., Plesiat P., Kahr A.L., Pechere J.C. Differential selection of multidrug efflux systems by quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1997;41:2540–2543. doi: 10.1128/aac.41.11.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Viswanadhan V.N., Ghose A.K., Revankar G.R., Robins R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Model. 1989;29:163–172. [Google Scholar]
- 42.Aleksić I., Šegan S., Andrić F., Zlatović M., Moric I., Opsenica D.M., Senerovic L. Long-chain 4-aminoquinolines as quorum sensing inhibitors in Serratia marcescens and Pseudomonas aeruginosa. ACS Chem. Biol. 2017;12:1425–1434. doi: 10.1021/acschembio.6b01149. [DOI] [PubMed] [Google Scholar]
- 43.Mikkelsen H., McMullan R., Filloux A. The Pseudomonas aeruginosa reference strain PA14 displays increased virulence due to a mutation in LadS. PLoS One. 2011;6 doi: 10.1371/journal.pone.0029113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zender M., Witzgall F., Kiefer A., Kirsch B., Maurer C.K., Kany A.M., Xu N., Schmelz S., Börger C., Blankenfeldt W., Empting M. Flexible fragment growing boosts potency of quorum-sensing inhibitors against Pseudomonas aeruginosa virulence. ChemMedChem. 2020;15:188–194. doi: 10.1002/cmdc.201900621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aleksic I., Jeremic J., Milivojevic D., Ilic-Tomic T., Šegan S., Zlatović M., Opsenica D.M., Senerovic L. N-benzyl derivatives of long-chained 4-amino-7-chloro-quionolines as inhibitors of pyocyanin production in Pseudomonas aeruginosa. ACS Chem. Biol. 2019;14:2800–2809. doi: 10.1021/acschembio.9b00682. [DOI] [PubMed] [Google Scholar]
- 46.Hossain M.A., Sattenapally N., Parikh H.I., Li W., Rumbaugh K.P., German N.A. Design, synthesis, and evaluation of compounds capable of reducing Pseudomonas aeruginosa virulence. Eur. J. Med. Chem. 2020;185:111800. doi: 10.1016/j.ejmech.2019.111800. [DOI] [PubMed] [Google Scholar]
- 47.Winter G., Waterman D.G., Parkhurst J.M., Brewster A.S., Gildea R.J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I.D., Sauter N.K., Evans G. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 2018;74:85–97. doi: 10.1107/S2059798317017235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kovalevskiy O., Nicholls R.A., Long F., Carlon A., Murshudov G.N. Overview of refinement procedures within REFMAC5: utilizing data from different sources. Acta Crystallogr. D Struct. Biol. 2018;74:215–227. doi: 10.1107/S2059798318000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Long F., Nicholls R.A., Emsley P., Graǽulis S., Merkys A., Vaitkus A., Murshudov G.N. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr. D Struct. Biol. 2017;73:112–122. doi: 10.1107/S2059798317000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liebschner D., Afonine P.V., Moriarty N.W., Poon B.K., Sobolev O.V., Terwilliger T.C., Adams P.D. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 2017;73:148–157. doi: 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Essar D.W., Eberly L., Hadero A., Crawford I.P. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J. Bacteriol. 1990;172:884–900. doi: 10.1128/jb.172.2.884-900.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Brien J., Wilson I., Orton T., Pognan F. Investigation of the alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.