

Abstract
T-cell receptor (TCR) gene transfer redirects T cells to target intracellular antigens. However, the potential autoreactivity generated by TCR mispairing and occurrence of graft-versus-host disease in the allogenic setting due to the retention of native TCRs remain major concerns. Natural killer T cells (NKTs) have shown promise as a platform for adoptive T-cell therapy in cancer patients. Here, we showed their utility for TCR gene transfer. We successfully engineered and expanded NKTs expressing a functional TCR (TCR-NKTs), showing HLA-restricted antitumor activity in xenogeneic mouse models in the absence of graft-versus-mouse reactions. We found that TCR-NKTs downregulated the invariant TCR (iTCR), leading to iTCR+TCR+ and iTCR–TCR+ populations. In-depth analyses of these subsets revealed that in iTCR–TCR+ NKTs, the iTCR, although expressed at the mRNA and protein levels, was retained in the cytoplasm. This effect resulted from a competition for binding to CD3 molecules for cell surface expression by the transgenic TCR. Overall, our results highlight the feasibility and advantages of using NKTs for TCR expression for adoptive cell immunotherapies. NKT low intrinsic alloreactivity that associated with the observed iTCR displacement by the engineered TCR represents ideal characteristics for “off-the-shelf” products without further TCR gene editing.
Keywords: Natural Killer T cells, transgenic TCR, adoptive T cell transfer, gene engineering, TCR mispairing
Introduction
The role played by cytotoxic T cells in tumor control has been recognized for years (1), but the clinical utility of their adoptive transfer has remained elusive due to the low frequency of high avidity, tumor-specific T cells naturally circulating in peripheral blood (2). Advances in gene engineering have facilitated the introduction of tumor-antigen specificity in polyclonal T cells via expression of ectopic T-cell receptors (TCRs)(3). Using this strategy, clinically relevant numbers of TCR-redirected T cells for adoptive transfer can be generated and have produced encouraging clinical results (4, 5).
Despite the clinical success, a major issue of the TCR technology remains the expression of both the ectopic and endogenous TCR chains within the same T cell. This phenomenon can cause competition for binding to CD3 molecules, resulting in a reduced expression of the transgenic TCR (6) and TCR mispairing between the endogenous and transgenic TCR chains, causing potential autoreactivity (7). The retention of functional endogenous TCRs may exacerbate graft-versus-host disease (GvHD) if these cells are used as an allogeneic product (8).
Several strategies have been developed to enhance TCR transgenic expression, like the inclusion of a codon-optimized sequence (9), the addition of extra disulfide bonds to the transgenic TCR chains (10), the introduction of murine residues in the TCR constant region domain (11), and the replacement of key amino acids in the framework of the variable region (3, 12). siRNA gene silencing (13) and TCR gene editing (14) have been explored to knockdown the endogenous receptor of T cells for replacement with a tumor-specific TCR. However, siRNA-mediated TCR knockdown is generally suboptimal, and the scale-up of gene editing for clinical use remains challenging and of unknown safety profile.
We thus have explored the use of natural killer T cells (NKTs) as a TCR-based cell platform to overcome the issues associated with TCR engineering in T cells. Type 1 NKTs are an evolutionarily conserved subset of innate lymphocytes characterized by expression of the invariant TCR α-chain Vα24-Jα18 and reactivity to glycolipids presented by the monomorphic MHC class I-like molecule CD1d (15). In humans, the invariant TCR (iTCR) Vα24-Jα18 chain is paired almost exclusively with TCR Vβ11 (TRBV25–1) chains (16). We reasoned that the limited repertoire of the iTCR and its CD1d restriction should minimize, if not eliminate, both the TCR mispairing phenomena and the risk of GvHD when NKTs express a tumor-specific TCR. Here, we showed the successful TCR engineering, expansion, and HLA-restricted antitumor activity of TCR-redirected NKTs, and we dissected the mechanism by which the transgenic TCR displaces the endogenous iTCR from the cell surface of engineered NKTs.
Material and Methods
Cell lines.
The tumor cell lines SK-MEL-5 and M14 (melanoma), T2 (TAP transporter deficient), SupT1 (CD3+TCR–), and 293T cells were obtained from the American Type Culture Collection (ATCC) between 2015 and 2018. The tumor cell line C8161 (melanoma) was obtained in 2016 from the tissue culture facility at University of North Carolina, Chapel Hill (TCF, UNC-CH). K562 cells we previously established as described from Quintarelli C. et al. (17). All cells were maintained in culture with the appropriate media, either RPMI 1640 (Gibco), DMEM (Gibco), or IMDM (Gibco) supplemented with 10% FBS (Sigma), 1% L-glutamine (Gibco), and 1% penicillin-streptomycin (Gibco) in a humidified atmosphere containing 5% CO2 at 37°C. The M14 cell line was transduced with a SFG gamma retroviral vector encoding the HLA-A2*0201 molecule (17) to obtain the M14-A2 cell line upon cell sorting. SK-MEL-5, M14-wild-type (wt), and M14-A2 cells were transduced with a SFG gamma retroviral vector encoding the firefly luciferase gene and the fusion protein enhanced GFP (eGFP-FFluc)(18). Cells were kept in culture for less than 6 consecutive months, after which aliquots from the original expanded vial were used. All tumor cell lines were routinely tested to exclude contamination with mycoplasma and assessed for the expression of transgenes (like HLA-A2, CD40L, CD80, OX40L, eGFP) and tumor markers by flow cytometry to confirm identity.
Retroviral constructs and generation of retroviral supernatants.
Retroviral supernatants were prepared by transient transfection of 293T cells (17) and used to transduce T cells and NKTs (isolated as stated below). The HLA-A2-restricted TCRs specific for Tyrosinase368–376 (Tyr-TCR recognizing the YMDGTMSQV peptide) and MART-126–35 (MART-1-TCR recognizing the ELAGIGILTV peptide) were obtained from Morgan R. (19) and were cloned into the SFG retroviral vector (20). The HLA-A2-restricted survivin-specific TCR (Sur-TCR recognizing the LMLGEFLKL peptide 96–104) and the HLA-A2-restricted PRAME-specific TCR (PRAME-TCR recognizing the ALYVDSLFFL peptide p300) are previously described (21). The human CD8α cDNA (NM_001768) was cloned into the SFG retroviral vector. The human CD3 complex (CD3δ-2A-CD3γ-2A-CD3ε-2A-CD3ζ) was design as previously reported (6) and synthetized from Geneart and cloned in the SFG retroviral vector.
NKT and T-cell isolation, transduction, and in vitro expansion.
Buffy coats from healthy volunteer blood donors were purchased from the Gulf Coast Regional Blood Center (Houston, Texas, USA). Peripheral blood mononuclear cells (PBMCs) were isolated by Lymphoprep (Accurate Chemical and Scientific Corporation) density gradient centrifugation, according to the manufacturer’s protocol. NKTs were purified from PBMCs using anti-iNKT microbeads (Miltenyi Biotech). NKTs were cultured in complete medium, consisting of 45% Click’s medium (Irvine Scientific), 45% RPMI 1640 (Hyclone), 10% FBS (Hyclone), 1% L-glutammine (Gibco), and 1% penicillin-streptomycin (Gibco). Upon NKT positive selection (LS column, Miltenyi Biotech), the negative fraction was used as feeder cells after irradiation (40 Gy, RS-2000 Biological System). For the first stimulation (S1), feeder cells were used once at day 0 at the PBMC:NKT ratio of 10:1. α-galactosylceramide (αGalCer, 100 ng/mL, Diagnocine LLC) was added at day 0 and IL2 (200 IU/mL, StemCell Technologies) was added at day 0, 2 and 4. NKTs were transduced in retronectin (Takara, 7µg/ml) 24-wells-coated plates (22) at day 6 when the NKT purity was more than 85%, and cells were re-stimulated with irradiated feeder cells, at the PBMC:NKT ratio of 5:1 (S2). The feeder cells were loaded with the peptide specific for each TCR (20 µM, YMDGTMSQV for the Tyr-TCR, ELAGIGILTV for the MART-1-TCR, or ALYVDSLFFL for the PRAME-TCR from Genemed Synthesis) and αGalCer (100 ng/mL) in the presence of IL2 (200 IU/mL). NKTs were then further expanded for 10 days in the presence of IL2 (200 IU/mL). In selected experiments, NKTs, CD8+, and unselected T cells were isolated from the same buffy coat. T cells were positively (LS column, Miltenyi Biotech) selected using CD8-specific microbeads (Miltenyi Biotech). Unselected T cells and CD8+ T cells were activated, transduced, and expanded in complete medium with IL7 (10 ng/mL, Peprotech) and IL15 (5 ng/mL, Peprotech) as previously reported (23). Briefly T cells were activated on anti-CD3 (1µg/ml, Miltenyi Biotech) and anti-CD28 (1µg/ml, BD Biosciences) 24-wells-coated plates. At day 2 T cells were transduced in retronectin (Takara, 7µg/ml) 24-wells-coated plates. On day 10, T cells were stimulated with peptide-loaded (20 µM) and irradiated (80 Gy) artificial antigen presenting cells (aAPCs) consisting of K562 cells expressing CD40L, CD80, OX40L, and the HLA-A*02 molecules (17) at an aAPC:T-cell ratio of 1:4, and further expanded using IL7 (10 ng/mL) and IL15 (5 ng/mL) for 10 days. In selected experiments NKTs were co-transduced with retroviral vectors encoding the CD8α or the CD3 complex.
Immunophenotyping and flow sorting.
T and NKT cells were stained with antibodies (Abs) against CD3 (APC-H7, clone SK7), CD62L (BV605, clone DREG-56), PD-1 (PE-Cy7, clone EH12.1), LAG3 (PE, clone T47–530), TIM3 (BV711, clone 7D3), CD45 (APC, clone 2D1), CD4 (PE-Cy7, clone SK3), and CD8 (Alexa Fluor 700, clone RPA-T8) from BD Biosciences. Tumor cells and aAPCs were stained with antibodies (Abs) against HLA-A2 (PE, clone BB7.2) and CD276 (BV421, clone 7–517) from BD Biosciences. The purity of NKTs was assessed by staining the cells with a tetramer specific for the iTCR (PE-conjugated human CD1d tetramer loaded with αGalCer; ProImmune) and the PE-conjugated Ab specific for TCR Vα24 chain (anti-iNKT, clone 6B11, from BD Biosciences). The expression of the Tyr-TCR in NKTs was assed using a Tyr-TCR-specific pentamer (Pro5 MHC Pentamer A*02:01 YMDGTMSQV; ProImmune) followed by the staining with the Pro5 Fluorotag APC (ProImmune). NKT transduction efficiency with ectopic TCRs was assessed with a FITC-conjugated Ab specific for the TCR β12 chain (for the Tyr-TCR and MART-1-TCR; Beckman Coulter) or the TCR β14 chain (for the PRAME-TCR; Beckman Coulter), or stained with a APC-conjugated Ab specific for the murine TCR constant β chain (for the Sur-TCR; BD Biosciences). Data acquisition was performed on a BD FACSCanto II or BD LSRFortessa using the BD FACS-Diva software. Data analyses were performed with the FlowJo software. To count the number of CD3 and iTCR molecules expressed from NKTs, we used the PE Fluorescence Quantitation Kit (BD Quantibrite™). In selected experiments, NKTs were co-stained with an anti-iTCR tetramer and anti-Tyr-TCR pentamer, and the iTCR+Tyr-TCR–, iTCR–Tyr-TCR+, and iTCR+Tyr-TCR+ NKTs were sorted using a FACSAria II at the Flow Cytometry Core Facility (UNC, Chapel Hill). After sorting cells (purity >96%), were kept in culture overnight in complete media and IL2 (200 IU/mL) before performing the functional assays.
51Cr-release assay.
The cytotoxic activity of NKTs and CD8+ T cells was evaluated using a standard 5-hour 51Cr release assay (17). In brief, 5 ×103 51Cr (Perkin Elmer)-labeled target cells (HLA-A2+ mismatched phytohemagglutinin (PHA) blasts (24), M14-wt, M14-A2, C8161 and SK-MEL-5) per well were plated in triplicate in a 96-well plate (v bottom) with different ratios (40:1, 20:1, 10:1 and 5:1) of effector cells and incubated for 5 hours at 37°C. The supernatant was collected and analyzed with a gamma-counter (Perkin Elmer). HLA-A2+ (PHA) blasts were incubated for 2 hours at 37°C with specific peptides (100nM, YMDGTMSQV for the Tyr-TCR or ELAGIGILTV for the MART-1-TCR, from Genemed Synthesis) and washed twice with complete medium. Target cells were incubated with medium alone or in 1% Triton X-100 (Sigma-Aldrich) to determine the spontaneous and maximum 51Cr release, respectively. The mean percentage of specific lysis of triplicate wells was calculated as follows: [(test counts - spontaneous counts) / (maximum counts - spontaneous counts)] x 100%.
Confocal microscopy.
SupT1 cells and NKTs were prepared according to the manufacturer’s instructions (Abcam immunofluorescence protocol). Briefly, cells were fixed with cytofix buffer (BD Biosciences) and permeabilized with 0.1% Triron X in PBS. Cells were then incubated with anti-Vα24Jα18 (clone 6B11, Novus biologicals) diluted at 1:800 in PBS/1% BSA. The Alexa Fluor 546 goat anti-mouse IgG1 (Invitrogen) diluted at 1:500 in PBS/1% BSA was used as a secondary Ab. Cells were mounted cover slips with one drop of ProLong Diamond Antifade Mountant with DAPI (Invitrogen). Data acquisition was performed on LSM700 Zeiss confocal microscopy using ZEN software (ZEISS Microscopy). Data analysis was performed with Fiji software.
TCR repertoire profiling/TCR amplicon sequencing.
DNA or RNA were extracted from sorted NKTs using the Qiagen DNeasy or RNeasy micro kits. NKT receptor β chain libraries were prepared using the Clontech SMARTer Human TCR α/β profiling kit, according to manufacturer’s protocol. Libraries were sequenced via 2×150 paired end on an Illumina MiSeq at the UNC High-Throughput Sequencing Facility (HTSF). TCR variable chain calling was performed using IMGT/HighV-QUEST, with subsequent analyses performed using the tcR R package (25).
Real-time Q-PCR.
iTCR and Tyr-TCR expression and integration were measured in NKTs by quantitative polymerase chain reaction (Q-PCR) with specific primers and probes and normalized to the 18S gene expression. Briefly, RNA was extracted from NKTs (RNeasy Plus Kit, Qiagen) and 1µg of RNA was used for reverse transcription (5X VILO Reaction Mix and 10X SuperScript Enzyme Mix from Invitrogen). For the Q-PCR reaction (TaqMan 2X Universal PCR Master Mix from Life Technologies) 20ng of cDNA were used in duplicates. The relative expression was calculated as follows: 2^- [(CTgene – CT18S) – CTVα24 in NT-NKTs]. DNA was extracted from NKTs (DNeasy Blood & Tissue Kit, Qiagen) and 500ng of DNA were used for the Q-PCR reaction. To calculate the number of copies of Tyr-TCR construct in NKTs post sorting, we created a standard curve with the SFG Tyr-TCR plasmid and the QuantStudio Real-Time PCR software automatically calculated the number of copies/100ng DNA. Data acquisition was performed on a QuantStudio 6 Flex from Life Technologies using the QuantStudio Real-Time PCR software. The primers and probe for the 18S (Hs03003631_g1) were purchased from Thermo Fisher Scientific. Va24 primers: Forward 5’-TGTCAAGCTGGTCGAGAAAAG-3’ Reverse: 5’-TCATGAGCAGATTAAACCCGG-3’ Probe 5’-CCACTTTCAGGAGGAGGATTCGGAAC-3’. Tyr-TRAC primers: Forward 5’-ACTTCGACAGCCAGATCAAC-3’ Reverse: 5’-TGTGAAGCTGGTCTGATTG −3’ Probe 5’-TGCTGGACATGAAGGCGATGGA-3’. Tyr-TRBC primers: Forward 5’-TGGGTCAACGGCAAAGAG-3’ Reverse: 5’-TGGCATCTGAAGTGGTTCC −3’ Probe 5’-CCGATCCCCAGGCCTACAAAGAG-3’.
Western blot.
Three millions of tumor cells (M14-wt, M14-A2, C8161 and SK-MEL-5) were lysate with 81µl of 2X Laemmil Sample Buffer (Bio Rad) and 9µl of 2-Mercaptoethanol (Fisher Scientific). Ten µl of total protein lysates were separated by Mini-PROTEAN TGX Stain-Free Gels (BIO RAD) electrophoresis. After protein transfer, membranes were incubated with 3µl of anti-tyrosinase (ThermoFisher, clone T311) or 15 µl of anti-MART-1 (Invitrogen, clone M2–7C10). After washes, membranes were incubated with 3µl of HRP-conjugated goat anti-mouse IgG (H+L) secondary antibody (Invitrogen). Staining for β-actin (Santa Cruz Biotechnolgy) was used as loading control. Membranes were developed with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific).
Cocultures and ELISAs.
NKTs and CD8+ T cells (105 cells/well) were cocultured with tumor cell lines (M14-wt, M14-A2, C8161 and SK-MEL-5) (105 cells/well) at an E:T ratio of 1:1 in 24-well plates, in complete medium, in the absence of cytokines. After 3 days of culture, cells were harvested and stained for CD3 (APC-H7, clone SK7 from BD Biosciences) and CD276 (BV421, clone 7–517 from BD Biosciences) monoclonal (m)Abs to detect T cells and tumor cells, respectively. Percentage of residual tumor cells in culture were enumerated by flow cytometry. NKTs (105 cells/well) were also cocultured with HLA-A2+ T cell blasts loaded with the YMDGTMSQV peptide (100 nM) at an E:T ratio of 1:1 or activated with 10 µL of anti-Vα24 (clone 6B11, from BD Biosciences) in 1 mL of complete medium in the absence of cytokines. Culture supernatants were harvested after 24 hours of culture and IFNγ measured in 100µl of supernatant with the DuoSet Human IFNγ ELISA kit (R&D System). Data acquisition was performed on a Synegrgy2 microplate reader (BioTek) using the Gen5 software.
ELISpot assay.
The IFNγ ELISpot assay was performed as previously described (17). Briefly, 2 × 104 NKTs or CD8+ T cells per well were plated in triplicate and then stimulated with 2 × 104 T2 cells loaded with specific peptides (YMDGTMSQV for the Tyr-TCR, ELAGIGILTV for the MART-1-TCR, or ALYVDSLFFL for the PRAME-TCR). NKTs and CD8+ T cells were stimulated with PMA (25 ng/mL), ionomycin (1 µg/mL)(both from Sigma-Aldrich), and IL2 (100 IU/mL) as a positive control. After 18 hours, IFNγ single-forming cells (SFCs) were counted using the ELIspot reader (AID Classic ELR07) and analyzed using the Elispot7.0 software.
Study approval.
All mouse experiments were performed in accordance with UNC Animal Husbandry and Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by UNC IACUC (IACUC ID: 49268).
Xenograft models.
Female and male NSG mice (7 – 9 weeks of age, obtained from the UNC Animal Core) were injected either subcutaneously (s.c.) with 0.5 × 106 SK-MEL-5 tumor cells or intravenously (i.v.) via tail injection with 0.5 – 1 × 106 FFluc-labeled M14-wt or M14-A2 tumor cells. In specific experiments, mice received low dose irradiation (100 cGy) 24 hours before receiving i.v. 1 × 106 FFluc-labeled M14-A2 cells. Seven days after tumor cell injection (day 0) and at days +5 and +12, mice were infused i.v. with 1 × 107 or 5 × 106 non transduced (NT)-NKTs or Tyr-TCR-NKTs (expressing or not eGFP-FFluc, and non-sorted for TCR expression). In selected experiments, non transduced (NT)-T cells and T cells expressing the Tyr-TCR (Tyr-TCR-Ts, and non-sorted for TCR expression) were also used. In selected experiments, recombinant human IL2 (1000 U/mouse) was administered intraperitoneally (i.p.) every two days for a total of 8 doses after the adoptive transfer of either NKTs or T cells. Melanoma tumor cell growth was monitored weekly either with caliper measurement for s.c. tumors, or by bioluminescence (BLI)(total phlux, photons/second) using the IVIS kinetic in vivo imaging system (Perkin Elmer) for the metastatic models. Mice were sacrificed according to UNC guidelines for either tumor growth or occurrence of sign of discomfort, including graft-versus-mouse disease (weight loss, fur loss, and lethargy). When mice were sacrificed, peripheral blood was collected from the heart and spleen, liver and tumor were smashed on cell strainers and washed with 2 ml of PBS. Peripheral blood, spleen, liver, and tumors were analyzed to detect the presence of NKTs or T cells [Stained with antibodies (Abs) against CD3 (APC-H7, clone SK7) and CD45 (APC, clone 2D1)] by flow cytometry using CountBright absolute counting beads (Invitrogen). In selected experiments, tumors were isolated, homogenized in a total of 1 mL of PBS, and 100µl of the supernatant were used to measure the human IFNγ using the DuoSet Human IFNγ ELISA kit (R&D System). Infiltrating NKTs were counted in the tumors by staining with antibodies (Abs) against CD3 (APC-H7, clone SK7) and CD45 (APC, clone 2D1)and counting beads (Invitrogen) and analyzing via flow cytometry.
Statistics.
Data were summarized as the mean ± SD. Student’s t test or two-way ANOVA were used to determine statistically significant differences between treatment groups, with Bonferroni’s correction for multiple comparisons when appropriate (Prism 6: GraphPad Software). Survival analysis was performed using the Kaplan-Meier method (Prism 6: GraphPad Software). All p values less than 0.05 were considered statistically significant.
Results
NKTs can be engineered to express an ectopic TCR
Isolated and cultured NKTs (Fig. 1A) robustly and consistently expanded ex vivo (fold-increase: 17±4 after the first stimulation, S1)(Fig. 1B). By day 5–7 of culture (before retroviral transduction), 93%±4% of the isolated cells expressed the iTCR, as assessed by flow cytometry using a specific tetramer (Fig. 1B). As a proof-of-concept, NKTs were transduced with a gamma retroviral vector to express an HLA-A2–restricted, tyrosinase-specific TCR (Tyr-TCR)(19). Tyr-TCR was detectable in 76%±8% NKTs (Tyr-TCR-NKTs), and the expression remained stable over the course of the entire culture period (Fig. 1C). The growth kinetics of NKTs was not hampered by the expression of the Tyr-TCR, as 1.2×108±8.5×107 cells were obtained at the end of the culture period (S2), a number that is sufficient for clinical adoptive transfer (Fig. 1D). Tyr-TCR-NKTs retained the phenotypic characteristics of non-transduced NKTs, including preservation of CD4 and CD8 composition and expression of CD62L and exhaustion markers (Fig. 1E). Tyr-TCR NKTs showed HLA class I–restriction, as they lysed HLA-A2+ PHA blasts loaded with the HLA-A*0201 tyrosinase YMD peptide (77%±28% lysis at the E:T ratio of 40:1), whereas control NKTs (NT-NKTs) showed negligible cytotoxic activity (Fig. 1F). Overall, NKTs could be successfully manipulated to express a functional MHC class I–restricted TCR.
Figure 1. NKTs can be genetically modified to express an HLA-A2–restricted Tyrosinase-specific TCR (Tyr-TCR).
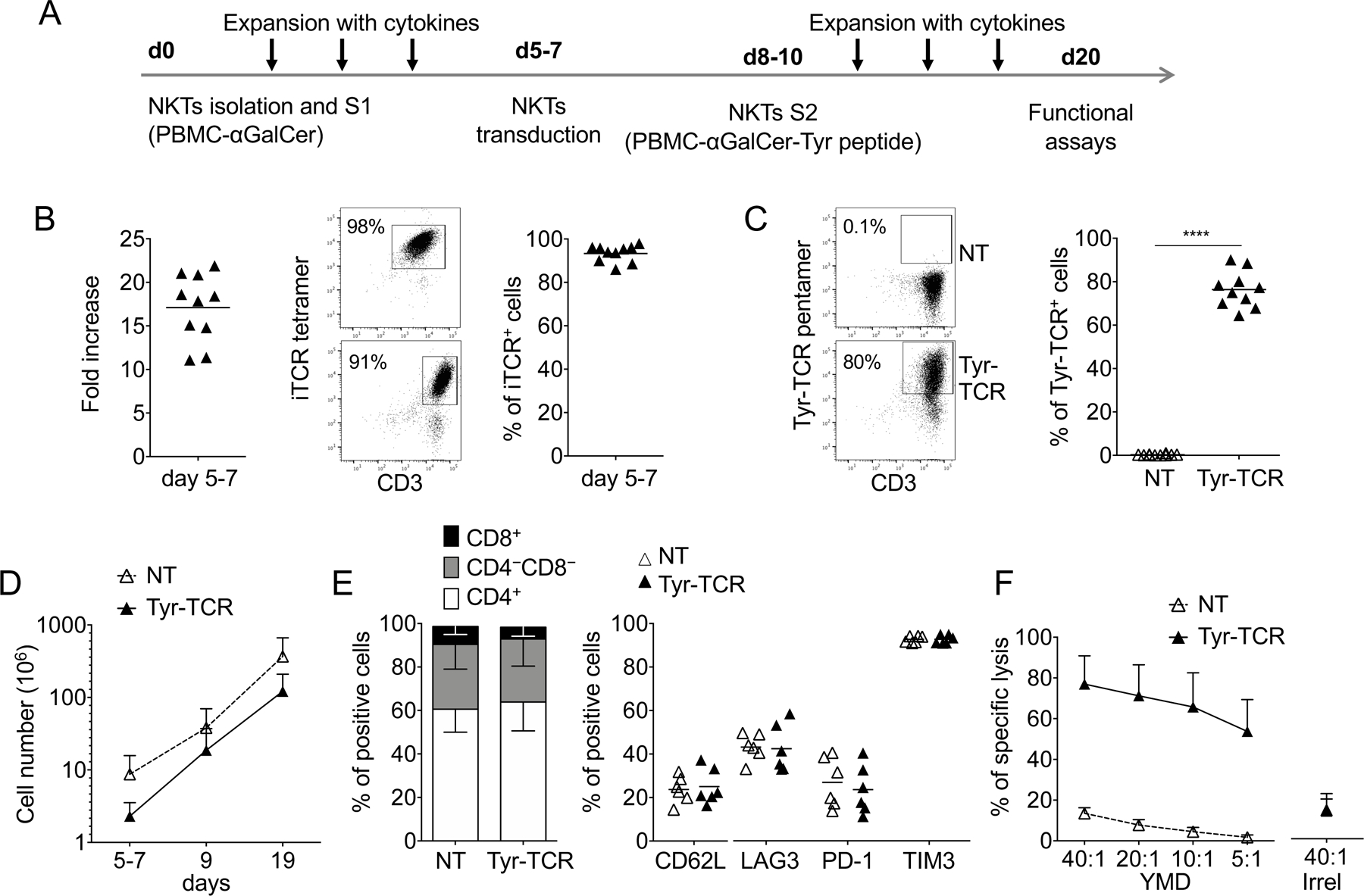
A. Schematic timeline of the protocol used to select, transduce, and expand NKTs. S1 and S2 indicate the first and second stimulation, respectively. B. Fold increase (left), representative flow cytometry plots (middle), and purity of NKTs (right) on day 5–7 after S1 (n=10, mean is shown). C. Representative flow cytometry plots (left) and summary (right) of Tyr-TCR expression by control (NKT-NT) and transduced (Tyr-TCR NKTs) NKTs assessed at day 19 of culture (n=10, mean is shown). ****p<0.0001, paired t test. D. Total cell numbers of NT-NKTs and Tyr-TCR NKTs at day 19 (n=10, mean and SD are shown). E. Phenotypic characterization of NT-NKTs and Tyr-TCR-NKTs at day 19–20 of culture (n=6, mean is shown). F. NT-NKTs and Tyr-TCR NKTs were tested in a 5-hour 51Cr-release assay against different ratios of HLA-A2+ PHA blasts loaded with either the specific tyrosinase peptide (YMD) or an irrelevant MART-1 peptide (Irrel., ELAGIGLTV), at the concentration of 100 nM (n=4, mean and SD are shown).
An ectopic TCR displaces the iTCR for expression on the cell surface in NKTs
As early as 3–4 days after retroviral transduction with the Tyr-TCR expressing vector, Tyr-TCR-NKTs consisted of two distinct subsets, namely CD3+iTCR+ (59%±18%) and CD3+iTCR– (41%±18%) cells (Fig. 2A). A similar downregulation of the iTCR was observed when NKTs were transduced with gamma retroviral vectors encoding other HLA-A2-restricted TCRs specific for survivin (Sur-TCR), PRAME (PRAME-TCR), and MART-1 (MART-1-TCR) antigens (Supplementary Fig. S1A), thus, excluding that the observed emergence of CD3+iTCR– NKTs was an exclusive phenomenon caused by the Tyr-TCR. Staining with iTCR and Tyr-TCR multimers revealed three distinct cell subsets: iTCR–Tyr-TCR+ (44%±15%), iTCR+Tyr-TCR+ (34%±11%), and iTCR+Tyr-TCR– (19%±11%) cells. These three subsets were also identified when using other combinations of antibodies (Vα24, Vβ11, and Vβ12 antibodies) recognizing Tyr-TCR and iTCR (Fig. 2B). Using SupT1 cells (CD3+TCR–)(26), we observed that ectopic TCRs outcompeted iTCRs for cell surface expression (Supplementary Fig. S1B) and that iTCRs were retained within the cytoplasm (Supplementary Fig. S1C). When iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ NKTs were sorted, the Vα24 chain of the iTCR was detectable in both subsets by confocal microscopy but was confined to the cytoplasm in iTCR–Tyr-TCR+ NKTs, whereas it was expressed on the cell surface in iTCR+Tyr-TCR+ NKTs (Fig. 2C). Based on these results, we hypothesized that in iTCR–Tyr-TCR+ NKTs, the Tyr-TCR displaced the iTCR for CD3 usage, and as a consequence, it was preferentially transferred to the cell surface. To corroborate this premise, we transduced NKTs with a retroviral vector encoding the CD3γδεζ genes (6) to provide an excess of CD3 to NKTs, and confirmed an increase of CD3 molecules on the cell surface (Fig. 2D). When Tyr-TCR– NKTs were transduced with the CD3 encoding vector, we observed an increase of the iTCR+Tyr-TCR+ subset (from 34%±14% to 72%±3%, gated on Tyr-TCR+ cells, p<0.0001)(Fig. 2E). Overall, these data indicated that the ectopic TCR can displace the iTCR from CD3 binding in NKTs.
Figure 2. Transgenic TCRs outcompete the iTCR for expression on the cell surface in NKTs.
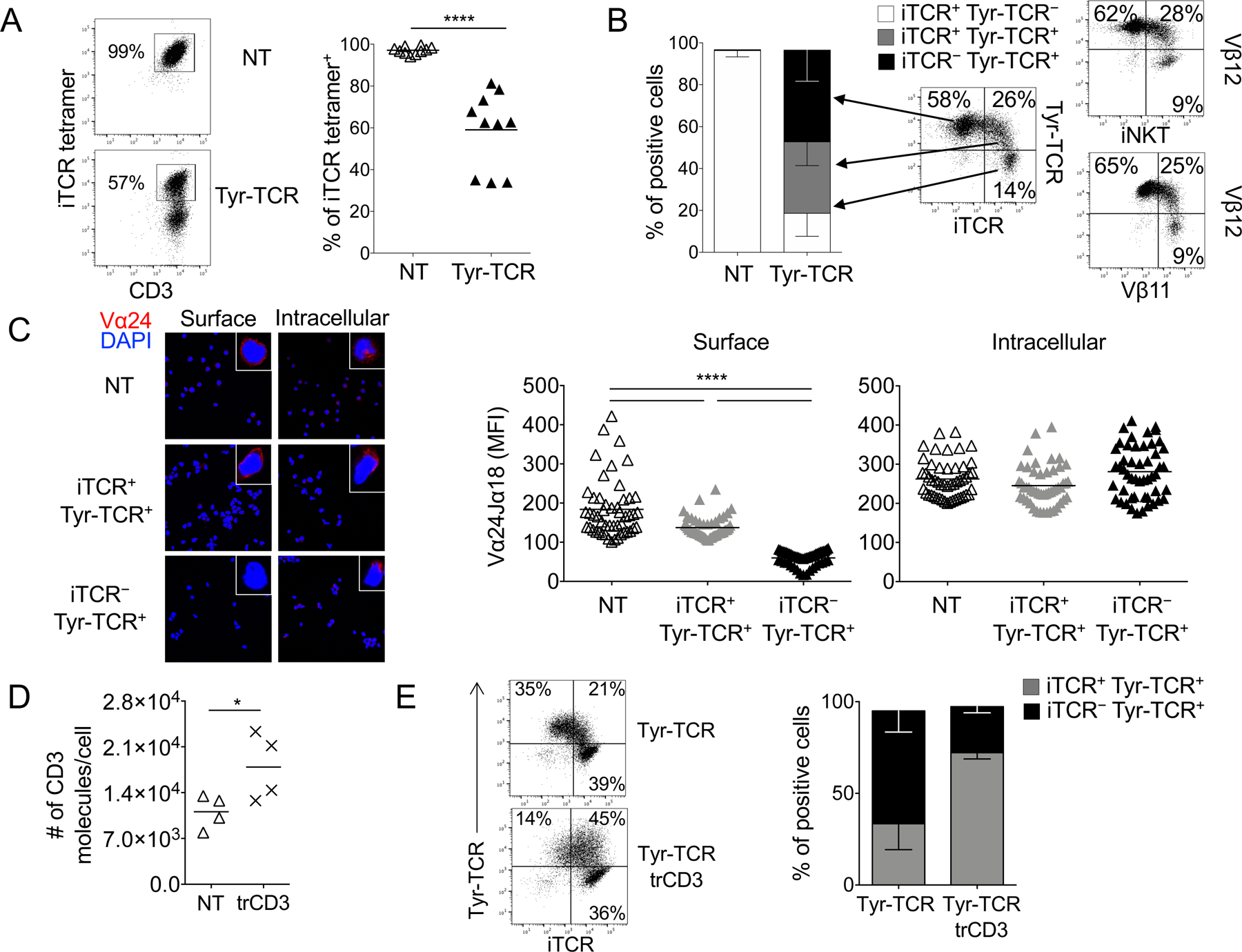
A. Representative flow cytometry plots (left panel) and summary (right panel) illustrating the expression of the iTCR in NT-NKTs and Tyr-TCR-NKTs 4 days after transduction (n=10, mean is shown). ****p<0.0001, paired t test. B. Subset composition of NT-NKTs and Tyr-TCR NKTs at the end of S2 (day 18–20) defined by expression of iTCR and Tyr-TCR detected by specific pentamers (n=10; left, SD is shown). Representative flow cytometry plots illustrating the expression of the iTCR (stained alternatively with the iTCR tetramer, anti-Vα24, or anti-Vβ11), and the Tyr-TCR (stained either with the Tyr-TCR pentamer or with anti-Vβ12)(right). C. Surface and intracellular staining of sorted iTCR+Tyr-TCR–, iTCR–Tyr-TCR+, and iTCR+Tyr-TCR+ NKT subsets with the Vα24Jα18 Ab (red staining). The blue staining indicates the DAPI. Shown are representative images of a single field of view taken via confocal microscopy (Magnification 63X). Graphs show the summary of the MFIs for surface and intracellular staining (n=50, right, mean is shown). MFI was calculated on NKTs obtained from 4 images for each condition. ****p<0.0001, unpaired t test. D. Number of CD3 molecules per cell in control NKTs (NT) and NKTs transduced with a vector encoding the CD3 complex (trd-CD3) (n=4, mean is shown). *p=0.0151, paired t test. E. Representative flow cytometry plots (left) and summary (right) illustrating the percentage of iTCR–Tyr-TCR+ and iTCR+Tyr-TCR+ cells gated on Tyr-TCR+ NKTs, in NKTs transduced with the Tyr-TCR encoding vector alone (Tyr-TCR NKTs), and in NKTs co-transduced with the vectors encoding the Tyr-TCR and CD3 (Tyr-TCR-CD3 NKTs)(n=5, SD is shown).
Higher transcripts of the ectopic TCR outcompetes the endogenous iTCR
We then studied how the transgenic TCR displaces the endogenous iTCR in NKTs. We first analyzed if iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ subsets showed differences in co-receptor expression, but both CD4 and CD8 were equally distributed within the two subsets (Fig. 3A). Although the Vα24-Jα18 chain is known to preferentially pair with Vβ11 (TRBV25–1) chains in human NKTs (16), we explored if combination with distinct Vβ chains correlated with the specific NKTs subsets. iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ were sorted using specific TCR multimers (Supplementary Fig. S2A) and subsequently analyzed for TCRβ using RNA amplicon sequencing. Although TRBV25–1 was the dominant detected TCR Vβ chain (range: 90.54–96.49%), a proportion of the repertoire was distributed across the other 47 Vβ chains (Fig. 3B). However, TCR Vβ chain distribution was conserved between iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ subsets, suggesting that the TCR Vβ repertoire differences did not segregate for iTCR expression. The majority of shared TCR clones (identified by the CDR3 sequence of the Vβ chain) was observed within each donor rather than within the iTCR+Tyr-TCR+ or iTCR–Tyr-TCR+ subsets (Supplementary Fig. S2B), indicating that the type of TCR Vβ chain associated with the Vα24-Jα18 was not affecting the binding with CD3, and thus, the iTCR displacement in iTCR–Tyr-TCR+ NKTs.
Figure 3. Tyr-TCR function in NKTs does not require the presence of the iTCR.
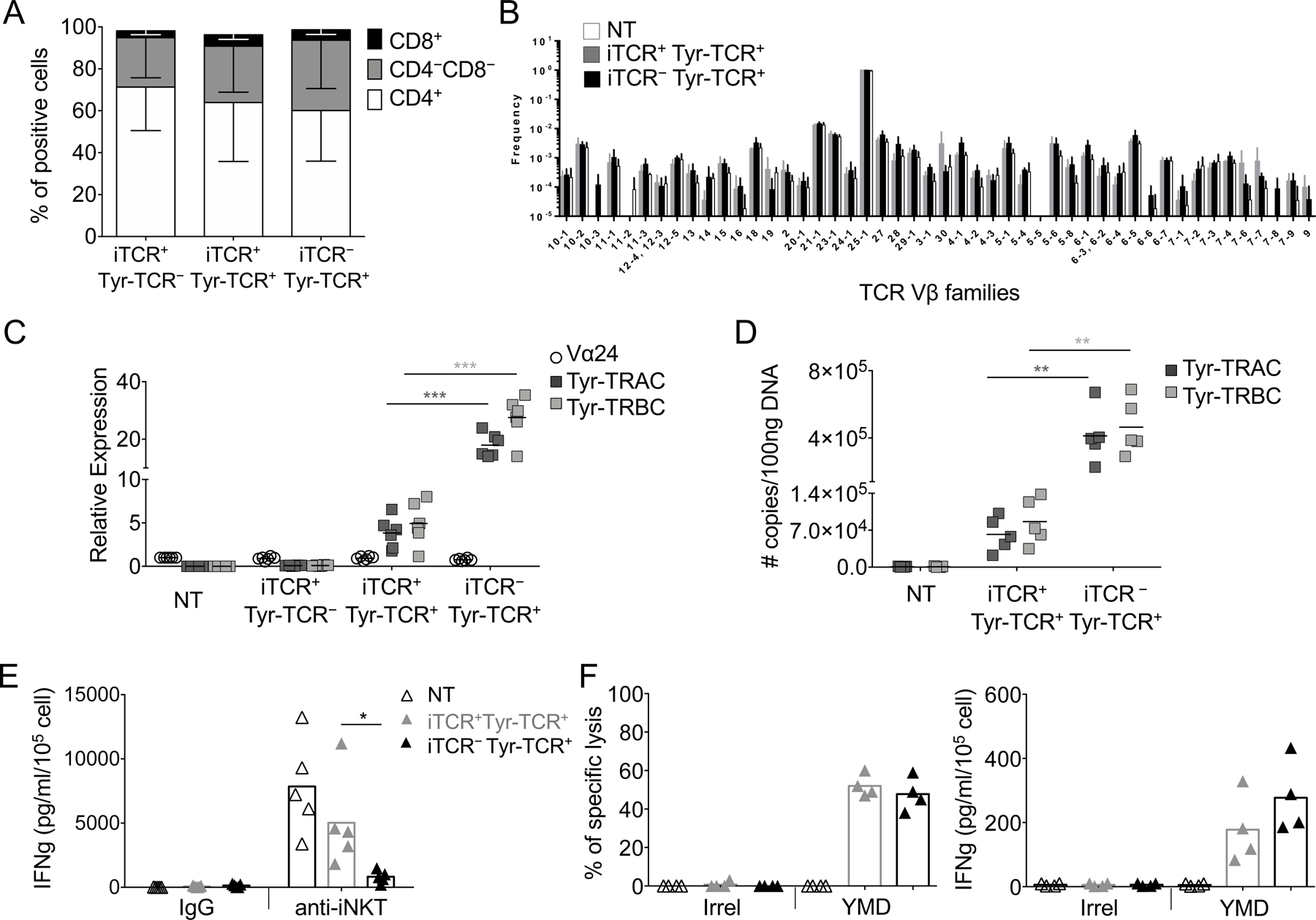
A. CD4/CD8 composition in each subset of Tyr-TCR-NKTs (n=3, SD is shown). B. Frequency of TCR Vβ chain RNA in NT-NKTs, iTCR+Tyr-TCR+ NKTs, and iTCR–Tyr-TCR+ NKTs obtained after sorting and TCR amplicon RNA-sequencing of each subset (SD is shown). C. Relative expression of the TCR Vα24 chain for the iTCR, and of TCRα (Tyr-TRAC) and TCRβ (Tyr-TRBC) chains for Tyr-TCR assessed by qPCR in NT-NKTs, iTCR+Tyr-TCR–, iTCR+Tyr-TCR+, and iTCR–Tyr-TCR+ NKT subsets (n=6, mean is shown). ***p=0.0004 for Tyr-TRAC and ***p=0.0002 for Tyr-TRBC, paired t test. D. Quantitation of TCRα (Tyr-TRAC) and TCRβ (Tyr-TRBC) copy number of integrated transgene via qPCR in NT-NKTs, iTCR+Tyr-TCR+, and iTCR–Tyr-TCR+ NKT subsets (n=5, mean is shown). **p=0.0087 for Tyr-TRAC and **p=0.0062 for Tyr-TRBC, paired t test. E. Quantification of IFNγ in supernatants collected after 24 hours from the activation of NT, iTCR-–Tyr-TCR+, and iTCR+Tyr-TCR+ NKTs with the Vα24 antibody. Isotype control was used as negative control (n=5, mean is shown). *p=0.0139, paired t test. F. iTCR–Tyr-TCR+ and iTCR+Tyr-TCR+ NKTs were tested in a 5-hour 51Cr-release assay (E:T=40:1) against HLA-A2+ PHA blasts loaded with an irrelevant MART-1 peptide (Irrel., ELAGIGLTV) or with the YMD peptide (100 nM)(left)(n=4, mean is shown). Quantification of IFNγ in supernatants collected after 24 hours from the co-cultures (E:T=1:1) of iTCR–Tyr-TCR+ or iTCR+Tyr-TCR+ NKTs with HLA-A2+ PHA blasts loaded with an irrelevant peptide (Irrel.) or with the YMN peptide (100 nM)(right)(n=4, mean is shown).
We next reasoned that the iTCR expression may be associated with transgene integrant copy number variations between the iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ subsets. We assessed the mRNA expressionof both Tyr-TRAC and Tyr-TRBC chains (transgenic TCR) and of Vα24 (iTCR) in both iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ subsets, and found the mRNA of both Tyr-TCR chains to be significantly higher than the mRNA expression of the iTCR chain (Fig. 3C). Both Tyr-TCR chains were more significantly expressed in iTCR–Tyr-TCR+ cells than in the iTCR+Tyr-TCR+ subset (p=0.0004)(Fig. 3C). The higher mRNA expression of Tyr-TRAC and Tyr-TRBC chains in iTCR–Tyr-TCR+ NKTs reflected the higher copy numbers of the transgene integrants (Fig. 3D). We then analyzed the functionality of sorted iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ NKTs and found that it was independent from the expression of the iTCR because only the iTCR+Tyr-TCR+ subset released IFNγ in response to the agonistic Vα24 mAb (clone 6B11)(Fig. 3E), but both iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ subsets lysed and produced IFNγ in response to HLA-A2+ PHA blasts loaded with the HLA-A*0201–restricted YMD peptide (Fig. 3F). Overall, these data suggest that the ectopic TCR outcompeted the iTCR for CD3 binding when the α- and β-TCR chains were expressed at higher levels than iTCR chains, so that the HLA-restricted cytotoxic activity of TCR-NKTs could be exploited regardless of the presence of the iTCR on the cell surface.
Tyr-TCR-NKTs acquire HLA-A2–restricted cytotoxic activity against tumor cells
To measure the antitumor activity of Tyr-TCR-NKTs against tumor cells expressing tyrosinase, we used the HLA-A2+Tyr+ melanoma cell line SK-MEL-5. The HLA-A2+Tyr– melanoma cell line C8161 was used as a negative control. As additional controls, we tested the melanoma cell line M14 that is HLA-A2–Tyr+ (M14-wt) and the same cell line genetically modified to express the HLA-A2 molecule (M14-A2)(Fig. 4A). Tyr-TCR-NKTs consistently lysed the HLA-A2+Tyr+ M14-A2 and SK-MEL-5 cells but not the HLA-A2–Tyr+ (M14-wt) or HLA-A2+Tyr– (C8161) tumor cells (Fig. 4B). Both iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ NKTs subsets lysed the M14-A2 and SK-MEL-5 cells (Supplementary Fig. S3A). We then cocultured Tyr-TCR-NKTs and control NT-NKTs with these melanoma cell lines for 3 days. Both M14-A2 and SK-MEL-5 cells were efficiently eliminated (<3% residual tumor cells) by Tyr-TCR-NKTs, whereas M14-wt and C8161 cells overgrew and accounted for more than 85% of the cells at the end of the 3-day culture (Fig. 4C). NT-NKTs did not eliminate any of these tumor cells. The antitumor activity of Tyr-TCR-NKTs was paralleled by IFNγ production, which was only detected when Tyr-TCR-NKTs were cocultured with M14-A2 and SK-MEL-5 cells (Fig. 4D). The IFNγ production in response to melanoma cell lines was comparable in both iTCR+Tyr-TCR+ and iTCR–Tyr-TCR+ NKTs subsets (Supplementary Fig. S3B). Overall, these data demonstrated the antitumor activity of NKTs genetically modified to express an HLA-A2–restricted TCR.
Figure 4. Tyr-TCR NKTs target HLA-A2+Tyr+ cells.
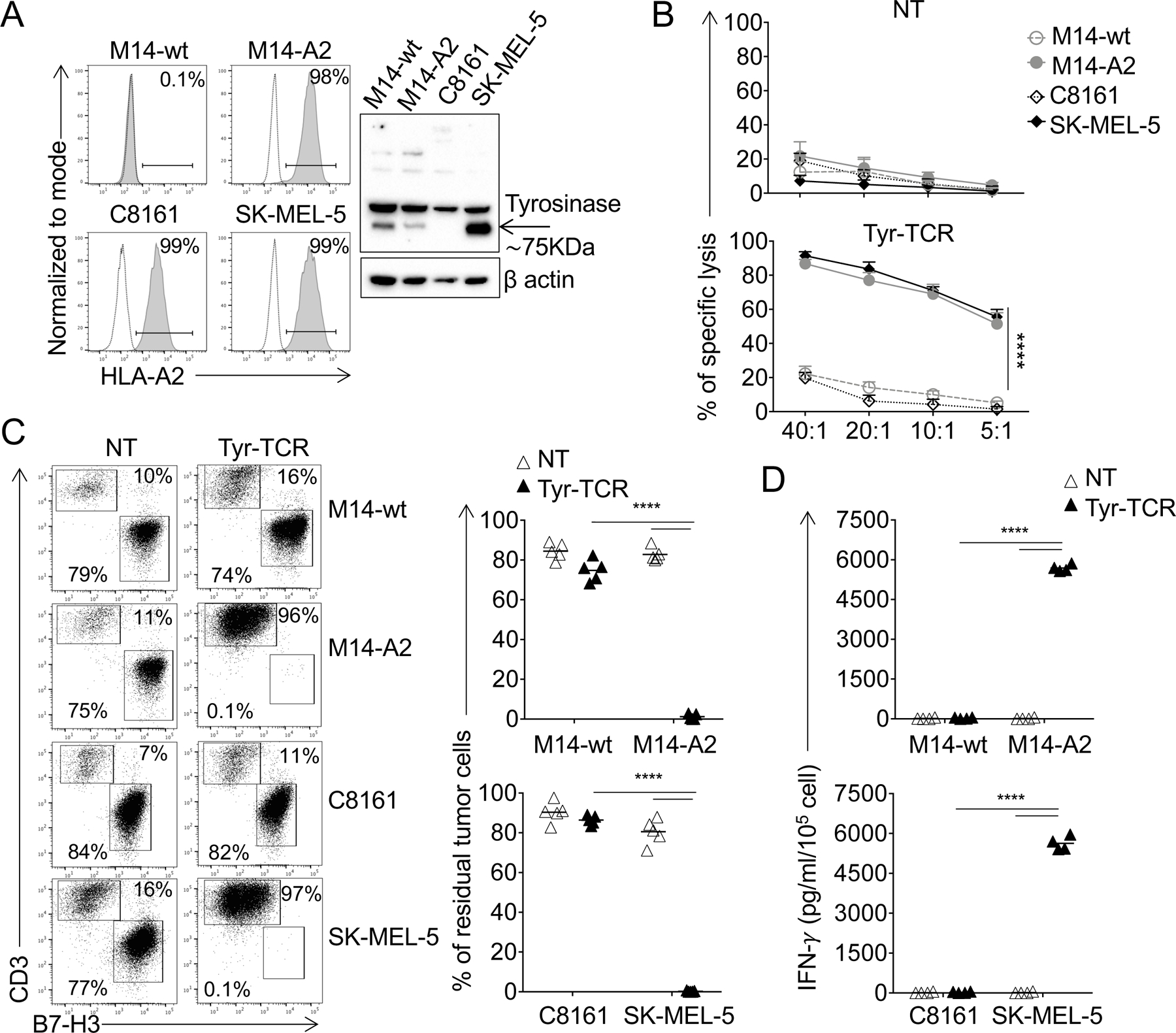
A. Expression of the HLA-A2 molecule in melanoma cell lines assessed by flow cytometry (left). Dotted and solid lines represent the isotype and HLA-A2 mAbs, respectively. The M14-wt cell line was modified to express the HLA-A2 molecule (M14-A2) via retroviral gene transfer. Detection of tyrosinase in lysates of the indicated melanoma cell lines assessed by Western blot (right). Staining for β-actin was used as a loading control. B. NT-NKTs (upper graph) and Tyr-TCR-NKTs (lower graph) were tested against the melanoma cell lines in a 5-hour 51Cr-release assay (n=4, mean and SD are shown). ****p<0.0001, 2-way ANOVA with Bonferroni correction. C. NT-NKTs and Tyr-TCR-NKTs were cocultured with melanoma cell lines (E:T=1:1) for 3 days. Cells were then collected and stained with the CD3 and CD276 mAbs to identify NKTs and melanoma cells, respectively, by flow cytometry. Representative flow plots (left) and summary of the quantification of residual tumor cells in the culture (right)(n=5, mean is shown). ****p<0.0001, paired t test. D. Quantification of IFNγ production in the supernatant collected after 24 hours of coculture of NT-NKTs and Tyr-TCR NKTs (E:T=1:1)(n=4, mean is shown). ****p<0.0001, paired t test.
High-functional avid TCRs are required to generate TCR-redirected NKTs
To assess if TCR avidity played a role in determining the antitumor activity of TCR-redirected NKTs, we used NKTs expressing an HLA-A2–restricted MART-1–specific TCR (MART-1-TCR), which is characterized by lower functional avidity compared to the Tyr-TCR (19). Transduction of NKTs with MART-1–TCR was comparable to that of Tyr-TCR (75%±13% vs 78%±10%, respectively)(Fig. 5A) and also in terms of expression (MART-1–TCR MFI=22377±1753 vs. Tyr-TCR MFI=19461±2336)(Supplementary Fig. S4A). However, MART-1–TCR NKTs produced less IFNγ spot forming cells (SFC) against T2 cells loaded with decreasing concentrations of the specific peptide compared to Tyr-TCR-NKTs (Fig. 5B), despite TCRs being expressed at comparable levels. Similarly, MART-1–TCR NKTs showed lower cytotoxic activity against HLA-A2+ PHA blasts loaded with the specific peptide than Tyr-TCR-NKTs (Fig. 5C). In contrast, when expressed in CD8+ T cells, both Tyr-TCR and MART-1–TCR (Fig. 5D) showed similar functionality to one another, assessed by IFNγ SFCs in response to peptide-loaded T2 cells (Fig. 5E) and cytotoxic activity against peptide-loaded HLA-A2+ PHA blasts (Fig. 5F). We also compared the antitumor activity of Tyr-TCR– and MART-1-TCR–expressing T cells and NKTs against the SK-MEL5 cell line, which expresses both tyrosinase and MART-1 (Fig. 5G). Although CD8+ T cells expressing either the Tyr-TCR or MART-1–TCR eliminated HLA-A2+Tyr+MART-1+ melanoma cells in coculture experiments (Fig. 5H), only Tyr-TCR-NKTs successfully eliminated these tumor cells (Fig. 5I). These data were confirmed using another low-affinity HLA-A2-restricted TCR specific for PRAME (PRAME-TCR). As observed for the MART-1-TCR, despite comparable transduction efficiency and MFI (Supplementary Fig. S4B), PRAME-TCR-NKT functions were inferior to that of PRAME-TCR-Ts, as the former produced less IFNγ against T2 cells loaded with decreasing concentrations of the specific peptide (Supplementary Fig. S4C) and showed an inferior cytotoxic activity against peptide-loaded HLA-A2+ PHA blasts (Supplementary Fig. S4D).
Figure 5: A high-affinity TCR is required to redirect NKT specificity.
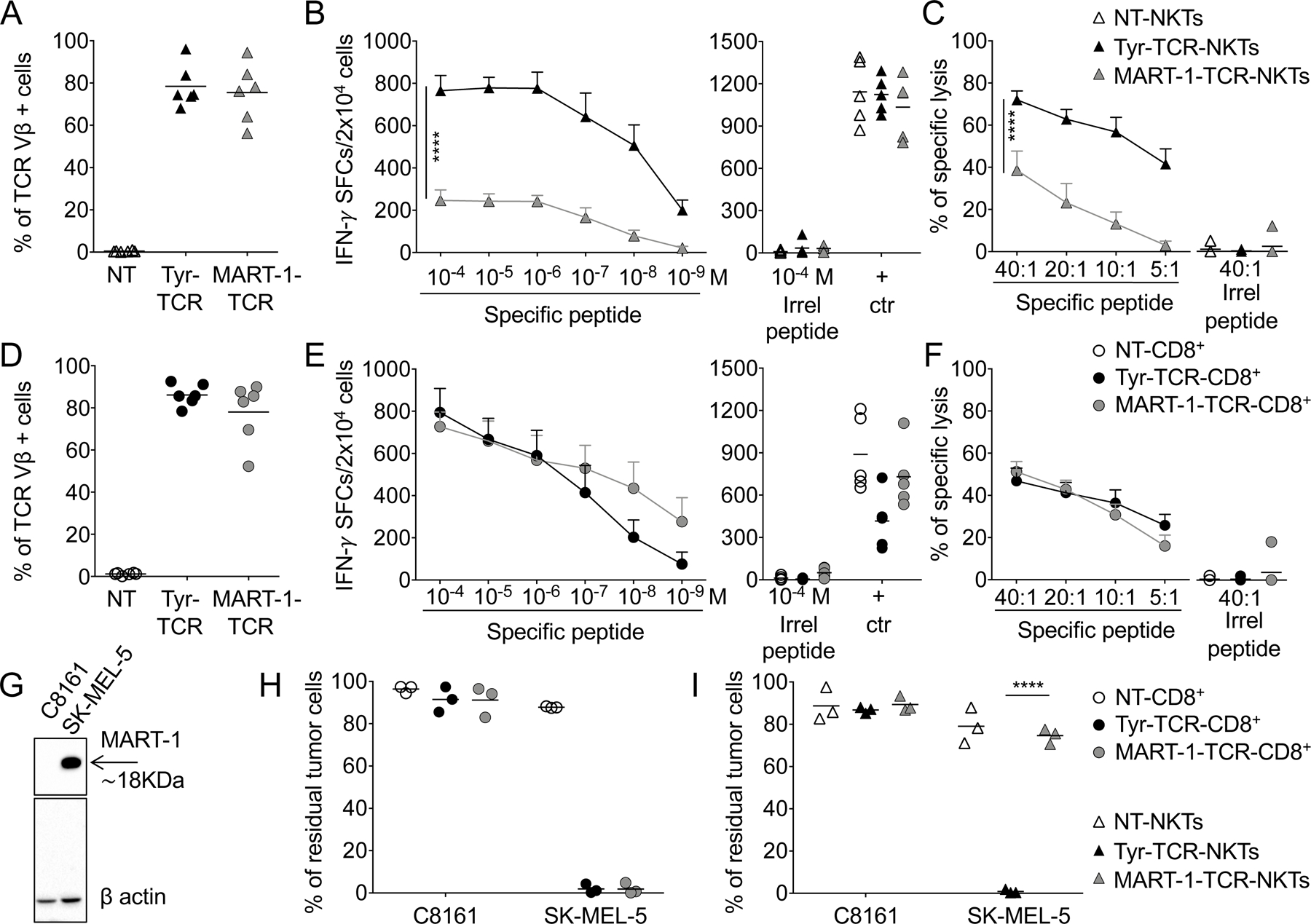
A,D. Expression of Tyr-TCR and MART-1-TCR in (A) NKTs and (D) CD8+ T cells, assessed with an antibody specific for the TCR Vβ12 chain, 3 days after transduction (n=6, mean is shown). NT-NKTs and NT-CD8+ T cells were also used as controls. B,E. TCR functional avidity assessed by IFNγ ELISpot assays for Tyr-TCR and MART-1-TCR in (B) NKTs and (E) CD8+ T cells against T2 cells loaded with decreasing concentrations of the specific peptide (left graphs). T2 cells loaded with an irrelevant peptide and PMA/ionomycin/IL2 (+ctr) were used as negative and positive controls, respectively (right graphs)(n=5, mean and SEM are shown). ****p<0.0001, 2-way ANOVA with Bonferroni correction. C,F. Cytotoxic activity assessed in a 5-hour 51Cr release assay for NT, Tyr-TCR, and MART-1-TCR (C) NKTs and (F) CD8+ T cells against HLA-A2+ PHA blasts pulsed with either the specific peptide or an irrelevant peptide (Irrel., ELAGIGLTV) at the concentration of 100 nM (n=5, mean and SD are shown). ****p<0.0001 by 2-way ANOVA with Bonferroni correction. G. Detection of MART-1 in lysates of melanoma cell lines assessed by Western blot. Staining for β-actin was used as a loading control. H,I. NT, Tyr-TCR, and MART-1-TCR (H) CD8+ T cells and (I) NKTs were cocultured with SK-MEL5 or C8161 (E:T=1:1) for 3 days. Cells were then collected and stained with the CD3 and B7-H3 mAbs to identify NKTs/CD8+ T cells and tumor cells, respectively, by flow cytometry. Quantification of the residual tumor cells in the culture (n=3, mean is shown). ****p<0.0001, paired t test.
Because CD8+ T cells expressing either MART-1-TCR or Tyr-TCR were equally effective, we explored if the functionality of MART-1-TCR NKTs, that were mostly CD4+ or CD4–CD8– cells, could be rescued by overexpressing the CD8α subunit, as previously reported for CD4+ T cells (27). Co-expression of both a MART-1-TCR and human CD8α (>60% CD8α+), this modification failed to enhance the functional avidity of MART-1-TCR-NKTs (Supplementary Fig. S5A–C). Thus, our data suggest that the intrinsic functional avidity of the TCR was critical to functionally redirect NKTs activity via HLA class I–restricted TCRs.
Tyr-TCR NKTs provide antitumor effects in vivo
Having shown that functional avid Tyr-TCR grants the antigen specificity of NKTs in vitro, we used a xenogeneic NSG melanoma mouse model to assess if Tyr-TCR NKTs controlled tumor growth in vivo. NKTs were administered intravenously (i.v.) in mice engrafted subcutaneously (s.c.) with the melanoma cell line SK-MEL-5 (Fig. 6A). Tyr-TCR NKTs exhibited superior control of tumor growth than NT-NKTs (317 mm3±124 mm3 vs. 1121 mm3±216 mm3, respectively at day 64, p<0.0001)(Fig. 6B, Supplementary Fig. S6A). None of the NKT-treated mice developed signs of graft-versus-mouse toxicity. In harvested tumors, we observed a higher number of infiltrating NKTs in mice treated with Tyr-TCR NKTs compared to NT-NKTs (997 NKTs±650 NKTs vs. 351 NKTs±401 NKTs, respectively) and higher detection of IFNγ release (183.5 pg/mL±179.1 pg/mL vs. 12.1 pg/mL±13.7 pg/mL, respectively)(Fig. 6C), suggesting that Tyr-NKTs were functional at the tumor site. In contrast, no difference in the numbers of Tyr-TCR NKTs and NT-NKTs was detected in peripheral blood, liver, and spleen and the expression of the Tyr-TCR was detectable in all the Tyr-TCR-NKTs isolated (Supplementary Fig. S6B–C).
Figure 6. Tyr-TCR-NKTs control the growth of HLA-A2+Tyr+ cells in vivo.
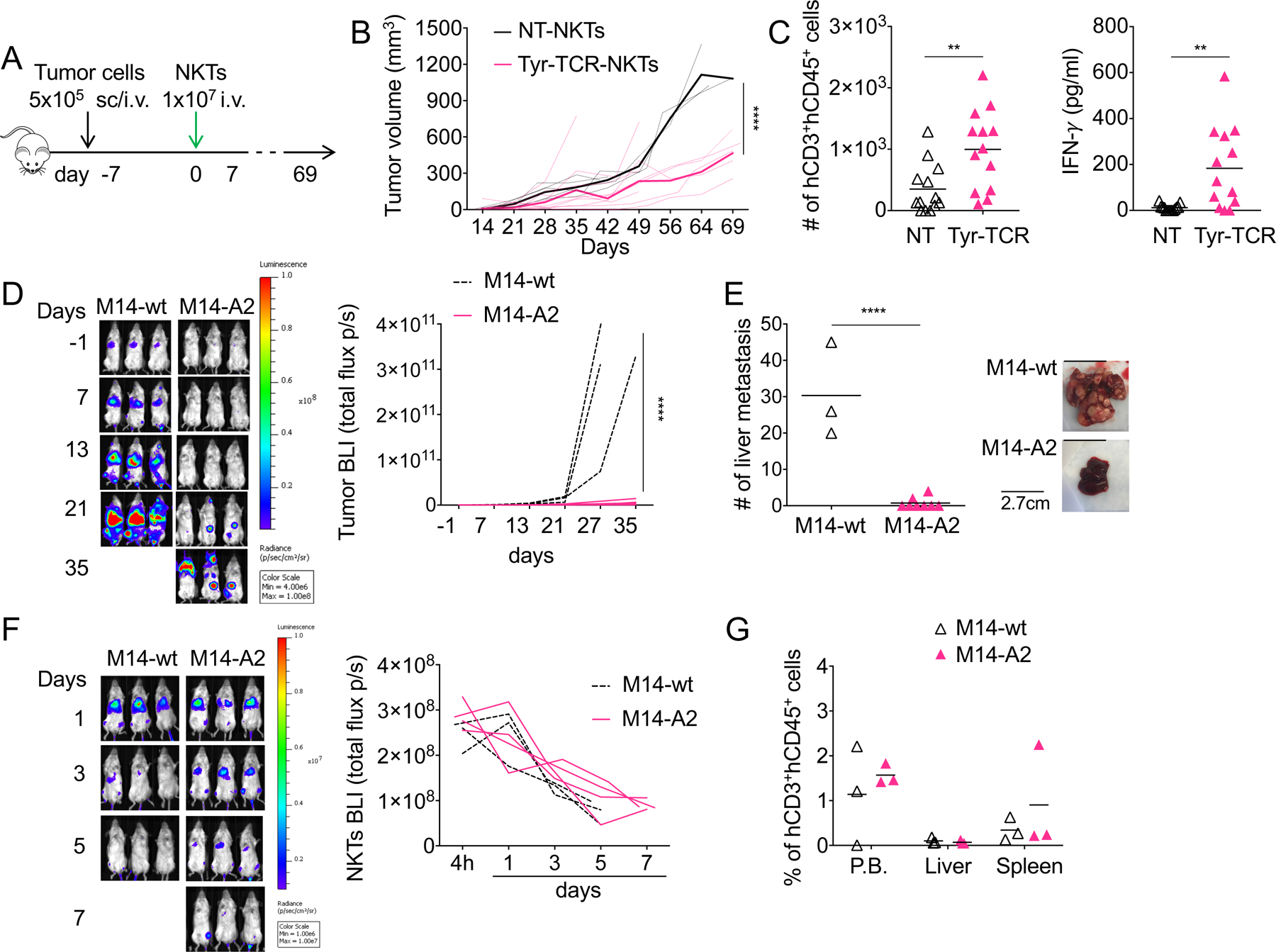
A. Schema of the xenograft tumor model in NSG mice engrafted subcutaneously (s.c.) with SK-MEL-5 cells or intravenously (i.v.) with M14 cells and treated one week later with NKTs. B. Tumor growth in mice engrafted s.c. with SK-MEL-5 tumor cells and treated with NT-NKTs (n=5) or Tyr-TCR NKTs (n=10). Dotted lines represent individual mice, bolded solid lines represent the mean for the group. ****p<0.0001, 2-way ANOVA with Bonferroni correction. C. Quantification of human CD3+CD45+ cells (left) and IFNγ (right) in each tumor harvested from mice engrafted with SK-MEL-5 tumor cells and treated with either NT-NKTs (n=13) or Tyr-TCR NKTs (n=13) (mean is shown). **p=0.0055 and **p=0.0021, respectively, unpaired t test. D. Representative tumor bioluminescence (BLI)(left)(Color scale: Min=4.00e6, Max=1.00e8) and BLI kinetics of all treated mice (right) engrafted intravenously (i.v.) with eGFP-FFLuc-labeled M14-wt (n=3) or M14-A2 (n=8) tumor cells. ****p<0.0001, 2-way ANOVA with Bonferroni correction. E. Mice engrafted with eGFP-FFLuc-labeled M14-wt (n=3) or M14-A2 (n=8) and treated with Tyr-TCR NKTs were euthanized and tumor liver metastases counted (left) (mean is shown). Representative images of the livers (right). ****p<0.0001, unpaired t test. F. Mice were infused i.v. with 1 × 106 M14-wt or M14-A2 cells, and after 7 days, they were injected with 1 × 107 Tyr-TCR NKTs labelled with eGFP-FFluc. Representative Tyr-TCR-NKTs BLI (left))(Color scale: Min=1.00e6, Max=1.00e7), and BLI kinetics of all mice (right) of M14-wt (n=3) or M14-A2 (n=4) tumor-bearing mice. G. Quantification of human CD3+CD45+ cells the in peripheral blood (P.B.), liver, and spleen of M14-wt (n=3) or M14-A2 (n=3) tumor bearing mice treated with Tyr-TCR NKTs (mean is shown).
To further assess in vivo the HLA-restriction of the antitumor responses mediated by Tyr-TCR NKTs, we also generated a metastatic murine model in which NSG mice were injected i.v. with either M14-wt or M14-A2 cells modified to express the enhanced green fluorescent protein (eGFP) and the firefly luciferase gene (FFLuc) (eGFP-FFLuc). Tumor bioluminescence (BLI) was reduced only in mice engrafted with M14-A2 cells and treated with Tyr-TCR NKTs (p<0.0001; Fig. 6D). When euthanized between day 41 and 63, the number of liver tumor metastases in M14-A2 tumor-bearing mice was significantly reduced compared to M14-wt tumor-bearing mice (1±2 vs. 30±13, p<0.0001)(Fig. 6E). In the same mouse model, we also assessed in vivo the persistence of Tyr-TCR NKTs using NKTs labelled with eGFP-FFLuc and unlabeled tumor cells. Tyr-TCR NKT BLI was detectable for 7 days after infusion (Fig. 6F). NKT persistence was also corroborated by phenotypic analyses in the peripheral blood and spleen (1.6%±0.2% and 0.9±1.2% hCD3+hCD45+ cells, respectively) of treated mice (Fig. 6G). To assess if Tyr-TCR NKTs produced superior antitumor control when their persistence was prolonged, we infused multiple doses of Tyr-TCR NKTs and compared the efficacy of Tyr-TCR-NKTs and Tyr-TCR-Ts generated from the same donors (Fig. 7A).Both Tyr-TCR NKTs and Tyr-TCR-Ts, which were detectable in the circulation up to day 40–60 (Fig. 7B), successfully controlled tumor growth (Fig. 7C–D) and produced an improved overall survival compared to NT-NKTs and NT-Ts (p<0.0001)(Fig. 7E). The number of liver tumor metastases was significantly reduced in mice treated with either the Tyr-TCR NKTs or the Tyr-TCR-Ts compared to NT-NKTs and NT-Ts (p<0.0001)( Supplementary Fig. S6D). Tyr-TCR NKTs and Tyr-TCR-Ts were detectable in both the liver and spleen of mice sacrificed between day 40 and 60 (Supplementary Fig. S6E). However, 11 of the 18 mice (61%) receiving Tyr-TCR-Ts developed lethal graft-versus-mouse disease characterized by weight and fur loss and lethargy (Fig. 7F). This graft-versus-mouse disease also confounded the observed long-term tumor control mediated from the Tyr-TCR-Ts. Finally, we used the same in vivo models, but decreased the number of T cells and NKTs injected (5 × 106 cells/injection)(Supplementary Fig.S7A). The antitumor activity of Tyr-TCR NKTs and Tyr-TCR-Ts treated mice was comparable (Supplementary Fig.S7B–D), and the Tyr-TCR-Ts continued to cause a lethal graft-versus-mouse effect (Supplementary Fig. S7E). Thus, our data indicated that TCR-NKTs could control tumor growth in vivo without causing reactivity against murine tissues.
Figure 7. Multiple infusions of Tyr-TCR NKTs control tumor growth, without inducing graft-versus-mouse effects.
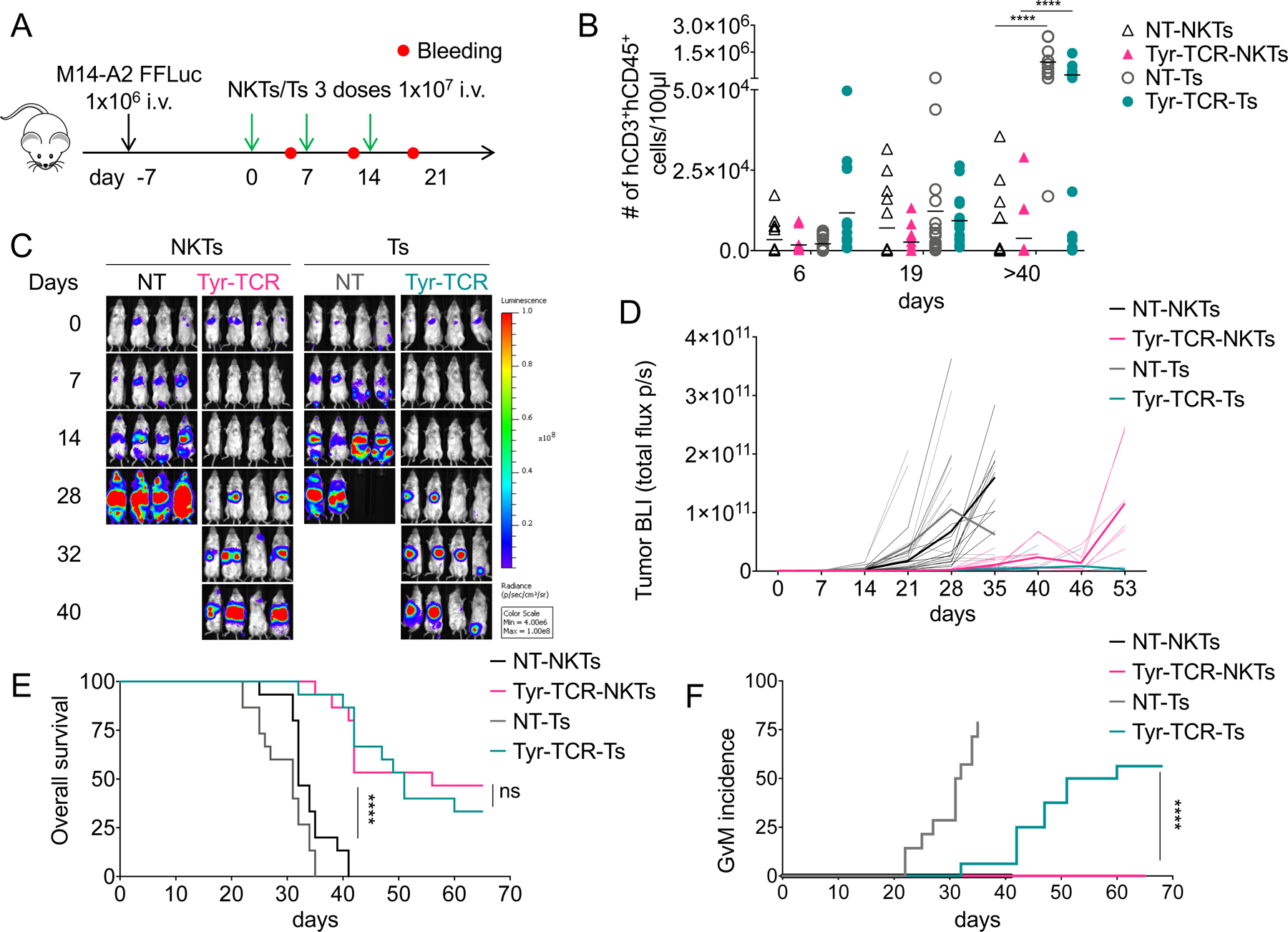
A. Schema of the xenograft tumor model in NSG mice inoculated intravenously (i.v.) with M14 cells and treated with 3 injections of NKTs or T cells (1×107 cells each). B. Quantification of human CD3+CD45+ cells in the peripheral blood collected 6–7 days after each NKT/T-cell infusion and at sacrifice (day>40) (mean is shown). ****p<0.0001, 2-way ANOVA with Bonferroni correction. C,D. Representative (C) tumor BLI (Color scale: Min=4.00e6, Max=1.00e8) and (D) BLI kinetics of all treated mice in four independent experiments engrafted with the eGFP-FFluc-labeled M14-A2 cells and treated with NT-NKTs (n=15), Tyr-TCR-NKTs (n=15), NT-Ts (n=15), or Tyr-TCR-Ts (n=15). Dotted lines represent individual mice, bolded solid lines represent the mean for the group. NT-NKTs vs. Tyr-TCR NKTs p<0.0001 at day 28 and 35, 2-way ANOVA with Bonferroni correction. Tyr-TCR-NKTs vs. Tyr-TCR Ts p<0.0001 at day 53, 2-way ANOVA with Bonferroni correction. E. Kaplan Meier survival curve (n=15 mice/group); ****p<0.0001, log-rank test. F. Graft-versus-mouse incidence curve (n=15 mice/group); ****p<0.0001, log-rank test.
Discussion
We showed that the antigen specificity of human NKTs could be redirected toward tumor antigens by inserting an ectopic TCR. Our study critically outlined the advantages of using this platform for the expression of ectopic TCRs, namely how the TCR could outcompete the iTCR for the cell surface expression without the need for additional manipulation to knockdown endogenous TCR chains as required in T cells.
Adoptive transfer of polyclonal T cells expressing high-affinity TCRs have shown promising clinical responses (28). However, current issues related to ectopic TCR expression in T cells suggest that exploring alternative platforms for TCR expression may be beneficial to further enhance clinical activity and safety of TCR-engineered cells. We previously demonstrated that NKTs can be engineered to express a chimeric antigen receptor for redirected tumor target capabilities (29). Jiang et al. previously showed that an HLA class I-restricted TCR targeting an epitope of the 38-kDa protein of the Mycobacterium tuberculosis can be expressed by NKTs and react against DCs pulsed with the 38-kDa protein (30). However, redirecting NKTs for the targeting of tumor cells via HLA class I restriction has remained so far unexplored.
Here, we have shown that NKTs expressing an ectopic HLA class I–restricted TCR can be generated in less than three weeks of culture, and in sufficient numbers for the clinical translation. We showed that the ectopic TCR can displace the iTCR on a proportion of NKTs by outcompeting for binding to the CD3 complex. This phenomenon occurred when the mRNA of the ectopic TCR chains was overexpressed compared to the endogenous iTCR, with higher transcript numbers dictating which receptor was expressed on the cell surface of NKTs. Interestingly, Sommermeyer et al. previously showed that a transgenic TCR can outcompete the endogenous TCR in CMV-specific T-cell clones (31). Our study, thus, underlines that the restricted TCR repertoire of polyclonal NTKs, compared to the broad TCR repertoire of polyclonal T lymphocytes, overcomes the hurdle of gene editing required in polyclonal T cells to knockdown the endogenous TCRs by granting the displacement of the iTCR.
Independently from the expression of the iTCR, TCR-NKTs proved to specifically recognize the peptide/HLA-A2 complex and exhibited a potent antitumor effect against HLA-A2+ tumor cells expressing the targeted antigen. TCR-NKTs showed fast killing of the target cells and activation as demonstrated by the production of IFNγ. TCR-NKTs maintained their specificity in vivo and mediated tumor regression leading to improved overall survival. In particular, TCR-NKTs could control tumor metastasis within the liver. It has been reported that murine NKTs can accumulate in the liver of mice bearing hepatocellular carcinomas and inhibit the tumor growth in both primary and metastatic liver models (32). It remains to be demonstrated how human NKTs in NSG mice causes a robust antitumor effect in liver metastases. Our antitumor effects were significant, considering that, because NKTs are well-known for orchestrating immune responses by inducing maturation of DCs, activation of T and B cells and transactivation of NK cells (33–35), our model likely underestimated the real potentials of TCR-NKTs.
Finally, one further advantage of using NKTs rather than polyclonal T cells for TCR expression lies in the reduced risk of GvHD when both ectopic and native TCRs are retained by transduced cells. Preclinical and clinical evidence suggests that NKTs may act as negative regulators of GvHD, making them relevant in the allogeneic context (36–39). Corroborating these clinical suggestions are previous observations that CAR-engineered human NKTs do not cause graft-versus-mouse – a surrogate of GvHD – in NSG mice (29, 37–40). Here, we ed that TCR-NKTs could not only be equally effective as TCR-expressing T cells in controlling tumor growth, but that, unlike T cells, they did not cause graft-versus-mouse in NSG mice. Thus, the manipulations needed for appropriate expression of a transgenic TCR and abrogation of the endogenous TCR in T cells appear dispensable in NKT cells. Overall, we showed that using NKTs as a cell platform for TCR expression in cancer immunotherapy is feasible, and provide significant advantages compared to T cells. Based on our current analysis, the only requirement for effective antitumor activity via HLA-restricted TCR by NKTs is the need for expressing TCRs characterized by high functional avidity.
Supplementary Material
Acknowledgments
The imaging core and UNC Flow Cytometry core facilities are supported in part by an NCI cancer core grant, P30-CA016086–40. This work was supported in part by Cell Medica. BS is supported in part by R01HL114564 and GD is supported in part by R01-CA193140.
Footnotes
Conflict of Interest Statement: G. Dotti reports receiving commercial research grants from Cell Medica and Bellicum Pharmaceutical and is a consultant/advisory board member for MolMed. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cieri N, Mastaglio S, Oliveira G, Casucci M, Bondanza A, and Bonini C. Adoptive immunotherapy with genetically modified lymphocytes in allogeneic stem cell transplantation. Immunol Rev. 2014;257(1):165–80. [DOI] [PubMed] [Google Scholar]
- 3.Morris EC, and Stauss HJ. Optimizing T-cell receptor gene therapy for hematologic malignancies. Blood. 2016;127(26):3305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol. 2017;35(29):3322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmadi M, King JW, Xue SA, Voisine C, Holler A, Wright GP, et al. CD3 limits the efficacy of TCR gene therapy in vivo. Blood. 2011;118(13):3528–37. [DOI] [PubMed] [Google Scholar]
- 7.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16(5):565–70, 1p following 70. [DOI] [PubMed] [Google Scholar]
- 8.Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood. 2017;130(5):606–18. [DOI] [PubMed] [Google Scholar]
- 9.Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. 2008;180(9):6116–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, and Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67(8):3898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, and Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66(17):8878–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sommermeyer D, and Uckert W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J Immunol. 2010;184(11):6223–31. [DOI] [PubMed] [Google Scholar]
- 13.Bunse M, Bendle GM, Linnemann C, Bies L, Schulz S, Schumacher TN, et al. RNAi-mediated TCR knockdown prevents autoimmunity in mice caused by mixed TCR dimers following TCR gene transfer. Mol Ther. 2014;22(11):1983–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh N, Shi J, June CH, and Ruella M. Genome-Editing Technologies in Adoptive T Cell Immunotherapy for Cancer. Curr Hematol Malig Rep. 2017;12(6):522–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brennan PJ, Brigl M, and Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol. 2013;13(2):101–17. [DOI] [PubMed] [Google Scholar]
- 16.Kronenberg M When less is more: T lymphocyte populations with restricted antigen receptor diversity. J Immunol. 2014;193(3):975–6. [DOI] [PubMed] [Google Scholar]
- 17.Quintarelli C, Dotti G, De Angelis B, Hoyos V, Mims M, Luciano L, et al. Cytotoxic T lymphocytes directed to the preferentially expressed antigen of melanoma (PRAME) target chronic myeloid leukemia. Blood. 2008;112(5):1876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frankel TL, Burns WR, Peng PD, Yu Z, Chinnasamy D, Wargo JA, et al. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J Immunol. 2010;184(11):5988–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossig C, Bollard CM, Nuchtern JG, Rooney CM, and Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99(6):2009–16. [DOI] [PubMed] [Google Scholar]
- 21.Arber C, Feng X, Abhyankar H, Romero E, Wu MF, Heslop HE, et al. Survivin-specific T cell receptor targets tumor but not T cells. J Clin Invest. 2015;125(1):157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savoldo B, Goss J, Liu Z, Huls MH, Doster S, Gee AP, et al. Generation of autologous Epstein-Barr virus-specific cytotoxic T cells for adoptive immunotherapy in solid organ transplant recipients. Transplantation. 2001;72(6):1078–86. [DOI] [PubMed] [Google Scholar]
- 25.Nazarov VI, Pogorelyy MV, Komech EA, Zvyagin IV, Bolotin DA, Shugay M, et al. tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinformatics. 2015;16:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hiasa A, Hirayama M, Nishikawa H, Kitano S, Nukaya I, Yu SS, et al. Long-term phenotypic, functional and genetic stability of cancer-specific T-cell receptor (TCR) alphabeta genes transduced to CD8+ T cells. Gene Ther. 2008;15(9):695–9. [DOI] [PubMed] [Google Scholar]
- 27.Willemsen RA, Sebestyen Z, Ronteltap C, Berrevoets C, Drexhage J, and Debets R. CD8 alpha coreceptor to improve TCR gene transfer to treat melanoma: down-regulation of tumor-specific production of IL-4, IL-5, and IL-10. J Immunol. 2006;177(2):991–8. [DOI] [PubMed] [Google Scholar]
- 28.Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21(5):1019–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. 2014;124(18):2824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang ZM, Luo W, Wen Q, Liu SD, Hao PP, Zhou CY, et al. Development of genetically engineered iNKT cells expressing TCRs specific for the M. tuberculosis 38-kDa antigen. J Transl Med. 2015;13:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sommermeyer D, Neudorfer J, Weinhold M, Leisegang M, Engels B, Noessner E, et al. Designer T cells by T cell receptor replacement. Eur J Immunol. 2006;36(11):3052–9. [DOI] [PubMed] [Google Scholar]
- 32.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujii S, Shimizu K, Smith C, Bonifaz L, and Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198(2):267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171(10):5140–7. [DOI] [PubMed] [Google Scholar]
- 35.Barral P, Eckl-Dorna J, Harwood NE, De Santo C, Salio M, Illarionov P, et al. B cell receptor-mediated uptake of CD1d-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc Natl Acad Sci U S A. 2008;105(24):8345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leveson-Gower DB, Olson JA, Sega EI, Luong RH, Baker J, Zeiser R, et al. Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism. Blood. 2011;117(11):3220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du J, Paz K, Thangavelu G, Schneidawind D, Baker J, Flynn R, et al. Invariant natural killer T cells ameliorate murine chronic GVHD by expanding donor regulatory T cells. Blood. 2017;129(23):3121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaidos A, Patterson S, Szydlo R, Chaudhry MS, Dazzi F, Kanfer E, et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood. 2012;119(21):5030–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malard F, Labopin M, Chevallier P, Guillaume T, Duquesne A, Rialland F, et al. Larger number of invariant natural killer T cells in PBSC allografts correlates with improved GVHD-free and progression-free survival. Blood. 2016;127(14):1828–35. [DOI] [PubMed] [Google Scholar]
- 40.Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell. 2018;34(4):596–610 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.