

Abstract

Ylide-substituted phosphines have been shown to be excellent ligands for C–N coupling reactions under mild reaction conditions. Here we report studies on the impact of the steric demand of the substituent in the ylide-backbone on the catalytic activity. Two new YPhos ligands with bulky ortho-tolyl (pinkYPhos) and mesityl (mesYPhos) substituents were synthesized, which are slightly more sterically demanding than their phenyl analogue but considerably less flexible. This change in the ligand design leads to higher selectivities and yields in the arylation of small primary amines compared to previously reported YPhos ligands. Even MeNH2 and EtNH2 could be coupled at room temperature with a series of aryl chlorides in high yields.
Introduction
Transition metal catalyzed cross-coupling reactions have developed into a powerful tool in modern synthetic chemistry, allowing the synthesis of complex molecules under relatively mild reaction conditions from usually readily available starting materials. The C—N coupling reaction (Buchwald–Hartwig amination) of aryl electrophiles with amines is one of the most important methods due to the ubiquity of amine moieties in many pharmaceuticals, natural products, agrochemicals, and fine chemicals used in materials chemistry and beyond.1 The Buchwald–Hartwig amination has experienced remarkable advances in the last 25 years, which are mainly connected with the development of new ancillary ligands. Electron-rich and sterically bulky monophosphines2 as well as N-heterocyclic carbenes3 have been found to be particularly suited in that chemistry to generate and stabilize low-coordinated palladium species that readily undergo oxidative addition of C—X bonds, including the cheaper but more challenging aryl chlorides.
Small unbranched primary alkyl amines such as methyl or ethylamine are some of the most challenging substrates in C—N coupling reactions. This is due to two inherent challenges connected with these substrates. Due to their small size, selectivity between the mono and diarylation product is often problematic.; thus, very sterically hindered ligands are required to allow for selective monoarylation. Alkyl amines are also prone to β-hydride elimination, which might lead to the formation of side products and thus requires a special ligand design to prevent the intramolecular C—H activation step. Because of these limitations, comparably few synthetic protocols for the coupling of these amines have been reported in the past years. In the case of palladium-catalyzed reactions, the first efficient protocol for methylamine coupling with aryl chlorides was described by Buchwald and co-workers in 2008 using palladium precatalysts with the biarylphosphine BrettPhos, A (Figure 1).4 Since then, a number of other ligands have found to be highly efficient in this transformation. For example, Hartwig described the use of CyPF-tBu (B) both for ethyl and methylamine with a series of aryl bromides and chlorides.5 Stradiotto and co-workers reported on the use of the DalPhos family of ligands (e.g., Me-DalPhos (C)6 and Mor-DalPhos (D)7) as well as a phosphine-functionalized NHC ligand8 as versatile ligands in a series of coupling reactions, including not only methylamine but also secondary amines. It must be noted that C—N coupling reactions including primary amines have recently also been reported with phosphine-ligated nickel complexes,9 including couplings under mild conditions10 as well as copper-catalyzed protocols; however, these require harsh reaction conditions or only allow for the amination of activated aryl electrophiles such as aryl iodides.11 Despite these advances made in past years, most of the catalysts still require higher reaction temperatures for the amination of aryl chlorides or make use of expensive ligands.
Figure 1.
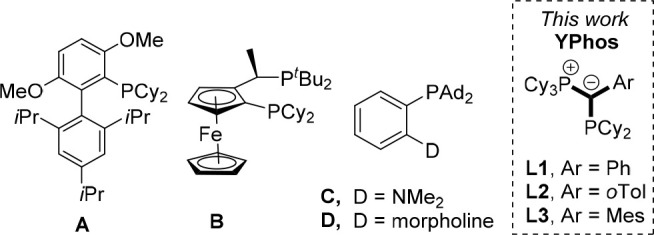
Phosphines used for the selective monoarylation of small primary alkylamines.
Recently, our group reported on the use of ylide-substituted phosphines (YPhos) as highly efficient ligands in gold catalysis12 as well as Pd-catalyzed C—N and C—C coupling reactions.13 YPhos ligands are, in general, electron-rich phosphines and easy to synthesise in few steps from cheap starting materials. Furthermore, the modification of the ylide backbone allowed for an additional tuning of the electronic and steric properties and hence of the catalytic activity of their metal complexes. For example, the replacement of the methyl group in the ligand Ph3PC(Me)PCy2 with a sulfonyl or cyano group led to an increase of the catalytic activity in gold catalyzed hydroaminations by orders of magnitude, thus allowing for catalysis with parts per million-level catalyst loadings.12 In palladium catalysis, the analogous PCy3-substituted YPhos ligand joYPhos (L1) with a phenyl group in the ylide-backbone proved to be highly effective in Buchwald–Hartwig aminations of aryl chlorides at room temperature, allowing for turnover frequencies greater than 10.000 h–1 with improved selectivities in comparison to its methyl-substituted analogue.14 However, diarylation was observed as a side product with small primary amines. To address this limitation of L1, we became interested in the impact of the steric demand of the backbone substituent on the selectivity in mono vs diarylation reactions. Therefore, we addressed the synthesis of the ortho-tolyl- (pinkYPhos, L2) and mesityl- (mesYPhos, L3) substituted YPhos ligands. Here, we show that this modification indeed leads to a coherent structure–selectivity relationship and enables the selective monoarylation of methyl and ethyl amine with aryl chlorides at room temperature.
Results and Discussion
Ligand Synthesis and Properties
The synthesis of the ligands L2 and L3 was attempted via the same protocol as used for the synthesis of ligand L1.14 For the ortho-tolyl ligand CyYoTolPCy2, the formation of the phosphonium salt 2a and subsequent deprotonation to L2 was revealed to be facile and allowed the isolation of the ligand as a colorless solid in 84% yield from 1a (Scheme 1). In contrast, the preparation of the mesityl ligand CyYMesPCy2 (L3) failed under the same reaction conditions. No complete conversion to the α-phosphino phosphonium salt 2b was observed when treating the phosphonium iodide 1b with butyllithium and Cy2PCl. We hypothesized that this might be due to an equilibrium between 1b and 2b as a consequence of the competing attack of the chloride at 2b. This results in the reformation of the starting material and hence mixtures of 2b and 1b. To prevent the attack of the halide, the chloride anion was replaced by the addition of NaBF4. Thus, α-phosphino phosphonium salt 2b could be isolated in 62% yield as a colorless solid, which was stable in solution. Due to the steric bulk of the mesityl group, deprotonation also proved to be difficult but was accomplished using potassium tert-butoxide at low temperatures. At higher temperatures, PCy3 elimination and the formation of the C—C coupled diphosphine (Mes(PCy2)(H)C)2 were observed (Supporting Information). Nonetheless, L3 could be isolated as a colorless solid and in a moderate 41% yield. The YPhos ligands L2 and L3 are characterized by two doublets in the 31P{1H} NMR spectra at −0.5 and 19.3 ppm (2JPP = 138.2 Hz) for L2 and 6.1 and 13.8 ppm (2JPP = 145.3 Hz) for L3.
Scheme 1. Preparation of Ligands L2 and L3.

Single crystals of both ligands could be obtained by the slow evaporation of their saturated hexane solutions (Figure 2). The molecular structures of L2 and L3 are similar to that reported for L1.14 All ligands show similar bond lengths in the central P—C—P linkage and similar P—C—P angles between 112.5(1)° (L2) and 113.7(1)° (L1). This is rather surprising since the YPhos ligands in general were found to sensitively respond to steric pressure by changes in the P—C—P angle. For example, changes of more than 15° in the P—C—P angle were observed for the methyl-substituted ligand (keYPhos) in different Pd complexes depending on the demand of other coligands at the metal.15 The similarity of the structures of L1–L3, however, is probably the result of a change in the orientation of the aryl substituent relative to the P—C—P plane. While the corresponding P2—C1—C2—C3 angle in L1 amounts to 75.3(1)°, it increases to 89.1(1)° in L3. Thus, the latter features an almost ideal perpendicular arrangement of the mesityl group relative to the P—C—P linkage. Furthermore, the C1—C2 distances to the aryl substituents are longer in L2 and L3 (approximately 1.510 Å) in comparison to that in L1 (1.489(2) Å), thus also slightly reducing the steric pressure.
Figure 2.

Molecular structures of CyYoTolPCy2 (L2) and CyYMesPCy2 (L3). Crystallographic details are provided in the Supporting Information.
The steric and electronic properties of L2 and L3 were measured by determinations of the Tolman electronic parameter (TEP) and the buried volume (%Vbur). The TEP value was derived from the CO stretching frequency in the corresponding L·Rh(acac)CO complexes in DCM. Crystals of [L2·Rh(CO)acac] were grown by cooling of a DCM solution of the complex to −30 °C and confirmed the formation of the rhodium complex for the tolyl ligand L2 (Figure 3). With TEPs of 2048.0 (L2) and 2048.4 cm–1 (L3), both ligands are slightly stronger donors than L1 (2050.1 cm–1)13a and similarly stronger than the commonly used N-heterocyclic carbenes IMes (TEP = 2050.7 cm–1) and IPr (TEP = 2051.5 cm–1).16
Figure 3.

Molecular structure of [L2·Rh(CO)acac], L2·AuCl, and L3·AuCl. Crystallographic details are provided in the Supporting Information.
To determine %Vbur, the corresponding L·AuCl complexes were prepared from the free ligands and (THT)AuCl (THT = tetrahydrothiophene) and isolated as colorless solids in moderate yields of approximately 55%. Both crystals were grown by diffusion of pentane into a THF solution of the gold complex. The crystal structure (Figure 3) of YoTolPCy2·AuCl yielded a buried volume of %Vbur = 49.4% for L2, while a slightly higher value of %Vbur = 50.7% was found for L3. Thus, both ligands cover approximately half the sphere around a metal center and are thus more sterically demanding than their phenyl analogue L1, which exhibits a buried volume of 47.9%. In the gold complex, L3 again shows an ideal perpendicular arrangement of the mesityl substituent relative to the P—C—P moiety, thus indicating an ideal protection of the ylidic carbon atom by the two ortho-methyl substituents. Interestingly, the tolyl ligand shows a disorder in the molecular structure of the free ligand as well as in the gold complex, which concerns the geometry around the ylidic carbon atom C1. While a planar geometry around C1 was found in all structures of the YPhos ligands and their metal complexes, L2 shows a slightly pyramidalized carbon atom in both crystal structures, with a sum of angles around C1 of approximately 355°. The pyramidalization always results in an opening of the pocket between the cyclohexyl groups to accommodate the ortho-methyl substituent. This flexibility is prevented in the mesityl ligand.
Pd-Catalyzed C–N Coupling of Small Alkyl Amines
With the successful synthesis of the ligands, we turned our attention toward the impact of the backbone substituent on the efficiency of the ligands in Pd-catalyzed amination reactions. We assumed that the formation of the active species with Pd2(dba)3 might be slow, particularly with the bulky mesityl ligand. Thus, at first the formation of the L·Pd(dba) complex was investigated to get an estimation of the time required for the catalyst preformation. To this end, the reaction of Pd2(dba)3 with an equivalent amount of ligand was followed by 31P NMR spectroscopy. Both dba complexes exhibit distinct NMR features, giving rise to two doublets in the 31P{1H} NMR spectra with coupling constants of approximately 90 Hz. Reaction monitoring revealed that L2 requires only 1 h reaction time to completely convert into the L·Pd(dba) complex, while 16 h were needed for L3 (Figures S8 and S9).
With this information in hand, the catalytic ability of the ligands was tested. We focused on the amination of aryl chlorides with small alkyl amines, which are usually difficult to selectively monoarylate. We selected the coupling of p-tolyl chloride with the primary amines MeNH2, EtNH2, nBuNH2, BnNH2 (Bn = benzyl), iPrNH2, and tBuNH2 as a test protocol. We also included three secondary amines (Et2NH, piperidine, and N-methylaniline) to examine whether these substrates can also be coupled. The reactions were conducted at room temperature with 0.5 mol % ligand and 0.25 mol % Pd2dba3·dba. The results obtained after 1 h of reaction time with L2 and L3 are given in Figure 4. Longer reaction times did not lead to a significant change of the obtained yields. The activity of pinkyPhos and mesYPhos was compared with joYPhos (L1) as well as the PtBu2 ligand trYPhos (L4) to gain insights into the structure–selectivity relationships. The comparison shows clear differences in the catalytic activity depending on the backbone substituent. While the smaller phenyl-substituted joYPhos (L1) is the most efficient ligand for secondary amines, it is less efficient for primary amines. In contrast, the ortho-tolyl and especially the mesityl-substituted ligands L2 and L3 are very efficient for the coupling of primary amines, with L3 giving superior results. To our delight, methylamine and ethylamine, which are particularly difficult substrates, could also be selectively monoarylated. Likewise, nBuNH2, BnNH2, and iPrNH2 were all fully converted into the corresponding aniline derivatives within only 1 h of reaction time. However, tert-butyl amine seems to be the limit in steric demand of primary amines and could not be coupled under these reaction conditions. The high selectivity for the monoarylation of small primary amines with L2 and L3 is reflected in the low conversions observed for secondary amines. Here, L3 led to considerably lower yields than joYPhos.
Figure 4.

Comparison of the catalytic activity of L1–L4. Reaction conditions are as follows: 0.5 mol % catalyst, RT, 1 h, and aryl chloride/amine 1:1.1. The yield was determined by GC FID analysis with tetradecane as an internal standard.
The results clearly demonstrate that the steric bulk of L2 and L3 is necessary to allow the selective monoarylation, particularly with MeNH2. Here, the smaller L1 delivers considerable amounts of the diarylation product (>10%). However, it is not only the steric bulk of the ligand that is important. This becomes clear from the fact that the tert-butyl ligand L4 (%Vbur = 51.3%), which is of similar size to L3, gives lower yields. This can be explained by the higher reactivity and lower stability of the L4-based palladium complexes, which were already observed in case of the α-arylation of ketones.13b In contrast to the methyl group in the backbone of L4, the ortho-tolyl and mesityl groups impart steric bulk but also the protection of the carbanionic center, which stabilizes the catalytically active species and thus hampers the decomposition of the catalyst.
Nonetheless, it is remarkable that a simple modification of the ligand backbone from phenyl to ortho-tolyl and mesityl leads to such an impact on the selectivity of the catalysts toward different substrates. Presumably, this selectivity difference is not only the result of the different steric bulk of the ligands—note that the %Vbur values of the ligands L1–L3 are within only 4%—but also results from differences in the flexibility of the ligands. Thus, the larger substituents in the backbone prevent large changes in the P—C—P angles, which are necessary to move the PCy3 moiety away from the metal to open the coordination sphere around the metal for larger substrates. While this flexibility is beneficial for fast catalysis, it leads to lower selectivities. Due to these structural features, joYPhos (L1) seems to be the ideal ligand for secondary amines, while mesYPhos (L3) is best for small unhindered primary amines.
Motivated by the excellent activity of ligand L3 for the coupling of small unhindered primary amines at room temperature, we tested the isolation of these compounds as well as a broader substrate scope. We were pleased to see that amines 5aa to 5ae could be isolated in good to excellent yields (Figure 5). Since methylamine and ethylamine are difficult substrates for which only a limited number of catalysts exist, we further focused on these substrates. Aryl chlorides with electron-withdrawing as well as electron-donating substituents could be coupled in good to high yields. The same holds true for somewhat more sterically demanding substrates with ortho-substituents (5ba, 5bb, and 5ea). Additionally, 2-chloropyridine could be successfully converted to the corresponding methyl- or ethylamines, although lower yields were observed with 0.5 mol % catalyst loading.
Figure 5.

Substrate scope of the C—N coupling of primary amines with L3. Reaction conditions are as follows: 0.5 mol % catalyst, RT, 90 min, aryl chloride/amine 1:1.1, isolated yields. X-ray crystallographic data for 5ea can be found in the Supporting Information. aGC yield. b1 mol % catalyst was used.
Conclusion
In conclusion, we reported on the preparation of two new YPhos ligands with a bulky o-tolyl (pinkYPhos) and mesityl substituent (mesYPhos) in the ylide backbone. This modification led to a slight increase of the steric demand and a more rigid ligand structure compared to the joYPhos ligand with a phenyl group in the backbone. A comparison of the activity of the different YPhos ligands in the C—N coupling of aryl chlorides with different primary and secondary amines revealed that the increased bulk and lower flexibility of the ligand structures allow for higher selectivities in the coupling of unhindered substrates. Particularly, mesYPhos gave high yields for the monoarylation of methyl- and ethylamine at room temperature. These results demonstrate that the backbone substituent in ylide-substituted phosphines not only controls the donor properties of these ligands but also provides a further handle to adjust the steric demand and particularly the flexibility of the ligand.
Experimental Section
General Methods
All experiments were carried out under a dry and oxygen-free argon atmosphere using standard Schlenk techniques. Involved solvents were dried using an MBraun SPS-800 (THF, DCM, toluene, acetonitrile, diethyl ether, and pentane) or in accordance with standard procedures. Deuterated solvents were stored over molecular sieves in an argon-filled glovebox. ClPCy2 was prepared according to published procedures.17 Pd2(dba)3·dba and (THT)AuCl were donated by UMICORE AG & Co.18 All other reagents were purchased from Sigma-Aldrich, ABCR, Rockwood Lithium, or Acros Organics and used without further purification. NMR spectra were recorded on Avance-400 spectrometers at 25 °C unless stated otherwise. All chemical shift values are in ppm in regard to the δ scale. All spin–spin coupling constants (J) are printed in Hertz (Hz). To display multiplicities and signal forms correctly, the following abbreviations were used: s = singlet, d = doublet, t = triplet, q = quartet, hept = heptet, m = multiplet, dd = doublet of doublet, ddd = doublet of doublet of doublet, and br = broad signal. Signal assignments were supported by HSQC (1H/13C) and HMBC (1H/13C, 1H/31P) correlation experiments for all ligands, their precursors, and their metal complexes. The isolated cross-coupling products were analyzed according to their shifts. Cyclohexyl groups were assigned according to the scheme below. Elemental analyses were performed on an Elementar vario MICRO-cube elemental analyzer. IR-Spectra were recorded on a Thermo Nicolet iS5 FT-IR spectrometer in the transmission mode with a Specac “Omni-cell” with KBr plates and a 0.1 mm spacer or with an ATR module at 22 °C. Column chromatography was performed on a Reveleris X2 (BÜCHI) flash chromatography system using Reveleris packed columns. Melting points were collected on a Stuart SMP 30 with a heat-up speed of 2 °C min–1.

Synthesis of Phosphonium Salt 1a
4.7 mL (5.0 g, 35.6 mmol, 1.0 equiv) of 1-(chloromethyl)-2-methylbenzene and 11.0 g (39.1 mmol, 1.1 equiv) of tricyclohexylphosphine were suspended in 60 mL of dry toluene and stirred at room temperature overnight. The precipitated solid was filtered through a Schlenk frit and washed two times with 7 mL of dry toluene. The solid was dried for 5 h, giving the product as a colorless solid (13.8 g, 32.8 mmol, 92%): 1H NMR (400 MHz, CD2Cl2) δ 7.36–7.28 (m, 1H, CH,oTol), 7.28–7.18 (m, 3H, CH,oTol), 4.14 (d, 2JHP = 14.3, 2H, P—CH2—Tol), 2.92–2.69 (m, 3H, CH,Cy,H1), 2.50 (d, J = 1.4 Hz, 3H, CH3), 2.05–1.93 (m, 6H, CH2,Cy,H2), 1.93–1.83 (m, 6H, CH2,Cy,H3), 1.82–1.72 (m, 3H, CH2,Cy,H4), 1.63–1.45 (m, 6H, CH2,Cy,H2), 1.46–1.35 (m, 6H, CH2,Cy,H3), 1.33–1.19 (m, 3H, CH2,Cy,H4); 31P{1H} NMR (162 MHz, CD2Cl2) δ 30.8. Further spectroscopic and physical properties match the literature report.19
Synthesis of 2-(Iodomethyl)-1,3,5-trimethylbenzene
Here we report an alternative synthesis route. A two-necked flask with 15 g (0.10 mol, 1.0 equiv) of 2,4,6-trimethylbenzyl alcohol was equipped with a dropping funnel. The solid was dissolved in 100 mL of DCM, and 8.0 mL (13 g, 0.11 mol, 1.1 equiv) of thionyl chloride was filled into the dropping funnel. The reagent was added dropwise under vigorous stirring, and the suspension was stirred for an additional hour. The reaction mixture was quenched with 50 mL of water, and the organic phase was extracted three times with 50 mL of water in a separating funnel. The organic phase was dried over magnesium sulfate, and the solvent was removed in vacuo. The successful formation of the intermediate 2-(chloromethyl)-1,3,5-trimethylbenzene was confirmed by NMR spectroscopy. The intermediate product (15.1 g, 0.09 mol, 1.0 equiv) and 14.8 mg (0.10 mol, 1.1 equiv) of sodium iodide were dissolved in 100 mL of acetonitrile, and the solution was refluxed with an oil bath overnight. The solid was filtered over a filter paper, and the solvent was removed in vacuo. The solid was dissolved in 100 mL of ethyl acetate and extracted three times with 50 mL of water in a separating funnel. The aqueous phase was extracted one more time with 100 mL of ethyl acetate. The organic phases were combined, and the solvent was removed at reduced pressure to yield the product as a light yellow solid (20.1 g, 0.8 mol, 78%): 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 2H, CH,Mes,meta), 4.46 (s, 2H, I—CH2—Mes), 2.32 (s, 6H, CH3,Mes,ortho), 2.26 (s, 3H, CH3,Mes,para). Further spectroscopic and physical properties match with the literature report.20
Synthesis of Phosphonium salt 1b
9.77 g (37.6 mmol, 1.05 equiv) of 2-(iodomethyl)-1,3,5-trimethylbenzene and 10 g (35.6 mmol, 1.0 equiv) of tricyclohexylphosphine were suspended in 120 mL of dry toluene and stirred at room temperature overnight. The precipitated solid was filtered through a Schlenk frit and washed two times with 20 mL of dry toluene. The solid was dried for 5 h, giving the product as a colorless solid (19.3 g, 35.6 mmol, 99%): 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 2H, CH,Mes,meta), 3.85 (d, 2JHP = 12.7 Hz, 2H, P—CH2—Mes), 2.73–2.49 (m, 3H, CH,Cy,H1), 2.39 (s, 6H, CH3,Mes,ortho), 2.24 (s, 3H, CH3,Mes,para), 1.93–1.82 (m, 12H, CH2,Cy,H2+H3), 1.82–1.69 (m, 3H, CH2,Cy,H4), 1.68–1.48 (m, 6H, CH2,Cy,H2), 1.50–1.21 (m, 9H, CH2,Cy,H3+H4); 13C{1H} NMR (101 MHz, CDCl3) δ 138.3 (d, 5JCP = 3.6 Hz, CMes,para), 137.1 (d, 3JCP = 4.5 Hz, CMes,ortho), 130.4 (d, 4JCP = 3.0 Hz, CHMes,meta), 123.5 (d, 2JCP = 8.5 Hz, CMes,ipso), 32.9 (d, 1JCP = 36.8 Hz, CHCy,C1), 27.6 (d, 2JCP = 4.4 Hz, CH2,Cy,C2), 26.9 (d, 3JCP = 11.6 Hz, CH2,Cy,C3), 25.4 (d, 4JCP = 1.8 Hz, CH2,Cy,C4), 22.1 (d, 4JCP = 1.2 Hz, CH3,Mes,ortho), 20.8 (d, 6JCP = 1.2 Hz, CH3,Mes,para), 19.9 (d, 1JCP = 40.7 Hz, P—CH2—Mes); 31P{1H} NMR (162 MHz, CDCl3) δ 31.6; IR (ATR) 2931 (s), 2849 (s), 1445 (s), 1397 (w), 1381 (w), 1179 (w), 1121 (w), 1045 (w), 1036 (w), 1009 (m), 887 (m), 869 (w), 851 (s), 828 (w), 791 (w), 740 (w), 564 (w), 532 (w), 521 (w); mp 210.8–215.2 °C.
Synthesis of Ligand L2
A Schlenk flask was filled with 4.0 g (9.5 mmol, 1.0 equiv) of phosphonium salt 1a, which was suspended in 60 mL of dry THF. To the solution was added 6.5 mL (9.5 mmol, 1.46 M in hexane, 1 equiv) of n-butyllithium dropwise. After the complete addition, a light-yellow solution formed. To this solution was added 2.2 mL (2.3 g, 10.0 mmol, 1.05 equiv) chlorodicyclohexylphosphine dropwise, and the solution was stirred overnight. The precipitated colorless solid was filtered through a glass frit and washed with 15 mL of dry THF. The solid was dried in vacuo, and the intermediate phosphonium salt was isolated as a colorless solid (5.2 g, 8.4 mmol, 88%). Next, 5.19 g (8.41 mmol, 1.0 equiv) of the intermediate phosphonium salt and 1.04 g (9.25 mmol, 1.1 equiv) of sodium tert-butoxide were added into a Schlenk flask and suspended in 80 mL of THF. After 1 h, a clear solution formed. The solvent was removed, and the solid was suspended in 60 mL of toluene. The solid was filtered off and washed with an additional 10 mL of toluene. The filtrated solvent was removed, and the remaining solid washed with 50 mL of dry acetonitrile. The product was obtained as a colorless solid (4.72 g, 8.13 mmol, 96%, yield of the two steps 84%): 1H NMR (400 MHz, C6D6) δ 7.55 (d, 3JHH = 7.6 Hz, 1H, CHTol,ortho′), 7.28 (d, 3JHH = 7.3 Hz, 1H, CHTol,meta), 7.20–7.13 (m, 1H, CHTol,meta ′), 7.09 (t, 3JHH = 7.3 Hz, 1H, CHTol,para), 2.71 (s, 3H, CH3), 2.58–2.36 (m, 2H, CH2,PCy2), 2.33–2.21 (m, 3H, CHPCy3,H1), 2.15–1.85 (m, 11H, CH2,PCy3,H2+PCy2 + CHPCy2,H1), 1.80–1.66 (m, 10H, CH2,PCy3,H3+PCy2), 1.63–1.43 (m, 13H, CH2,PCy3,H2+H4+PCy2), 1.41–1.22 (m, 7H, CH2,PCy2), 1.17–0.92 (m, 9H, CH2,PCy3,H3+H4); 13C{1H} NMR (101 MHz, C6D6) δ 143.7 (dd, 2JCP = 9.9 Hz, 2JCP = 2.6 Hz, CTol,ipso), 141.4 (CTol,ortho), 137.8 (d, 3JCP = 3.2 Hz, CHTol,ortho′), 130.6 (CHTol,meta), 124.5 (d, 4JCP = 2.0 Hz, CHTol,meta′), 124.1 (d, 5JCP = 2.1 Hz, CHTol,para), 40.1 (d, 1JCP = 14.0 Hz, CHPCy2,C1), 36.9 (dd, 1JCP = 48.1 Hz, 3JCP = 7.7 Hz, CHPCy3,C1), 36.9–36.4 (m, CHPCy2,C1), 33.7 (d, 2JCP = 23.4 Hz, CH2,PCy2,C2), 32.9 (d, 2JCP = 20.6 Hz, CH2,PCy2,C2), 31.2 (d, 2JCP = 11.7 Hz, CH2,PCy2,C2), 29.8 (CH2,PCy2,C3), 29.0 (d, 2JPP = 14.6 Hz, CH2,PCy2,C2), 28.8 (m, CH2,PCy3,C2), 28.1 (m, CH2,PCy2,C3), 27.7 (m, CH2,PCy3,C3+PCy2,C3), 27.1 (m, CH2,PCy2,C4), 26.6 (CH2,PCy3,C4), 22.4 (CH3), 17.9 (dd, 1JCP = 99.0 Hz, 1JCP = 29.7 Hz, P—C–—P); 31P{1H} NMR (162 MHz, C6D6) δ 19.3 (d, 2JPP = 138.2 Hz, PCy3), −0.5 (d, 2JPP = 138.2 Hz, PCy2); Anal. Calcd. for C38H62P2 C 78.58, H 10.76; found C 78.38, H 10.46; IR (ATR) 2915 (s), 2846 (s), 1590 (w), 1474 (w), 1445 (s), 1326 (w), 1282 (w), 1265 (w), 1217 (m), 1175 (w), 1129 (w), 1108 (w), 1074 (w), 1050 (w), 1006 (m), 974 (m), 899 (s), 885 (s), 846 (m), 813 (w), 791 (w), 747 (w), 725 (s), 569 (w), 543 (s), 523 (w), 512 (w); mp 157.8–160.9 °C.
Synthesis of Ligand L3
Phosphonium salt 1b (5.0 g, 9.3 mmol, 1.0 equiv) was suspended in 70 mL of toluene, and 5.82 mL of n-butyllithium (1.59 M in hexane, 1.0 equiv) was added dropwise. The remaining solid was filtered off and washed with 10 mL of toluene. Half the solvent was removed at reduced pressure, and 2.1 mL (2.2 g, 1.0 equiv) of chlorodicyclohexylphosphine was added. The solution was stirred for 3 days at room temperature, and the resulting colorless solid was filtered off and washed with pentane (2 × 10 mL) and dried in vacuo, thus giving the intermediate phosphonium salt (4.3 g, 5.8 mmol, 63%). Next, 0.64 mg (5.8 mmol, 1.0 equiv) of NaBF4 was added to the phosphonium salt, and the mixture was redissolved in 50 mL of acetonitrile and stirred overnight at room temperature. The resulting solid was filtered off and washed several times with MeCN (3 × 5 mL), and the solvent was removed at reduced pressure. The oily residue was suspended in 80 mL of diethyl ether, and the suspension was stirred overnight until a white solid precipitated from the solution. The colorless BF4 salt was filtered off and dried in vacuo (4.0 g, 5.7 mmol, 98%). Then, 0.50 g (0.7 mmol, 1.0 equiv) of the BF4 salt was suspended in 40 mL of toluene, and 0.081 g (0.7 mmol, 1.0 equiv) of potassium tert-butoxide was dissolved in a second flask in 40 mL of toluene. Both solutions were cooled to −78 °C (dry ice/acetone bath) and stirred for 30 min at that temperature. The potassium tert-butoxide solution was transferred to the suspension, and the mixture was allowed to slowly warm to room temperature overnight. The residue was filtered off, and the solvent was removed in vacuo. The solid was washed with 20 mL of acetonitrile and dried in vacuo to yield the ligand as a colorless solid (0.29 g, 0.5 mmol, 66%, overall yield: 41%): 1H NMR (400 MHz, C6D6) δ 6.99 (s, 2H, CHMes,meta), 2.76 (d, 5JHH = 1.4 Hz, 6H, CH3,Mes,ortho), 2.49–2.32 (m, 2H, CH2,PCy2,H2), 2.21 (s, 3H, CH3,Mes,para), 2.22–2.12 (m, 6H, CH2,PCy3,H2), 2.14–4.98 (m, 5H, CHPCy2,H1+PCy3,H1), 1.98–1.80 (m, 4H, CH2,PCy2,H2+H3), 1.82–1.65 (m, 10H, CH2,PCy3,H3 + CH2, PCy2,H3+H4), 1.66–1.40 (m, 15H, CH2,PCy3,H2+H4 + CH2,PCy2,H2+H3), 1.38–1.22 (m, 4H, CH2,PCy2,H3+H4), 1.20–0.91 (m, 9H, CH2,PCy3,H3+H4); 13C{1H} NMR (101 MHz, C6D6) δ 142.7 (d, 4JCP = 4.6 Hz, CHMes,meta), 140.8 (d, 2JCP = 9.5 Hz, CMes,ipso), 133.4 (d, 5JCP = 2.7 Hz, CMes,para), 129.0 (d, 3JCP = 1.9 Hz, CMes,ortho), 41.4 (dd, 1JCP = 17.8 Hz, 3JCP = 6.2 Hz, CH2,PCy2,C1), 39.6 (dd, 1JCP = 46.5 Hz, 3JCP = 6.3 Hz, CH2,PCy3,C1), 35.0 (d, 2JCP = 24.8 Hz, CH2,PCy2,C2), 32.1 (d, 2JCP = 3.6 Hz, CH2,PCy2,C2), 29.5 (d, 3JCP = 15.4 Hz, CH2,PCy2,C3), 29.3 (dd, 2JCP = 5.8 Hz, 4JCP = 3.6 Hz, CH2,PCy3,C2), 29.0 (d, 3JCP = 4.3 Hz, CH2,PCy2,C3), 28.2 (d, 3JCP = 10.4 Hz, CH2,PCy3,C3), 27.6 (CH2,PCy2,C4), 26.9 (CH2,PCy3,C4), 24.1 (CH3,Mes,ortho), 21.0 (CH3,Mes,para), 14.5 (dd, 1JCP = 103.4 Hz, 1JCP = 30.2 Hz, P—C–—P); 31P{1H} NMR (162 MHz, C6D6) δ 13.8 (d, 2JPP = 145.3 Hz, PCy3), 6.1 (d, 2JPP = 145.3 Hz, PCy2); CHNS Anal. Calc. for C40H66P2 C 78.90, H 10.93; found C 78.68, H 10.88; IR (ATR) 2922 (s), 2849 (m), 1444 (m), 1262 (w), 1216 (w), 1202 (w), 1154 (w), 1105 (w), 1071 (w), 1048 (w), 1005 (w), 969 (s), 942 (m), 897 (w), 883 (w), 870 (m), 851 (m), 808 (w), 742 (w), 730 (m), 693 (w), 569 (m), 519 (w), 502 (w); mp 177.0–181.5 °C.
Synthesis of L2·AuCl
To ligand L2 (150 mg, 0.27 mmol, 1.05 equiv) and (THT)AuCl (82.7 mg, 0.26 mmol, 1 equiv) was added 5 mL of pentane. The suspension was stirred for 3 days at room temperature. The solid was filtered and washed with 5 mL of pentane. The solid was dried at 50 °C with an oil bath in vacuo. The product was obtained as a colorless solid (116 mg, 0.14 mmol, 55%): 1H NMR (400 MHz, CD2Cl2) δ 7.40 (d, 3JHH = 7.5 Hz, 1H, CHTol,ortho′), 7.20 (d, 3JHH = 7.5 Hz, 1H, CHTol,meta), 7.10 (t, 3JHH = 7.5 Hz, 1H, CHTol,meta′), 7.02 (t, 3JHH = 7.5 Hz, 1H, CHTol,para), 2.56–2.85 (m, 3H, CHPCy3,H1), 2.50 (s, 3H, CH3), 2.41–2.32 (m, 1H, CH2,PCy2,H2), 2.32–2.23 (m, 1H, CH2,PCy2,H2), 2.17–2.00 (m, 6H, CH2,PCy3,H2), 1.99–1.41 (m, 26H, CH2,PCy3,H2+H3+H4+PCy2,H2+H3+H4 + CHPCy2,H1), 1.35–1.02 (m, 18H, CH2,PCy3,H3+H4+PCy2,H2+H3+H4); 13C{1H} NMR (101 MHz, CD2Cl2) δ 143.7 (dd, 3JCP = 5.7 Hz, 3JCP = 2.9 Hz, CTol,ortho), 140.9 (d, 3JCP = 2.6 Hz, CHTol,ortho′), 139.5 (dd, 2JCP = 4.8 Hz, 2JCP = 3.0 Hz, CTol,ipso), 131.4 (dd, 4JCP = 1.9 Hz, 4JCP = 1.9 Hz, CHTol,meta), 126.9 (d, 5JCP = 2.2 Hz, 5JCP = 2.2 Hz, CHTol,para), 125.3 (dd, 4JCP = 2.3 Hz, 4JCP = 2.3 Hz, CHTol,meta′), 42.0 (d, 1JCP = 37.0 Hz, CHPCy2,C1), 40.5 (dd, 1JCP = 38.8 Hz, 3JCP = 3.8 Hz, CHPCy2,C1), 38.4 (dd, 1JCP = 47.7 Hz, 3JCP = 1.8 Hz, CHPCy3,C1), 34.1 (d, 2JCP = 2.5 Hz, CH2,PCy2,C2), 34.0 (d, 2JCP = 2.8 Hz, CH2,PCy2,C2), 31.2 (CH2,PCy2,C2), 30.6 (CH2,PCy2,C2), 29.6 (dd, 2JCP = 8.7 Hz, 4JCP = 3.3 Hz, CH2,PCy3,C2), 28.5 (d, 3JCP = 13.8 Hz, CH2,PCy2,C3), 28.2 (d, 3JCP = 11.4 Hz, CH2,PCy3,C3), 28.0–27.6 (m, CH2,PCy2,C3), 26.9–26.4 (m, CH2,PCy3,C4+PCy2,C4), 22.7 (CH3), 14.8 (dd, 1JCP = 97.8 Hz, 1JCP = 60.4 Hz); 31P{1H} NMR (162 MHz, CD2Cl2) δ 34.4 (d, 2JPP = 60.5 Hz, PCy3), 25.7 (d, 2JPP = 60.5 Hz, PCy2); Anal. Calcd. for C38H62P2ClAu C 56.12, H 7.68; found: C 55.63, H 7.82; IR (ATR) 2917 (s), 2846 (s), 1739 (s), 1447 (s), 1365 (m), 1228 (s), 1217 (s), 1205 (s), 1108 (w), 1009 (s), 990 (s), 919 (m), 888 (s), 846 (m), 738 (m), 729 (m), 545 (s), 510 (m); mp 214.1–219.3 °C (decomposition).
Synthesis of L3·AuCl
To ligand L3 (70 mg, 1.05 eq., 0.12 mmol) and (THT)AuCl (35.1 mg, 1 eq., 0.11 mmol) was added 5 mL of THF, and the colorless suspension was stirred for 2 days at room temperature. To the solution was added toluene (5 mL), and the suspension was stirred for another 30 min. The resulting solid was filtered and washed with pentane (3 × 5 mL) to yield the gold complex as a colorless solid (50 mg, 0.06 mmol, 54%): 1H NMR (400 MHz, CD2Cl2) δ 6.88 (s, 2H, CHMes,meta), 2.75–2.52 (m, 3H, CHPCy3,H1), 2.51 (s, 6H, CH3,Mes,para), 2.41–2.24 (m, 8H, CH2,PCy3,H2 + CH2,PCy2,H2), 2.22 (s, 3H, CH3,Mes,para), 1.96–1.84 (m, 2H, CHPCy2,H1), 1.90–1.73 (m, 9H, CH2,PCy3,H3+H4), 1.73–1.65 (m, 4H, CH2,PCy2,H3+H4), 1.64–1.53 (m, 4H, CH2,PCy2,H3+H4), 1.52–1.38 (m, 2H, CH2,PCy2,H2), 1.36–1.00 (m, 23H, CH2,PCy3,H2+H3+H4 + CH2,PCy2,H2+H3); 13C{1H} NMR (101 MHz, CD2Cl2) δ 145.6–142.7 (m, CMes,ortho), 137.6–136.3 (m, CMes,ipso), 136.2–135.5 (m, CMes,para), 129.7 (t, 3JCP = 2.1 Hz, CHMes,meta), 40.5 (dd, 1JCP = 37.0 Hz, 3JCP = 1.7 Hz, CHPCy2,C1), 40.4–38.7 (br, CHPCy3,C1), 35.4 (d, 2JCP = 3.9 Hz, CH2,PCy2,C2), 30.5 (CH2,PCy2,C2), 29.6 (CH2,PCy3,C2), 28.3 (d, 3JCP = 14.4 Hz, CH2,PCy2,C3), 28.0 (d, 3JCP = 11.0 Hz, CH2,PCy3,C3), 27.8 (d, 3JCP = 11.3 Hz, CH2,PCy2,C3), 26.7 (d, 4JCP = 1.6 Hz, CH2,PCy2,C4), 26.6 (d, 4JCP = 1.7 Hz, CH2,PCy3,C4), 24.3 (d, 4JCP = 1.5 Hz, CH3,Mes,ortho), 20.8 (CH3,Mes,para), 12.5 (dd, 1JCP = 99.5 Hz, 1JCP = 62.3 Hz, P—C–—P); 31P{1H} NMR (162 MHz, CD2Cl2) δ 37.5 (d, 2JPP = 64.7 Hz, PCy3), 22.2 (d, 2JPP = 64.7 Hz, PCy2); Anal. Calcd. for C40H66P2ClAu C 57.10, H 7.94; found C 57.37, H 7.95; IR (ATP) 2922 (s), 2849 (m), 1444 (m), 1323 (w), 1268 (w), 1198 (m), 1172 (w), 1108 (w), 1072 (w), 1004 (m), 1004 (s), 952 (m), 852 (s), 816 (w), 742 (m), 595 (m), 566 (m); mp 224.5–227.8 °C (decomposition).
Preparation of L2·Pd(dba)
For this reaction, 10.0 mg (0.02 mmol, 1 equiv) of ligand L2 and 11.7 mg (0.02 mmol, 1 equiv) of Pd2dba3·dba were dissolved in 0.6 mL of THF-d8, and the solution was shaken for 1 h. The reaction progress was monitored by 31P{1H} NMR spectroscopy, and the solution was applied for further applications: 1H NMR (THF-d8, 400 MHz) δ 7.95–6.80 (m, 34H, CHdba+Tol), 2.42 (s, 3H, CH3), 2.37–2.16 (m, 3H, CHPCy3), 2.18–0.73 (m, 52H, CH2,PCy3 + CH,PCy2 + CH2,PCy2); 31P{1H} NMR (162 MHz, THF-d8) δ 25.2 (d, 2JPP = 89.4 Hz, PCy3), 22.6 (d, 2JPP = 89.4 Hz, PCy2).
Preparation of L3·Pd(dba)
For this reaction, 10.0 mg (0.02 mmol, 1 equiv) of ligand L3 and 11.2 mg (0.02 mmol, 1 equiv) of Pd2dba3·dba were dissolved in 0.6 mL of THF-d8, and the solution was shaken for 19 h. The reaction progress was monitored by 31P{1H} NMR spectroscopy and the solution was applied for further reactions: 1H NMR (THF-d8, 400 MHz) δ 8.07–7.05 (m, 14H, CHdba), 6.79 (s, 2H, CHMes,meta), 2.45 (s, 6H, CH3,Mes,ortho), 2.15 (s, 3H, CH3,Mes,para), 3.09–0.51 (m, 55H, CHPCy3 + CH2,PCy3 + CHPCy2 + CH2,PCy2); 31P{1H} NMR (162 MHz, THF-d8) δ 30.6 (d, 2JPP = 91.8 Hz, PCy3), 20.0 (d, 2JPP = 91.8 Hz, PCy2).
Procedure of the C–N Coupling Reaction Screening
A 5 mL vial with a rubber cap and stirring bar was charged with 142.2 mg (1.27 mmol, 1.5 equiv) of potassium tert-butoxide and 143.0 mg (0.85 mmol, 1.0 equiv) of 1,3,5-trimethoxy benzene (NMR standard) in the glovebox. Outside, 0.10 mL (107.0 mg, 1.0 equiv) of 4-chlorotoluene and 0.92 mmol (1.1 equiv) of a primary amine were added to the vial via syringe. The mixture was filled to a volume of 4 mL with THF. A second vial was charged with 4.22 μmol (0.005 equiv) ligand L2 or L3 and 2.88 mg (4.22 μmol, 0.005 equiv) of Pd2dba3·dba. Next, 0.5 mL of THF was added to the vial, and the mixture was stirred for 1 h (L2) or 16 h (L3). The catalyst solution was added to the first vial. For reaction monitoring, 0.1 mL of the reaction solution was quenched with 0.1 mL of water after a certain period. After extraction, the organic phase was dried in a flow of pressurized air. The residue was dissolved in CDCl3, and solution was filtered into an NMR tube to remove the remaining salt.
Procedure for Compound Isolation
A Schlenk tube was charged with 712.5 mg (6.35 mmol, 1.5 equiv) of potassium tert-butoxide in the glovebox, and 4.23 mmol (1.0 equiv) chloroarene and 4.62 mmol (1.1 equiv) primary amine were added into the tube. The mixture was filled to 20 mL of THF. A 5 mL vial was charged in the glovebox with 12.9 mg (0.02 mmol, 0.005 equiv) of L3 and 14.4 mg (0.02 mmol, 0.005 equiv) of Pd2dba3·dba. The catalyst mixture was dissolved in 2.5 mL of THF, and the mixture was stirred overnight at room temperature. The catalytic solution was added to the Schlenk tube. After 90 min of reaction time, the mixture was quenched with 5 mL of a saturated NaCl solution and poured into a separating funnel. To the mixture was added 10 mL of ethyl acetate, and the organic phase was extracted three times with 1 mL HCl (37% solution) in 10 mL distilled water. The aqueous phases were combined, and the solution was neutralized with Na2CO3 until reaching pH 8. Then, the aqueous phase was extracted with three portions of 10 mL of ethyl acetate. The organic phases were combined, and the solvent was removed in vacuo. The purity was checked by NMR; if not pure, the crude product was purified via column chromatography (4 g silica-packed weld column, 0–30% EtOAc in hexane).
Isolation of 5aa
A yellow oil (403 mg, 3.3 mmol, 79%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.19–6.88 (m, 2H), 6.76–6.37 (m, 2H), 3.77–3.47 (br, 1H, NH), 2.82 (s, 3H, CH3,NMe), 2.25 (s, 3H, CH3,Tol); 13C{1H} NMR (CDCl3, 101 MHz) δ 147.3 (CTol), 129.8 (CHTol), 126.7 (CTol), 112.8 (CHTol), 31.3 (CH3,NMe), 20.5 (CH3,Tol). Spectral data obtained for the compound are in good agreement with the reported data.21
Isolation of 5ab
A light yellow oil (570 mg, 4.2 mmol, 99%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.00 (d, 2H, 3JHH = 8.3 Hz, CHTol), 6.56 (d, 2H, 3JHH = 8.3 Hz, CHTol), 3.62–3.29 (br, 1H, NH), 3.15 (q, 2H, 3JHH = 7.2 Hz, CH2,NEt), 2.25 (s, 3H, CH3,Tol), 1.26 (t, 3H, 3JHH = 7.2 Hz, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 146.3 (CTol), 129.8 (CHTol), 126.6 (CTol), 113.1 (CHTol), 39.0 (CH2,NEt), 20.5 (CH3,Tol), 15.1 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.22
Isolation of 5ac
A light yellow oil (685 mg. 4.2 mmol, 99%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.01 (d, 2H, 3JHH = 8.5 Hz, CHTol), 6.56 (d, 2H, 3JHH = 8.5 Hz, CHTol), 3.61–3.38 (br, 1H, NH), 3.11 (t, 2H, 3JHH = 7.1 Hz, CH2,Bu), 2.27 (s, 3H, CH3,Tol), 2.02–1.54 (m, 2H, CH3,NBu), 1.53–1.27 (m, 2H, CH2,NBu), 1.12–0.82 (m, 3H, CH3,NBu); 13C{1H} NMR (CDCl3, 101 MHz) δ 146.4 (CTol), 129.8 (CHTol), 126.4 (CTol), 113.0 (CHTol), 44.2 (CH2,Bu), 31.9 (CH2,Bu), 20.5 (CH2,Bu), 20.4 (CH3,Tol), 14.0 (CH3,Bu). Spectral data obtained for the compound are in good agreement with the reported data.23
Isolation of 5ad
A light yellow oil (561 mg, 3.8 mmol, 89%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 6.99 (d, 2H, 3JHH = 8.1 Hz, CHTol), 6.53 (d, 2H, 3JHH = 8.1 Hz, CHTol), 3.61 (sept, 1H, 3JHH = 6.3 Hz, CHPiPr), 3.32–3.03 (br, 1H, NH), 2.24 (s, 3H, CH3,Tol), 1.21 (d, 6H, 3JHH = 6.3 Hz, CH3,NiPr); 13C{1H} NMR (CDCl3, 101 MHz) δ 145.4 (CTol), 129.9 (CHTol), 126.4 (CTol), 113.7 (CHTol), 44.7 (CHiPr), 23.2 (CH3,iPr), 20.5 (CH3,Tol). Spectral data obtained for the compound are in good agreement with the reported data.24
Isolation of 5ae
A light yellow oil (805, 4.1 mmol, 97%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.48–7.34 (m, 4H, CHNBz), 7.33–7.27 (m, 1H, CHNBz), 7.02 (d, 2H, 3JHH = 8.2 Hz, CHTol), 6.60 (d, 2H, 3JHH = 8.2 Hz, CHTol), 4.34 (s, 2H, CH2,NBz), 4.05–3.89 (br, 1H, NH), 2.27 (s, 3H, CH3,Tol); 13C{1H} NMR (CDCl3, 101 MHz) δ 146.0 (CTol), 139.7 (CNBz), 129.8 (CHTol), 128.6 (CHNBz), 127.5 (CHNBz), 127.2 (CHNBz), 126.8 (CTol), 113.1 (CHTol), 48.7 (CH2,NBz), 20.4 (CH3,Tol). Spectral data obtained for the compound are in good agreement with the reported data.25
Isolation of 5ba
A light yellow oil (475 mg, 3.9 mmol, 93%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.20–7.13 (m, 1H, CHTol), 7.13–6.93 (m, 1H, CHTol), 6.79–6.65 (m, 1H, CHTol), 6.65–6.58 (m, 1H, CHTol), 3.93–3.58 (br, 1H, NH), 2.90 (s, 3H, CH3,NMe), 2.15 (s, 3H, CH3,Tol); 13C{1H} NMR (CDCl3, 101 MHz) δ 147.3 (CTol), 130.1 (CHTol), 127.3 (CHTol), 122.1 (CTol), 117.1 (CHTol), 109.4 (CHTol), 31.0 (CH3,NMe), 17.5 (CH3,Tol). Spectral data obtained for the compound are in good agreement with the reported data.26
Isolation of 5bb
A light yellow oil (350 mg, 2.6 mmol, 61%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.14 (m, 1H, CHTol), 7.07 (m, 1H, CHTol), 6.78–6.60 (m, 2H, CHTol), 3.66–3.30 (br, 1H, NH), 3.22 (q, 3JHH = 7.1 Hz, 2H, CH2,NEt), 2.15 (s, 3H, CH3,Tol), 1.32 (t, 3JHH = 7.1 Hz, 3H, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 146.5 (CTol), 130.2 (CHTol), 127.3 (CHTol), 121.9 (CTol), 116.9 (CHTol), 109.8 (CHTol), 38.6 (CH2, NEt), 17.6 (CH3,Tol), 15.1 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.22
Isolation of 5ca
The reaction was performed with 25.8 mg (0.04 mmol, 0.01 equiv) of L3 and 28.8 mg (0.04 mmol, 0.01 equiv) of Pd2dba3·dba. A dark yellow oil (683 mg, 4.1 mmol, 97%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 5.89 (t, 4JHH = 2.2 Hz, 1H, CHarom,para), 5.81 (d, 4JHH = 2.2 Hz, 2H, CHarom,ortho), 3.93–3.79 (br, 1H, NH), 3.76 (s, 6H, CH3,OMe), 2.81 (s, 3H, CH3,NMe); 13C{1H} NMR (CDCl3, 101 MHz) δ 161.9 (Carom), 151.3 (Carom), 91.5 (CHarom,ortho), 89.9 (CHarom,para), 55.3 (OCH3), 30.9 (NHCH3). Spectral data obtained for the compound are in good agreement with the reported data.27
Isolation of 5cb
The reaction was performed with 25.8 mg (0.04 mmol, 0.01 equiv) of L3 and 28.8 mg (0.04 mmol, 0.01 equiv) of Pd2dba3·dba. A dark yellow oil (637 mg, 3.5 mmol, 83%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 5.88 (t, 4JHH = 2.2 Hz, 1H, CHarom,para), 5.81 (d, 4JHH = 2.2 Hz, 2H, CHarom,ortho), 3.79–3.71 (br, 1H, NH), 3.75 (s, 6H, CH3,OMe), 3.13 (q, 3JHH = 7.1 Hz, 2H, CH2,NEt), 1.24 (t, 3JHH = 7.1 Hz, 3H, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 161.9 (Carom), 150.4 (Carom), 91.8 (CHarom,ortho), 89.8 (CHarom,para), 55.3 (CH3,OMe), 38.7 (CH2, NEt), 14.9 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.28
Isolation of 5db
A redish oil (630 mg, 3.6 mmol, 84%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.26–7.17 (m, 2H, CHarom), 6.63–6.55 (m, 2H, CHarom), 4.05–3.40 (br, 1H, NH), 3.15 (q, 3JHH = 7.1 Hz, 2H, CH2,NEt), 1.29 (s, 9H, CH3,tBu), 1.25 (t, 3JHH = 7.1 Hz, 3H, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 146.2 (Carom), 140.3 (Carom), 126.1 (CHarom), 112.8 (CHarom), 39.0 (CH2,NEt), 34.0 (CtBu), 31.7 (CH3,tBu), 15.1 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.29
Isolation of 5ea
A light yellow oil (806 mg, 4.1 mmol, 97%) was isolated, which crystallized upon standing overnight: 1H NMR (CDCl3, 400 MHz) δ 7.45–7.37 (m, 2H, CHarom), 7.37–7.29 (m, 3H, CHarom), 7.25–7.16 (m, 1H, CHarom), 6.92–6.58 (m, 2H, CHarom), 4.09–3.78 (br, 1H, NH), 2.95 (s, 3H, CH3,NMe), 2.04 (s, 3H, CH3,Me); 13C{1H} NMR (CDCl3, 101 MHz) δ 147.6 (Carom), 142.8 (Carom), 142.6 (Carom), 129.6 (CHarom), 128.1 (CHarom), 126.7 (CHarom), 126.5 (CHarom), 119.5 (Carom), 119.2 (CHarom), 108.4 (CHarom), 31.2 (CH3,NMe), 14.3 (CH3,Me); Anal. Calcd. for C14H15N C 85.24, H 7.66, N 7.10; found: C 85.12, H 7.67, N 7.18; IR (ATR) 3445 (w), 3051 (w), 2993 (w), 2905 (w), 2819 (w), 1904 (w), 1587 (m), 1570 (m), 1511 (m), 1490 (m), 1470 (s), 1441 (m), 1428 (m), 1377 (w), 1324 (m), 1287 (s), 1193 (m), 1167 (m), 1121 (w), 1072 (m), 1057 (m), 1028 (m), 1000 (m), 986 (m), 920 (w), 845 (w), 803 (w), 789 (s), 759 (s), 720 (s), 703 (s), 609 (m); mp 56.6–58.3 °C.
Isolation of 5fa
A dark brown oil (653 mg, 4.2 mmol, 98%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.92–7.75 (m, 2H, CHarom), 7.51–7.34 (m, 3H, CHarom), 7.30–7.22 (m, 1H, CHarom), 6.75–6.40 (m, 1H, CHarom), 4.62–4.46 (br, 1H, NH), 3.04 (s, 3H, CH3,NMe); 13C{1H} NMR (CDCl3, 101 MHz) δ 144.6 (Carom), 134.4 (Carom), 128.8 (CHarom), 126.8 (CHarom), 125.8 (CHarom), 124.8 (CHarom), 123.6 (Carom), 119.9 (CHarom), 117.5 (CHarom), 104.0 (CHarom), 31.2 (CH3,NMe). Spectral data obtained for the compound are in good agreement with the reported data.30
Isolation of 5ga
The reaction was performed with 25.8 mg (0.04 mmol, 0.01 equiv) of L3 and 28.8 mg (0.04 mmol, 0.01 equiv) of Pd2dba3·dba. A dark yellow oil (543 mg, 4.0 mmol, 94%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 6.86–6.76 (m, 2H, CHarom), 6.65–6.57 (m, 2H, CHarom), 3.75 (s, 3H, CH3,OMe), 3.73–3.26 (br, 1H, NH), 2.81 (s, 3H, CH3,NMe); 13C{1H} NMR (CDCl3, 101 MHz) δ 152.3 (Carom), 143.7 (Carom), 115.1 (CHarom), 113.9 (CHarom), 56.0 (CH3,OMe), 31.8 (CH3,NMe). Spectral data obtained for the compound are in good agreement with the reported data.31
Isolation of 5gb
The reaction was performed with 25.8 mg (0.04 mmol, 0.01 equiv) of L3 and 28.8 mg (0.04 mmol, 0.01 equiv) of Pd2dba3·dba. A dark yellow oil (532 mg, 3.5 mmol, 83%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 6.83–6.74 (m, 2H, CHarom), 6.64–6.55 (m, 2H, CHarom), 3.75 (s, 3H, CH3,OMe), 3.50–3.33 (br, 1H, NH), 3.12 (q, 3JHH = 7.1 Hz, 2H, CH2,NEt), 1.24 (t, 3JHH = 7.1 Hz, 3H, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 152.3 (Carom), 142.8 (Carom), 115.0 (CHarom), 114.3 (CHarom), 56.0 (CH3,OMe), 39.7 (CH2,NEt), 15.1 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.22
Isolation of 5hb
The reaction was performed with 25.8 mg (0.04 mmol, 0.01 equiv) of L3 and 28.8 mg (0.04 mmol, 0.01 equiv) of Pd2dba3·dba. A light yellow oil (445 mg, 3.2 mmol, 76%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 6.94–6.83 (m, 2H, CHarom), 6.59–6.49 (m, 2H, CHarom), 3.62–3.36 (br, 1H, NH), 3.12 (q, 3JHH = 7.1 Hz, 2H, CH2,NEt), 1.25 (t, 3JHH = 7.1 Hz, 3H, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 155.9 (d, 1JCP = 234.5 Hz, Carom), 144.9 (Carom), 115.8 (d, JCF = 22.3 Hz, CHarom), 113.7 (d, JCF = 7.4 Hz, CHarom), 39.3 (CH2,NEt), 15.0 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.32
Isolation of 5ib
The reaction was performed with 25.8 mg (0.04 mmol, 0.01 equiv) of L3 and 28.8 mg (0.04 mmol, 0.01 equiv) of Pd2dba3·dba. A light yellow oil (241 mg, 1.7 mmol, 41%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 7.06–6.91 (m, 2H, CHarom), 6.76–6.66 (m, 1H, CHarom), 6.65–6.55 (m, 1H, CHarom), 3.84–3.74 (br, 1H, NH), 3.19 (q, 3JHH = , 2H, CH2,NEt), 1.29 (t, 3JHH = , 3H, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 151.7 (d, 1JCF = 238.0 Hz, Carom), 137.1 (d, JCF = 11.5 Hz, Carom), 124.7 (d, JCF = 3.4 Hz, CHarom), 116.5 (d, JCF = 7.0 Hz, CHarom), 114.4 (d, JCF = 18.4 Hz, CHarom), 112.1 (d, JCF = 3.6 Hz, CHarom), 38.3 (CH2,NEt), 15.0 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.32
Isolation of 5ja
The reaction was performed with 25.8 mg (0.04 mmol, 0.01 equiv) of L3 and 28.8 mg (0.04 mmol, 0.01 equiv) of Pd2dba3·dba. A light yellow oil (405 mg, 3.7 mmol, 89%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 8.09 (dd, 1H, JHH = 5.1 Hz, JHH = 1.9 Hz, CHarom), 7.43 (ddd, 1H, JHH = 8.7 Hz, JHH = 7.1 Hz, JHH = 1.9 Hz, CHarom), 6.57 (dd, 1H, JHH = 7.1 Hz, JHH = 5.1 Hz, CHarom), 6.38 (d, 1H, JHH = 8.4 Hz, CHarom), 4.65–4.33 (br, 1H, NH), 2.92 (d, 3H, 3JHH = 3.8 Hz, CH3,NMe); 13C{1H} NMR (CDCl3, 101 MHz) δ 159.6 (CHarom), 148.1 (CHarom), 137.4 (CHarom), 112.7 (CHarom), 106.2 (CHarom), 29.1 (CH3,NMe). Spectral data obtained for the compound are in good agreement with the reported data.33
Isolation of 5jb
A light yellow oil (235 mg, 3.3 mmol, 46%) was isolated: 1H NMR (CDCl3, 400 MHz) δ 8.05 (ddd, JHH = 5.1 Hz, JHH = 1.9 Hz, JHH = 1.0 Hz, 1H, CHarom), 7.38 (ddd, JHH = 8.4 Hz, JHH = 7.2 Hz, JHH = 1.9 Hz, 1H, CHarom), 6.52 (ddd, JHH = 7.2 Hz, JHH = 5.1 Hz, JHH = 1.0 Hz, 1H, CHarom), 6.34 (d, JHH = 8.4 Hz, 1H, CHarom), 4.79–4.41 (br, 1H, NH), 3.60–3.08 (m, 2H, CH2,NEt), 1.22 (t, 3JHH = 7.2 Hz, 3H, CH3,NEt); 13C{1H} NMR (CDCl3, 101 MHz) δ 159.0 (Carom), 148.2 (Carom), 137.4 (CHarom), 112.7 (CHarom), 106.4 (CHarom), 36.9 (CH2,NEt), 14.9 (CH3,NEt). Spectral data obtained for the compound are in good agreement with the reported data.34
Acknowledgments
Funded by the European Research Council (Starting Grant YlideLigands 677749). We also thank UMICORE for donating chemicals and for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c01771.
The authors declare the following competing financial interest(s): The authors have filed patent WO2019030304 covering the YPhos ligands and precatalysts discussed, which is held by UMICORE AG & Co. KG and products will be made commercially available from.
Supplementary Material
References
- a Ruiz-Castillo P.; Buchwald S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dorel R.; Grugel C. P.; Haydl A. The Buchwald–Hartwig Amination After 25 Years. Angew. Chem., Int. Ed. 2019, 58, 17118–17129. 10.1002/anie.201904795. [DOI] [PubMed] [Google Scholar]; c Buchwald S. L.; Mauger C.; Mignani G.; Scholz U. Industrial-Scale Palladium-Catalyzed Coupling of Aryl Halides and Amines – A Personal Account. Adv. Synth. Catal. 2006, 348, 23–39. 10.1002/adsc.200505158. [DOI] [Google Scholar]
- a Dennis J. M.; White N. A.; Liu R. Y.; Buchwald S. L. Breaking the Base Barrier: An Electron-Deficient Palladium Catalyst Enables the Use of a Common Soluble Base in C–N Coupling. J. Am. Chem. Soc. 2018, 140, 4721–4725. 10.1021/jacs.8b01696. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Park N. H.; Vinogradova E. V.; Surry D. S.; Buchwald S. L. Design of New Ligands for the Palladium-Catalyzed Arylation of α-Branched Secondary Amines. Angew. Chem., Int. Ed. 2015, 54, 8259–8262. 10.1002/anie.201502626. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ruiz-Castillo P.; Blackmond D. G.; Buchwald S. L. Rational Ligand Design for the Arylation of Hindered Primary Amines Guided by Reaction Progress Kinetic Analysis. J. Am. Chem. Soc. 2015, 137, 3085. 10.1021/ja512903g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lundgren R. J.; Peters B. D.; Alsabeh P. G.; Stradiotto M. A P,N-ligand for Palladium-Catalyzed Ammonia Arylation: Coupling of Deactivated Aryl Chlorides, Chemoselective Arylations, and Room Temperature Reactions. Angew. Chem., Int. Ed. 2010, 49, 4071–4074. 10.1002/anie.201000526. [DOI] [PubMed] [Google Scholar]; e Hill L. L.; Moore L. R.; Huang R.; Craciun R.; Vincent A. J.; Dixon D. A.; Chou J.; Woltermann C. J.; Shaughnessy K. H. Bulky Alkylphosphines with Neopentyl Substituents as Ligands in the Amination of Aryl Bromides and Chlorides. J. Org. Chem. 2006, 71, 5117–5125. 10.1021/jo060303x. [DOI] [PubMed] [Google Scholar]; f Rataboul F.; Zapf A.; Jackstell R.; Harkal S.; Riermeier T.; Monsees A.; Dingerdissen W.; Beller M. New Ligands for a General Palladium-Catalyzed Amination of Aryl and Heteroaryl Chlorides. Chem. - Eur. J. 2004, 10, 2983–2990. 10.1002/chem.200306026. [DOI] [PubMed] [Google Scholar]; g Shen Q.; Ogata T.; Hartwig J. F. Highly Reactive, General and Long-Lived Catalysts for Palladium-Catalyzed Amination of Heteroaryl and Aryl Chlorides, Bromides, and Iodides: Scope and Structure–Activity Relationships. J. Am. Chem. Soc. 2008, 130, 6586–6596. 10.1021/ja077074w. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Ackermann L.; Born R. Modular Diamino- and Dioxophosphine Oxides and Chlorides as Ligands for Transition-Metal-Catalyzed C-C and C-N Couplings with Aryl Chlorides. Angew. Chem., Int. Ed. 2005, 44, 2444–2447. 10.1002/anie.200462371. [DOI] [PubMed] [Google Scholar]
- a Marion N.; Navarro O.; Mei J.; Stevens E. D.; Scott N. M.; Nolan S. P. Modified (NHC)Pd(allyl)Cl (NHC = N-Heterocyclic Carbene) Complexes for Room-Temperature Suzuki–Miyaura and Buchwald–Hartwig Reactions. J. Am. Chem. Soc. 2006, 128, 4101–4111. 10.1021/ja057704z. [DOI] [PubMed] [Google Scholar]; b Organ M. G.; Abdel-Hadi M.; Avola S.; Dubovyk I.; Hadei N.; Kantchev E. A. B.; O'Brien C. J.; Sayah M.; Valente C. Pd-Catalyzed Aryl Amination Mediated by Well Defined, N-Heterocyclic Carbene (NHC)–Pd Precatalysts, PEPPSI. Chem. - Eur. J. 2008, 14, 2443–2452. 10.1002/chem.200701621. [DOI] [PubMed] [Google Scholar]; c Marion N.; Ecarnot E. C.; Navarro O.; Amoroso D.; Bell A.; Nolan S. P. (IPr)Pd(acac)Cl: An Easily Synthesized, Efficient, and Versatile Precatalyst for C-N and C-C Bond Formation. J. Org. Chem. 2006, 71, 3816–3821. 10.1021/jo060190h. [DOI] [PubMed] [Google Scholar]
- Fors B. P.; Watson D. A.; Biscoe M. R.; Buchwald S. L. A Highly Active Catalyst for Pd-Catalyzed Amination Reactions: Cross-Coupling Reactions Using Aryl Mesylates and the Highly Selective Monoarylation of Primary Amines Using Aryl Chlorides. J. Am. Chem. Soc. 2008, 130, 13552–13554. 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R. A.; Hartwig J. F. Palladium-Catalyzed Amination of Aryl Chlorides and Bromides with Ammonium Salts. Org. Lett. 2014, 16, 4388–4391. 10.1021/ol501739g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren R. J.; Sappong-Kumankumah A.; Stradiotto M. A Highly Versatile Catalyst System for the Cross-Coupling of Aryl Chlorides and Amines. Chem. - Eur. J. 2010, 16, 1983–1991. 10.1002/chem.200902316. [DOI] [PubMed] [Google Scholar]
- Tardiff B. J.; McDonald R.; Ferguson M. J.; Stradiotto M. Rational and Predictable Chemoselective Synthesis of Oligoamines via Buchwald–Hartwig Amination of (Hetero)Aryl Chlorides Employing Mor-DalPhos. J. Org. Chem. 2012, 77, 1056–1071. 10.1021/jo202358p. [DOI] [PubMed] [Google Scholar]
- Wheaton C. R.; Bow J.-P. J.; Stradiotto M. New Phosphine-Functionalized NHC Ligands: Discovery of an Effective Catalyst for the Room-Temperature Amination of Aryl Chlorides with Primary and Secondary Amines. Organometallics 2013, 32, 6148–6161. 10.1021/om400684n. [DOI] [Google Scholar]
- a Green R. A.; Hartwig J. F. Nickel-Catalyzed Amination of Aryl Chlorides with Ammonia or Ammonium Salts. Angew. Chem., Int. Ed. 2015, 54, 3768–3772. 10.1002/anie.201500404. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wolfe J. P.; Buchwald S. L. Nickel-catalyzed amination of aryl chlorides. J. Am. Chem. Soc. 1997, 119, 6054–6058. 10.1021/ja964391m. [DOI] [Google Scholar]; c Park N. H.; Teverovskiy G.; Buchwald S. L. Development of an air-stable nickel precatalyst for the amination of aryl chlorides, sulfamates, mesylates, and triflates. Org. Lett. 2014, 16, 220–223. 10.1021/ol403209k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Clark J. S. K.; Lavoie C. M.; MacQueen P. M.; Ferguson M. J.; Stradiotto M. A Comparative Reactivity Survey of Some Prominent Bisphosphine Nickel(II) Precatalysts in C–N Cross-Coupling. Organometallics 2016, 35, 3248–3254. 10.1021/acs.organomet.6b00650. [DOI] [Google Scholar]; b Gatien A. V.; Lavoie C. M.; Bennett R. N.; Ferguson M. J.; McDonald R.; Johnson E. R.; Speed A. W. H.; Stradiotto M. Application of Diazaphospholidine/Diazaphospholene-Based Bisphosphines in Room-Temperature Nickel-Catalyzed C(sp2)–N Cross-Couplings of Primary Alkylamines with (Hetero)aryl Chlorides and Bromides. ACS Catal. 2018, 8, 5328–5339. 10.1021/acscatal.8b01005. [DOI] [Google Scholar]
- a Wang D.; Kuang D.; Zhang F.; Yang C.; Zhu X. Room-Temperature Copper-Catalyzed Arylation of Dimethylamine and Methylamine in Neat Water. Adv. Synth. Catal. 2015, 357, 714–718. 10.1002/adsc.201400785. [DOI] [Google Scholar]; b Jiao J.; Zhang X.-R.; Chang N.-H.; Wang J.; Wei J.-F.; Shi X.-Y.; Chen Z.-G. A Facile and Practical Copper Powder-Catalyzed, Solvent- and Ligand-Free Ullmann Amination of Aryl Halides. J. Org. Chem. 2011, 76, 1180–1183. 10.1021/jo102169t. [DOI] [PubMed] [Google Scholar]; c Bhunia S.; Pawar G. G.; Kumar S. V.; Jiang Y. W.; Ma D. W. Selected copper-based reactions for C-N, C-O, C-S, and C-C bond formation. Angew. Chem., Int. Ed. 2017, 56, 16136–16179. 10.1002/anie.201701690. [DOI] [PubMed] [Google Scholar]
- a Schwarz C.; Scherpf T.; Rodstein I.; Weismann J.; Feichtner K.; Gessner V. H. Ylide-Functionalization via Metalated Ylides: Synthesis and Structural Properties. ChemistryOpen 2019, 8, 621–626. 10.1002/open.201900094. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Scherpf T.; Schwarz C.; Scharf L. T.; Zur J.-A.; Helbig A.; Gessner V. H. Ylide-functionalized phosphines: Strong Donor Ligands for Homogenous Catalysis. Angew. Chem., Int. Ed. 2018, 57, 12859–12864. 10.1002/anie.201805372. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schwarz C.; Handelmann J.; Baier D. M.; Ouissa A.; Gessner V. H. Mono- and diylide-substituted phosphines (YPhos): impact of the ligand properties on the catalytic activity in gold(I)-catalysed hydroaminations. Catal. Sci. Technol. 2019, 9, 6808–6815. 10.1039/C9CY01861A. [DOI] [Google Scholar]; d Scherpf T.; Rodstein I.; Paassen M.; Gessner V. H. Group 9 and 10 Metal Complexes of an Ylide-Substituted Phosphine: Coordination versus Cyclometalation and Oxidative Addition. Inorg. Chem. 2019, 58, 8151–8161. 10.1021/acs.inorgchem.9b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Weber P.; Scherpf T.; Rodstein I.; Lichte D.; Scharf L. T.; Gooßen L. J.; Gessner V. H. A Highly Active Ylide-Functionalized Phosphine for Palladium-Catalyzed Aminations of Aryl Chlorides. Angew. Chem., Int. Ed. 2019, 58, 3203–3207. 10.1002/anie.201810696. [DOI] [PubMed] [Google Scholar]; b Hu X.-Q.; Lichte D.; Rodstein I.; Weber P.; Seitz A.-K.; Scherpf T.; Gessner V. H.; Gooßen L. J. Ylide-Functionalized Phosphine (YPhos)–Palladium Catalysts: Selective Monoarylation of Alkyl Ketones with Aryl Chlorides. Org. Lett. 2019, 21, 7558–7562. 10.1021/acs.orglett.9b02830. [DOI] [PubMed] [Google Scholar]; c Scherpf T.; Steinert H.; Großjohann A.; Dilchert K.; Tappen J.; Rodstein I.; Gessner V. H. Efficient Pd-Catalyzed Direct Coupling of Aryl Clorides with Alkyllithium Reagents. Angew. Chem. 2020, 10.1002/ange.202008866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappen J.; Rodstein I.; McGuire K.; Großjohann A.; Löffler J.; Scherpf T.; Gessner V. H. Palladium Complexes Based on Ylide-Functionalized Phosphines (YPhos): Broadly Applicable High-Performance Precatalysts for the Amination of Aryl Halides at Room Temperature. Chem. - Eur. J. 2020, 26, 4281–4288. 10.1002/chem.201905535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf L. T.; Rodstein I.; Schmidt M.; Scherpf T.; Gessner V. H. Unraveling the High Activity of Ylide-Functionalized Phosphines in Palladium-Catalyzed Amination Reactions: A Comparative Study with CyJohnPhos and PtBu3. ACS Catal. 2020, 10, 999–1009. 10.1021/acscatal.9b04666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nelson D. J.; Nolan S. P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. 10.1039/c3cs60146c. [DOI] [PubMed] [Google Scholar]; b Dorta R.; Stevens E. D.; Scott N. M.; Costabile C.; Cavallo L.; Hoff C. D.; Nolan S. P. Steric and Electronic Properties of N-Heterocyclic Carbenes (NHC): A Detailed Study on Their Interaction with Ni(CO)4. J. Am. Chem. Soc. 2005, 127, 2485–2495. 10.1021/ja0438821. [DOI] [PubMed] [Google Scholar]
- Voskuil W.; Arens J.F. CHLORODIISOPROPYLPHOSPHINE. Org. Synth. 1968, 48, 47. 10.15227/orgsyn.048.0047. [DOI] [Google Scholar]
- Weber P.; Biafora A.; Doppiu A.; Bongard H.-J.; Kelm H.; Gooßen L. J. A Comparative Study of Dibenzylideneacetone Palladium Complexes in Catalysis. Org. Process Res. Dev. 2019, 23, 1462–1470. 10.1021/acs.oprd.9b00214. [DOI] [Google Scholar]
- Pews-Davtyan A.; Jackstell R.; Spannenberg A.; Beller M. Zwitterionic Phosphonium Ligands: Synthesis, Characterization and Application in Telomerization of 1,3-Butadiene. Chem. Commun. 2016, 52 (48), 7568–7571. 10.1039/C6CC02747D. [DOI] [PubMed] [Google Scholar]
- Langer P.; Yang L.; Pfeiffer C. R.; Lewis W.; Champness N. R. Restricting Shuttling in Bis(Imidazolium)··· pillar[5]arene Rotaxanes Using Metal Coordination. Dalton Trans. 2019, 48 (1), 58–64. 10.1039/C8DT04096F. [DOI] [PubMed] [Google Scholar]
- González I.; Mosquera J.; Guerrero C.; Rodríguez R.; Cruces J. Selective Monomethylation of Anilines by Cu(OAc)2-Promoted Cross-Coupling with MeB(OH)2. Org. Lett. 2009, 11 (8), 1677–1680. 10.1021/ol802882k. [DOI] [PubMed] [Google Scholar]
- Nacario R.; Kotakonda S.; Fouchard D. M. D.; Tillekeratne L. M. V.; Hudson R. A. Reductive Monoalkylation of Aromatic and Aliphatic Nitro Compounds and the Corresponding Amines with Nitriles. Org. Lett. 2005, 7 (3), 471–474. 10.1021/ol047580f. [DOI] [PubMed] [Google Scholar]
- Watanabe Y.; Tsuji Y.; Ige H.; Ohsugi Y.; Ohta T. Ruthenium-Catalyzed N-Alkylation and N-Benzylation of Aminoarenes with Alcohols. J. Org. Chem. 1984, 49 (18), 3359–3363. 10.1021/jo00192a021. [DOI] [Google Scholar]
- Bernardi P.; Dembech P.; Fabbri G.; Ricci A.; Seconi G. A General and Convenient Procedure for the Synthesis of N -Alkylarylamines and N -Alkylheteroarylamines by Electrophilic Amination of Cuprates with N -Alkylhydroxylamines. J. Org. Chem. 1999, 64 (2), 641–643. 10.1021/jo981412h. [DOI] [Google Scholar]
- Tanaka M.; Kobayashi T. Simple and High Yield Synthesis of Aldimines via Palladium Complex-Catalyzed Reduction of Imidoyl Chlorides. Synthesis 1985, 1985 (10), 967–969. 10.1055/s-1985-31406. [DOI] [Google Scholar]
- Sun N.; Wang S.; Mo W.; Hu B.; Shen Z.; Hu X. A Facile Protocol for the Synthesis of Mono-N-Methyl Anilines via Formimidate Intermediates. Tetrahedron 2010, 66 (35), 7142–7148. 10.1016/j.tet.2010.06.091. [DOI] [Google Scholar]
- Fors B. P.; Watson D. A.; Biscoe M. R.; Buchwald S. L. A Highly Active Catalyst for Pd-Catalyzed Amination Reactions: Cross-Coupling Reactions Using Aryl Mesylates and the Highly Selective Monoarylation of Primary Amines Using Aryl Chlorides. J. Am. Chem. Soc. 2008, 130 (41), 13552–13554. 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher J.-P.; Baudin G.; Wendeborn F.; Adam J.-M.; Lehmann U.; Birbaum J.-L.. High-Capacity Optical Storage Media. WO 2006/018352 A1, 2006.
- Wang D.; Zhao K.; Xu C.; Miao H.; Ding Y. Synthesis, Structures of Benzoxazolyl Iridium(III) Complexes, and Applications on C–C and C–N Bond Formation Reactions under Solvent-Free Conditions: Catalytic Activity Enhanced by Noncoordinating Anion without Silver Effect. ACS Catal. 2014, 4 (11), 3910–3918. 10.1021/cs5009909. [DOI] [Google Scholar]
- MacLellan P.; Clayden J. Enantioselective Synthesis of Tertiary Thiols by Intramolecular Arylation of Lithiated Thiocarbamates. Chem. Commun. 2011, 47 (12), 3395. 10.1039/c0cc04912c. [DOI] [PubMed] [Google Scholar]
- Lundgren R. J.; Sappong-Kumankumah A.; Stradiotto M. A Highly Versatile Catalyst System for the Cross-Coupling of Aryl Chlorides and Amines. Chem. - Eur. J. 2010, 16 (6), 1983–1991. 10.1002/chem.200902316. [DOI] [PubMed] [Google Scholar]
- Borisova N. E.; Ivanov A. V.; Matveev P. I.; Smirnova A. A.; Belova E. V.; Kalmykov S. N.; Myasoedov B. F. Screening of the Structure of Americium Extractants Based on a 2,2’-Bipyridyl Scaffold: A Simple Way to a N2,O2-Tetradentate Ligands Library for Rational Design of An/Ln Extractants. ChemistrySelect 2018, 3 (7), 1983–1989. 10.1002/slct.201702741. [DOI] [Google Scholar]
- Londregan A. T.; Jennings S.; Wei L. General and Mild Preparation of 2-Aminopyridines. Org. Lett. 2010, 12 (22), 5254–5257. 10.1021/ol102301u. [DOI] [PubMed] [Google Scholar]
- Singh O. M.; Singh S. J.; Kim S.-N.; Lee S.-G. Reaction of Lithioamines with Alkyl Halides: A Convenient Direct Synthesis of N-Alkylaminopyridines. Bull. Korean Chem. Soc. 2007, 28 (1), 115–117. 10.5012/bkcs.2007.28.1.115. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.