

Abstract

The complex phosphorylation pattern of natural and modified pentaphosphorylated magic spot nucleotides is generated in a highly efficient way. A cyclic pyrophosphoryl phosphoramidite (cPyPA) reagent is used to introduce four phosphates on nucleosides regioselectively in a one-flask key transformation. The obtained magic spot nucleotides are used to develop a capillary electrophoresis UV detection method, enabling nucleotide assignment in complex bacterial extracts.
Introduction
Magic spot nucleotides (MSN) are central bacterial alarmones, mainly known as governors of the highly conserved stringent response (SR), which is a global stress adaption mechanism. MSN constitute a class of densely 5′,3′-phosphorylated nucleotides, consisting of tetraphosphorylated, pentaphosphorylated, and other diversely phosphorylated species (see Figure 1).1,2
Figure 1.

General structure and nomenclature of tetra- and penta-phosphorylated magic spot nucleotides (MSN) with a typical 5′,3′-substitution pattern.
ppGpp (1) and pppGpp (3) are formed by (p)ppGpp synthase/hydrolase enzymes (Rel/Spot-homologue-enzymes (RSH)) when bacteria are confronted with harmful conditions, such as starvation, heat, or extreme pH values.3 During the SR, these alarmones lead to the reallocation of bacterial resources by the induction of global transcriptional alterations4 and by direct control of molecular targets.5,6 ppGpp (1) and pppGpp (3) are often generalized as (p)ppGpp, with a focus on the more abundant ppGpp (1). However, there is significant experimental evidence for their differential roles in bacteria.7,8 For example, the guanosine-5′-triphosphate,3′-diphosphate pyrophosphatase enzyme GppA in Escherichia coli (E. coli) is balancing ratios of ppGpp (1) and pppGpp (3).9,10 Exopolyphosphatase PPX,11 nucleosidase PpnN,12 or even RelA itself13 are regulated more potently by pppGpp (3) as compared to its tetraphosphorylated sibling.
Accordingly, Steinchen et al. state the following: “The complex framework of alarmone synthesis [is] [ . . .] communicating fundamentally different stress signals through two similar but not identical alarmones.”14
In contrast to (p)ppGpp, the existence and role of adenosine MSN in bacteria are controversially discussed since the 1970s.15,16 More recent reports from Bruhn-Olszewska et al. demonstrated in vitro how pppApp (4) has opposite regulatory effects compared to ppGpp (1) when binding to E. coli RNA polymerase. Furthermore, pppApp (4) is a stronger effector than ppApp (2).10 In a subsequent study, Sobala et al. described pppApp synthesis in wild-type E. coli.17 In 2019, Laub et al. discovered a type VI secretion system effector (Tas 1) from Pseudomonas aeruginosa that is injected into competing bacterial cells and converts the ADP/ATP pool into (p)ppApp, leading to massive growth inhibition and cell death.18 Consequently, it is now clear that pppApp (4) and ppApp (2) can be considered as physiologically relevant compounds, whose differential effects are understudied.
Organic synthetic approaches, especially for pentaphosphorylated MSN, are still limited, despite their obvious importance to provide tool and reference compounds for analytical purposes on scale.6,8,19 Since the 1970s, enzymatic methods based on 3′-pyrophosphortransferases were developed and have since dominated the field to access pppNpp structures (Figure 2A).20,21 However, enzyme preparation, high substrate specificity, and difficult removal of byproducts interfere with the application of these methods to access large amounts of modified MSN. In 1985, Schattenkerk et al. developed the first chemical synthesis of pppGpp (3) based on a complex protecting group approach in 19 steps and 4% yield (Figure 2B).22 In 2019, our group presented a chemoenzymatic approach toward pppGpp (3) based on sequential bisphosphorylations using a fluorenylmethyl modified phosphoramidite 5 (Fm–P-amidite) in combination with commercially available Ribonuclease T2 (Figure 2C).23
Figure 2.

Overview of synthetic procedures targeting pentaphosphorylated MSN: PG, protecting group; B, base.
In the first part of this manuscript, we present a synthetic method to obtain pentaphosphorylated MSN and analogues with an unprecedented step economy24,25 (Figure 2D). The synthetic core element relies on a simultaneous and regioselective one-flask introduction of four phosphates using a cyclic pyrophosphoryl phosphoramidite (cPyPA, 6) that has been used previously in polyphosphate and nucleotide synthesis,26−28 in combination with regioselective enzymatic cyclophosphate hydrolysis by RNase T2. This key step, followed by chemoselective phosphoric anhydride construction,29 enables access to a diverse array of important MSN tool compounds, which are then used in the second part of this study to develop a new analytical platform to study MSN.
Given the high charge density and instability of many MSN, they are particularly challenging to extract and analyze without decomposition. Method development is aggravated by the limited availability of MSN as reference compounds. A widely applied method to analyze MSN in biological samples is thin-layer chromatography (TLC) of 32P-labeled analytes.17 High-performance liquid chromatography (HPLC)-based methods have also been developed toward this goal, including anion-exchange chromatography (AEX),30 ion-paired reverse-phase chromatography (IPRP),31 and hydrophilic interaction chromatography (HILIC).32 In light of the comparably poor sensitivity of UV detection, large amounts of biological samples are usually required for monitoring the abundance of MSN in vivo.33 Mass spectrometry facilitates the detection of much lower quantities,34,35,19 but AEX- and IPRP-based separation methods have limited compatibility with MS detectors. Moreover, AEX methods exhibit low column efficiency and suffer from unstable retention and analysis time.18,30,33 IPRP Chromatography is more efficient, but the resolution of ppGpp (1) is poor. Concomitantly, the life span of the column is significantly shortened when processing complex biological samples.30,33 Considering the above limitations, we describe the development of capillary electrophoresis (CE) using our synthetic reference compounds into a competitive separation platform for MSN in complex samples, with benefits regarding column efficiency, sample consumption, analytical costs, and analysis time. Our method is inspired by previous CE-based approaches for the separation of nucleotides with mono-, di-, and triphosphates,36 relying on a fused silica capillary and a background electrolyte (BGE) that is in principle compatible with MS analysis.
Results and Discussion
Synthesis of Modified Pentaphosphorylated MSN
Adenosine (7) and 2N-(Fmoc)-guanosine (8) served as synthetic starting points. The latter was used, as guanosine has only limited solubility in organic solvents compatible with P-Amidite chemistry, such as DMF and MeCN. It is readily accessible in one step following a procedure by Quaedflieg et al.37
The synthesis (see Scheme 1A) commenced with the bisphosphitylation of nucleosides 7 and 8 using an excess of cPyPA (6) in the presence of 5-(ethylthio)-1H-tetrazole (ETT) as an acidic activator. The bisphosphitylation occurred unselectively, generating a mixture of 5′,3′- and 5′,2′-phosphitylated species. Overreaction toward trisphosphitylated products was not observed. Oxidation of the intermediate bis-deoxycyclotriphosphate esters with mCPBA led to the corresponding bis-cyclotriphosphate esters (9, Scheme 1A). Subsequent treatment of 9 with amine-nucleophiles induced a reaction cascade as shown in Scheme 1C. Nucleophilic amines efficiently ring-open cyclotriphosphates 9 under the exclusive formation of linearized amido-triphosphates (16, Scheme 1C).27,28,38 In the particular case described here, amines play a dual role by additionally enabling the nucleophilic attack of the 2′- or 3′-OH group toward the adjacent phosphate chain. This base-assisted 5-exocyclization results in the formation of corresponding 2′,3′-cyclophosphate 17 in a regioconvergent manner (see S-96). The transformation of 9 to 17 could also be explained by an alternative, concerted mechanism. In the case of Fmoc-guanosine (8), amine addition in step c, moreover, induces the removal of Fmoc, which at this stage is beneficial for the overall strategy. The generated 2′,3′-cyclophosphates (10, Scheme 1A) were regioselectively hydrolyzed with commercially available Ribonuclease T2 to give 3′-monophosphates 11–15 exclusively, underlining the substrate promiscuity of this enzyme. The resulting tetraphosphorylated amido-pppGp and pppAp derivatives 11–15 were isolated as sodium salts after RP-plug filtration or SAX purification in yields of 79–54% after four or five steps, respectively. The reaction sequence is compatible with the application of primary and secondary amines, providing the opportunity to access a high structural diversity with great simplicity. Notably, the first three (four) reaction steps can be performed in one flask and in less than a 90 min overall reaction time, while the purification of intermediates is not required. To increase solubility for the next steps, the cations of the amido-pppNp compounds 11–14 were exchanged from sodium to tetrabutylammonium (TBA) (Scheme 2). The resulting TBA salts were subjected to a P(III)–P(V) anhydride synthesis protocol using Fm–P–Amidite 5(29) in the presence of ETT. 31P NMR analysis of the reaction mixture revealed highly selective phosphitylation of the 3′-monophosphate in the presence of amidotriphosphate and other unprotected nucleophilic groups. Oxidation with mCPBA followed by Fm removal using DBU delivered the corresponding amido-pppNpp structures 18–21 in yields of 72–41% after SAX-purification. A yield of 41% was obtained due to solubility issues of the corresponding TBA salts, leading to the recovery of unconsumed starting material.
Scheme 1. Regioselective Tetraphosphorylation of Nucleosides Using cPyPA (6) and RNase T2.
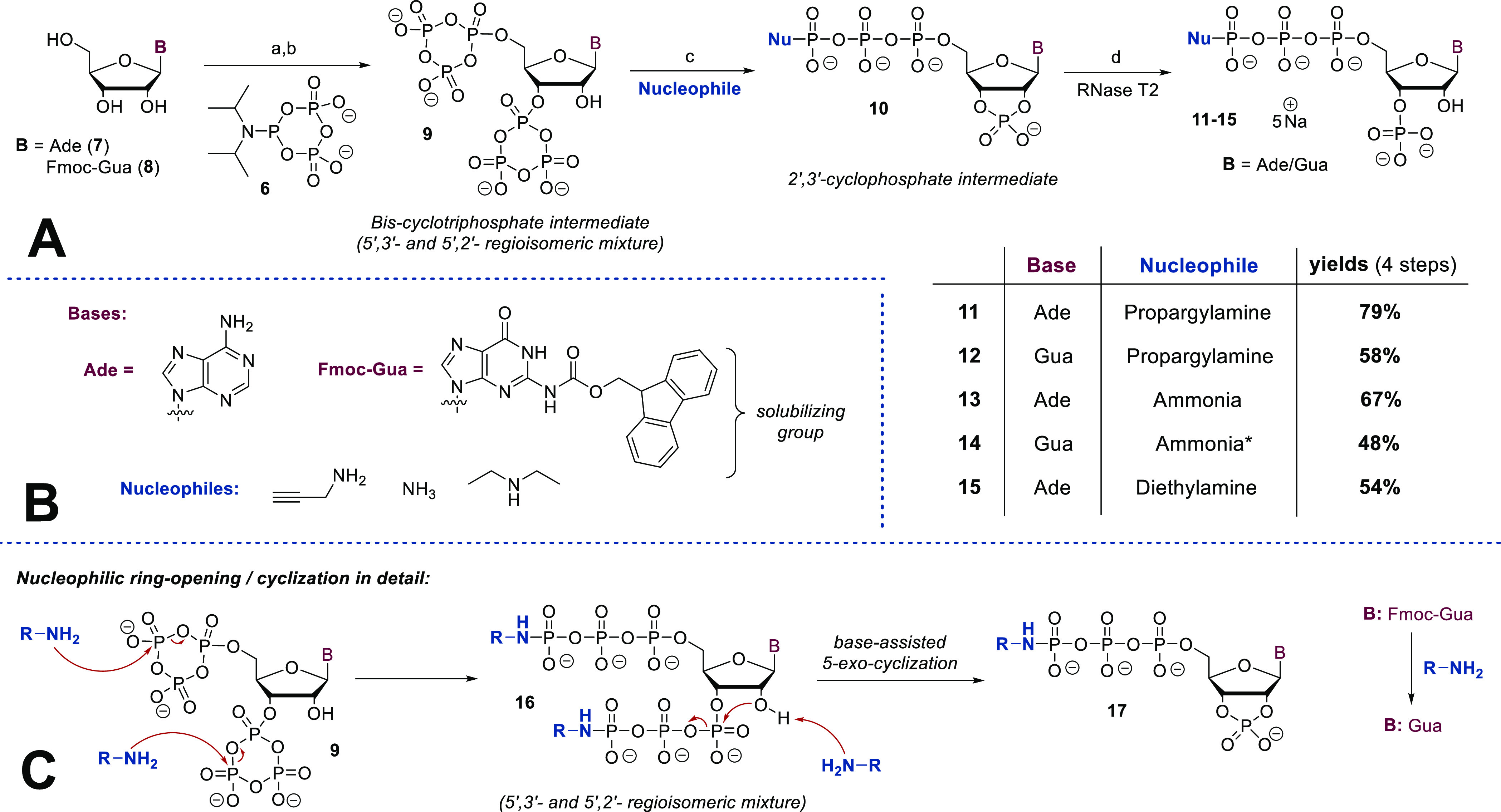
Synthetic details: (a) 6 (5.0 equiv), ETT (12 equiv), DMF, rt, 45 min; (b) mCPBA (7.5 equiv), 0 °C, 10 min; (c) amine (150 equiv); (d) RNase T2, H2O, 37 °C, 3–48 h. (*) In the case of 14, a mixture of ammonia and diazabicycloundecen (DBU) was applied.
Scheme 2. Chemoselective Phosphorylation toward Amido-pppNpp Structures.
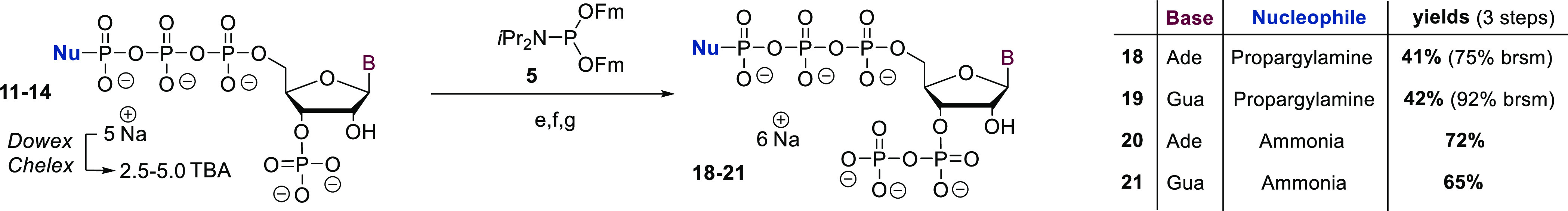
Synthetic details: (e) 5 (1.7 equiv), ETT (3.5 equiv), DMF, rt, 15 min; (f) mCPBA (2.1 equiv), 0 °C, 10 min; (g) DBU (10 vol %), rt, 30 min.
The versatility of the corresponding amido-MSN analogues as a platform for further structural diversification was studied, focusing on propargylamido-variants 18 and 19 (see Scheme 3). The conversion of 18 and 19 into their parent natural MSN was achieved by applying an aqueous solution of 18 and 19 to a Dowex-H+ plug filtration. This slightly acidic environment smoothly induced P–N-bond hydrolysis,39 affording pppApp (4) and pppGpp (3). After precipitation, 3 and 4 were isolated in total yields of 26% and 18% with respect to the starting materials 7 and 8 after eight and nine steps and only two intermediates that required purification. Treatment of propargylamido-pppApp (18) with H218O led to 18O-incorporation (93%) and consequently to a “heavy” isotopomer 22 of natural pppApp (4). Such compounds are useful for absolute MSN quantification based on MS technologies and recovery experiments and to study potential phosphate transfer.19,40 Furthermore, the biotinylated pppApp analogue 23 was accessible in 98% yield using a CuI-catalyzed 1,3-dipolar cycloaddition. Biotinylated MSN can principally be applied for pull-down experiments to identify unknown receptors.6 A Cy3-fluorophore was introduced using a similar approach in 85% yield. Such fluorescently labeled nucleotides enable receptor interaction quantification using methods such as fluorescence polarization assays41 or microscale thermophoresis.42 In contrast to 18, amido-pppApp 20 was more stable toward PN-bond hydrolysis, requiring HClO4 at pH 3 to induce complete turnover in 1 h (see Experimental Section).
Scheme 3. Follow-up Chemistry: Derivatization of Amido-pppNpp Structures.
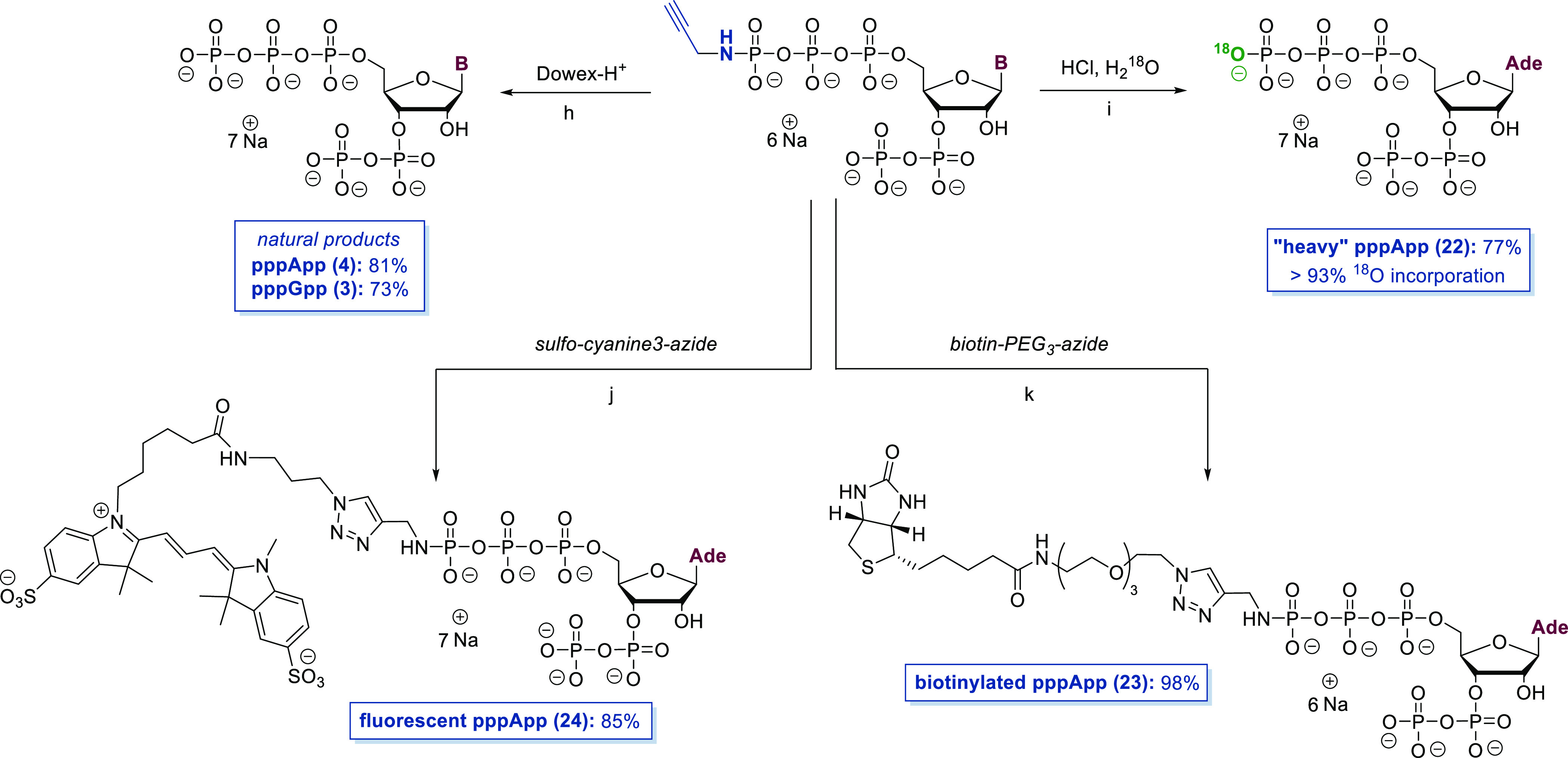
Synthetic details: (h) Dowex-H+ column; (i) HCl, H218O, pH 3, rt, 1 h; (j) sulfo-cyanine-3-azide (1.7 equiv), Na-ascorbate (2.5 equiv), CuSO4 × 5 H2O (35 mol %), TEAA–buffer (pH 7, 200 mM), rt, 30 min; (k) biotin-PEG3-azide (2.5 equiv), Na-ascorbate (2.5 equiv), CuSO4 × 5 H2O (35 mol %), TEAA–buffer (pH 7, 200 mM), rt, 30 min.
CE-UV Analysis of Magic Spot Nucleotides
The development of a UV detection-based nucleotide pool analysis of bacterial samples requires separation of the most common nucleotides. For the first time, a CE method for the analysis of nucleoside tetraphosphate and pentaphosphate using a fused silica capillary is developed. Baseline separation of adenosine and guanosine monophosphate (AMP, GMP), diphosphate (ADP, GDP), triphosphate (ATP, GTP), tetraphosphate (ppApp, ppGpp), and pentaphosphate (pppApp, pppGpp) was achieved by employing a background electrolyte (BGE) consisting of 40 mM ammonium bicarbonate at pH 10.1 (Figure 3). The migration order is nucleoside monophosphate, diphosphate, triphosphate, pentaphosphate, and tetraphosphate, e.g., GMP, GDP, GTP, pppGpp, and ppGpp. The significant differences in migration time of MSN compared to the most common NMPs, NDPs, and NTPs is beneficial for the identification of MSN in complex samples. The reversal of migration order regarding ppGpp and pppGpp is puzzling and could be a result of the different hydrodynamic radii.
Figure 3.
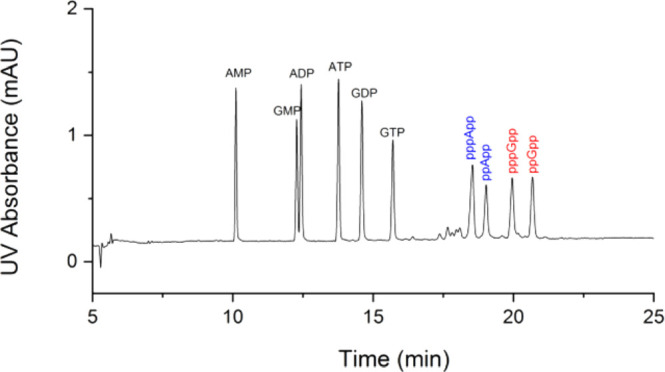
CE-UV analysis of several nucleotides. Baseline separation of AMP, GMP, ADP, ATP, GDP, GTP, pppApp, ppApp, pppGpp, and ppGpp. Composition of the running buffer: 40 mM ammonium bicarbonate titrated by ammonium hydroxide solution to pH 10.1. Separation voltage: 18 kV. UV detection at 250 nm. Temperature: 30 °C. Solute concentration: 40 μM each. Injection: 25 mbar, 3 s (2.2 nL).
The average column efficiency for the ten nucleotide standards is 2.9 × 105 theoretical plates per meter. The resolution between pppApp, ppApp, pppGpp, and ppGpp is 2.3, 4.6, and 3.5, respectively. Meanwhile, the average symmetry factor of 10 nucleotides is 1.05. The peak shape in this CE method is superior to that obtained in AEX with peak tailing.18 For optimization, BGEs with various concentrations (30–60 mM) of ammonium bicarbonate (Figure S1) and pH (9.8–10.2) (Figure S2) were investigated. Under all nine different conditions tested, ppGpp, pppGpp, ppApp, and pppApp were baseline separated, pointing toward a facile reproducibility also in other laboratories. With 30 mM ammonium bicarbonate in the BGE, baseline separation of the ten nucleotide standards is performed in 18 min, which compares favorably to any reported HPLC-based method.30,31,18
To study the applicability of the method to bacterial samples, we monitored ppGpp changes in two types of bacterial extracts. E. coli cells were grown in the minimal medium under limited amino acid starvation conditions in the absence or presence of serine hydroxamate (SHX). SHX is a small molecule that enhances amino acid starvation and induces the stringent response, which triggers (p)ppGpp accumulation.43 The established CE-UV method enabled monitoring of the induced accumulation of ppGpp after SHX treatment (Figure S3) by 4.2-fold as compared to the untreated sample. This ppGpp increase upon SHX treatment is in line with results reported by other groups.19
Conclusion
In summary, a new synthetic method toward pentaphosphorylated MSN was presented. A regioselective tetraphosphorylation using cPyPA as a key step in combination with RNase T2 provides access to pppGpp and pppApp in a highly step economical fashion. In addition to the natural alarmones, fluorescent, biotinylated, and isotopomeric analogues were synthesized, allowing for diverse possible applications in future chemical biology experiments. We are confident that simplified synthetic access to MSN and analogues will be a key driver for future research in this field.
We demonstrate the utility of the synthetic material by developing a CE-UV method with main benefits regarding column efficiency, sample consumption, resolution, and time required for analysis. The simultaneous analysis of ppGpp, pppGpp, ppApp, pppApp, and other cellular nucleotides has been performed for the first time using this method. Even with a UV detector, monitoring ppGpp levels in biological samples is feasible. Yet, the reported CE separation conditions are compatible with mass spectrometry detectors, which may allow its translation to an even more sensitive CE-MS method. CE-MS implementation for identifying novel MSN and for quantifying less abundant MSN in other organisms is currently under development in our laboratory.
Experimental Section
General Procedures
Reactions were carried out in flame-dried glassware under an atmosphere of dry Ar unless noted otherwise. Air- and moisture-sensitive liquids and solutions were transferred via syringe or stainless steel cannula. Reagents were purchased from commercial suppliers (Acros, Aldrich, Fluka, TCI) and used without further purification, unless noted otherwise. Solvents were obtained in analytical grade and used as received for extractions, precipitation, and solid washing. Dry DMF and MeCN for reactions were purchased in a dry form from Sigma and stored over molecular sieves as well as under an atmosphere of dry Ar. Ribonuclease T2 from Aspergillus oryzae (50 ku) was purchased from Worthington Biochemical Corporation as lyophilized powder and dissolved in a storage buffer [glycerol/NaH2PO4 (10 mM, pH 6.8), 1:1]. The stock solution was stored at −20 °C. Deuterated solvents for NMR and reactions were obtained from Armar Chemicals, Switzerland, and Euriso-top, Germany, in the indicated purity grade and used as received for NMR spectroscopy. Strong ion-exchange chromatography was performed using an automated Äkta system. Q-Sepharose was purchased from Aldrich. Buffer solutions were produced manually using ultrapure H2O. Lyophilizations were done with Christ Freeze-Dryer Alpha 1–4 LDplus and Christ Freeze-Dryer Alpha 1–2 LDplus. 1H NMR spectra were recorded on Bruker 300 MHz, Bruker 400 MHz (with cryoprobe), and Bruker 500 MHz spectrometers in the indicated deuterated solvent. Data are reported as follows: chemical shift (δ, ppm), multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br s, broad signal), coupling constant(s) (J, Hz), integration. All signals were referenced to the internal solvent signal as a standard (D2O, δ 4.70). 13C{1H} NMR spectra were recorded with 1H decoupling on Bruker 126 MHz and Bruker 101 MHz (with cryoprobe) spectrometers at 298 K in the indicated deuterated solvent. 31P{1H} NMR spectra and 31P NMR spectra were recorded with 1H decoupling or 1H coupling, respectively, on Bruker 202 MHz, 162 MHz (with cryoprobe), and 122 MHz spectrometers in the indicated deuterated solvent. All signals were referenced to an internal standard (PPP). Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. Mass spectra were recorded by C. Warth (mass spectrometry service of the University of Freiburg) on a Thermo LCQ Advantage [spray voltage, 2.5–4.0 kV; spray current, 5 μA; ion transfer tube, 250 (150) °C; evaporation temperature, 50–400 °C].
CE-UV Measurements
The measurements were performed on an Agilent 7100 capillary electrophoresis system equipped with a diode-array detector (Agilent, Germany). Data were processed using the Agilent CE ChemStation Software. Fused silica capillaries (G1600–61232) were obtained from Agilent Technologies. Electrophoretic separations were carried out in a fused silica capillary (50 μM i.d. × 365 μm o.d.) with an effective column length of 56 cm (64.5 cm total length). Running buffers consisting of different concentrations of ammonium bicarbonate with different pH’s were freshly prepared and filtered before use. The separation voltage was 18 kV, and the applied detection wavelength was 250 nm.
Bacterial Growth Conditions
Precultures were freshly inoculated from cryo-stocks into 5 mL of LB medium and were grown overnight at 37 °C. Cells were then pelleted by centrifugation (5000 rpm for 10 min at 4 °C), resuspended, and used to inoculate the main culture in 50 mL methionine- and cysteine-depleted M63 medium (glycerol 20 g/L; KH2PO4 13.6 g/L; (NH4)2SO4 2.0 g/L; FeSO4 0.5 mg/L; Mg2SO4 0.2 g/L; 18 amino acids each at 0.1 mM; thiamine 10 μg/mL; pH 7.0) to a final OD600 of 0.2. The cultures were grown at 37 °C and 180 rpm until they reached an OD600 of 0.8. Subsequently, 40 × 108 cells were treated with 2 M formic acid, flash-frozen in liquid N2, and stored at −80 °C until further purification. When indicated, cells were treated with 625 ng/mL serine hydroxamate (SHX) for 1 h before formic acid treatment and storage.
Extraction and Purification of Nucleotides
Frozen formic acid-treated cell extracts were thawed at 37 °C and subsequently stored at 4 °C. Nucleotides were extracted by repeated vortexing of the formic acid containing cells extracts over 30 min at 4 °C. Samples were then centrifuged at 7000g for 10 min at 4 °C, and the nucleotides in the supernatant were extracted as previously described by Ihara et al. in 2015 and employed for bacteria as well.34,44 For solid-phase extraction (SPE) purification, 3 cm3 OASIS WAX Vac cartridges with 60 mg of sorbent and 30 μm particle size (Waters Corp. USA) were used. The OASIS WAX column was equilibrated first with 1 mL of methanol, followed by 1 mL of 50 mM ammonium acetate, pH 4.5. After centrifuging, the supernatant was loaded onto the column and washed with the ammonium acetate solution and then methanol. The bound nucleotides were finally eluted with a mixture of methanol/water/NH4OH (20:70:10). Eluted extracts were lyophilized overnight and resuspended in 200 μL of water for CE-MS analysis.
Synthetic Procedures
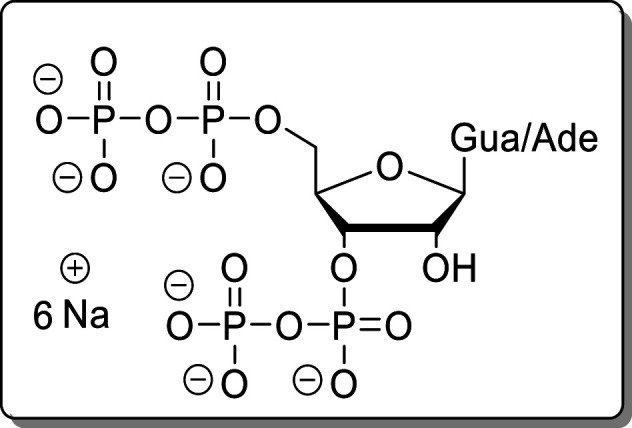
ppNpp (1/2) were synthesized according to literature.23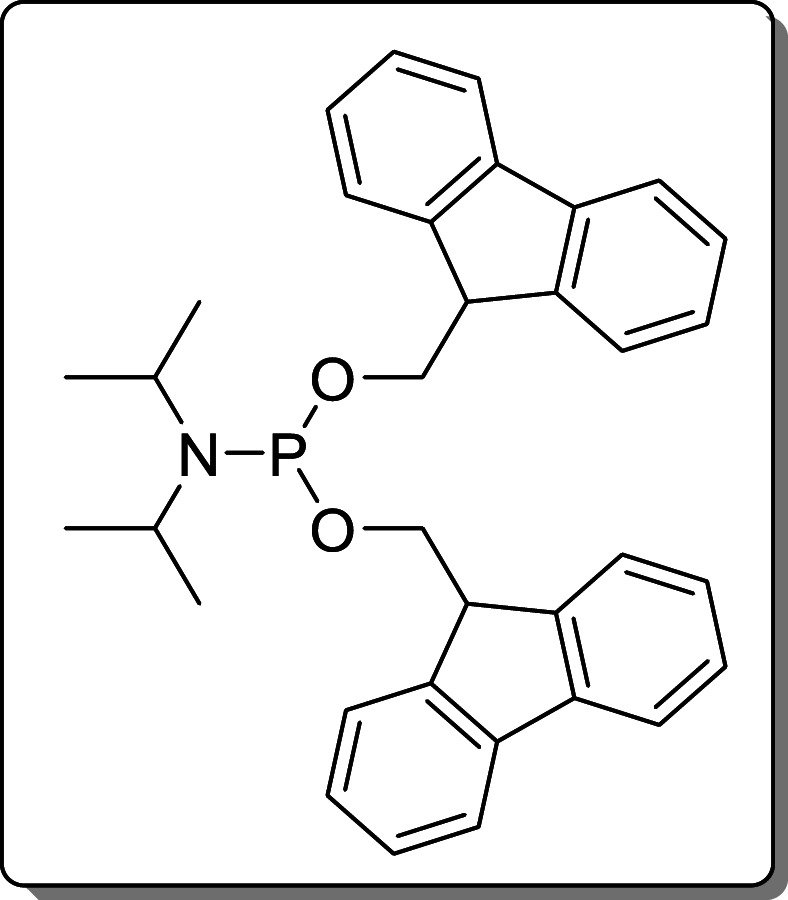
(FmO)2P–N(iPr)2 (5) was synthesized according
to literature29 and stored at −20
°C.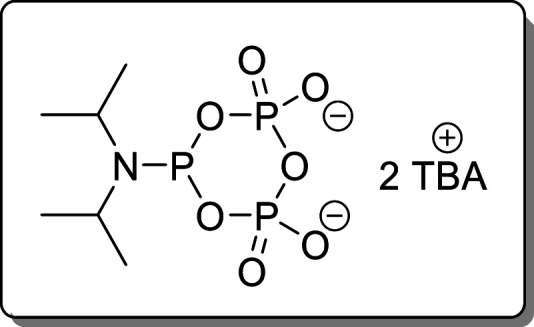
Cyclic pyrophosphoryl-P-amidite (cPyPA,6) was synthesized according to literature27 and stored over molecular sieves at −20 °C.
2-N-(9-Fluorenylmethoxycarbonyl)guanosine (8) was synthesized in one step according to Quaedflieg et al. Analytical data were identical to literature.37
General Procedure A
For the synthesis of amido-pppNp compounds, nucleoside and ETT (12 equiv) were coevaporated separately with dry MeCN (2 × 3.0 mL). The nucleoside was dissolved in dry DMF (40 mM), and a solution of ETT in dry DMF (1.0 M) was added. Then cPyPA (6, 150 mM in DMF, 5.0 equiv) was added dropwise within 30 s, and the resulting solution was stirred for 45 min at rt. The solution was cooled to 0 °C, and mCPBA (77%, 7.5 equiv) was added. The mixture was stirred at 0 °C for 10 min before amine (150 equiv) was added. The resulting solution was stirred for 30 min at rt before being precipitated by the addition of Et2O (4 vol). The precipitate was separated by centrifugation (7700g, 5 min), washed with Et2O (2 × 2 vol), and dried under a high vacuum. The resulting solid was dissolved in H2O (8.0 mM), and RNase T2 (500 u) was added. The solution was incubated for 3–48 h at 37 °C.
Purification Method 1
TEAA–buffer (1.0 M, 0.1 vol) was added to the crude product solution, and the mixture was applied to a plug of C18-AQ-RP-silica (preconditioned with 100 mM TEAA). The product was eluted with TEAA–buffer (100 mM). The product-containing fractions were combined and precipitated using a 4-fold volume of NaClO4 solution (−20 °C, 500 mM in acetone). The resulting solid was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 10 mL), and dried under a high vacuum.
Purification Method 2
The crude product was purified
by SAX (Q-Sepharose, NaClO4–buffer gradient). Product
containing fractions were combined and precipitated using a 4-fold
volume of NaClO4 solution (−20 °C, 500 mM in
acetone). The resulting solid was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 10 mL), and dried
under a high vacuum.
Adenosine-5′-γ-P-propargylamido-triphosphate-3′-phosphate (11)
The reaction was performed according to General Procedure A using adenosine (7, 40.0 mg, 140 μmol) and propargylamine. The crude product was purified using Purification Method 1. The product (11, 82.0 mg, 111 μmol, 79%) was isolated as a white solid.
1H NMR* (400 MHz, D2O): δ 8.50 (s, 1H), 8.19 (s, 1H), 6.10 (d, J = 7.2 Hz, 1H), 4.54–4.46 (m, 1H), 4.23–4.12 (m, 2H),
3.50 (d, J = 9.8 Hz, 2H), 2.30** (dd, J = 2.6 Hz, 1H). 31P{1H} NMR (162 MHz, D2O): δ 4.06, −2.78 (d, J = 21.0
Hz), −11.31 (d, J = 18.9 Hz), −22.68
(dd, J = 20.7, 19.0 Hz). 13C{1H} NMR (101 MHz, D2O): δ 155.7 (dd), 152.9, 149.5,
140.0, 118.7, 86.4, 84.2 (dd, J = 9.3, 5.3 Hz), 74.4
(d, J = 3.3 Hz), 73.7 (d, J = 4.7
Hz), 65.8, 30.9. HRMS (ESI): m/z calcd for C13H18N6O15P4 [M – H2]2–, 310.9896;
found, 310.9895. (*) 2′H and 3′H were hidden under the
HDO peak. (**) The acetylenic proton integral does not fit the theoretical
value. This is due H/D-exchange in D2O. The existence of
the PN-bond is proven by 1H–31P HMBC
(see Supporting Information).
Guanosine-5′-γ-P-propargylamido-triphosphate-3′-phosphate (12)
The reaction was performed according to General Procedure A using 2-N-Fmoc-guanosine (8, 50.0 mg, 99.0 μmol) and propargylamine. The crude product was purified using Purification Method 1 and Purification Method 2 subsequently. The product (12, 43.1 mg, 57.1 μmol, 58%) was isolated as a white solid.
1H NMR (400 MHz,
D2O): δ 8.07 (s, 1H), 5.88 (d, J = 7.3 Hz, 1H), 4.83–4.75 (m, 1H), 4.53–4.37 (m, 1H),
4.16 (dd, J = 4.3 Hz, 2H), 3.52 (d, J = 10.0 Hz, 2H), 2.37 (s, 1H). 31P{1H} NMR
(162 MHz, D2O): δ 4.03, −2.73 (d, J = 20.6 Hz), −11.28 (d, J = 18.2
Hz), −22.64 (dd, J = 20.5, 17.8 Hz). 13C{1H} NMR (101 MHz, D2O): δ 159.4,
154.2, 152.1, 137.9, 116.5, 86.6, 83.9 (dd, J = 9.4,
5.5 Hz), 73.5 (d, J = 4.6 Hz), 73.5 (d, J = 3.2 Hz), 65.8 (d, J = 5.5 Hz), 30.8. HRMS (ESI): m/z calcd for C13H18N6O16P4 [M – H2]2–, 318.9870; found, 318.9869.
Adenosine-5′-γ-P-amido-triphosphate-3′-phosphate (13)
The reaction was performed according to General Procedure A using adenosine (7, 30.0 mg, 105 μmol) and conc ammonia. Instead of the Et2O precipitation, precipitation using NaClO4 solution (500 mM in acetone) was performed. The crude product was purified using Purification Method 1 and Purification Method 2 subsequently. The product (13, 48.9 mg, 70.3 μmol, 67%) was isolated as a white solid.
1H NMR (400 MHz, D2O): δ
8.48 (s, 1H), 8.18 (s, 1H), 6.10 (d, J = 7.1 Hz,
1H), 4.76–4.71 (m, 1H), 4.69–4.64 (m, 1H), 4.53–4.47
(m, 1H), 4.24–4.10 (m, 2H). 31P{1H} NMR
(162 MHz, D2O): δ 4.07, −0.86 (d, J = 19.1 Hz), −11.20 (d, J = 19.1
Hz), −22.35 (dd, J = 19.1 Hz). 13C{1H} NMR (101 MHz, D2O): δ 155.7, 152.9,
149.4, 139.9, 118.6, 86.4, 84.1 (dd, J = 9.1, 5.1
Hz), 74.4 (d, J = 3.6 Hz), 73.7 (d, J = 4.5 Hz), 65.8 (d, J = 5.7 Hz). HRMS (ESI): m/z calcd for C10H17N6O15P4 [M – H]−, 584.9708; found, 584.9715.
Guanosine-5′-γ-P-amido-triphosphate-3′-phosphate (14)
The reaction was performed according to General Procedure A using 2-N-Fmoc-guanosine (8, 70.0 mg, 139 μmol) and an NH3/DBU mixture (1:1). The crude product was purified using Purification Method 1 and Purification Method 2 subsequently. The product (14, 46.9 mg, 65.8 μmol, 48%) was isolated as a white solid.
1H NMR (400 MHz, D2O): δ 8.07 (s, 1H),
5.89 (d, J = 7.2 Hz, 1H), 4.78–4.72 (m, 1H),
4.69–4.64 (m, 1H), 4.48–4.42 (m, 1H), 4.21–4.12
(m, 2H). 31P{1H} NMR (162 MHz, D2O): δ 3.88, −0.92 (d, J = 19.0 Hz),
−11.27 (d, J = 19.2 Hz), −22.45 (dd, J = 19.1 Hz). 13C{1H} NMR (101 MHz,
D2O): δ 159.2, 154.0, 152.1, 137.9, 116.3, 86.6,
83.9 (dd, J = 9.2, 5.2 Hz), 73.5 (d, J = 3.6 Hz), 65.8 (d, J = 5.6 Hz). HRMS (ESI): m/z calcd for C10H15N6Na2O16P4 [M –
H]−, 644.9296; found, 644.9286.
Adenosine-5′-γ-P-diethylamido-triphosphate-3′-phosphate (15)
The reaction was performed according to General Procedure A using adenosine (7, 30.0 mg, 105 μmol) and diethylamine. The crude product was purified using Purification Method 1 and Purification Method 2 subsequently. The product (15, 42.3 mg, 56.2 μmol, 54%) was isolated as a white solid.
1H NMR (400 MHz, D2O): δ 8.49 (s, 1H), 8.18 (s, 1H), 6.08 (d, J = 7.1 Hz, 1H), 4.76 (dd, J = 7.2, 4.9 Hz, 1H), 4.47 (d, J = 2.8 Hz, 1H), 4.14 (t, J = 4.2 Hz, 2H), 2.84 (dq, J = 11.4, 7.1 Hz, 4H), 0.86 (t, J = 7.1 Hz, 6H). 31P{1H} NMR (162 MHz, D2O): δ 4.08, −0.35 (d, J = 24.8 Hz), −11.18 (d, J = 17.2 Hz), −22.49 (dd, J = 24.5, 17.5 Hz). 13C{1H} NMR (101 MHz, D2O): δ 155.7, 152.9, 149.5, 140.0, 118.6, 86.4, 84.1 (dd, J = 9.3, 5.3 Hz), 74.2 (d, J = 3.2 Hz), 73.7 (d, J = 4.6 Hz), 65.8 (d, J = 5.6 Hz), 40.4 (d, J = 3.7 Hz), 13.7 (d, J = 3.8 Hz). HRMS (ESI): m/z calcd for C14H25N6O15P4 [M – H]−, 641.0334; found, 641.0340.
Cation-Exchange Procedure 1
Nucleoside phosphates were applied to a Q-Sepharose column, eluted with NH4HCO3–buffer, and lyophilized. The resulting solid was redissolved in H2O, TBA(OH) was added, and the mixture was again lyophilized.
Cation-Exchange Procedure 2
Nucleoside phosphates were applied to a Chelex 100 column preconditioned with TBA(Br) (500 mM). UV-active fractions were combined and lyophilized.
General Procedure B
For the synthesis of amido-pppNpp
compounds, nucleoside tetraphosphate TBA salts were dissolved in DMF
(3.5 mM). ETT (3.5 equiv) was added, followed by a solution of (FmO)2P–N(iPr)2 (5, 1.7 equiv) in DMF (15 mM). The resulting solution was stirred for
15 min and cooled to 0 °C before mCPBA (2.1
equiv) was added. The solution was stirred for 10 min at 0 °C,
and DBU (10 vol %) was added. The mixture was stirred for 30 min at
rt and precipitated with Et2O (4 vol). The precipitate
was separated by centrifugation (7700g, 5 min), washed
with Et2O (2 × 15 mL), and dried under a high vacuum.
The crude product was dissolved in H2O and purified using
using Purification Method 2. Starting
material and cyclized products were recovered as mixtures and treated
with RNaseT2 for 3 h at 37 °C. Subsequent application of Cation-Exchange Procedure 1 regenerated the
corresponding starting materials.
Adenosine-5′-γ-P-propargylamido-triphosphate-3′-diphosphate (18)
The cation of 11 was exchanged using Cation-Exchange Procedure 1. The reaction was performed according to General Procedure B using 11 × 2.4 TBA (19.0 mg, 15.8 μmol). The product (18, 5.35 mg, 6.40 μmol, 41% [75% brsm]) was isolated as a white solid.
1H NMR (400 MHz, D2O): δ 8.48 (s, 1H), 8.19 (s, 1H), 6.13 (d, J = 6.3 Hz, 1H), 4.93–4.82 (m, 1H), 4.79–4.74 (m, 1H),
4.56–4.45 (m, 1H), 4.26–4.14 (m, 2H), 3.49 (dd, J = 9.9, 2.3 Hz, 2H), 2.30 (dd, J = 2.6
Hz, 1H). 31P{1H} NMR (162 MHz, D2O): δ −2.66 (d, J = 20.6 Hz), −5.54
(d, J = 22.1 Hz), −10.75 (d, J = 22.1 Hz), −11.23 (d, J = 17.9 Hz), −22.42
(dd, J = 20.6, 18.4 Hz). 13C{1H} NMR (101 MHz, D2O): δ 155.7, 152.9, 149.3, 140.0,
118.7, 86.7, 87.2–83.3 (m), 74.7 (d, J = 5.1
Hz), 73.7 (d, J = 4.2 Hz), 65.4 (d, J = 5.5 Hz), 30.8. HRMS (ESI): m/z calcd for C13H16N6Na6O18P5 [M + Na6 – H5]+, 836.8590; found, 836.8583.
Guanosine-5′-γ-P-propargylamido-triphosphate-3′-diphosphate (19)
The cation of 12 was exchanged using Cation-Exchange Procedure 1. The reaction was performed according to General Procedure B using 12 × 2.3 TBA (36.0 mg, 30.1 μmol). The product (19, 10.8 mg, 12.6 μmol, 42% [92% brsm]) was isolated as a white solid.
1H NMR (400 MHz, D2O): δ 7.97 (s, 1H), 5.84 (d, J = 6.2
Hz, 1H), 4.86–4.76 (m, 1H), 4.71 (dd, J =
5.6 Hz, 1H), 4.43–4.33 (m, 1H), 4.18–4.03 (m, 2H), 3.44
(dd, J = 10.1, 2.4 Hz, 2H), 2.30 (dd, J = 2.5 Hz, 1H). 31P{1H} NMR (162 MHz, D2O): δ −2.63 (d, J = 21.0 Hz),
−5.55 (d, J = 22.1 Hz), −10.75 (d, J = 22.2 Hz), −11.22 (d, J = 18.2
Hz), −22.43 (dd, J = 20.2, 18.5 Hz). 13C{1H} NMR (101 MHz, D2O): δ 159.3,
154.1, 152.0, 137.9, 116.4, 86.9, 83.2 (dd, J = 9.2,
4.1 Hz), 74.7 (d, J = 5.5 Hz), 73.0 (d, J = 4.2 Hz), 71.0, 65.5 (d, J = 5.1 Hz), 30.2. HRMS
(ESI): m/z calcd for C13H19N6O19P5 [M –
H2]2–, 358.9702; found, 358.9700.
Adenosine-5′-γ-P-amido-triphosphate-3′-diphosphate (20)
Cation of 13 was exchanged using Cation-Exchange Procedure 2. The reaction was performed according to General Procedure B using 13 × 5.1 TBA (18.0 mg, 9.92 μmol). The product (20, 5.67 mg, 7.11 μmol, 72%) was isolated as a white solid.
1H NMR (400 MHz, D2O):
δ 8.46 (s, 1H), 8.18 (s, 1H), 6.13 (d, J =
6.3 Hz, 1H), 4.87 (ddd, J = 8.5, 3.9 Hz, 1H), 4.78–4.74
(m, 1H), 4.55–4.50 (m, 1H), 4.26–4.13 (m, 2H). 31P{1H} NMR (162 MHz, D2O): δ −0.81
(d, J = 19.0 Hz), −5.46 (d, J = 22.1 Hz), −10.68 (d, J = 21.9 Hz), −11.20
(d, J = 18.7 Hz), −22.25 (dd, J = 18.8 Hz). 13C{1H} NMR (101 MHz, D2O): δ 155.6, 152.9, 149.3, 139.9, 118.7, 86.7, 83.4 (dd, J = 9.4, 4.4 Hz), 74.8 (d, J = 5.2 Hz),
73.8 (d, J = 4.2 Hz), 65.4 (d, J = 5.8 Hz). HRMS (ESI): m/z calcd
for C10H17N6NaO18P5 [M + Na – H2]−, 686.9191;
found, 686.9197.
Guanosine-5′-γ-P-amido-triphosphate-3′-diphosphate (21)
The cation of 14 was exchanged using Cation-Exchange Procedure 2. The reaction was performed according to General Procedure B using 14 × 8.0 TBA (46.0 mg, 18.1 μmol). The product (21, 9.70 mg, 11.7 μmol, 65%) was isolated as a white solid.
1H NMR (400 MHz, D2O):
δ 8.05 (s, 1H), 5.92 (d, J = 6.3 Hz, 1H), 4.87
(ddd, J = 8.5, 5.1, 3.2 Hz, 1H), 4.77 (ddd, J = 6.4, 5.0, 1.3 Hz, 1H), 4.51–4.44 (m, 1H), 4.26–4.11
(m, 2H). 31P{1H} NMR (162 MHz, D2O): δ −0.84 (d, J = 19.0 Hz), −5.60
(d, J = 22.1 Hz), −10.73 (d, J = 22.1 Hz), −11.24 (d, J = 19.1 Hz), −22.31
(dd, J = 19.1 Hz). 13C{1H}
NMR (101 MHz, D2O): δ 159.2, 154.0, 151.9, 137.9,
116.3, 86.9, 83.2 (dd, J = 9.0, 4.4 Hz), 74.8 (d, J = 5.2 Hz), 73.1 (d, J = 4.1 Hz), 65.6
(d, J = 5.6 Hz). HRMS (ESI): m/z calcd for C10H17N6O19P5 [M – H2]2–, 339.9624; found, 339.9622.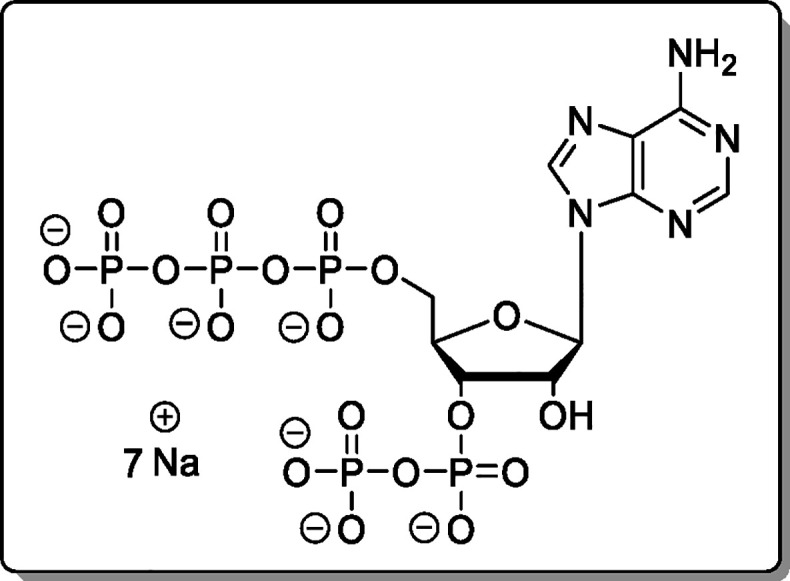
Adenosine-5′-triphosphate-3′-diphosphate (pppApp, 4)
Propargylamido-pppApp (18, 1.00 mg, 1.20 μmol) was dissolved in H2O and applied to a Dowex-H+ column and eluted with H2O. UV-active fractions (TLC) were combined and precipitated using 4 vol of NaClO4 solution (−20 °C, 500 mM in acetone). The precipitate was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 5.0 mL), and dried under a high vacuum. The product (4, 796 μg, 969 nmol, 81%) was isolated as a white solid.
Moreover, 4 was prepared from amido-pppApp (20); 20 (1.30 mg, 1.63 μmol) was dissolved in H2O (500 μL). The solution was acidified to pH 3 using HClO4 and incubated for 1 h at room temp. The product was precipitated using 4 vol of NaClO4 solution (−20 °C, 500 mM in acetone). The precipitate was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 3.0 mL), and dried under a high vacuum. The product (4, 1.14 mg, 1.43 μmol, 88%) was isolated as a white solid.
1H NMR (400 MHz, D2O): δ
8.53 (s, 1H),
8.24 (s, 1H), 6.14 (d, J = 7.0 Hz, 1H), 4.91 (br
s, 1H), 4.82 (t, J = 5.9 Hz, 1H), 4.59 (br s, 1H),
4.30–4.13 (m, 2H). 31P{1H} NMR (162 MHz,
D2O): δ −10.36 (d, J = 20.4
Hz), −10.64 (d, J = 19.0 Hz), −11.29
(d, J = 18.7 Hz), −11.67 (d, J = 19.9 Hz), −22.83 (br s). 13C{1H}
NMR (101 MHz, D2O): δ 154.3, 151.0, 149.2, 140.5,
118.6, 86.4, 83.7 (dd, J = 9.0, 2.8 Hz), 75.5 (d, J = 5.5 Hz), 73.9 (d, J = 5.1 Hz), 65.5
(d, J = 5.7 Hz). HRMS (ESI): m/z calcd for C10H17N5O19P5 [M + H]+, 667.9357; found, 667.9354.
Guanosine-5′-triphosphate-3′-diphosphate (pppGpp, 3)
Propargylamido-pppGpp (19, 2.60 mg, 3.05 μmol) was dissolved in H2O and applied to a Dowex-H+ column and eluted with H2O. UV-active fractions (TLC) were combined and precipitated using 4 vol of NaClO4 solution (−20 °C, 500 mM in acetone). The precipitate was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 5.0 mL), and dried under a high vacuum. The product (3, 1.87 mg, 2.23 μmol, 73%) was isolated as a white solid.
NMR data were in accordance with the literature. HRMS (ESI): m/z calcd for C10H16N5O20P5 [M – H2]2–, 340.4544; found, 340.4540.

18O-Adenosine-5′-triphosphate-3′-diphosphate (18O-pppApp, 22)
Propargylamido-pppApp (18, 500 μg, 598 nmol) was dissolved in H218O (50 μL). The solution was adjusted to pH 3 using HCl solution (conc) and incubated at rt for 1 h. The product was precipitated with NaClO4 solution (−20 °C, 500 mM in acetone, 5.0 mL). The precipitate was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 5.0 mL), and dried under a high vacuum. The product (22, 377 mg, 459 nmol, 77%, >93% 18O) was isolated as a white solid.
NMR data was in accordance
with data from 4. HRMS (ESI): m/z calcd for C10H16N5O1818OP5 [M – H2]2–, 333.4590; found, 333.4628.
Biotin-PEG3-triazolmethanamido-pppApp (23)
Propargylamido-pppApp (18, 1.06 mg, 1.27 μmol) was dissolved in TEAA–buffer (200 mM, 1.0 mL). A solution of biotin-PEG3-azide (1.41 mg, 3.18 μmol, 2.5 equiv) in DMSO (50 μL) was added. The solution was degassed for 10 min using argon. Afterward, sodium ascorbate (628 μg, 3.18 μmol, 2.5 equiv) and CuSO4 × 5 H2O (111 μg, 444 nmol, 35 mol %) were added, and the solution was stirred for 30 min at rt. The complete turnover was determined by HPLC. The product was precipitated with NaClO4 solution (−20 °C, 500 mM in acetone, 10 mL). The precipitate was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 5.0 mL), and dried under a high vacuum. The product (23, 1.61 mg, 1.26 μmol, 98%) was isolated as a white solid.
1H NMR* (400 MHz, D2O): δ 8.42 (s, 1H),
8.13 (s, 1H), 7.66 (s, 1H), 6.04 (d, J = 6.2 Hz,
1H), 4.88 (br s, 1H), 4.50 (t, J = 6.4 Hz, 2H), 4.36
(br s, 2H), 4.31 (dd, J = 7.9, 4.5 Hz, 1H), 4.28–4.16
(m, 2H), 3.91 (d, J = 9.2 Hz, 2H), 3.85–3.76
(m, 2H), 3.60–3.43 (m, 10H), 3.29 (dd, J =
5.3 Hz, 2H), 3.25–3.17 (m, 1H), 2.89 (dd, J = 13.1, 5.0 Hz, 1H), 2.68 (d, J = 13.0 Hz, 1H),
2.18 (d, J = 7.4 Hz, 2H), 1.72–1.41 (m, 4H),
1.36–1.23 (m, 2H). 31P{1H} NMR (162 MHz,
D2O): δ −2.39 (d, J = 20.2
Hz), −5.23 to −6.36 (m), −10.52 to −11.10
(m), −11.28 (d, J = 17.8 Hz), −22.01
to −23.05 (m). HRMS (ESI): m/z calcd for C31H51N12O23P5S [M – H2]2–, 573.0805;
found, 573.0815. The product was also characterized by HPLC-MS (see Supporting Information). (*) One proton (2′H
or 3′H) is hidden under the HDO peak.
Sulfo-cyanine-3-triazolmethanamido-pppApp (24)
Propargylamido-pppApp (1, 1.12 mg, 1.34 μmol) was dissolved in TEAA–buffer (200 mM, 1.0 mL). A solution of sulfo-cyanine3-azide (1.68 mg, 2.28 μmol, 1.7 equiv) in H2O (50 μL) was added. The solution was degassed for 10 min using argon. Afterward, sodium ascorbate (663 μg, 3.35 μmol, 2.5 equiv) and CuSO4 × 5 H2O (117 μg, 468 nmol, 35 mol %) were added, and the solution was stirred for 30 min at rt. The complete turnover was determined by HPLC. The crude product was applied to automated SAX (Äkta pure, Q-Sepharose, NaClO4–buffer). The product containing fractions were precipitated using a 4-fold volume of NaClO4 solution (−20 °C, 500 mM in acetone). The precipitate was separated by centrifugation (7700g, 5 min), washed with acetone (3 × 10 mL), and dried under a high vacuum. The product (24, 1.78 mg, 1.14 μmol, 85%) was isolated as a violet solid.
1H NMR* (400 MHz, D2O): δ 8.42 (s, 1H), 8.41 (dd, J = 13.6 Hz, 1H), 8.02 (s, 1H), 7.82 (d, J = 1.7 Hz, 1H), 7.81 (d, J = 1.3 Hz, 1H), 7.79 (dd, J = 4.1, 1.8 Hz, 1H), 7.77 (dd, J = 4.2, 1.8 Hz, 1H), 7.74 (br s, 1H), 7.32–7.28 (m, 2H), 6.24 (d, J = 13.5 Hz, 1H), 6.18 (d, J = 13.5 Hz, 1H), 6.03 (d, J = 6.5 Hz, 1H), 4.70 (br s, 1H), 4.50 (s, 1H), 4.29–4.15 (m, 4H), 4.06–3.91 (m, 4H), 3.49 (s, 3H), 2.99 (dd, J = 6.5 Hz, 2H), 2.09 (dd, J = 7.2 Hz, 2H), 1.96–1.87 (m, 2H), 1.75 (dd, J = 7.8 Hz, 2H), 1.68 (s, 6H), 1.66 (s, 3H), 1.66 (s, 3H), 1.56–1.45 (m, 2H), 1.34–1.23 (m, 2H). HRMS (ESI): m/z calcd for C46H64N12O25P5S2 [M]+, 1403.2230; found, 1403.2218. The product was also characterized by HPLC-MS (see Supporting Information). (*) One proton (2′H or 3′H) is hidden under the HDO peak.
Acknowledgments
We thank Dr. Manfred Keller from MagRes of the University of Freiburg for a significant amount of time for NMR spectroscopy and Christoph Warth for HRMS measurements. We thank Dr. M. Cashel for insightful comments. This study was supported by the Deutsche Forschungsgemeinschaft (DFG) under Germany’s Excellence Strategy (CIBBS, EXC-2189, Project ID 390939984). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 864246, to H.J.J.). Moreover, this study was supported by grants from the DFG (ID 403222702, SFB1381 to H.G.K,. and GRK2202 to C.J.T. and H.G.K.). We gratefully acknowledge financial support from the Studienstiftung des deutschen Volkes and the Brigitte-Schlieben-Lange-Programm.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c00841.
Detailed 1D and 2D NMR including structural assignments; HRMS and HPLC-MS characterization data; supplementary CE-UV data; influence of the salt concentration and pH on the separation of nucleotide standards; electropherogram of E. coli cell extracts (PDF)
Author Contributions
# T.M.H. and D.Q. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Potrykus K.; Cashel M. (p)ppGpp: still magical?. Annu. Rev. Microbiol. 2008, 62, 35–51. 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- Gaca A. O.; Kudrin P.; Colomer-Winter C.; Beljantseva J.; Liu K.; Anderson B.; Wang J. D.; Rejman D.; Potrykus K.; Cashel M.; Hauryliuk V.; Lemos J. A. From (p)ppGpp to (pp)pGpp: Characterization of Regulatory Effects of pGpp Synthesized by the Small Alarmone Synthetase of Enterococcus faecalis. J. Bacteriol. 2015, 197, 2908–2919. 10.1128/JB.00324-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalebroux Z.; Swanson M. S. ppGpp: magic beyond RNA polymerase. Nat. Rev. Microbiol. 2012, 10, 203–212. 10.1038/nrmicro2720. [DOI] [PubMed] [Google Scholar]
- Haugen S. P.; Ross W.; Gourse R. L. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 2008, 6, 507–519. 10.1038/nrmicro1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zbornikova E.; Rejman D.; Gerdes K. Novel (p)ppGpp Binding and Metabolizing Proteins of Escherichia coli. mBio 2018, 9, e02188–17 10.1128/mBio.02188-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Dai P.; Ding D.; Del Rosario A.; Grant R. A.; Pentelute B. L.; Laub M. T. Affinity-based capture and identification of protein effectors of the growth regulator ppGpp. Nat. Chem. Biol. 2019, 15, 141–150. 10.1038/s41589-018-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechold U.; Potrykus K.; Murphy H.; Murakami K. S.; Cashel M. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 2013, 41, 6175–6189. 10.1093/nar/gkt302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syal K.; Chatterji D. Differential binding of ppGpp and pppGpp to E. coli RNA polymerase: photo-labeling and mass spectral studies. Genes Cells. 2015, 20, 1006–1016. 10.1111/gtc.12304. [DOI] [PubMed] [Google Scholar]
- Keasling J. D.; Bertsch L.; Kornberg A. Guanosine pentaphosphate phosphohydrolase of Escherichia coli is a long-chain exopolyphosphatase. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 7029–7033. 10.1073/pnas.90.15.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn-Olszewska B.; Molodtsov V.; Sobala M.; Dylewski M.; Murakami K. S.; Cashel M.; Potrykus K. Structure-function comparisons of (p)ppApp vs (p)ppGpp for Escherichia coli RNA polymerase binding sites and for rrnB P1 promoter regulatory responses in vitro. Biochim. Biophys. Acta, Gene Regul. Mech. 2018, 1861, 731–742. 10.1016/j.bbagrm.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda A.; Murphy H.; Cashel M.; Kornberg A. Guanosine Tetra- and Pentaphosphate Promote Accumulation of Inorganic Polyphosphate in Escherichia coli. J. Biol. Chem. 1997, 272, 21240–21243. 10.1074/jbc.272.34.21240. [DOI] [PubMed] [Google Scholar]
- Zhang Y. E.; Baerentsen R. L.; Fuhrer T.; Sauer U.; Gerdes K.; Brodersen D. E. (p)ppGpp Regulates a Bacterial Nucleosidase by an Allosteric Two-Domain Switch. Mol. Cell 2019, 74, 1239–1249. 10.1016/j.molcel.2019.03.035. [DOI] [PubMed] [Google Scholar]
- Kudrin P.; Dzhygyr I.; Ishiguro K.; Beljantseva J.; Maksimova E.; Oliveira S. R. A.; Varik V.; Payoe R.; Konevega A. L.; Tenson T.; Suzuki T.; Hauryliuk V. The ribosomal A-site finger is crucial for binding and activation of the stringent factor RelA. Nucleic Acids Res. 2018, 46, 1973–1983. 10.1093/nar/gky023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinchen W.; Schuhmacher J. S.; Altegoer F.; Fage C. D.; Srinivasan V.; Linne U.; Marahiel M. A.; Bange G. Catalytic mechanism and allosteric regulation of an oligomeric (p)ppGpp synthetase by an alarmone. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 13348–13353. 10.1073/pnas.1505271112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhaese H. J.; Hoch J. A.; Groscurth R. Studies on the control of development: isolation of Bacillus subtilis mutants blocked early in sporulation and defective in synthesis of highly phosphorylated nucleotides. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 1125–1129. 10.1073/pnas.74.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikehara K.; Ando H.; Takada Y.; Sugae K. Presence of Guanosine 5′-Diphosphate 3′-Diphosphate in Bacillus subtilis Vegetative Cells. J. Biochem. 1981, 89, 511–516. 10.1093/oxfordjournals.jbchem.a133226. [DOI] [PubMed] [Google Scholar]
- Sobala M.; Bruhn-Olszewska B.; Cashel M.; Potrykus K. Methylobacterium extorquens RSH Enzyme Synthesizes (p)ppGpp and pppApp in vitro and in vivo, and Leads to Discovery of pppApp Synthesis in Escherichia coli. Front. Microbiol. 2019, 10, 859. 10.3389/fmicb.2019.00859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S.; Wang B.; Walker M. D.; Tran H.-K. R.; Stogios P. J.; Savchenko A.; Grant R. A.; McArthur A. G.; Laub M. T.; Whitney J. C. An interbacterial toxin inhibits target cell growth by synthesizing (p)ppApp. Nature 2019, 575, 674–678. 10.1038/s41586-019-1735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patacq C.; Chaudet N.; Létisse F. Absolute Quantification of ppGpp and pppGpp by Double-Spike Isotope Dilution Ion Chromatography-High-Resolution Mass Spectrometry. Anal. Chem. 2018, 90, 10715–10723. 10.1021/acs.analchem.8b00829. [DOI] [PubMed] [Google Scholar]
- Oki T.; Yoshimoto A.; Sato S.; Takamatsu A. Purine nucleotide pyrophosphotransferase from Streptomyces morookaensis, capable of synthesizing pppApp and pppGpp. Biochim. Biophys. Acta 1975, 410, 262–272. 10.1016/0005-2744(75)90228-4. [DOI] [PubMed] [Google Scholar]
- Krohn M.; Wagner R. A procedure for the rapid preparation of guanosine tetraphosphate (ppGpp) from Escherichia coli ribosomes. Anal. Biochem. 1995, 225, 188–190. 10.1006/abio.1995.1138. [DOI] [PubMed] [Google Scholar]
- Schattenkerk C.; Wreesmann C. T.; Van der Marel G. A.; Van Boom J. H. Synthesis of riboguanosine pentaphosphate ppprGpp (Magic Spot II) via a phosphotriester approach. Nucleic Acids Res. 1985, 13, 3635–3649. 10.1093/nar/13.10.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas T. M.; Ebensperger P.; Eisenbeis V. B.; Nopper C.; Dürr T.; Jork N.; Steck N.; Jessen-Trefzer C.; Jessen H. J. Magic spot nucleotides: Tunable Target-specific Chemoenzymatic Synthesis. Chem. Commun. 2019, 55, 5339–5342. 10.1039/C9CC01688K. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res. 2008, 41, 40–49. 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- Schwan J.; Christmann M. Enabling strategies for step efficient syntheses. Chem. Soc. Rev. 2018, 47, 7985–7995. 10.1039/C8CS00399H. [DOI] [PubMed] [Google Scholar]
- Singh J.; Steck N.; De D.; Hofer A.; Ripp A.; Captain I.; Keller M.; Wender P. A.; Bhandari R.; Jessen H. J. A Phosphoramidite Analog of Cyclotriphosphate Enables Iterative Polyphosphorylations. Angew. Chem., Int. Ed. 2019, 58, 3928–3933. 10.1002/anie.201814366. [DOI] [PubMed] [Google Scholar]
- Singh J.; Ripp A.; Haas T. M.; Qiu D.; Keller M.; Wender P. A.; Siegel J. S.; Baldridge K. K.; Jessen H. J. Synthesis of modified nucleoside oligophosphates simplified: Fast, pure, and protecting group free. J. Am. Chem. Soc. 2019, 141, 15013–15017. 10.1021/jacs.9b08273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezold D.; Dürr T.; Singh J.; Jessen H. J. Cyclotriphosphate: A Brief History, Recent Developments, and Perspectives in Synthesis. Chem. - Eur. J. 2020, 26, 2298–2308. 10.1002/chem.201904433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremosnik G. S.; Hofer A.; Jessen H. J. Iterative Synthesis of Nucleoside Oligophosphates with Phosphoramidites. Angew. Chem., Int. Ed. 2014, 53, 286–289. 10.1002/anie.201306265. [DOI] [PubMed] [Google Scholar]
- Varik V.; Oliveira S. R. A.; Hauryliuk V.; Tenson T. HPLC-based quantification of bacterial housekeeping nucleotides and alarmone messengers ppGpp and pppGpp. Sci. Rep. 2017, 7, 11022. 10.1038/s41598-017-10988-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckstein M. H.; He J.; Rubin H. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol. 2008, 190, 718–726. 10.1128/JB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbornikova E.; Knejzlik Z.; Hauryliuk V.; Krasny L.; Rejman D. Analysis of nucleotide pools in bacteria using HPLC-MS in HILIC mode. Talanta 2019, 205, 120161. 10.1016/j.talanta.2019.120161. [DOI] [PubMed] [Google Scholar]
- Jin H.; Lao Y. M.; Zhou J.; Zhang H. J.; Cai Z. H. A rapid UHPLC-HILIC method for algal guanosine 5′-diphosphate 3′-diphosphate (ppGpp) and the potential separation mechanism. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2018, 1096, 143–153. 10.1016/j.jchromb.2018.08.009. [DOI] [PubMed] [Google Scholar]
- Bartoli J.; Citerne S.; Mouille G.; Bouveret E.; Field B.. Quantification of guanosine tetraphosphate and other nucleotides in plants and algae using stable isotope-labelled internal standards. bioRxiv 2019. [DOI] [PubMed] [Google Scholar]
- Ihara Y.; Ohta H.; Masuda S. A highly sensitive quantification method for the accumulation of alarmone ppGpp in Arabidopsis thaliana using UPLC-ESI-qMS/MS. J. Plant Res. 2015, 128, 511–518. 10.1007/s10265-015-0711-1. [DOI] [PubMed] [Google Scholar]
- a Drouin N.; Pezzatti J.; Gagnebin Y.; González-Ruiz V.; Schappler J.; Rudaz S. Effective mobility as a robust criterion for compound annotation and identification in metabolomics: Toward a mobility-based library. Anal. Chim. Acta 2018, 1032, 178–187. 10.1016/j.aca.2018.05.063. [DOI] [PubMed] [Google Scholar]; b Soga T.; Igarashi K.; Ito C.; Mizobuchi K.; Zimmermann H.-P.; Tomita M. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal. Chem. 2009, 81, 6165–6174. 10.1021/ac900675k. [DOI] [PubMed] [Google Scholar]; c Liu J.-X.; Aerts J. T.; Rubakhin S. S.; Zhang X.-X.; Sweedler J. V. Analysis of endogenous nucleotides by single cell capillary electrophoresis-mass spectrometry. Analyst 2014, 139, 5835–5842. 10.1039/C4AN01133C. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Friedecký D.; Tomková J.; Maier V.; Janost’áková A.; Procházka M.; Adam T. Capillary electrophoretic method for nucleotide analysis in cells: application on inherited metabolic disorders. Electrophoresis 2007, 28, 373–380. 10.1002/elps.200600262. [DOI] [PubMed] [Google Scholar]
- Quaedflieg P. J. L. M.; Van der Heiden A. P.; Koole L. H.; Coenen A. J. J. M.; van der Wal S.; Meijer E. M. Synthesis and Conformational Analysis of Phosphate-Methylated RNA Dinucleotides. J. Org. Chem. 1991, 56, 5846–5859. 10.1021/jo00020a028. [DOI] [Google Scholar]
- Thilo E. The Structural Chemistry of Condensed Inorganic Phosphates. Angew. Chem., Int. Ed. Engl. 1965, 4, 1061–1071. 10.1002/anie.196510611. [DOI] [Google Scholar]
- Ermert S.; Marx A.; Hacker S. M. Phosphat-Modified Nucleotides for Monitoring Enzyme Activity. Top Curr. Chem. 2017, 375, 28. 10.1007/s41061-017-0117-8. [DOI] [PubMed] [Google Scholar]
- Müller A. C.; Giambruno R.; Weißer J.; Májek P.; Hofer A.; Bigenzahn J. W.; Superti-Furga G.; Jessen H. J.; Bennett K. L. Identifying Kinase Substrates via a Heavy ATP Kinase Assay and Quantitative Mass Spectrometry. Sci. Rep. 2016, 6, 28107. 10.1038/srep28107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A. M.; Taylor C. W. Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc. 2011, 6, 365–387. 10.1038/nprot.2011.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laventie B.-J.; Sangermani M.; Estermann F.; Manfredi P.; Planes R.; Hug I.; Jaeger T.; Meunier E.; Broz P.; Jenal U. A Sufrace-Induced Asymmetric Program Promotes Tissue Colonization by Pseudomonas aeruginosa. Cell Host Microbe 2019, 25, 140–152. 10.1016/j.chom.2018.11.008. [DOI] [PubMed] [Google Scholar]
- Ferullo D. J.; Lovett S. T. The Stringent Response and Cell Cycle Arrest in Escherichia coli. PLoS Genet. 2008, 4, e1000300 10.1371/journal.pgen.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juengert J. R.; Borisova M.; Mayer C.; Wolz C.; Brigham C. J.; Sinskey A. J.; Jendrossek D. Appl. Environ. Microbiol. 2017, 83, e00755–17 10.1128/AEM.00755-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.