

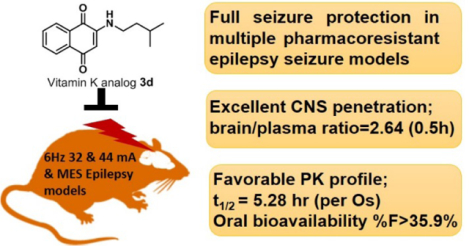
Abstract
Despite the availability of more than 25 anti-seizure drugs on the market, approximately 30% of patients with epilepsy still suffer from seizures. Thus, the epilepsy therapy market has a great need for a breakthrough drug that will aid pharmacoresistant patients. In our previous study, we discovered a vitamin K analog, 2h, which displayed modest anti-seizure activity in zebrafish and mouse seizure models. However, there were limitations for this compound due to its pharmacokinetic profile. In this study, we developed a new series of vitamin K analogs by modifying the structure of 2h. Among these, compound 3d shows full protection in a rodent pharmacoresistant seizure model with limited rotarod motor toxicity and favorable PK properties. Furthermore, the brain/plasma concentration ratio of 3d indicates its excellent permeability to the brain. The resulting data shows 3d can be further developed as a potential anti-seizure drug in the clinic.
Graphical Abstract
Introduction
Epilepsy is one of the most common neurological disorders and affects an estimated 70 million people worldwide.1 Many people with epilepsy have more than one type of seizure and may have additional neurological issues. Anti-seizure drugs (ASDs) are the mainstay treatment for epilepsy, effectively controlling seizures for around 70% patients with epilepsy. Driven by the limited efficacy and toxicity of the established anti-seizure agents, development of ASD has exploded in the past quarter-century, with more than 25 ASDs on the market.2 Nevertheless, around 30% of patients have inadequate control of their seizures, a percentage unchanged since 1881.3–4 Currently, there are two main challenges for the treatment of epilepsy; the first and the most important one is drug resistance, or pharmacoresistance, which refers to patients who do not achieve adequate seizure control after trials of at least two appropriate ASDs. Clinically, there are limited therapeutic intervention for pharmacoresistant epilepsy, but only for patients with disabling focal epilepsy.4 The second major concern for ASDs is tolerability, as people with epilepsy often require lifelong drug treatment.5 However, none of the ASDs currently on the market are free from side effects. In fact, these side-effects have been found to harm the quality of life of epilepsy patients more than the seizures themselves, and can lead to low adherence and discontinuation of treatment.6 New third-generation ASDs are marketable due to lowered toxicity and adverse drug interactions; however, they are only marginally (if at all) more efficacious than older drugs.7 Thus, the epilepsy therapy market is in great need of a breakthrough drug that would aid medication-resistant patients and be better tolerated.8–9 This leaves room for new drugs and drug combinations to gain market share quickly.
Pentylenetetrazol (PTZ), a chemical convulsant, induces seizures in various animal models of epilepsy, including worms, flies, frogs, zebrafish and mice,10–11 among which, the zebrafish PTZ seizure model is widely used for the initial screen of ASDs, since it matches well with rodent models and is amenable to higher-throughput screening.12 As epilepsy comes in many forms, it is also prudent that efficacy of the leading candidate is tested in a wide range of rodent models, particularly in animal models of medication-resistant epilepsy.7, 13 For over 40 years, the National Institute of Neurological Disorders and Stroke (NINDS) has supported the Epilepsy Therapy Screening Program (ETSP), which plays a key role in the identification of novel anti-seizure agents to address the unmet needs in epilepsy. The current workflow focuses on pharmacoresistant and etiologically relevant models (https://panache.ninds.nih.gov/CurrentModels.aspx).14–15 Here in this article, our compounds were screened for acute seizure models including the mouse 6 Hz seizure, the maximal electroshock (MES) seizure, and subcutaneous PTZ seizure threshold (s.c. PTZ) models. The 6 Hz models are believed to model partial seizures observed in humans which are used to screen compound’s ability to block psychomotor seizures. This model exhibits pharmacoresistance when the stimulation intensity is increased from 22 to 44 mA.16–17 The 6 Hz 44 mA model is resistant to ASDs that target voltage-gated sodium channels. What’s more, although levetiracetam and valproic acid are effective in this model, they are significantly less potent at this stimulation intensity.18–19 Therefore, the 6 Hz 44 mA model provides a high fidelity threshold assay early in the drug identification process. MES is a model for generalized tonic-clonic seizures and used for screening a compound’s ability to prevent seizure spread when all neuronal circuits in the brain are maximally active. These seizures are highly reproducible and are electrophysiologically consistent with human seizures (https://panache.ninds.nih.gov/CurrentModels.aspx). The mouse 6 Hz and MES seizure models are included in ETSP workflow for pharmacoresistant epilepsy. The s.c. PTZ-induced seizure model detects the test compound’s ability to protect animals from exhibiting clonic, forebrain seizures.
In our previous study, we discovered compound 2h, which was effective in the 6 Hz 32 mA mouse seizure model, but had limitations in the pharmacokinetic (PK) profile due to short t1/2 and limited central nervous system exposure (Figure 1). Moreover, 2h did not show any protection in mice when given the higher 44 mA stimulation intensity in the 6 Hz model.20 Therefore, in this study, we modify the structure of 2h to develop a new series of Vitamin K (VK) analogs. To accelerate the discovery process, a two-tier whole organism screening process was first set up using the zebrafish PTZ model, which is amenable to higher-throughput examination to establish structure activity relationship that correlates well with higher animal models.11 After establishing the structure-activity relationship (SAR) in the zebrafish model, top candidates were examined for in vivo anti-seizure activity and PK profiles in mice. Compound 3d shows titratable full protection in the 6 Hz seizure model at 32 and 44 mA, with limited motor toxicity and favorable PK properties, making 3d a leading ASD candidate.
Figure 1.
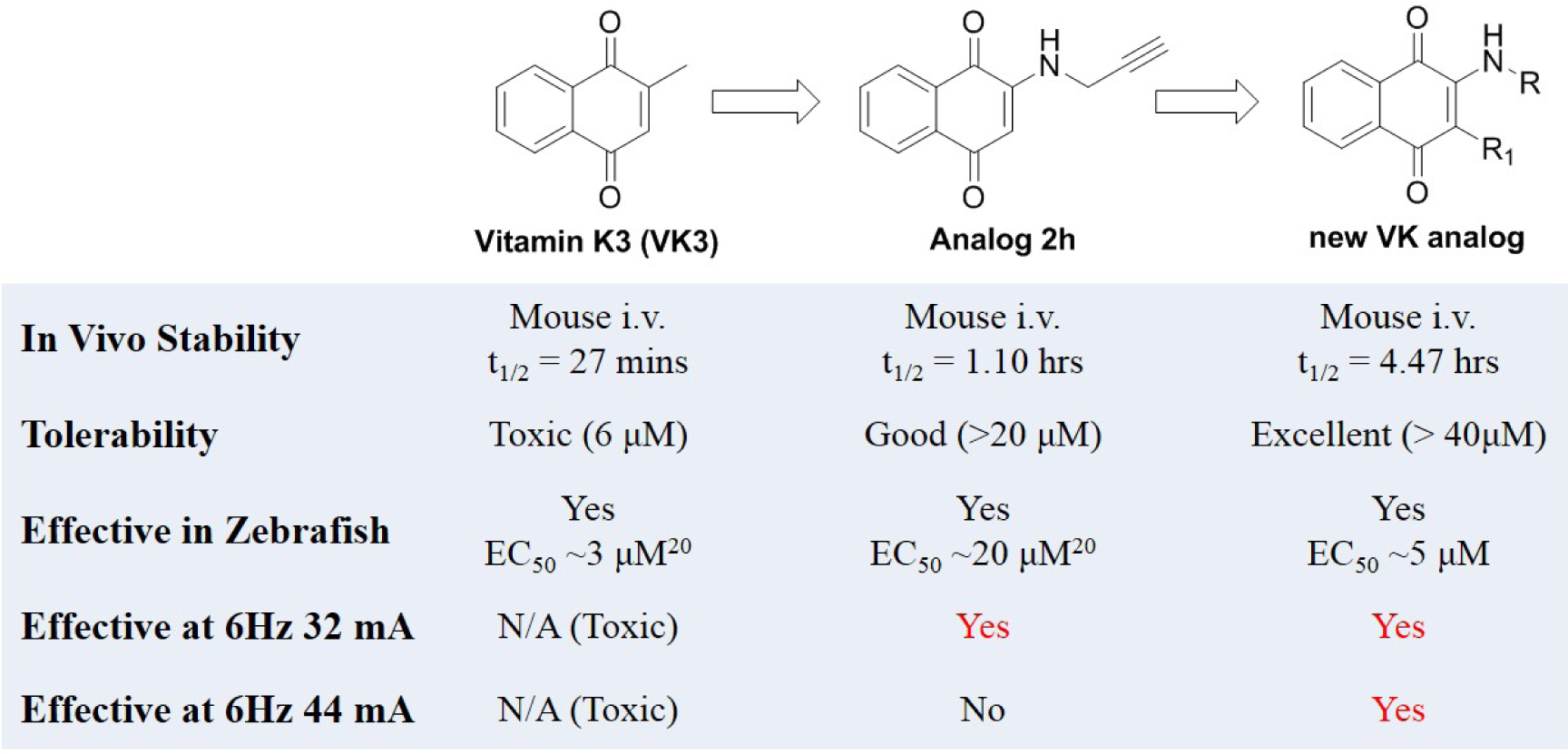
Evolution of a new VK analogs with excellent seizure protection and PK profile. VK3 and Analog 2h data are previously published.20
Chemistry
Scheme 1 shows the synthesis of 3a-3w. 2-bromonaphthalene-1,4-dione (1) was treated with various fatty amines (3a-3n), diamines (3o-3s) and alcohol amine (3t-3w) to yield desired compounds. 2-methylnaphthalene-1,4-dione (4) was reacted with 3,3-dimethylbutan-1-amine or 3-methylbutan-1-amine to obtain 5a or 5b, respectively ( Scheme 2).
Scheme 1:
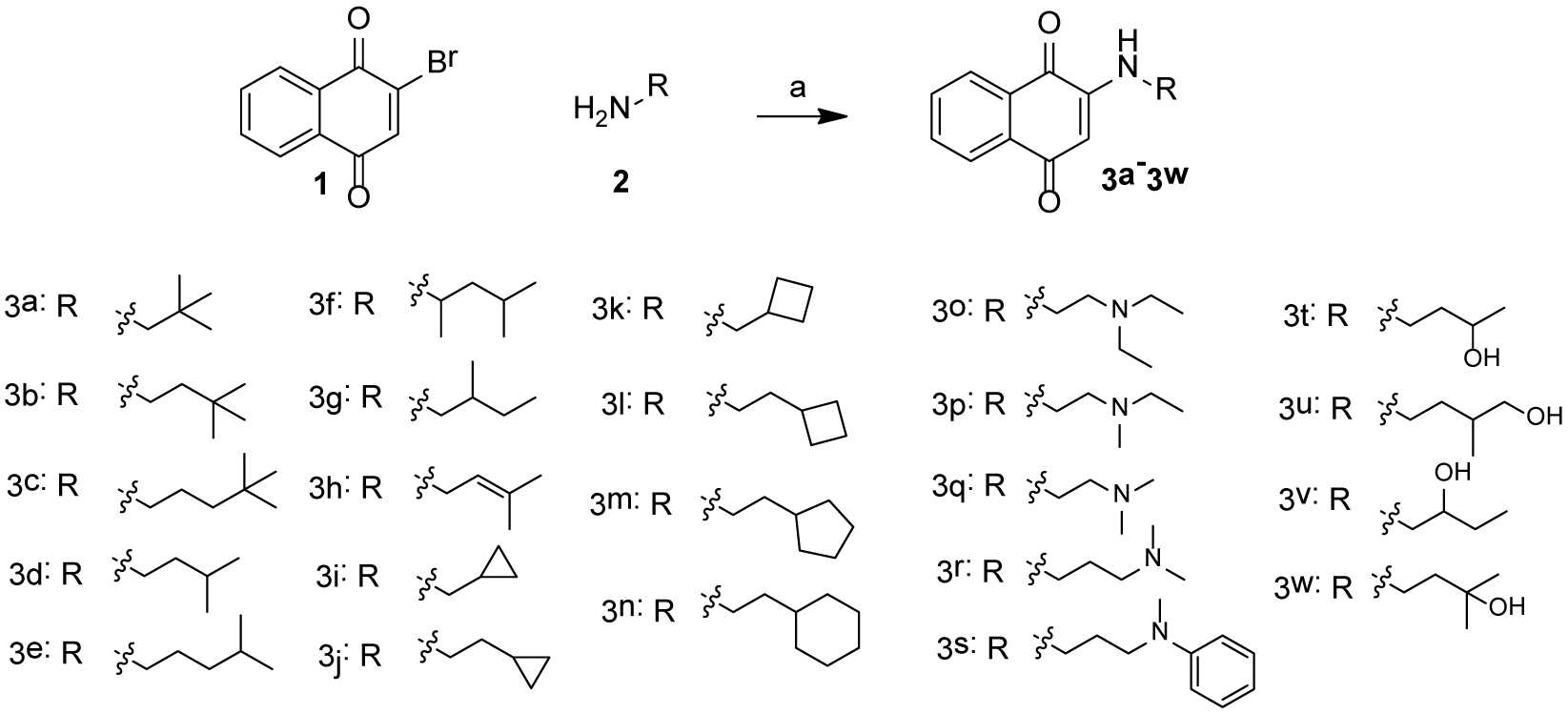
Reagent and condition: (a), different saturated and unsaturated fatty amines, ethanol, room temperature, overnight, 50–65% yield.
Scheme 2:
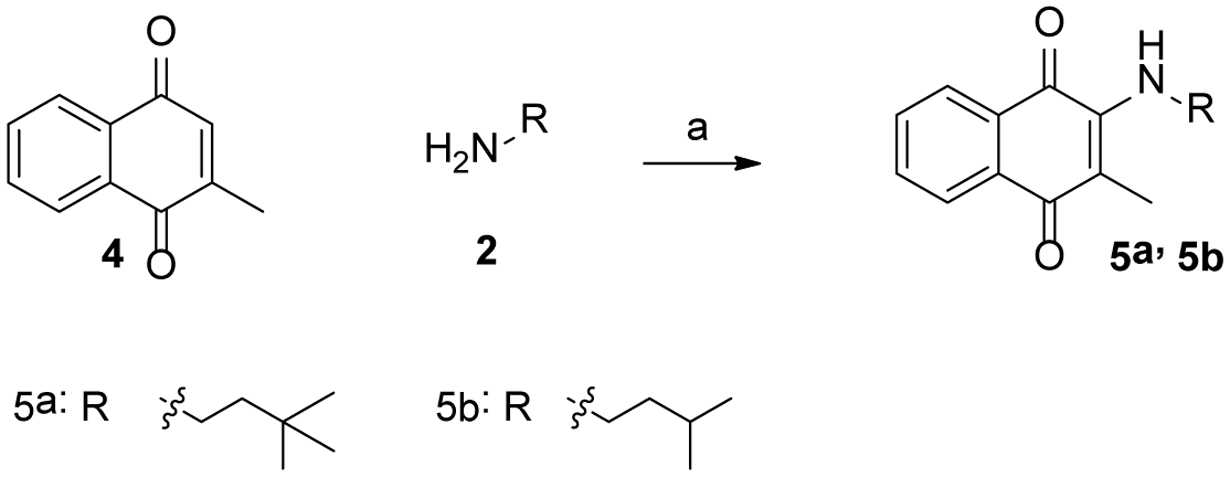
Reagent and condition: (a), 3-methylbutan-1-amine or 3,3-dimethylbutan-1-amine, methanol and dichloromethane, room temperature, overnight, 58–65% yield.
Results and Discussion
Discovery of A New VK Analog with Excellent Seizure Protection and PK Profile
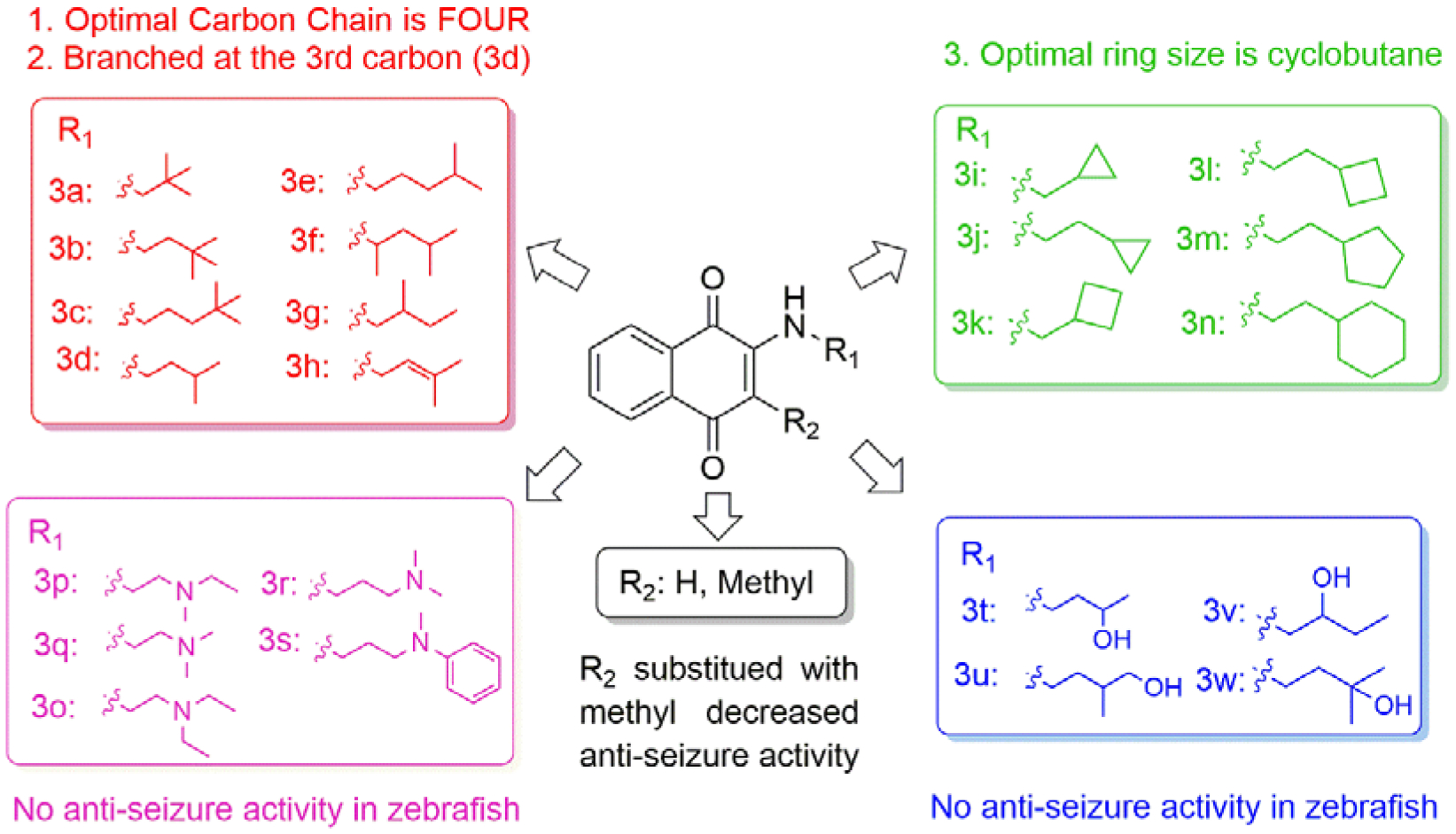
In our previous study, we tested VK1, VK2, and VK3, and found that the smallest VK, VK3, was protective and could reduce PTZ-induced locomotor activity in zebrafish. However, the lethal dose of VK3 in zebrafish is 7 μM, which is similar to its effective does (3–6 μM), therefore, VK3 shows significant toxicity.20 Further modification achieved VK analog 2h, which exhibited clear anti-seizure activity in zebrafish at the concentration of 20 μM with no observable toxicity. In 6 Hz 32 mA mouse models, 2h displayed full protection at 15 min; however, further testing in the higher intensity 6 Hz 44 mA model showed that 2h was no longer protective.20 The short half-life (1.06 h) of 2h correlates to its acute but short protective duration in the mouse seizure model (effective within 15 min but the protection drops off sharply in less than a half-hour). While the compound is stable, displaying no catabolic conversion even after two hours in mouse serum, its plasma concentration rapidly diminishes in vivo, suggesting either rapid hepatic metabolism or diminished renal reuptake. This finding is not surprising, as alkynes are known to be oxidized and excreted rapidly by the liver and kidney; therefore, we modified alkynes of 2h to produce a new series of VK analogs, and at the same time, also modified 2-position (R1 group) (Figure 1). In detail, substitution of alkynes with aliphatic chains yields compounds 3a-3h, while with cyclic aliphatic group yields 3i-3j. The compounds 3o-3s and 3t-3u were the structures containing tertiary amine and oxhydryl, respectively. Substitution of hydrogen of 3b and 3d with methyl yields 5a and 5b, respectively (Scheme 1).
Anti-seizure Activity of VK Analogs in Zebrafish
Previous studies showed that zebrafish displays analogous electrophysiological and molecular features associated with mammalian seizure models when exposed to the convulsant agent PTZ.11 The level of seizure severity in the brains of zebrafish, as determined by field potential measurements, corresponds with increased swimming activity, which is measured as distance traveled. The zebrafish assay can be used to screen greater than 500,000 mutagenized fish for seizure-resistant genes,21 and was further validated against rodent models of epilepsy with 13 current ASDs.12 Therefore, we initially used the zebrafish PTZ-induced seizure model to screen our compounds. Zebrafish larvae (7 days post-fertilization, dpf) were treated with synthesized candidates at concentrations of 10 μM and 5 μM for 1 h prior to seizure induction with 15 mM PTZ, 5 mM and 10 mM VPA were used as positive controls. Total swimming distance in 15 min was recorded after PTZ-induced seizures.20 PTZ treatment induced a 7-fold increase in the total distance traveled compared to the average control zebrafish (Figure 2a and 2b). The level of seizure protection by compounds at 10 μM are shown in Figure 2a. Previous lead compound 2h did not significantly reduce distance traveled at the lower 10 μM concentration; we had previously observed seizure protection at 20 μM in our earlier study.20 Among compounds with aliphatic chains (3a-3h), the compounds 3b, 3d, 3e, 3f, 3g and 3h protect against PTZ-induced seizures by reducing the total distance traveled by more than 50%. It should be noted that 10 μM 3d, 3f and 3l display better activity than 10 mM VPA, which was used as positive control. Compound 3b with substitution of 2,2-dimethylbutyl shows better suppressive activity than 3a with one carbon shorter and 3c with one carbon longer, which indicates 2 carbons is the optimal length. This is also consistent with the activity of 3d and 3e. Within compounds bearing cyclic aliphatic group, 3l and 3m, with cyclobutyl and cyclopentyl respectively, reduce distance traveled greater than 50% as compared to zebrafish treated with PTZ only. The activity difference between 3k and 3l is also consistent with the conclusion drawn from 3a-3c. Unfortunately, compounds with tertiary amine (3o-3s), oxhydryl (3t-3w) as well as compounds 5a and 5b with methyl group in 2-position show no protection against PTZ-induced seizures. Among all of the tested compounds, 3d and 3l display the best activity in zebrafish, by reducing the distance traveled by 77.4% and 89.0%, respectively. 3d and 3l also show the best activity at 5 μM (Figure 2b, Table 1). The mean distance traveled per 1 min of the most active compounds, 3d and 3l, is also shown in Figure 2c, from which we can observe that these two compounds significantly reduced distance traveled in every minute. The SAR is summarized in Figure 3.
Figure 2.
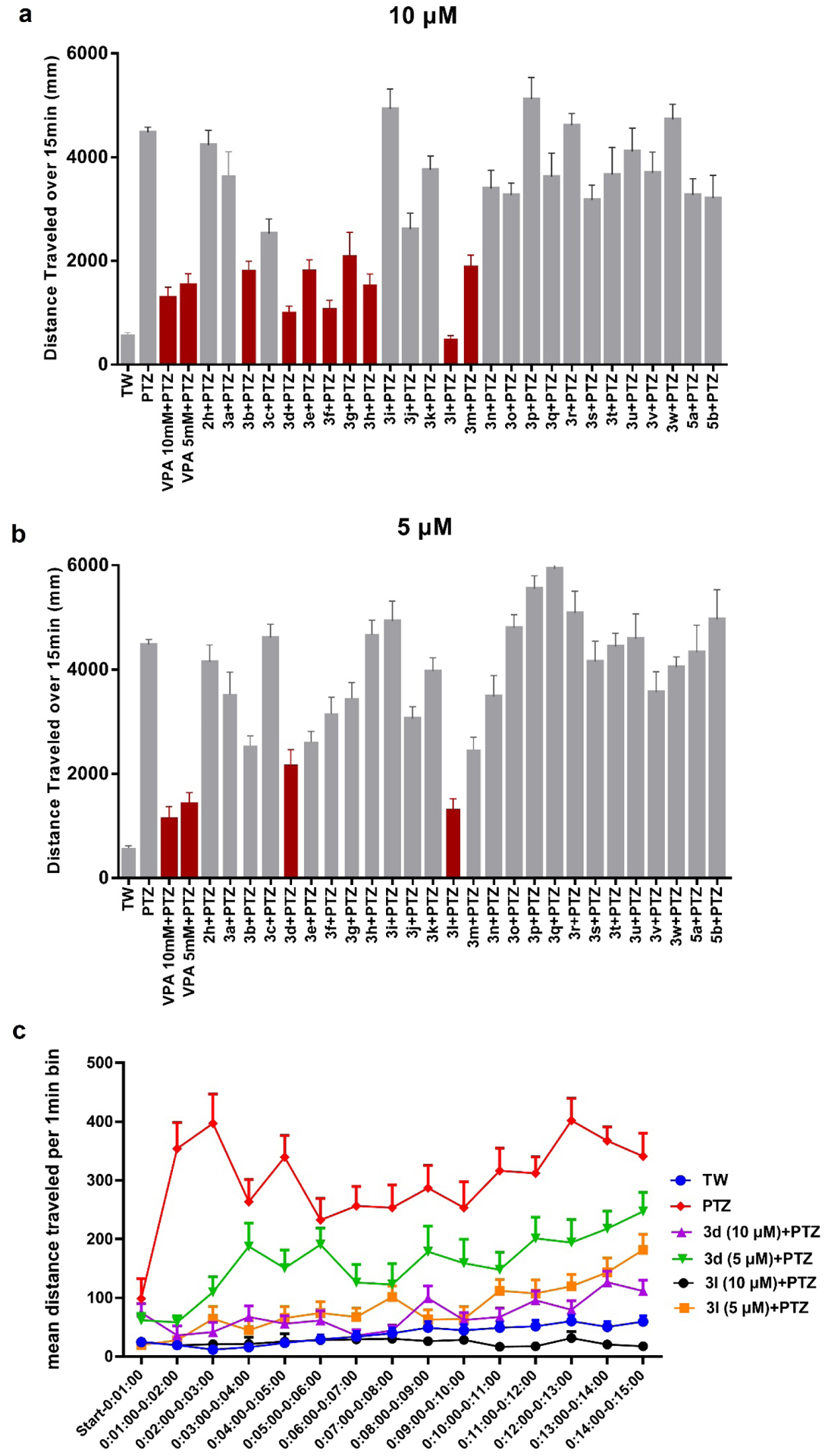
(a) and (b) Total mean distance traveled over 15 min. Synthesized compounds were tested at 10 μM and 5 μM, respectively. 5 mM and 10 mM VPA were used as positive controls. Data represents the mean distance traveled, error bar represents SEM, n = 8 for each group. The red color shows > 50% percentage of reduced travel distance (c) Mean distance traveled per 1 min after treatment of 3d and 3l, data are shown as mean distance traveled, error bar represents SEM, n = 8 for each group. With the exception of 3d at 10 μM + PTZ, all time points are significantly different from the positive PTZ run p < 0.01. TW = tank water only control.
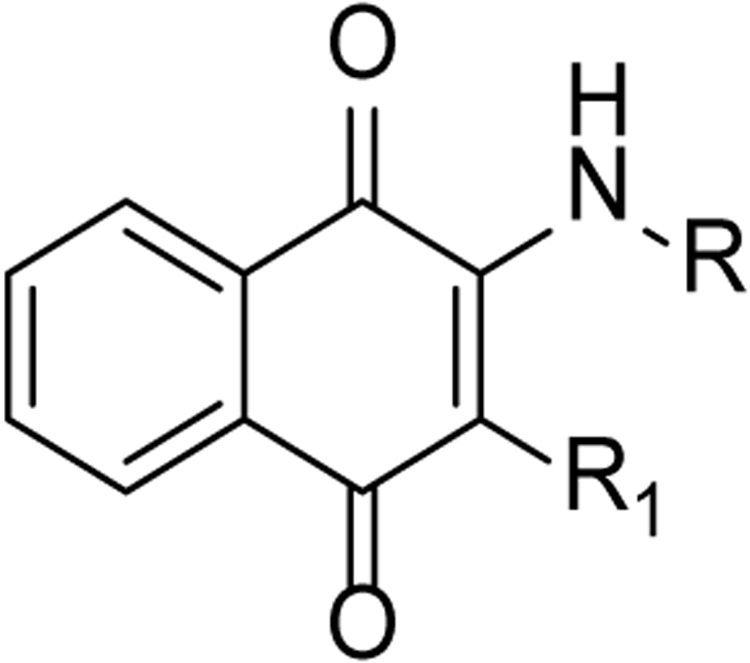
Table 1.
Reduction of PTZ-induced distance traveled after treatment of designed compounds at the concentration of 10 μM and 5 μM.
 | ||||
|---|---|---|---|---|
| Compounds No. | R | R1 | Percentage of reduced travel distance (%)a | |
| 10 μM | 5 μM | |||
| 2h | 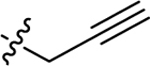 |
H | 5.4 ± 0.37 | 7.4 ± 0.58 |
| 3a | 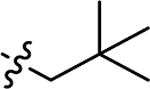 |
H | 18.4 ± 2.30 | 21.2 ± 2.53 |
| 3b | 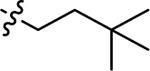 |
H | 59.2 ± 5.52 | 43.4 ± 3.30 |
| 3c | 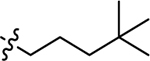 |
H | 43.5 ± 4.81 | −3.0 ± 0.17 |
| 3d | 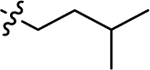 |
H | 77.4 ± 8.97 | 51.5 ± 6.79 |
| 3e | 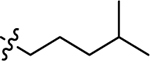 |
H | 59.1 ± 6.22 | 41.6± 3.20 |
| 3f | 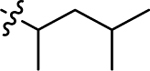 |
H | 75.8 ± 8.66 | 29.6 ± 2.97 |
| 3g | 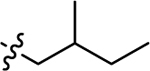 |
H | 52.9 ± 11.10 | 23.0 ± 2.01 |
| 3h | 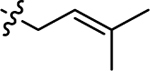 |
H | 65.6 ± 8.83 | −4.4 ± 0.26 |
| 3i | 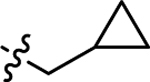 |
H | −10.1 ± 0.79 | −10.1 ± 0.79 |
| 3j | 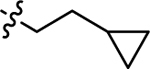 |
H | 41.6 ± 4.84 | 31.7 ± 2.38 |
| 3k | 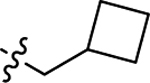 |
H | 16.1 ± 1.14 | 11.4 ± 0.73 |
| 3l |  |
H | 89.0 ± 11.34 | 70.4 ± 10.50 |
| 3m | 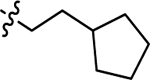 |
H | 57.4 ± 6.29 | 45.0 ± 4.75 |
| 3n | 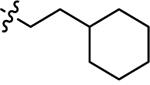 |
H | 24.1 ± 2.47 | 22.2 ± 2.58 |
| 3o | 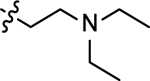 |
H | 27.0 ± 1.93 | −7.2 ± 0.38 |
| 3p | 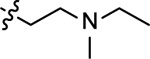 |
H | −14.3 ± 1.18 | −23.9 ± 1.08 |
| 3q | 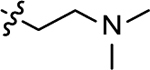 |
H | 19.1 ± 2.40 | −32.4 ± 2.01 |
| 3r | 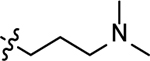 |
H | −2.90 ± 0.15 | −13.4 ± 1.13 |
| 3s | 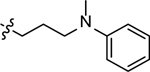 |
H | 29.1 ± 2.65 | 7.3 ± 0.69 |
| 3t | 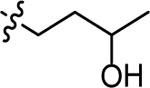 |
H | 18.2 ± 2.64 | 0.78 ± 0.04 |
| 3u | H | 8.2 ± 0.90 | −2.4 ± 0.26 | |
| 3v | 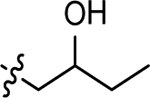 |
H | 17.4 ± 1.90 | 20.4 ± 2.28 |
| 3w | 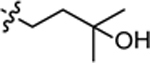 |
H | −5.6 ± 0.34 | 9.5 ± 0.46 |
| 5a | 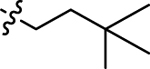 |
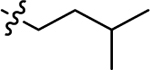 |
26.9 ± 2.55 | 3.3 ± 0.40 |
| 5b | 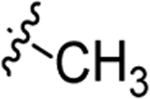 |
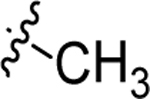 |
28.3 ± 3.88 | −10.8 ± 1.25 |
Assays were performed using eight zebrafish for each group. Values are the mean of ± SEM. n = 8 for each group. The negative number indicates increased distance traveled after treatment of designed compounds.
Figure 3.
Structure and activity relationship study of VK analogs.
Toxicity of VK Analogs in HT-22 Neurons
As most epilepsy patients need to use ASDs throughout their lives, safety is one of the major concerns of ASDs. Therefore, we tested the toxicity of these compounds in HT-22 neuronal cells (Table 2). At the concentration of 50 μM, compounds 3o-3r with tertiary amine exhibit the highest toxicity with about 100% growth inhibition. Even when the concentration was decreased to 10 μM, 3o-3r still inhibit growth by 74.5%−86.2%. It is interesting that when methyl group of N-substituent of 3r was changed to phenyl (3s), the percentage of inhibition is reduced to 10.25%. Compounds 3o-3s with oxhydryl groups also show high toxicity at a concentration of 50 μM with growth inhibition of 59.7%−76.7%, while inhibition drops to 19.4%−38.6% when the concentration is reduced to 10 μM. Compounds 3i, 3j and 3k with cyclic aliphatic groups show modest toxicity at 50 μM (54.1%−62.3%) which also decrease at the concentration of 10 μM. Compounds 3d and 3l, with the highest seizure protection in the PTZ-induced zebrafish seizure model, do not display obvious toxicity in HT-22 cells (Table 2).
Table 2.
Toxicity of VK analogs in HT-22-neurons.
| Compounds No. | Percentage of inhibition (%)a | |
|---|---|---|
| 50 μM | 10 μM | |
| 3a | 22.42 ± 6.63 | 10.78 ± 1.94 |
| 3b | 16.97 ± 10.55 | 11.91 ± 2.21 |
| 3c | 6.75 ± 1.29 | 3.81 ± 5.08 |
| 3d | 17.78 ± 1.41 | 11.86 ± 9.27 |
| 3e | 21.34 ± 3.42 | 28.77 ± 5.90 |
| 3f | 15.01 ± 2.87 | 2.09 ± 1.41 |
| 3g | 41.21 ± 2.98 | 14.81 ± 4.24 |
| 3h | 38.26 ± 0.84 | 14.96 ± 2.48 |
| 3i | 54.10 ± 6.32 | 16.83 ± 4.98 |
| 3j | 56.37 ± 0.79 | 34.69 ± 5.28 |
| 3k | 62.34 ± 0.07 | 33.70 ± 5.31 |
| 3l | 36.29 ± 4.49 | 27.21 ± 0.82 |
| 3m | 43.76 ± 2.83 | 35.37 ± 4.70 |
| 3n | 2.09 ± 2.06 | 0.07 ± 0.03 |
| 3o | 100.05 ± 0.21 | 83.84 ± 0.33 |
| 3p | 100.96 ± 0.52 | 86.21 ± 0.26 |
| 3q | 99.78 ± 0.16 | 82.63 ± 3.28 |
| 3r | 96.69 ± 0.69 | 74.53 ± 0.62 |
| 3s | 10.25 ± 6.61 | 11.53 ± 1.71 |
| 3t | 64.21 ± 6.55 | 19.39 ± 9.48 |
| 3u | 76.65 ± 3.41 | 25.66 ± 2.59 |
| 3v | 69.02 ± 2.45 | 38.61 ± 0.01 |
| 3w | 59.67 ± 0.11 | 25.52 ± 2.66 |
| 5a | 17.54 ± 2.83 | 10.15 ± 1.34 |
| 5b | 21.39 ± 3.72 | 11.24 ± 2.56 |
Note: Values are the mean ± SEM, three experiments were performed for each data (n = 3).
In Vivo Anti-seizure Activity in Rodent Seizure Models
The 6 Hz limbic seizure test using a stimulus intensity of 44 mA, is an acute model for pharmacoresistant seizures. Therefore, the most potent compounds 3d and 3l in zebrafish were tested for their anti-seizure activity in the 6 Hz rodent seizure models. Initial screening assays were performed using the lower stimulus intensities: 22 mA and 32 mA. Unfortunately, the most potent compound, 3l, in the PTZ-induced zebrafish seizure model shows limited efficacy in the 6 Hz mouse seizure model at 22 mA, without full seizure protection up to 400 mg/kg (Supporting Information Table S1). Thus, we did not continue testing 3l in the 6 Hz seizure model at 32 mA and 44 mA. First, a time course study was performed to establish a time to peak seizure protection of 1 h in 6 Hz models following ip administration (Supporting Information Table S2). Then, a dose-response quantitation study was performed at the 1 h peak response time to determine the ED50. Compound 3d is highly effective at both 32 mA and 44 mA, with full protection at the concentration of 400 mg/kg without obvious motor/sedative effects. EC50 values of 3d in the 6 Hz model at 32 mA and 44 mA are 152.7 and 263.7 mg/kg, respectively (Figure 4a, Table 3). Previously, 2h did not show any protection in 6 Hz 44 mA model,20 therefore, 3d is more potent than both 3l and 2h. This is consistent with the established pharmacoresistant seizures; most drugs lose seizure protection effectiveness with increased mA, thus making this increased 44 mA voltage intensity a model of pharmacoresistance. Furthermore, only four FDA approved anti-seizure medications to date (valproic acid, levetiracetam, felbamate and cannabidiol) have shown activity in this particular model and are all used clinically to treat refractory or hard-to-control seizures seizures.22–23 We also compared the EC50 and TD50 (median toxic dose, a toxicology term that relates to the median toxic dose of a substance in which toxicity occurs in 50% of a species) of 3d with these four drugs (Table 4). The reported EC50 value of levetiracetam is over 1000 mg/kg in this model and it is one of the most commonly prescribed medicine for refractory seizures, compound 3d’s EC50 value is comparable to all other anti-seizure medicines with EC50 values ranging from 100 mg/kg to 300 mg/kg. It should be noted that 3d is well tolerated with a TD50 value > 800 mg/kg, which is much higher than the other FDA approved drugs. Then 3d was further screened in MES model and produced full protection at 350 mg/kg with EC50 value of 108.12 mg/kg (Figure 4b, Table 3). Result shows 3d blocks seizures in the mouse 6 Hz and MES models in a dose-dependent manner, reaching full seizure protection at doses that do not produce motor impairment, which indicates its potential in the treatment of pharmacoresistant seizures. To provide more data of 3d’s efficacy against clonic seizures, the ED50 value was also determined in the s.c. PTZ mouse model at the 1h peak point. 3d protects 6 out of 8 animals at the concentration of 450 mg/kg with ED50 value of 349.22 mg/kg (Figure 4c, Table 3). Of note, 3d does not produce major motor impairment at the time of seizure testing, as the rotarod assay was performed immediately prior to 6 Hz stimulation (1 h after administration). However, other effects of treatment are noted following administration with 3d that will be the subject of ongoing investigation. Namely, 3d administration is associated with writhing (abdominal constriction) both at the time of treatment and during the ~1 h observation period that followed. In addition, some animals also show discolored urine and lethargy following treatment (data not shown). Subsequent studies also reveal that higher doses of 3d (e.g. greater than 500 mg/kg) are lethal in some animals after electric stimulation after 24h (data not shown). The mechanisms contributing to the lethality will be the subject of future studies. Nonetheless, 3d does not show signs of major motor impairment during the acute (~1h) observation period with a treatment dose up to 800 mg/kg in mice (mouse rotarod assay) (Supporting Information Table S2).
Figure 4.
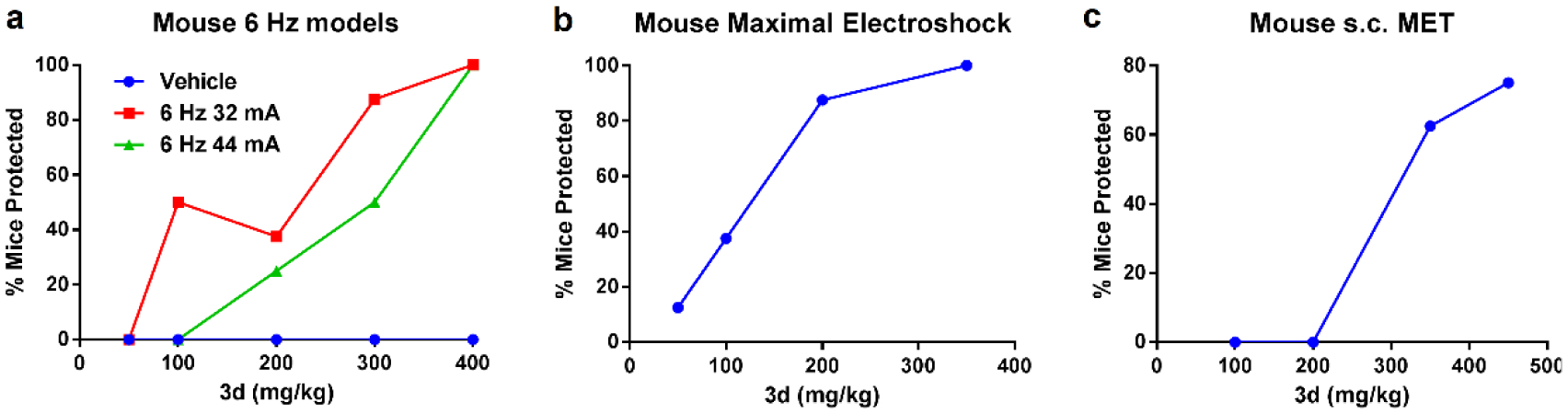
Compound 3d in the acute seizure models, 3d was injected with ip administration 1 h prior to electrical stimulation or injection of Metrazol. The percentage of mice protected is shown for each of the four dose levels for 3d (n = 8). (a) Dose-dependent effect of 3d in mouse 6 Hz 32 mA and 44 mA seizure models. (b) Dose-dependent effect of 3d in the mouse MES seizure model. (c) Dose-dependent effect of 3d in the s.c. MET seizure model.
Table 3.
Effect of 3d in 6 Hz mouse seizure models.
| Seizure model | Time of test | ED50 value (mg/kg) | 95% confidence interval (mg/kg) |
|---|---|---|---|
| 6 Hz 32mA mouse model | 1 h | 152.7 | 98.6–211.0 |
| 6 Hz 44mA mouse model | 1 h | 263.7 | 198.9–321.8 |
| MES model | 1 h | 108.1 | 70.4–152.9 |
| s.c. PTZ mouse model | 1 h | 349.2 | 256.0–432.1 |
Table 4.
Comparison of 3d and four FDA approved ASDs in the 6 Hz 44 mA seizure model.
Vitamin K Analogs Increase ATP Levels
The exact molecular mechanisms of seizures are unknown and may involve multiple different factors. The 6 Hz 44 mA seizure model is particularly resistant to ASDs that target voltage gated sodium and potassium channels. To eliminate that our inhibitors are targeting these ion channels, recurrent epileptiform discharges (REDs) in brain slices derived from the kainite-induced status epilepticus model of temporal lobe epilepsy were examined in vitro.24 Compound 3d does not attenuate spontaneous REDs (Supporting Information Figure S1), which is consistent with the hypothesis that it has novel mechanism of action other than voltage gated sodium and potassium ion channel inhibitors. Importantly, neurons have high metabolic energy demands, as well as a low capacity to store ATP. A sudden change in ATP levels in neurons can impair Na/K ATPase activity and decrease neuronal membrane potential, which can give rise to an increase in neuronal excitability resulting in seizures.25 In addition, heightened neuronal excitability impairs calcium sequestration resulting in an increased glutamate release into synaptic clefts, which can result in neuronal injury.25 The ketogenic diet and valproic acid (VPA) are commonly used in combination for relapse seizures and the mechanism of action is hypothesized to be involved in modulating mitochondrial activity.26–29 In the 12 marked ASDs tested in the pharmacoresistant seizure models, only VPA and levetiracetam displayed protection and both have mitochondrial effects.30 VK is also known to play a role in the mitochondrial electron transfer chain and is capable of maintaining mitochondrial homeostasis. What’s more, in our previous study, VK3 and its analog 2h can increase ATP levels in HT-22 cells at the concentration of 5 μM.20 Hence, effects of 3d and 3l on ATP levels were determined for both zebrafish and neuronal cells (HT-22). In zebrafish, 3d and 3l increase ATP levels by 19.0% and 21.4% at the concentration of 1 μM, respectively (Figure 5). In HT-22 cells, 3d and 3l increase ATP levels by 25.2% and 11.0% at the concentration of 0.5 μM, respectively (Figure 5). Therefore, the most potent compounds at preventing seizures in zebrafish, 3d and 3l, significantly increase ATP levels in zebrafish as well as HT-22 cells, which indicates that maintaining mitochondrial homeostasis and ATP levels are likely to play a role in its mechanism of action. The VK analogs’ molecular target likely involves proteins in the quinone oxidation/reduction process in the electron transport chain (ETC). VK in combination with vitamin C has been used in the clinic to enhance the oxidative phosphorylation process for the treatment of rare mitochondrial diseases with complex III mutations.31–32 VK has also been shown to play a key role as an alternative electron carrier that can rescue pink1 knock-out phenotype in Drosophila, and the VK synthase is a dominant enhancer of the phenotype.33 VK is also involved in maintaining calcium ion homeostasis in cells through modulating γ-glutamyl carboxylation of VK-dependent proteins. Proteins such as growth arrest specific gene 6 (Gas-6) and protein S regulate calcium ion signaling in the central nervous system. Gas-6 activation prevents amyloid beta protein and phospholipase A(2)-IIA induced calcium ion influx and attenuates neuronal death.34–35 Calcium ions, in turn, play a key role in the induction of seizures. Thus, the ability of VK to modulate the levels of calcium ion in neurons could be a major contributing factor in its ability to attenuate seizures.
Figure 5.
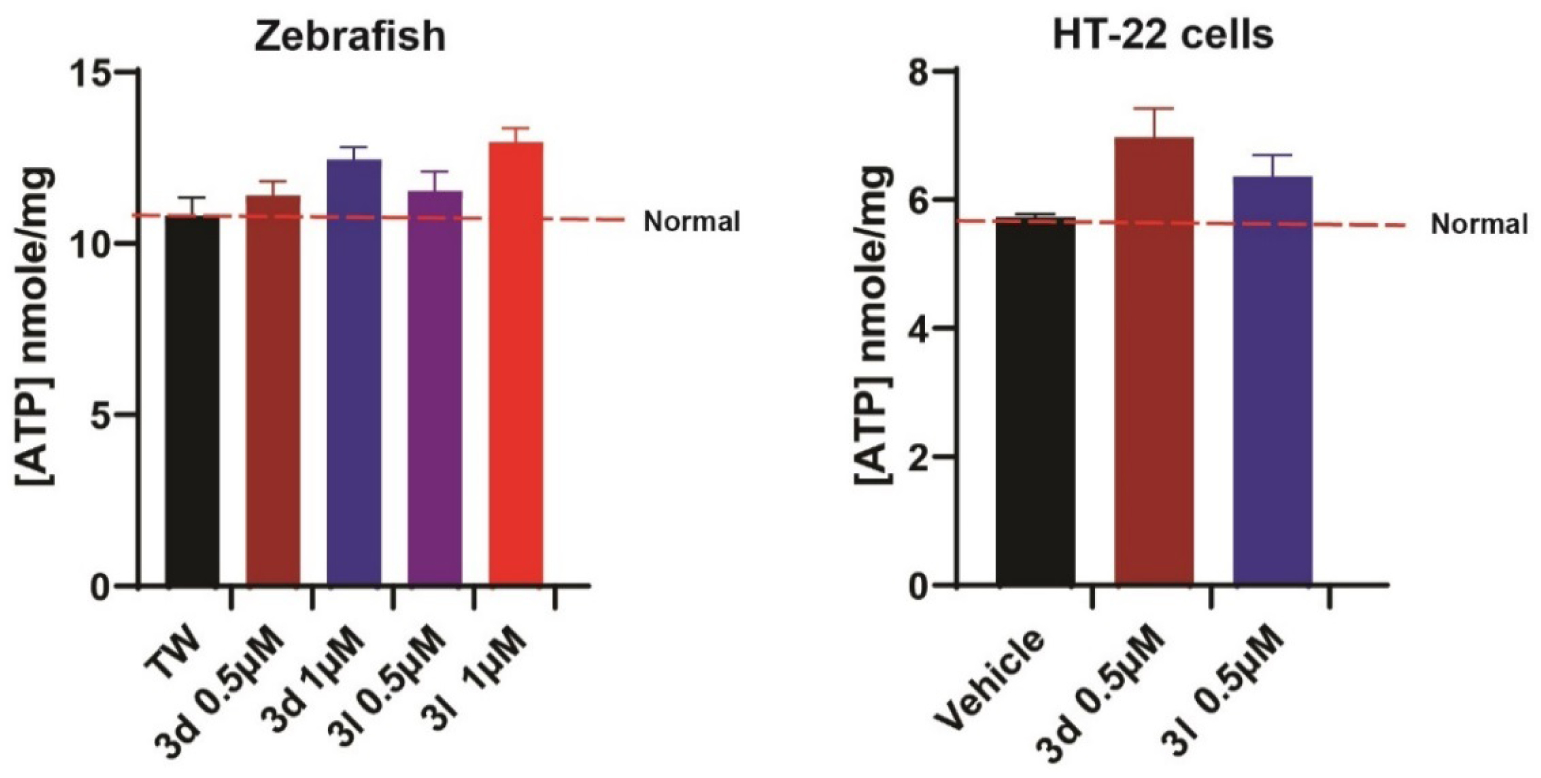
3d and 3l significantly increase ATP concentrations (nmole/mg) in zebrafish and HT-22 cells. Mean concentrations are plotted ± SEM, n = 7–13. Treatment with compounds significantly increased ATP concentrations compared to controls (p < 0.05).
Tissue Distribution and PK Profile of 3d
Compound 3d displays promising activity in rodent models and was chosen for further evaluation. As epilepsy is a neurological disorder, effective drug candidates need to cross the blood-brain barrier. 3d was expected to have excellent lipophilicity and blood-brain barrier permeation as shown by its predicted cLogP of 3.11 and a tPSA of 46.17 (molinspiration software http://www.molinspiration.com/). To verify the blood-brain barrier permeation of our compounds, 3d was assessed for brain/plasma concentration ratio at the 0.5, 1, 2, 4, and 8 h time points, respectively. The brain/plasma ratio for 3d is 2.64 at 0.5 h, and the ratio declines to 0.816 at 8 h, which indicates the excellent permeability of 3d (Figure 6). The brain/plasma concentration ratio of 2h is 0.325 at 0.5 h (Supporting information Table S3), which is much lower than 3d.
Figure 6.
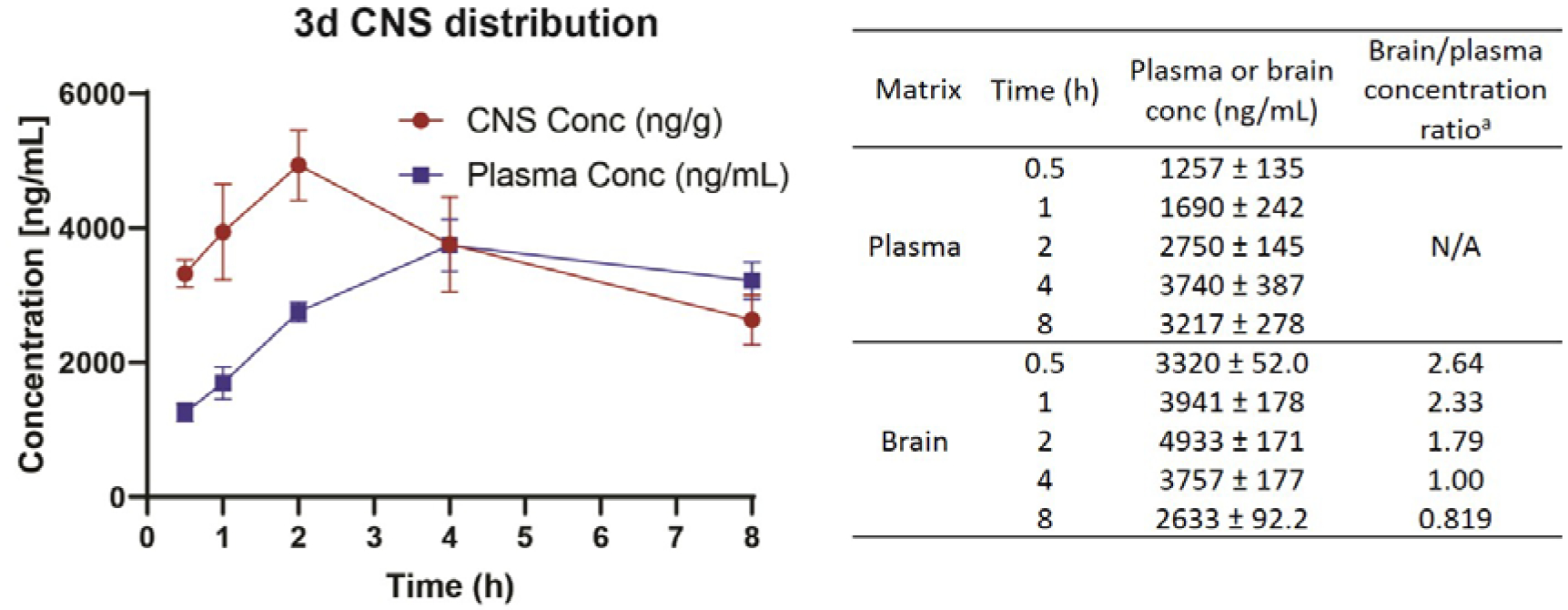
Brain/plasma concentration ratio of 3d with i.p. administration at concentration of 400 mg/kg.
Improvement of the PK profile of 2h is one of our goals to develop the new VK analogs as potential ASDs; therefore, we accessed the PK of 3d in mice. As we used ip administration in rodent models, we initially studied the PK profile of 3d with ip administration and compared with 2h. Compound 2h rapidly reaches its mean peak plasma concentration (Cmax) within 15 min. After that, its plasma concentration declines quickly with a short t1/2 of 1.10 h (Table 5, Figure 7a). Unlike 2h, 3d reaches its Cmax within 60 min, and its plasma concentration declines more slowly with a much longer t1/2 of 3.38 h (Table 5, Figure 7b). The area under the plasma concentration versus time curve (AUC) of 3d is 1,726 hr*ng/mL, which is 1.8 times higher than 2h (978 hr*ng/mL) (Table 5). Therefore, 3d displays a more favorable PK profile with longer t1/2 and higher AUC. A detailed PK study of 3d was also conducted by i.v. and oral administration; parameters are shown in Figure 8 and Table 6. With i.v. and oral administration, t1/2 of 3d are 4.47 and 5.28 h, respectively, and oral bioavailability of 3d is 35.9%. The PK profile indicates 3d is also suitable for oral administration.
Table 5.
PK parameters of 2h and 3d.a
| Cpd No. | Administration | Dose (mg/kg) | t1/2 (h) | tmax (h) | Cmax (ng/kg) | AUClast (hr*ng/mL) |
|---|---|---|---|---|---|---|
| 2h | ip | 20 | 1.10 ± 0.08 | 0.25 | 1490 ± 79.3 | 978 ± 111.5 |
| 3d | ip | 20 | 3.38 ± 0.16 | 1.00 | 320 ± 91.7 | 1716 ± 264.9 |
Note: ip intraperitoneal administration. Three mice were used for each compound.
Figure 7.
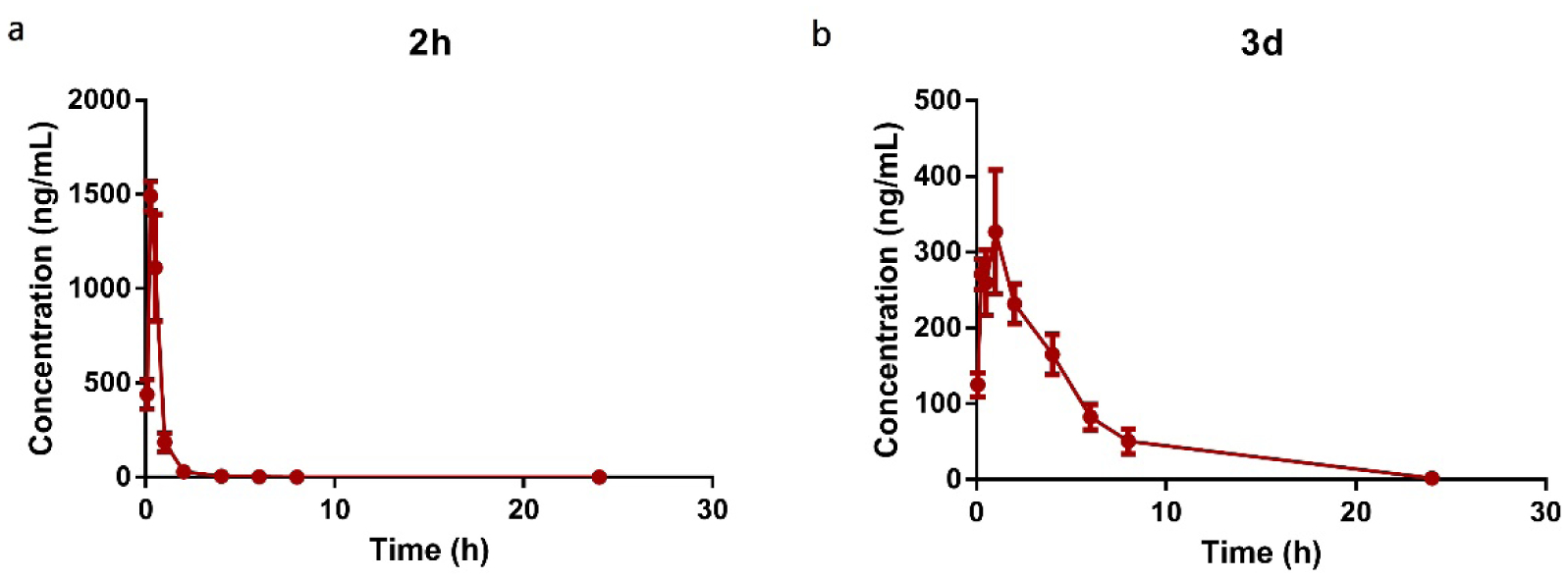
Comparison of PK profile of 2h and 3d by ip administration at 20mg/kg. 3 mice were used for each compound.
Figure 8.
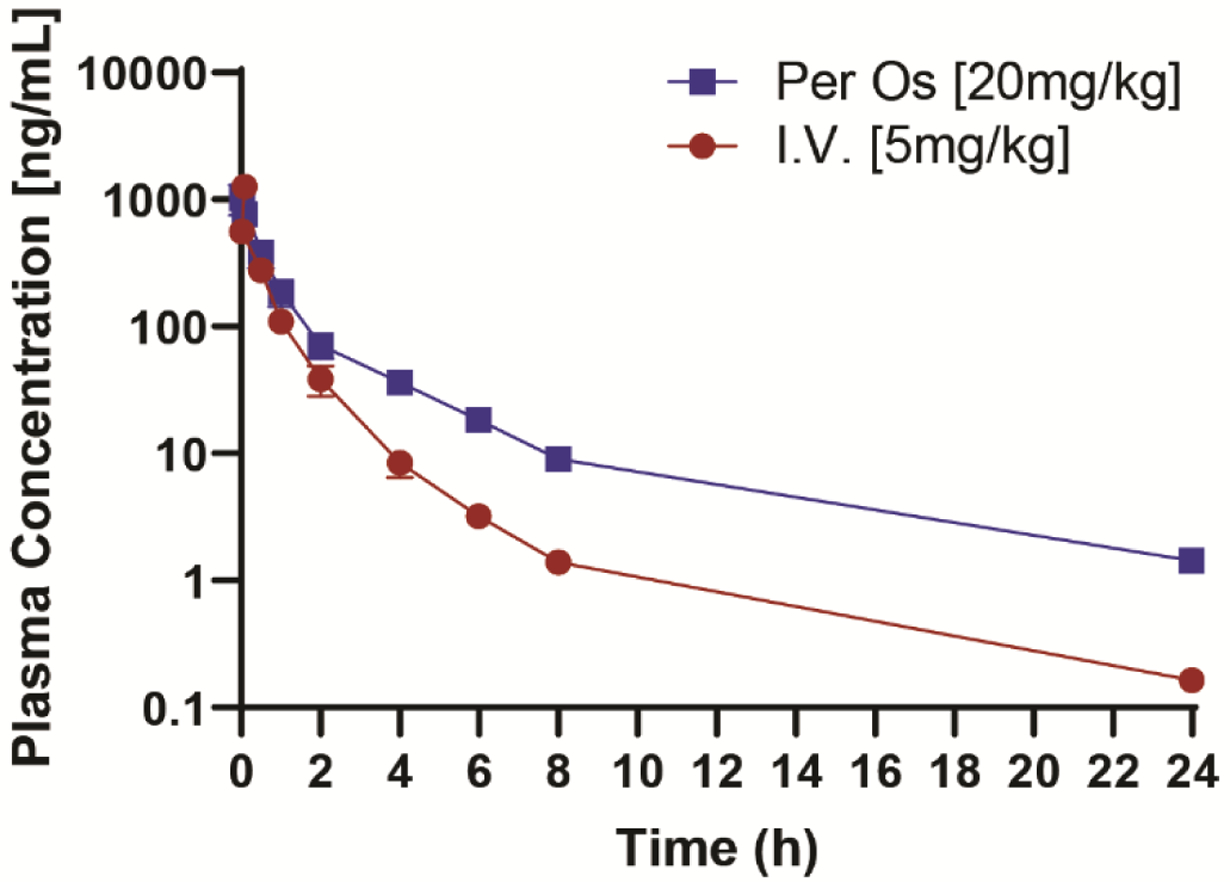
PK profile of 3d with iv and oral administration three mice were used for each compound.
Table 6.
PK parameters of 3d with iv and oral administration.a
| Administration | Dose (mg/kg) | t1/2 (h) | Cmax (ng/kg) | AUClast (hr*ng/mL) | F (%) |
|---|---|---|---|---|---|
| iv | 5 | 4.47±0.54 | 1907±84.65 | 636±25.5 | ND |
| po | 20 | 5.28±0.55 | 1025±273.3 | 903±175.5 | 35.9±6.7 |
Note: iv: intravenous injection administration; po: oral administration. Three mice were used for each experiment.
CYP450 Isoform and hERG Inhibition
Cytochrome P450 (CYP450) isozymes play an important role in metabolism of drugs. Assessment of the potential of a compound to inhibit a specific CYP450 enzyme allows prediction of potential drug-to-drug interactions that could occur in humans as the compound may alter the metabolism of a co-administered drug. Among all of the CYP450 isoforms, there are six major CYP450s that metabolize 90 percent of drugs. The two most significant are CYP3A4 and CYP2D6.36 According to the FDA Drug-Drug Interaction (DDI) Studies Guidance for Industry (www.fda.gov/media/108130/), we evaluated compound 3d’s potential to inhibit CYP isoforms including CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Among all the tested isoforms, only CYP1A2 is moderately inhibited by 3d with IC50 of 5.57 μM, and other CYP450 isoforms are all weakly inhibited by 3d with IC50 > 10 μM (Figure 9). Therefore, 3d does not significantly influence CYP450 enzymatic activity, which indicates it can be safely co-administered with other drugs, with specific attention paid to potential CYP1A2 interaction. We have also examined 3d’s inhibitory activity against hERG (human ether-à-go-go-related gene, a key potassium ion channel in the heart) to determine potential cardiac-toxicity. hERG inhibitory IC50 of 3d is higher than 30 μM comparing to the positive control verapamil at 400 nM (Supporting Information Figure S2). As plasma tissue binding ratio of 3d in mouse is 98.5% (Supporting Information Table S3), the free EC50 in mice is approximately 123 nM according to the PK study (Supporting Information Figure S3) and give a free safety margin of 243 folds.
Figure 9.
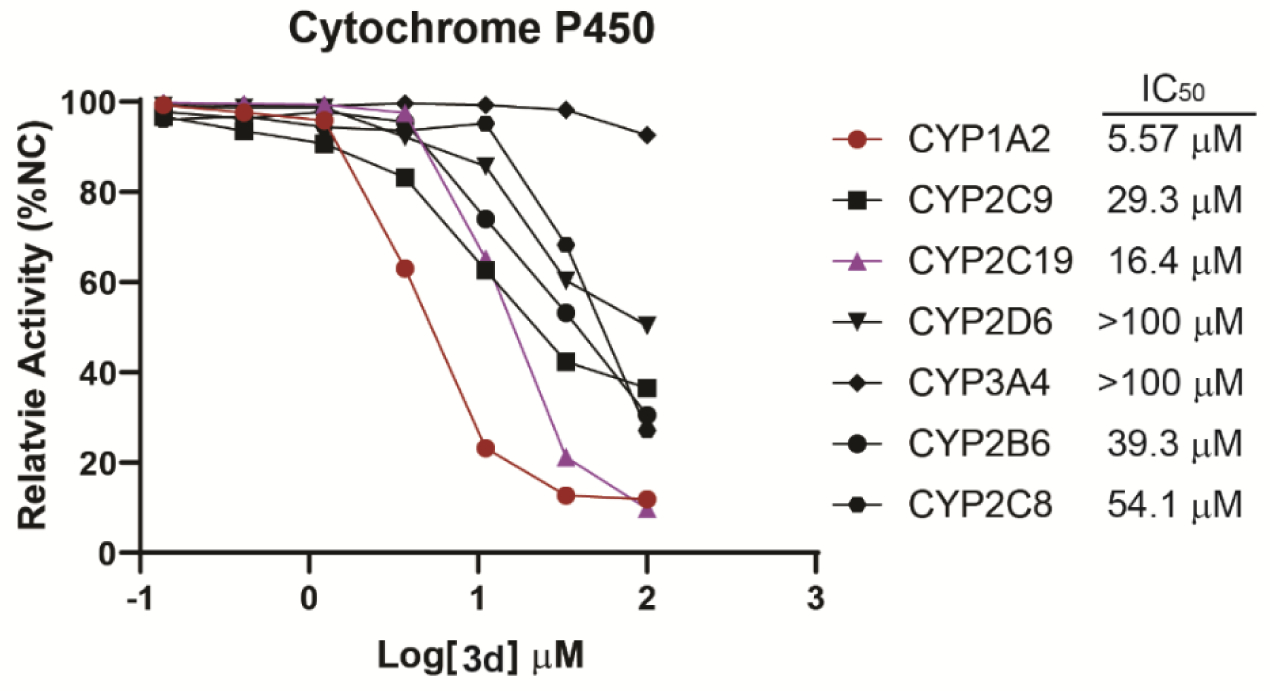
IC50 curves of 3d in the inhibition of CYP450 isoforms.
PAINS Exclusion Assay
Promiscuous compounds appear as “frequent hitters” and display misleading assay readouts. Thus, many of these compounds have been classified as Pan Assay Interference Compounds (PAINS).37 The quinone structure of VK3 contains the potential reactive “Michael acceptor” and can be considered as a promiscuous PAINS compound.37–38 Although, pharmacology studies above have shown that our analogs are not promiscuous compounds (weak and differential CYP inhibition, little hERG inhibition, and clear SAR in a whole organism), since 3d is a VK3 analog, we tested 3d for its ability for Michael reaction. VK3 and 3d were subjected to 2-mercaptoethanol, respectively, VK3 rapidly reacts with 2-mercaptoethanol, while our lead compound 3d does not react with thiol, even over a 24 h period (Supporting Information Figure S4), indicating 3d is not a PAINS compound. The deactivation of the “Michael acceptor” in 3d may be due to amidation of the naphthoquinone. In addition, unlike VK3, amidated naphthoquinone does not increase oxygen consumption in cells unlike electron donors such as VK3 and methylene blue from our previous publication,39 further indicating that an amidated naphthoquinone ring is also less likely to undergo redox cycle under physiological conditions.
Conclusions
In this study, we discover a new series of VK analogs with anti-seizure activity by the modification of VK3 analog 2h in the group of alkynes. Among these, the best compounds, 3d and 3l, as assessed by the zebrafish PTZ-induced seizure model showed significantly improved seizure protection compared to our previous lead compound 2h at the 10 μM and 5 μM concentrations. Compound 3d attenuates seizures in a dose-dependent manner in the mouse 6 Hz and MES models. Full seizure protection is also attained at doses that do not produce motor impairment in mice, which indicates its potential in the treatment of pharmacoresistant epilepsy. Moreover, 3d is also effective in the s.c. PTZ mouse model with an ED50 of 349.22 mg/kg, which indicates its efficacy against clonic seizures. 3d is well tolerated without obvious toxicity at the tested concentrations. Further PK studies show 3d has a longer t1/2 and higher AUC than previous lead compound 2h with ip administration. With iv and oral administration, t1/2 of 3d are 4.47 and 5.28 h, respectively; oral bioavailability of 3d is 35.9%, which indicates 3d is suitable for oral administration. In addition to the in vivo anti-seizure efficacy and favorable PK profile, 3d also shows excellent brain penetration.
For the clinical safety and tolerability concern, 3d does not show obvious toxicity at concentration of 50 μM in HT-22 neuronal cells. In mice seizure models, the LD50 for 3d is greater than 800 mg/kg, which is extremely high for a small molecule and has sufficient safety index even for its EC50 at approximately 200 mg/kg. What is more, 3d did not unduly influence CYP450 enzyme activity, it is expected to be safe with a low incidence of potential drug-to-drug interaction when co-administered with other therapeutic agents. Furthermore, 3d has a predictive free hERG-safety margin of 243 folds suggesting a high cardiac-safety index. All of our results indicate that 3d can be further developed as a potential ASD with good tolerability for patients with pharmacoresistant epilepsy.
Experimental Section
Material and Methods
All chemical starting materials including reagents and solvents were purchased from Sigma-Aldrich, Fisher Scientific, or other chemical vendors and used as received, unless otherwise noted. 1H and 13C NMR spectras were characterized using a Bruker Nanobay 400 MHz instrument in DMSO-d6 with TMS as an internal standard. Chemical shifts (δ) in parts per million, and coupling constants (J) in hertz (Hz) are reported. Mass spectral were determined using Thermo LCQ Fleet mass spectrometer via electrospray ionization. Column purification was performed via Teledyne Isco Combiflash 200 on prepacked C18-Aq columns. All target compounds’ purities were at least 95% detected at 254 nM via ESI-LCMS on an Agilent 1100 HPLC instrument using an ODS HYPERSIL column (5 μm, 4.6 mm × 250 mm) with water/methanol gradient plus 0.1% formic acid (3a-3w: 0–13 mins from 0% to 100% methanol, 13–14 mins: from 100% to 0% methanol. 5a and 5b: 0–3minutes from 0% to 90% methanol, 3–17 mins from 90% to 100% methanol, and 17–18 mins from 100% to 0% methanol).
General Procedure A:
2-bromonaphthalene-1,4-dione (1, 0.24g, 1mmol) was suspended in 20 mL anhydrous ethanol, then different fatty amine was added (2 mmol). The mixture was stirred overnight at room temperature. The ethanol solvent was removed via rotovap in vacuum, and the crude product was purified by recrystallization or combi-flash column.
Synthesis of 3a-3x
2-(neopentylamino)naphthalene-1,4-dione (3a)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 2,2-dimethylpropan-1-amine gave 3c as an orange-yellow solid, 56% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.99 (dd, J = 7.7, 1.3 Hz, 1H), 7.93 (dd, J = 7.6, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.18 (t, J = 6.8 Hz, 1H), 5.83 (s, 1H), 3.03 (d, J = 6.9 Hz, 2H), 0.94 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 182.00, 181.93, 149.68, 135.31, 133.60, 132.58, 130.77, 126.38, 125.73, 100.09, 53.10, 34.06, 27.91 . ESI-MS m/z: 244.25 [M + H]+.
2-((3,3-dimethylbutyl)amino)naphthalene-1,4-dione (3b)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 3,3-dimethylbutan-1-amine gave 3d as an orange-yellow solid, 53% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.3 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.52 (t, J = 6.0 Hz, 1H), 5.62 (s, 1H), 3.24 – 3.09 (m, 2H), 1.55 – 1.44 (m, 2H), 0.94 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 182.01, 181.54, 148.79, 135.29, 133.69, 132.56, 130.84, 126.33, 125.79, 99.45, 40.99, 38.97, 30.19, 29.58 . ESI-MS m/z: 258.25 [M + H]+.
2-((4,4-dimethylpentyl)amino)naphthalene-1,4-dione (3c)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 4,4-dimethylpentan-1-amine gave 3e as an orange-yellow solid, 63% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.6, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.55 (t, J = 6.2 Hz, 1H), 5.66 (s, 1H), 3.15 (q, J = 6.9 Hz, 2H), 1.58–1.51 (m, 2H), 1.22–1.18 (m, 2H), 0.86 (s, 10H). 13C NMR (101 MHz, DMSO-d6) δ 182.03, 181.63, 148.94, 135.26, 133.69, 132.54, 130.85, 126.32, 125.77, 99.59, 43.09, 41.15, 30.42, 29.63, 23.20. ESI-MS m/z: 272.25 [M + H]+.
2-(isopentylamino)naphthalene-1,4-dione (3d)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 3-methylbutan-1-amine gave 3a as an orange-yellow solid, 63% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (ddd, J = 14.0, 7.7, 1.3 Hz, 2H), 7.82 (td, J = 7.6, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.3 Hz, 1H), 7.56 (t, J = 6.1 Hz, 1H), 5.65 (s, 1H), 3.18 (q, J = 6.9 Hz, 2H), 1.68–1.51 (m, 1H), 1.50–1.45 (m, 2H), 0.91 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.03, 181.60, 148.90, 135.28, 133.67, 132.55, 130.85, 126.33, 125.77, 99.56, 40.66, 36.44, 25.97, 22.82. ESI-MS m/z: 244.25 [M + H]+.
2-((4-methylpentyl)amino)naphthalene-1,4-dione (3e)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 4-methylpentan-1-amine gave 3b as an orange-yellow solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.54 (t, J = 6.1 Hz, 1H), 5.66 (s, 1H), 3.15 (q, J = 6.8 Hz, 2H), 1.61–1.49 (m, 3H), 1.23–1.17 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.04, 181.62, 148.95, 135.25, 133.69, 132.53, 130.85, 126.31, 125.76, 99.60, 42.56, 36.12, 27.69, 25.67, 22.89 . ESI-MS m/z: 258.25 [M + H]+.
2-((4-methylpentan-2-yl)amino)naphthalene-1,4-dione (3f)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 4-methylpentan-2-amine gave 3f as an orange-yellow solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.6, 1.4 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.18 (d, J = 8.9 Hz, 1H), 5.71 (s, 1H), 3.64–3.57 (m, 1H), 1.68–1.55 (m, 2H), 1.34–1.25 (m, 1H), 1.15 (d, J = 6.3 Hz, 3H), 0.90–0.80 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.15, 181.72, 148.18, 135.30, 133.61, 132.55, 130.85, 126.35, 125.73, 99.43, 46.34, 44.65, 25.10, 23.02, 22.85, 20.02. ESI-MS m/z: 258.25 [M + H]+.
2-((2-methylbutyl)amino)naphthalene-1,4-dione (3g)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 2-methylbutan-1-amine gave 3g as an orange-yellow solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.6, 1.4 Hz, 1H), 7.93 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.3 Hz, 1H), 7.71 (td, J = 7.5, 1.3 Hz, 1H), 7.58 (t, J = 6.3 Hz, 1H), 5.65 (d, J = 4.8 Hz, 1H), 3.15–2.93 (m, 2H), 1.81–1.73 (m, 1H), 1.47 – 1.34 (m, 1H), 1.17 – 1.07 (m, 1H), 0.94 – 0.78 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.02, 181.62, 149.16, 135.27, 133.66, 132.54, 130.84, 126.33, 125.76, 99.76, 48.24, 33.43, 27.16, 17.55, 11.57. ESI-MS m/z: 244.25 [M + H]+.
2-((3-methylbut-2-en-1-yl)amino)naphthalene-1,4-dione(3h)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 3-methylbut-2-en-1-amine gave 3h as an orange-yellow solid, 60% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.90 (dd, J = 7.7, 1.3 Hz, 1H), 7.87 (dd, J = 7.6, 1.3 Hz, 1H), 7.75 (td, J = 7.5, 1.4 Hz, 1H), 7.65 (td, J = 7.5, 1.4 Hz, 1H), 7.54 (t, J = 6.0 Hz, 1H), 5.52 (s, 1H), 5.16–5.12 (m, 1H), 3.72 (t, J = 6.3 Hz, 2H), 1.64 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.06, 181.62, 148.78, 135.43, 135.26, 133.64, 132.58, 130.84, 126.30, 125.79, 120.17, 100.10, 40.50, 25.80, 18.36 . ESI-MS m/z: 242.17 [M + H]+.
2-((cyclopropylmethyl)amino)naphthalene-1,4-dione (3i)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and cyclopropylmethanamine gave 3i as an orange-yellow solid, 54% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.8, 1.3 Hz, 1H), 7.94 (dd, J = 7.6, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.52 (t, J = 6.1 Hz, 1H), 5.72 (s, 1H), 3.07 (t, J = 6.4 Hz, 2H), 1.18–1.09 (m, 1H), 0.50–1.46 (m, 2H), 0.29–0.25 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.08, 181.71, 148.95, 135.30, 133.65, 132.59, 130.81, 126.34, 125.78, 99.88, 46.58, 10.04, 3.98. ESI-MS m/z: 228.17 [M + H]+.
2-((2-cyclopropylethyl)amino)naphthalene-1,4-dione (3j)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 2-cyclopropylethanamine gave 3j as an orange-yellow solid, 55% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.87 (ddd, J = 13.8, 7.7, 1.3 Hz, 2H), 7.73 (td, J = 7.5, 1.4 Hz, 1H), 7.63 (td, J = 7.5, 1.4 Hz, 1H), 7.42 (t, J = 6.1 Hz, 1H), 5.60 (s, 1H), 3.19–3.14 (m, 2H), 1.44–1.38 (m, 2H), 0.70–0.60 (m, 1H), 0.35–0.31 (m, 2H), 0.08–0.05(m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.03, 181.63, 148.93, 135.27, 133.68, 132.54, 130.82, 126.32, 125.77, 99.65, 42.54, 32.79, 9.07, 4.65. ESI-MS m/z: 242.25 [M + H]+.
2-((cyclobutylmethyl)amino)naphthalene-1,4-dione (3k)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and cyclobutylmethanamine gave 3k as an orange-yellow solid, 57% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.93 (dd, J = 7.6, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.50 (t, J = 6.2 Hz, 1H), 5.68 (s, 1H), 3.21 (t, J = 6.7 Hz, 2H), 2.71–2.60 (m, 1H), 2.05–1.97 (m, 2H), 1.887–1.80 (m, 2H), 1.77–1.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.03, 181.65, 149.16, 135.27, 133.65, 132.55, 130.83, 126.32, 125.75, 99.81, 47.52, 33.76, 26.01, 18.35. ESI-MS m/z: 242.25 [M + H]+.
2-((2-cyclobutylethyl)amino)naphthalene-1,4-dione (3l)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 2-cyclobutylethanamine gave 3l as an orange-yellow solid, 49% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.95 (ddd, J = 12.6, 7.7, 1.3 Hz, 2H), 7.82 (td, J = 7.5, 1.3 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.49 (t, J = 6.2 Hz, 1H), 5.63 (s, 1H), 3.09 (q, J = 6.8 Hz, 2H), 2.35–2.25 (m, 1H), 2.09–1.99 (m, 2H), 1.88–1.73 (m, 2H), 1.70–1.57 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 182.01, 181.57, 148.90, 135.26, 133.68, 132.53, 130.83, 126.31, 125.77, 99.54, 40.49, 34.68, 33.69, 28.14, 18.64. ESI-MS m/z: 256.25 [M + H]+.
2-((2-cyclopentylethyl)amino)naphthalene-1,4-dione (3m)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 2-cyclopentylethanamine gave 3m as an orange-yellow solid, 51% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.55 (t, J = 6.1 Hz, 1H), 5.65 (s, 1H), 3.21–3.16 (m, 2H), 1.87–1.74 (m, 3H), 1.62–1.45 (m, 6H), 1.14–1.04 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.05, 181.59, 148.89, 135.28, 133.69, 132.55, 130.86, 126.33, 125.78, 99.56, 41.77, 37.82, 33.86, 32.58, 25.13. ESI-MS m/z: 270.25 [M + H]+.
2-((2-cyclohexylethyl)amino)naphthalene-1,4-dione (3n)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 2-cyclohexylethanamine gave 3n as an orange-yellow solid, 57% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.8, 1.3 Hz, 1H), 7.94 (dd, J = 7.6, 1.3 Hz, 1H), 7.83 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.53 (t, J = 6.0 Hz, 1H), 5.64 (s, 1H), 3.19 (q, J = 6.7 Hz, 2H), 1.74–1.60 (m, 5H), 1.51–1.45 (m, 2H), 1.36–1.04 (m, 4H), 0.96–0.87 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.05, 181.60, 148.90, 135.29, 133.70, 132.56, 130.87, 126.34, 125.78, 99.57, 35.36, 35.03, 33.06, 26.52, 26.19. ESI-MS m/z: 284.25 [M + H]+.
2-((2-(diethylamino)ethyl)amino)naphthalene-1,4-dione (3o)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and N1,N1-diethylethane-1,2-diamine gave 3t as an orange-yellow solid, 61% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.83 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.22 (t, J = 5.7 Hz, 1H), 5.69 (s, 1H), 3.21 (q, J = 6.3 Hz, 2H), 2.63 (t, J = 6.6 Hz, 2H), 2.51 (q, J = 7.1 Hz, 5H), 0.96 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 181.94, 181.70, 148.69, 135.36, 133.66, 132.63, 130.74, 126.36, 125.83, 99.95, 50.21, 46.79, 12.34. ESI-MS m/z: 273.25 [M + H]+.
2-((2-(ethyl(methyl)amino)ethyl)amino)naphthalene-1,4-dione (3p)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and N1-ethyl-N1-methylethane-1,2-diamine gave 3u as an orange-yellow solid, 68% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.83 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.19 (t, J = 5.6 Hz, 1H), 5.69 (s, 1H), 3.24 (q, J = 6.3 Hz, 2H), 2.56 (t, J = 6.5 Hz, 2H), 2.42 (q, J = 7.1 Hz, 2H), 2.19 (s, 3H), 0.99 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.93, 181.72, 148.68, 135.35, 133.64, 132.64, 130.75, 126.35, 125.83, 100.02, 54.17, 51.20, 41.54, 39.92, 12.61. ESI-MS m/z: 259.17 [M + H]+.
2-((2-(dimethylamino)ethyl)amino)naphthalene-1,4-dione (3q)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and N1,N1-dimethylethane-1,2-diamine gave 3v as an orange-yellow solid, 69% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.6, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.17 (t, J = 5.7 Hz, 1H), 5.69 (s, 1H), 3.24 (q, J = 6.2 Hz, 2H), 2.49–2.48 (m, 2H), 2.19 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 181.93 , 181.74 , 148.69 , 135.35 , 133.62 , 132.64 , 130.75 , 126.35 , 125.83 , 100.04 , 56.59 , 45.48 , 40.02 . ESI-MS m/z: 245.17 [M + H]+
2-((3-(dimethylamino)propyl)amino)naphthalene-1,4-dione (3r)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and N1,N1-dimethylpropane-1,3-diamine gave 3w as an orange-yellow solid, 61% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.93 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 2H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 5.67 (s, 1H), 3.21 (q, J = 6.5 Hz, 2H), 2.28 (t, J = 6.6 Hz, 2H), 2.15 (s, 6H), 1.71 (p, J = 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.01, 181.62, 149.09, 135.26, 133.71, 132.54, 130.84, 126.31, 125.78, 99.56, 57.42, 45.63, 41.22, 25.56. ESI-MS m/z: 259.17 [M + H]+
2-((3-(methyl(phenyl)amino)propyl)amino)naphthalene-1,4-dione (3s)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and N1-methyl-N1-phenylpropane-1,3-diamine gave 3x as an orange-yellow solid, 64% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.3 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.64 (t, J = 6.1 Hz, 1H), 7.16 – 7.07 (m, 2H), 6.75 – 6.65 (m, 2H), 6.58 (t, J = 7.2 Hz, 1H), 5.70 (s, 1H), 3.39 (s, 2H), 3.22 (q, J = 6.8 Hz, 2H), 2.87 (s, 3H), 1.82 (p, J = 7.1 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.00, 181.70, 149.43, 149.03, 135.25, 133.66, 132.57, 130.89, 129.41, 126.33, 125.77, 116.11, 112.50, 99.85, 49.91, 38.34, 24.95. ESI-MS m/z: 321.17 [M + H]+
2-((3-hydroxybutyl)amino)naphthalene-1,4-dione (3t)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 4-aminobutan-2-ol gave 3o as an orange-yellow solid, 54% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.6, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.59 (t, J = 5.9 Hz, 1H), 5.67 (s, 1H), 4.70 (d, J = 4.7 Hz, 1H), 3.75–3.68 (m, 1H), 3.24 (q, J = 6.7 Hz, 2H), 1.76–1.51 (m, 2H), 1.10 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.01, 181.62, 148.92, 135.27, 133.70, 132.54, 130.83, 126.31, 125.79, 99.58, 64.83, 39.91, 36.81, 24.20. ESI-MS m/z: 246.25 [M + H]+.
2-((4-hydroxy-3-methylbutyl)amino)naphthalene-1,4-dione (3u)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 4-amino-2-methylbutan-1-ol gave 3p as an orange-yellow solid, 57% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.56 (t, J = 6.0 Hz, 1H), 5.66 (s, 1H), 4.53 (t, J = 5.2 Hz, 1H), 3.28–3.14 (m, 4H), 1.70–1.55 (m, 2H), 1.40–1.31 (m, 1H), 0.88 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.04, 181.60, 148.89, 135.27, 133.69, 132.54, 130.84, 126.31, 125.78, 99.60, 66.40, 40.56, 33.80, 31.43, 17.22. ESI-MS m/z: 260.25 [M + H]+.
2-((2-hydroxybutyl)amino)naphthalene-1,4-dione (3v)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 1-aminobutan-2-ol gave 3q as an orange-yellow solid, 53% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 7.7, 1.3 Hz, 1H), 7.94 (dd, J = 7.7, 1.3 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.24 (t, J = 6.0 Hz, 1H), 5.74 (s, 1H), 4.94 (d, J = 5.2 Hz, 1H), 3.67–3.60 (m, 1H), 3.2–3.02 (m, 2H), 1.53–1.30 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.99, 181.79, 149.08, 135.32, 133.63, 132.61, 130.78, 126.34, 125.79, 100.14, 69.51, 48.47, 28.10, 10.41. ESI-MS m/z: 246.25 [M + H]+.
2-((3-hydroxy-3-methylbutyl)amino)naphthalene-1,4-dione (3w)
Using the General procedure A, 2-bromonaphthalene-1,4-dione and 4-amino-2-methylbutan-2-ol gave 3r as an orange-yellow solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.95 (ddd, J = 9.3, 7.7, 1.3 Hz, 2H), 7.82 (td, J = 7.5, 1.3 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.63 (t, J = 5.7 Hz, 1H), 5.65 (s, 1H), 4.58 (s, 1H), 3.28–3.23 (m, 2H), 1.71–1.68 (m, 2H), 1.16 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.00, 181.57, 148.82, 135.27, 133.74, 132.53, 130.83, 126.30, 125.80, 99.47, 69.12, 38.96, 29.78. ESI-MS m/z: 260.25 [M + H]+.
General procedure B:
2-methylnaphthalene-1,4-dione (3, 0.24g, 1mmol) was dissolved in an mixed solution of 10 mL methanol and 10 mL dichloromethane, then 3-methylbutan-1-amine (0.19g, 2mmol) or 3,3-dimethylbutan-1-amine (0.20g, 2mmol) was added. The mixture was allowed to stir overnight at room temperature. After the reaction finished, ethanol was removed under vacuum, and the rude product was purified by combi-flash column.
2-((3,3-dimethylbutyl)amino)-3-methylnaphthalene-1,4-dione (5a)
Using the General procedure B, 2-methylnaphthalene-1,4-dione and 3,3-dimethylbutan-1-amine gave 5b as an orange-yellow solid, 64% yield. 1H NMR (400 MHz, Chloroform-d) δ 8.08 (dt, J = 7.8, 1.5 Hz, 1H), 7.98 (dq, J = 7.7, 1.5 Hz, 1H), 7.67 (tq, J = 7.7, 1.3 Hz, 1H), 7.56 (tq, J = 7.5, 1.3 Hz, 1H), 5.70 (d, J = 5.6 Hz, 1H), 3.59–3.54 (m, 2H), 2.25 (d, J = 1.8 Hz, 3H), 1.77–1.66 (m, 1H), 1.56–1.51 (m, 2H), 0.95 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 183.55, 182.54, 146.18, 134.31, 133.55, 131.77, 130.29, 126.19, 125.96, 111.98, 43.78, 39.82, 25.72, 22.50, 11.25. ESI-MS m/z: 272.17 [M + H]+
2-(isopentylamino)-3-methylnaphthalene-1,4-dione (5b)
Using the General procedure B, 2-methylnaphthalene-1,4-dione and 3-methylbutan-1-amine gave 5b as an orange-yellow solid, 60% yield. 1H NMR (400 MHz, Chloroform-d) δ 8.08 (dd, J = 7.8, 1.0 Hz, 1H), 7.98 (dd, J = 7.8, 1.1 Hz, 1H), 7.67 (td, J = 7.6, 1.4 Hz, 1H), 7.56 (td, J = 7.5, 1.3 Hz, 1H), 5.65 (s, 1H), 3.59–3.54 (m, 2H), 2.25 (s, 3H), 1.57–1.53 (m, 2H), 0.97 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 183.52, 182.56, 146.21, 134.29, 133.57, 131.75, 130.31, 126.19, 125.96, 111.93, 44.91, 42.08, 30.04, 29.53, 11.23. ESI-MS m/z: 258.17 [M + H]+
Induction and Recording of Activity in Zebrafish
Zebrafish (AB strain) were from the Zebrafish International Resource Center (supported by P40 RR012546 from NIH-NCRR). All zebrafish studies have been approved by the Medical University of South Carolina Institutional Animal Care and Use Committee (IACUC-2018–00278) and performed in accordance with the guidelines. All experimental comparisons were from 7 dpf fish from the same clutch. The method used here is a slightly modified version of the one characterized by Baraban et al.,11 which indicates that the total distance traveled by zebrafish after PTZ induction of seizures quantitatively reflects seizure activity. 48-well Nunc plates (Cat # 12–565-322, FisherSci, Pittsburg, PA) were used to position a single zebrafish per well. Compounds were dissolved in DMSO and further diluted in E3 buffer (0.33 mM CaCl2, 5 mM NaCl, 0.33 mM MgSO4, and 0.17 mM KCl). The larvae are treated at 37°C for 1 h in sublethal concentrations of the compounds. After 1 h, PTZ (15 mM in 500 μL total volume per well) was added immediately to each well. The plate was immediately loaded into the Daniovision instrument (Noldus Information Technology) to track zebrafish movements over 15 min. At termination, zebrafish were monitored visually for overt toxicity including failure to startle and increased mortality. The Ethovision XT software (Noldus) recorded live video images. The total movement in millimeters was measured in one-minute time bins and compared between groups using one-way ANOVA statistical analyses by mean ± SEM (GraphPad Prism Software).
HT-22 cell Toxicity Study
HT-22 neuronal cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM/high glucose) with 10% fetal bovine serum (FBS) and 1% of antibiotic-antimycotic (Amphotericin B, Penicillin, and Streptomycin; Invitrogen) at 37 °C in 5% CO2. 2000 cells per well were plated in a clear 96-well plate, 12 h later, serially diluted compounds were added and incubated for 48 h. CellTiter-Blue (resazurin cell viability assay reagent) was added at a final concentration of 0.125 mg/mL to each well. The cell viability was determined after 2 h via resorufin fluorescence intensity at ex 560 nm/em 590 nm using a Tecan M200 Pro spectrophotometer. Data were processed and fitted via Prism (GraphPad) by subtracting the background and normalizing to the control wells.
6 Hz 32mA and 44mA Mouse Test
Investigational compounds were examined for their ability to attenuate psychomotor seizures via a low-frequency (6 Hz), long-duration (3 sec) electric shock delivered through corneal electrodes.16 8 male CF-1 mice were used per group per concentration. Experiments were conducted at stimulus intensities of 32 mA and 44 mA. Typically, the seizure was characterized by a brief stun followed immediately by jaw clonus, forelimb clonus, twitching of the vibrissae, and Straub tail lasting for at least 1 second. Animals not displaying this behavior were considered “protected”. 3d was dissolved in miglyol840/DMSO (95%/5%) and was dosed via ip administration. Quantification of the ED50 value was conducted at the time of peak effect (TPE). To determine the TPE, mice were treated with the investigational compound and tested at 0.25 h, 0.5 h, 1.0 h, 2.0 h 4.0 h, or based on time-points from previous studies. Groups of n = 8 mice were tested with various doses of the investigational compound. The data for each condition were presented as N/F, where N = number of animals protected and F = number of animals tested. The ED50, 95% confidence interval, the slope of the regression line, and the SEM of the slope are calculated by Probit analysis.40
Maximal Electroshock (MES) Test
An alternating current at 60 Hz 50 mA was delivered via corneal electrodes for 2s to mice. An electrolyte solution containing the anesthetic agent (0.5% tetracaine HCl) was applied to the eyes before the electric shock. 8 male CF-1 mice were used per group per concentration. 3d was suspended in 0.5% hydroxypropyl methylcellulose (HPMC) and was dosed via ip administration. If no hindlimb tonic extension was observed during the seizure, the animal was considered “protected” from the MES-induced seizures.19
Subcutaneous (s.c.) Pentylenetetrazole (PTZ) Test
The s.c. PTZ model determines the ability of potential anti-seizure compounds to attenuate a clonic forebrain seizure in mice.41 1 h after ASD-treatment, 85 mg/kg PTZ was injected subcutaneously into the midline of the mouse neck. The mice were observed for the next 30 min to determine if clonic spasms of the fore and/or hindlimbs, jaws, or vibrissae had occurred lasting 3–5s in an isolation cage to minimize stress and other variables. 8 male CF-1 mice were used per group per concentration. 3d was suspended in 0.5% HPMC and was dosed via ip administration. Mice without spasms or twitching were considered protected.19
Behavioral Toxicity in Mice
To assess the toxicity of compounds, all animals were visually inspected for signs of movement impairment due to neurological or muscular dysfunction. The rotarod procedure was used to evaluate motor impairment of each mouse tested.16 To examine whether the mouse can maintain its normal equilibrium and movement for a defined period of time, it is placed on a knurled rod that rotates at a constant speed of 6 rpm. If the mouse fell three times within one minute, it was considered impaired. The rotarod assessment was performed on the same animals prior to the 6 Hz assay.
Fluorometric ATP Assay
ATP content was measured by Biovision’s ATP Fluorometric Assay Kit (K354–100, Biovision, Milpitas, CA) as per our previous studies, with minor modifications (PMID:26519465). Briefly, one 7 dpf zebrafish was placed in each well of a 48-well plate. Fish were kept in a 28.5°C incubator and treated for 24 h with 3d and 3l. Stock solutions were dissolved in DMSO. Solutions were further diluted in E3 buffer (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4) to final concentrations of 0.5 μM and 1 μM in a total volume of 500 μL per well. DMSO was used in the control wells. Twelve 7 dpf fish per condition were collected in one microfuge tube and flash frozen in liquid nitrogen. Contents were stored at −80°C for later use. Fish were resuspended in 200 μL ATP buffer, homogenized, and centrifuged at 12,000 × g for 5 min. The supernatant fractions were retained for ATP measurements following the manufacturer’s protocol.
HT-22 hippocampal neuronal cells were maintained in DMEM/high glucose supplemented with 10% FBS (Hyclone, GE Healthcare, Chicago, IL), 1% antibiotic-antimycotic (Amphotericin B, Penicillin, and Streptomycin; Invitrogen) and 1% sodium pyruvate in a 37°C incubator with 5% CO2. HT-22 were seeded in 2 mL volume of media on 6 well plates (Grenier Bio-one, Monroe, NC) at a density of 1.5 × 105 cells per well for 24 h. In the same growth medium, the cells were treated for an additional 24 hours with 0.5 μM of 3d and 3l. The treated plates were maintained in a 37°C, 5% CO2 incubator. The cells were transferred to 15 mL falcon conical tubes after harvest, and centrifuged at 1000 × g for 5 min. Cell pellets resuspended in 6 mL cold Hank’s Balanced Salt Solution (HBSS) (Hyclone, GE Healthcare, Chicago, IL) and centrifuged for another 5 min. The HBSS solution was removed and lysates were transferred to an −80°C freezer until further use. Cell lysates were dissolved in 100 μL ATP Buffer as listed in the original protocol.
Zebrafish supernatant fractions and cell lysates were measured in a Tecan M200 Pro spectrophotometer plate reader at ex 535 nm/em 587 nm. ATP content was normalized by total protein using the Pierce BCA protein assay kit (#23255, Thermo Fisher Scientific, Waltman, MA).
Brain/Plasma Concentration Ratio Test
Male CF-1 mice were housed in individual cages on a 12 h light and 12 h dark cycle, with the room temperature at 22 ± 3°C. Before the PK studies, mice were fasted overnight. Water was provided ad libitum throughout the study and food was returned after the 6 h blood sample time. 3d was dissolved in 95% Miglyol 840: 5% DMSO. Animals were dosed via ip injections and blood samples were collected at the indicated time. Acetonitrile containing internal standard was added to plasma (3:1 ratio) to precipitate out the proteins. The resulting supernatants were isolated via centrifugation at 3000 g for 10 min. Each sample supernatant was analyzed via LCMS/MS. Serial dilution of the calibration standards and quality controls (1 mg/mL) were diluted in methanol : water (1:1, v/v) and spiked into blank plasma to give the standard curve from 1 ng/mL to 10 μg/mL for the compound and the quality control samples at three different concentration levels.
For the tissue sample PK analysis, PBS buffer and tissue sample (3:1) homogenized to obtain each tissue homogenate sample. Subsequently, acetonitrile with the internal standard was added to each tissue homogenate at 3:1 ratio, and the mixture was vortexed, centrifuged (3000 g for 10 min) and supernatant isolated for analysis by LC-MS/MS. Calibration standards curves were established similarly as described above. For each drug candidate, LC-MS/MS analysis was performed utilizing the multiple reaction monitoring protocol to detect their characteristic ions. Plasma and tissue homogenate concentration were measured as described above. The brain/plasma concentration ratio was obtained by mean tissue concentration (ng/g) mean plasma concentration (ng/mL).
Pharmacokinetics
Male CD-1 Mice were maintained as described in above. The dosing solution of each test compound was prepared in a desired oral or intravenous formulation using 20% DMA:40% PEG300:40% H2O; 5% DMSO:95% Neobee was used for ip administration. Three animals are dosed via gavage needle for oral and ip administration at 20 mg/kg (20 mL/kg) or via tail vein injection for iv administration at 5 mg/kg (5 mL/kg). Blood samples (30–50 μL per sample) were drawn via saphenous, jugular, or submandibular vein at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h after dosing. Blood samples were collected in Greiner MiniCollect K2EDTA tubes and centrifuged at 15,000g for 5 min to obtain plasma samples. Three volumes of acetonitrile containing internal standard was added to one volume of plasma and samples were centrifuged at 3000 g for 10 min. The resulting supernatants were removed for analysis by LC-MS/MS to determine the targeted compound concentration. Calibration standard and quality control curves were determined as described in the above section and treated identically as the PK samples. The targeted compound and the internal standard were identified using LC-MS/MS under multiple reaction monitoring mode to detect their characteristic ions. Plasma concentrations at each time point were then plotted against the time for the analysis. Linear trapezoidal method was used to calculate AUC. PK parameters were fitted using the non-compartmental analysis. For intravenous administration PK parameters, t1/2, C0, AUC, volume of distribution at steady-state (Vss), total plasma clearance (CLp), and mean residence time (MRT) were determined and reported. For extravascular administration, t1/2, Cmax, tmax, AUC, mean residence time (MRT), and bioavailability (F%) were determined and reported. All parameters were calculated for each animal as well as the combined mean, standard deviation, and coefficient of variation for the group.
CYP450 Inhibition Study
The CYP450 inhibition study was conducted by Touchstone Biosciences. 0.2 mg/mL mix-gender, pooled human liver microsomes (HLM), 100 mM potassium phosphate buffer, and a 2 mM NADPH cofactor were used in the inhibition assay. Individual CYP450 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP2B6, CYP2C8) were investigated where isoform-specific probe substrates (at concentrations near the specific enzyme’s Km) were incubated individually with HLM and compound 3d or a corresponding positive control inhibitor. The organic solvent was methanol (DMSO if compounds were not soluble in methanol) which was maintained at a final concentration no greater than 1% in the assay. After incubation at 37°C for 15–30 min, the reaction was stopped by adding two volumes of acetonitrile containing the internal standard. The resulting samples were centrifuged and the supernatants were collected and analyzed for metabolites of the probe substrates by LC-MS/MS at each of the tested concentrations. A decrease in the formation of the metabolites compared to vehicle control was used to calculate percent inhibition at each concentration or an IC50 value (test compound concentration which produces 50% inhibition). The percent inhibition was calculated based on a decrease in the formation of the metabolite of each specific substrate compared to vehicle control (negative control), whereas an IC50 was calculated based on a sigmoidal does-response model with variable slope. Bottom and top parameter constraints were fixed to 0 and 100, respectively. Compounds can be generally categorized as follows:
Potent inhibitor: IC50 < 1 μM
Moderate inhibitor: 1 μM ≤ IC50 ≤ 10 μM
Weak- or non-inhibitor: IC50 > 10 μM
Supplementary Material
Acknowledgements
All synthesis and experiments were performed in the United States. Chemical synthesis was performed at Neuroene Therapeutics. Animal experiments were performed at Medical University of South Carolina (zebrafish) and University of Utah (rodents). Pharmacokinetics and in vitro pharmacology were performed by Touchstone Biosciences. Research reported in this publication was supported by NINDS of the National Institutes of Health under award number R44NS097047 and South Carolina Research Authority Commercialization matching fund.
Abbreviations used
- ASDs
Anti-seizure drugs
- PTZ
Pentylenetetrazol
- NINDS
Neurological Disorders and Stroke
- ETSP
Epilepsy Therapy Screening Program
- MES
maximal electroshock seizure
- s.c. PTZ
Subcutaneous PTZ seizure threshold
- VK
Vitamin K
- TD50
median toxic dose
- tPSA
Topological molecules polar surface area
- Cmax
mean peak plasma concentration
- SEM
Mean and standard error of mean
- TPE
Time of peak effect
- HPMC
Hydroxypropyl methylcellulose
- MRT
Mean residence time
- F%
Bioavailability
- RED
Rapid equilibrium dialysis
Footnotes
Competing interests
C.J.C. and S.S.L.C. are the co-founders of Neuroene Therapeutics. The other authors declare no competing financial interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information:
The Supporting Information is available free of charge at https://pubs.acs.org. Time course experiment and toxicity 3l in the acute, electrically evoked mouse seizure models; time course experiment and toxicity of 3d in mouse; experiment method and result of plasma protein binding ratio of 3d in mouse and human; Experiment method and result of spontaneous REDs assay and hERG inhibition assay; The reaction of VK3 and 3d with 2-mercaptoethanol; Pharmacokinetic Data of 3d at concentration of 400 mg/kg and 200 mg/kg with ip administration; 1H-NMR and 13C-NMR spectrum of all target compounds (PDF); HPLC traces and purity of the target compounds.
Molecular Formula Strings (CSV).
Reference
- (1).Devinsky O; Vezzani A; O’Brien TJ; Jette N; Scheffer IE; de Curtis M; Perucca P Epilepsy. Nat. Rev. Dis Primers 2018, 4, 18024. [DOI] [PubMed] [Google Scholar]
- (2).Rho JM; White HS Brief history of anti-seizure drug development. Epilepsia open. 2018, 3, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chen Z; Brodie MJ; Liew D; Kwan P Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA. Neurol 2018, 75, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Löscher W; Klitgaard H; Twyman RE; Schmidt D New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov 2013, 12, 757–776. [DOI] [PubMed] [Google Scholar]
- (5).Wang Y; Chen Z An update for epilepsy research and antiepileptic drug development: Toward precise circuit therapy. Pharmacol. Ther 2019, 201, 77–93. [DOI] [PubMed] [Google Scholar]
- (6).Franco V; French JA; Perucca E Challenges in the clinical development of new antiepileptic drugs. Pharmacol. Res 2016, 103, 95–104. [DOI] [PubMed] [Google Scholar]
- (7).Löscher W; Schmidt D, Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011, 52, 657–678. [DOI] [PubMed] [Google Scholar]
- (8).Twyman RE, Strengthening the case for epilepsy drug development: bridging experiences from the alzheimer’s disease field-an opinion. Neurochem. Res 2017, 42, 2099–2115. [DOI] [PubMed] [Google Scholar]
- (9).Gohil K; Enhoffer D Modest growth seen in epilepsy market. Pharm. Ther 2014, 39, 786–787. [PMC free article] [PubMed] [Google Scholar]
- (10).Hansen SL; Sperling BB; Sánchez C Anticonvulsant and antiepileptogenic effects of GABAA receptor ligands in pentylenetetrazole-kindled mice. Prog Neuropsychopharmacol Biol Psychiatry. 2004, 28, 105–113. [DOI] [PubMed] [Google Scholar]
- (11).Baraban S; Taylor M; Castro P; Baier H Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience. 2005, 131, 759–768. [DOI] [PubMed] [Google Scholar]
- (12).Afrikanova T; Serruys A-SK; Buenafe OE; Clinckers R; Smolders I; de Witte PA; Crawford AD; Esguerra CV Validation of the zebrafish pentylenetetrazol seizure model: locomotor versus electrographic responses to antiepileptic drugs. PloS one. 2013, 8, e54166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Loscher W Animal Models of Drug-resistant Epilepsy In Novartis 243: Mechanisms of Drug Resistance in Epilepsy-Lessons from Oncology, 1st ed.; Bock G, Goode J, Eds.; John Wiley: Chichester, 2002; pp 149–166. [PubMed] [Google Scholar]
- (14).Wilcox KS; West PJ; Metcalf CS The current approach of the epilepsy therapy screening program contract site for identifying improved therapies for the treatment of pharmacoresistant seizures in epilepsy. Neuropharmacology. 2020, 166, 107811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kehne JH; Klein BD; Raeissi S; Sharma S The national institute of neurological disorders and stroke (NINDS) epilepsy therapy screening program (ETSP). Neurochem. Res 2017, 42, 1894–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Barton ME; Klein BD; Wolf HH; White HS Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [DOI] [PubMed] [Google Scholar]
- (17).Metcalf CS; West PJ; Thomson KE; Edwards SF; Smith MD; White HS; Wilcox KS Development and pharmacologic characterization of the rat 6 Hz model of partial seizures. Epilepsia. 2017, 58, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Leclercq K; Matagne A; Kaminski R Low potency and limited efficacy of antiepileptic drugs in the mouse 6 Hz corneal kindling model. Epilepsy Res. 2014, 108, 675–683. [DOI] [PubMed] [Google Scholar]
- (19).Klein BD; Jacobson CA; Metcalf CS; Smith MD; Wilcox KS; Hampson AJ; Kehne JH Evaluation of cannabidiol in animal seizure models by the epilepsy therapy screening program (ETSP). Neurochem. Res 2017, 42, 1939–1948. [DOI] [PubMed] [Google Scholar]
- (20).Rahn JJ; Bestman JE; Josey BJ; Inks ES; Stackley KD; Rogers CE; Chou CJ; Chan SS Novel vitamin K analogs suppress seizures in zebrafish and mouse models of epilepsy. Neuroscience. 2014, 259, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Baraban SC Emerging epilepsy models: insights from mice, flies, worms and fish. Curr. Opin. Neurol 2007, 20, 164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Patra PH; Barker-Haliski M; White HS; Whalley BJ; Glyn S; Sandhu H; Jones N; Bazelot M; Williams CM; McNeish AJ Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia. 2019, 60, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Barton ME; Klein BD; Wolf HH; White HS, Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [DOI] [PubMed] [Google Scholar]
- (24).West PJ; Saunders GW; Billingsley P; Smith MD; White HS; Metcalf CS; Wilcox KS Recurrent epileptiform discharges in the medial entorhinal cortex of kainate-treated rats are differentially sensitive to antiseizure drugs. Epilepsia. 2018, 59, 2035–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bindoff LA; Engelsen BA Mitochondrial diseases and epilepsy. Epilepsia. 2012, 53, 92–97. [DOI] [PubMed] [Google Scholar]
- (26).Lyczkowski DA; Pfeifer HH; Ghosh S; Thiele EA Safety and tolerability of the ketogenic diet in pediatric epilepsy: effects of valproate combination therapy. Epilepsia. 2005, 46, 1533–1538. [DOI] [PubMed] [Google Scholar]
- (27).Gano LB; Patel M; Rho JM Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res 2014, 55, 2211–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sitarz KS; Elliott HR; Karaman BS; Relton C; Chinnery PF; Horvath R Valproic acid triggers increased mitochondrial biogenesis in POLG-deficient fibroblasts. Mol. Genet. Metab 2014, 112, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Komulainen T; Lodge T; Hinttala R; Bolszak M; Pietila M; Koivunen P; Hakkola J; Poulton J; Morten KJ; Uusimaa J Sodium valproate induces mitochondrial respiration dysfunction in HepG2 in vitro cell model. Toxicology. 2015, 331, 47–56. [DOI] [PubMed] [Google Scholar]
- (30).Gibbs JE; Walker MC; Cock HR Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia. 2006, 47, 469–478. [DOI] [PubMed] [Google Scholar]
- (31).McCord JM; Fridovich I The utility of superoxide dismutase in studying free radical reactions. II. The mechanism of the mediation of cytochrome c reduction by a variety of electron carriers. J. Biol. Chem 1970, 245, 1374–1377. [PubMed] [Google Scholar]
- (32).Eleff S; Kennaway NG; Buist NR; Darley-Usmar VM; Capaldi RA; Bank WJ; Chance B 31P NMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle. Proc. Natl. Acad. Sci. U. S. A 1984, 81, 3529–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vos M; Esposito G; Edirisinghe JN; Vilain S; Haddad DM; Slabbaert JR; Van Meensel S; Schaap O; De Strooper B; Meganathan R; Morais VA; Verstreken P Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012, 336, 1306–1310. [DOI] [PubMed] [Google Scholar]
- (34).Yagami T; Ueda K; Asakura K; Sakaeda T; Nakazato H; Kuroda T; Hata S; Sakaguchi G; Itoh N; Nakano T; Kambayashi Y; Tsuzuki H Gas6 rescues cortical neurons from amyloid beta protein-induced apoptosis. Neuropharmacology. 2002, 43, 1289–1296. [DOI] [PubMed] [Google Scholar]
- (35).Yagami T; Ueda K; Asakura K; Okamura N; Sakaeda T; Sakaguchi G; Itoh N; Hashimoto Y; Nakano T; Fujimoto M Effect of Gas6 on secretory phospholipase A(2)-IIA-induced apoptosis in cortical neurons. Brain Res. 2003, 985, 142–149. [DOI] [PubMed] [Google Scholar]
- (36).Lynch T; Price A The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [PubMed] [Google Scholar]
- (37).Aldrich C; Bertozzi C; Georg GI; Kiessling L; Lindsley C; Liotta D; Merz KM Jr.; Schepartz A; Wang S The ecstasy and agony of assay interference compounds. J. Med. Chem 2017, 60, 2165–2168. [DOI] [PubMed] [Google Scholar]
- (38).Baell J; Walters MA. Chemistry: chemical con artists foil drug discovery. Nature. 2014, 513, 481–483. [DOI] [PubMed] [Google Scholar]
- (39).Rahn JJ; Bestman JE; Josey BJ; Inks ES; Stackley KD; Rogers CE; Chou CJ; Chan SS Novel vitamin K analogs suppress seizures in zebrafish and mouse models of epilepsy. Neuroscience. 2014, 259, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Finney DJ Probit analysis: A Statistical Treatment of The Sigmoid Response Curve, 2nd ed. Cambridge University Press, Cambridge, 1952: pp 8–159. [Google Scholar]
- (41).White H; Johnson M; Wolf H; Kupferberg H The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital. J. Neurol. Sci 1995, 16, 73–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.