

Abstract
Epidemiological studies show that the incidence of Wilms tumor (WT) in East‐Asian children is half of that in Caucasian children. Abnormalities of WT1,CTNNB1,WTX, and IGF2 were reported to be involved in Wilms tumorigenesis in Caucasians, although none of the studies simultaneously evaluated the four genes. WTX forms the β‐catenin degradation complex; however, the relationship between WTX abnormality and CTNNB1 mutation was uncertain in WTs. We examined abnormalities of the four genes in 114 Japanese with WTs to clarify the relationship between genetic and epigenetic factors and the incidence of WTs. We found that abnormalities of WTX and CTNNB1 were mutually exclusive, and that although CTNNB1 mutation was frequent in WTs with WT1 abnormality, but rare in WTs without, the incidences of WTX abnormality were similar between WTs with or without WT1 abnormality. These findings were consistent with those reported in Caucasian populations, and indicate multiple roles of WTX abnormality. Abnormalities of WT1,WTX and CTNNB1, and loss of IGF2 imprinting (LOI) were detected in 31.6%, 22.8%, 26.3%, and 21.1% of the 114 WTs, respectively. When we selected 101 sporadic WTs, the incidences of WT1,CTNNB1, or WTX abnormality were generally comparable between the two populations, whereas the incidence of IGF2 LOI was lower in Japanese than that of IGF2 LOI reported in Caucasians (P = 0.04). This is the first comprehensive study of the four genes, and the results supported the hypothesis that the lower incidence of IGF2 LOI contributes to the lower incidence of WTs in Japanese children. (Cancer Sci 2012; 103: 1129–1135)
The rates of various types of adult cancers, including prostate, lung, breast, liver and gastric cancers, differ among ethnic populations, which are thought to be caused by different environmental factors.1 In contrast, most pediatric tumors such as neuroblastomas and retinoblastomas show similar rates among ethnic populations. An exception is Wilms tumor (WT), a common pediatric tumor of the kidney, which is known to have different incidence rates between East‐Asian and Caucasian populations.2
Wilms tumor accounts for 8% of childhood cancers and occurs in 1 in 10 000 Caucasian children, though its incidence in East‐Asian populations is half of that.2 In Hawaii and Britain, the incidence of WT in people of Asian descent is about half to two‐thirds of that in Caucasians,3, 4 suggesting that environmental factors play little part in the lower incidence.
IGF2 is an imprinting gene expressed from the paternally inherited allele, and encodes a fetal polypeptide growth factor. Loss of imprinting (LOI) of IGF2 has been reported in various tumors, including pediatric tumors. There have been conflicting reports on the rates of IGF2 LOI in Japanese with WTs; one study reported IGF2 LOI in none of 21 tumors, whereas we previously reported it in seven of 27 tumors.5, 6 These results prompted us to analyze the IGF2 imprinting status in a substantial number of Japanese children with WTs.
Various abnormalities of the WT1 gene have been found in 13.0–17.6% of sporadic WTs.7, 8 WT1 is predominantly expressed in the embryonic kidney, and has a pivotal role in its development. We previously reported that half of 36 WTs with WT1 abnormality showed either LOI or paternal uniparental disomy (UPD) of IGF2.9 Other investigators reported that mice with a combination of Wt1 deletions and upregulated Igf2 expression, but not either factor alone, developed WT.10, 11, 12 These studies indicated that simultaneous alterations of the two genes are involved in the tumorigenesis of some WTs.
Wnt signaling plays a critical role in multiple cellular events that occur during development of the mammalian kidney, and β‐catenin encoded by CTNNB1 has been implicated as an integral component of this pathway. CTNNB1 mutations occur in 50–75% of WTs with WT1 abnormality, although the incidence is much lower in WTs without this abnormality.13, 14, 15 WTX is a candidate tumor suppressor gene isolated from the Xq11 region, and is also known to be involved in the Wnt signaling pathway.16, 17 Recent studies reported that WTX deletions/mutations occur in 8–24% of WTs.7, 8, 18, 19, 20
Thus, abnormalities of all four genes, WT1, CTNNB1, WTX, and IGF2, are interrelated and implicated in the tumorigenesis of WTs. To clarify which genetic and epigenetic alterations contribute to the different incidences of WT in East‐Asian and Caucasian children, and whether WTX and CTNNB1 or WT1 abnormalities coexist in WT, we performed mutational analyses in WT1, WTX and CTNNB1, and allelic expression and methylation analyses of IGF2 in 114 Japanese WTs. This is the first comprehensive study reporting the frequency of four genetic alterations responsible for WTs.
Materials and Methods
Patients and samples
One hundred and fourteen WTs were obtained from 111 Japanese infants or children, including 50 males and 61 females; three pairs of tumors were obtained from three syndromic patients with bilateral WTs. The age at diagnosis ranged from 2 months to 15 years with a median of 2 years and 9 months. In all cases, the diagnosis of WT was made as previously described.9 Pathologists in each institution verified that each sample for molecular genetic analysis contained 70% or more tumor cells. Normal samples were obtained from either peripheral blood or normal renal tissue adjacent to the tumor. The ethics committee of Saitama Cancer Center approved the study design.
Copy number and loss of heterozygosity analysis using single‐nucleotide polymorphisms arrays
High‐resolution single‐nucleotide polymorphisms (SNP) arrays, Affymetrix Mapping 50K‐Xba and 250K‐Nsp arrays (Affymetrix, Santa Clara, CA, USA), were used to analyze the chromosomal copy number and loss of heterozygosity (LOH) status in 114 tumors as described previously.9 Copy numbers and LOH were calculated using Copy Number Analyser for GeneChip® and allele‐specific copy‐number analysis using anonymous references programs (www.genome.umin.jp/CNAGtop2.html) with paired or anonymous references as controls.21, 22
Analysis of WT1 and CTNNB1 abnormalities and IGF2 LOI
We examined WT1 abnormalities using Southern blotting and/or an SNP array and sequencing as previously described.9 In addition, the combined bisulfite restriction assay (COBRA) and bisulfite sequencing was performed to confirm the methylation status of the WT1 promoter region. A part of the promoter region was amplified using the primers listed in Table S1. The polymerase chain reaction (PCR) products were digested with MboI (Takara Bio, Otsu, Japan). CTNNB1 mutations and LOI, UPD and LOH of IGF2 were analyzed as previously described.9
Deletion and mutation analysis of WTX on the active X chromosome
We screened all WTs for WTX deletions using SNP arrays and quantitative PCR (qPCR) with TaqMan probes on the 3′ and 5′ ends of WTX and MOCS2 at 5q11 where deletion rarely occurred in WT, as a reference gene. The primer sequences of WTX and MOCS2 are listed in Table S1.
The value of the number of WTX alleles was defined as the ratio of WTX/MOCS2. The reference values were <0.55 and 1.60 in male and female patients, respectively. To identify whether WTX deletions occurred on an active X chromosome, the methylation status of the promoter region of WTX or the androgen receptor (AR) gene was examined by both COBRA and bisulfite sequencing. Primers used for WTX and AR are listed in Table S1. In WTs having an active X chromosome with no WTX deletion, we performed a sequencing analysis for the all coding region of WTX as previously described.16 We classified nucleotide changes as mutations if the changes were not found in peripheral blood or normal kidney tissue.
Fluorescence in situ hybridization analysis for WTX
Fixed cells were prepared from fresh tumor tissues. A BAC clone, RP11‐1105P13, containing WTX was obtained from Roswell Park Cancer Institute (Buffalo, NY, USA). The clone is 179.3 kb in size, and covers the entire WTX of 15.7 kb. The copy number of the X chromosome was examined using specific alpha satellite DNA generated by PCR.23
Expression analysis of WTX mRNA
Total RNA was extracted from 49 WTs and four normal kidney tissues adjacent to WT or congenital mesoblastic nephroma. Total RNA from fetal kidney tissues at 6 weeks or 12 weeks (Virogen, Watertown, MA, USA) was used to confirm WTX mRNA levels in the developing kidney. First strand cDNA synthesis was performed as described previously.9 Primers derived from exons 1 and 2 are listed in Table S1. This assay recognized two isoforms of WTX, and ACTB was used as an endogenous gene expression control. Expression of WTX in WTs and fetal and normal kidneys was normalized to the average value of WTX mRNA in four normal kidneys.
Statistical analysis
Differences in the incidence of genetic and epigenetic characteristics in WT between any two ethnic populations were examined by the χ2 test using the 2 × 2 or 2 × 3 table.
Results
Alterations of the four genes, WT1, CTNNB1, WTX, and IGF2, were successfully examined in all 114 WTs.
WT1 abnormalities
We found WT1 abnormalities in 36 of 114 WTs (31.6%), including 35 reported previously (Table S2). All 13 syndromic WTs contained WT1 abnormalities in both alleles. Twenty‐three of 101 sporadic WTs (22.8%) also showed WT1 abnormalities in both alleles (Table 1). One sporadic WT with a hemizygous WT1 deletion (No. 31) showed complete promoter methylation of WT1 in the remaining allele and no WT1 mRNA expression.9 It further assumed that tumors holding no WT1 mutation with a hemizygous WT1 deletion or UPD at 11p13 might have promoter methylation of WT1. However, of 21 sporadic WTs with a hemizygous WT1 deletion or UPD at 11p13 without a WT1 mutation, COBRA showed an unmethylated WT1 promoter in 20, and partial promoter methylation in one (No. 45, data not shown); RNA was not available to analyze WT1 mRNA expression.
Table 1.
Incidence rates of WT1 or CTNNB1 abnormality in five series of Wilms tumor (WT)
| References | Present study | Ruteshouser et al.18 | Fukuzawa et al.7 | Corbin et al.8 | Wegert et al.20 |
|---|---|---|---|---|---|
| Total number | 114 | 125a | 59 | 57 | 429 |
| WT1 abnormality (%) | 36 (31.6) | 41 (32.8)a | 10 (16.9) | 14 (24.6) | 31/314 (9.9) |
| P‐valueb | 0.84 | 0.04 | 0.34 | <0.01 | |
| CTNNB1 mutation (%) | 30 (26.3) | 28 (22.4)a | 13 (22.0) | 18 (31.6) | 43 (10.0)c |
| Exon 3 (%) | 29 (25.4) | 10 (16.9) | 17 (29.8) | 43 (10.0) | |
| Exons 7 and 8 | 1 | 3 | 1 | Not done | |
| P‐valued | 0.48 | 0.54 | 0.47 | <0.01 | |
| Syndromic tumors with WT1 abnormality | 13 | 14 | 2 | 5 | |
| CTNNB1 mutation (%) | 9 (69.2) | 6 (42.9) | 2 (100) | 5 (100) | |
| Exon 3 | 9 | 1 | 4 | ||
| Exons 7 and 8 | 0 | 1 | 1 | ||
| P‐valued | 0.17 | 0.36 | 0.16 | ||
| Total number of sporadic tumors | 101 | 93 | 54 | 51 | |
| Sporadic tumors with CTNNB1 mutation (%) | 21 (20.8) | 18 (19.4) | 9 (16.6) | 12 (23.5) | |
| P‐valued | 0.80 | 0.54 | 0.70 | ||
| Sporadic tumors with WT1 abnormality (%) | 23 (22.8) | 26 (27.9) | 8 (13.0) | 9 (17.6) | |
| P‐valued | 0.41 | 0.24 | 0.66 | ||
| CTNNB1 mutation (%) | 15 (65.2) | 17 (65.4) | 6 | 7 (77.8) | |
| Exon 3 | 15 | 6 | 7 | ||
| Exons 7 and 8 | 0 | 0 | 0 | ||
| P‐valued | 0.99 | 0.61 | 0.49 | ||
| Sporadic tumors without WT1 abnormality | 78 | 67 | 46 | 42 | |
| CTNNB1 mutation (%) | 6 (7.7) | 1 (1.5) | 3 (10.2) | 5 (11.9) | |
| Exon 3 | 5 | 2 | 5 | ||
| Exons 7 and 8 | 1 | 1 | 0 | ||
| P‐valued | 0.08 | 0.81 | 0.45 |
The study was enriched for tumors with WT1 mutations and probably also with CTNNB1 mutation.
P‐value indicates the difference in the incidence of tumors with WT1 abnormality between the present and previous series of WTs.
Non‐hotspot mutations were not examined.
P‐value indicates the difference in the incidence of tumors with CTNNB1 mutation between the present and previous series of WTs.
CTNNB1 mutations
We identified CTNNB1 mutations in 30 tumors (26.3% of the 114 tumors), including 19 reported in a previous study (Tables 1 and S2).9 All mutations except one were located in the hotspot region. Non‐hotspot mutations in exons 7 and 8 of CTNNB1 previously reported by other investigators were not found in the present series; however, we found a single nucleotide change (c. 1375T>A/N387K) that has not been reported as an SNP or missense mutation in the National Center for Biotechnology Information (NCBI) database. CTNNB1 mutations were frequent in syndromic and sporadic WTs with WT1 abnormalities (9/13, 69.2% and 15/23, 65.2%, respectively), but infrequent in sporadic WTs with no WT1 abnormality (6/78, 7.7%).
Deletion and mutation of WTX
Quantitative PCR analysis showed a loss of the WTX locus in nine of 51 male tumors, and monoallelic and biallelic losses of WTX in 19 and three, respectively, of 63 female tumors (Fig. 1a and Table S2). Of the 19 female tumors with monoallelic WTX loss, three (Nos. 4, 67, and 68) showed a loss of the entire X chromosome identified by the SNP array analysis and the unmethylated promoter regions of the AR and WTX genes identified by the methylation analysis, indicating a complete loss of the inactive X chromosome and no loss of the active WTX (Fig. 1b,c). The other 16 tumors with monoallelic WTX had two X chromosomes; all 16 tumors had the unmethylated promoter region of AR, and one of the 16 also had the unmethylated promoter region of WTX and a deletion in only the 3′ site of WTX (No. 51, Fig. 1a), indicating a WTX deletion. Thus, 25 of 114 tumors were identified to have WTX deletions (Table 2).
Figure 1.
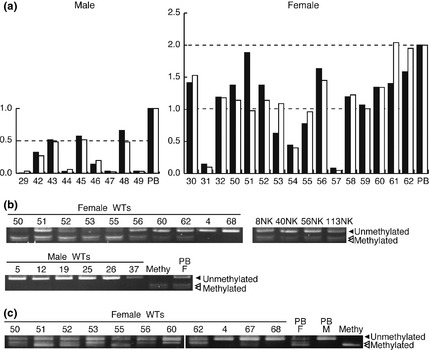
WTX deletion analysis. (a) WTX deletion was detected in Wilms tumors (WTs) from male (the left) and female (the right) patients by quantitative polymerase chain reaction (qPCR). Control male and female values were set to 1.0 and 2.0, respectively. Black and white bars indicate DNA values of the 5′and 3′ends of WTX, respectively. NK, normal kidney DNA from male or female patients used as a control. Methylation analysis on the promoter regions of the WTX (b) and AR (c) genes by COBRA. Bisulfite‐modified PCR products were digested with ClaI for WTX and TaqI for AR. Numbers above lanes indicate the tumor number. PB, lymphocyte DNA; Methy, control methylated DNA; M, male; F, female.
Table 2.
WTX deletion/mutation reported in the present and previous series of Wilms tumor (WT)
| Present study | Ruteshouser et al.18 | Perotti et al.19 | Fukuzawa et al.7 | Corbin et al.8 | Wegert et al.20 | |
|---|---|---|---|---|---|---|
| Total number of tumors | 114 | 125 | 102 | 59 | 57 | 399 |
| WTX abnormality (%) | 26 (22.8) | 20 (16.0) | 8 (7.8) | 14 (23.7) | 5 (8.8) | 68 (17.0)a |
| P‐valueb | 0.19 | 0.02 | 0.7 | 0.03 | 0.26 | |
| Deletion (%) | 25 (21.9) | 16 (12.8) | 7 (6.9) | 10 (16.9) | 2 (3.5) | 68/399 (17) |
| Mutation (%)c | 1 (0.8) | 4 (3.2) | 1 (1) | 4 (6.8) | 3 (5.3) | 2/104 (1.9) |
| E557GfsX22/wt ex | 1007ins4/fs378X | |||||
| R353X | ||||||
| Tumors with WT1 abnormality | 36 | 41 | 10 | 14 | ||
| WTX deletion | 4 | 4 | 0 | 0 | ||
| WTX mutationc | 0 | 3 | 3 | 0 | ||
| 367del/fs368X | R353X/mut ex | |||||
| S210X (2 tumors) | R358X/mut ex | |||||
| A29T/wt ex | ||||||
| Total number of sporadic tumors | 101 | 93 | 54 | 51 | ||
| Sporadic tumors with WTX abnormality (%) | 26 (25.7) | 18 (19.4) | 12 (22.2) | 5 (9.8) | ||
| P‐valueb | 0.29 | 0.63 | 0.02 | |||
| Sporadic tumors with WT1 abnormality | 23 | 26 | 8 | 9 | ||
| WTX deletion (%) | 4 (17.4) | 4 (30.8) | 0 | 0 | ||
| WTX mutation | 0 | 2 | 2 | 0 | ||
| Sporadic tumors without WT1 abnormality | 78 | 67 | 46 | 42 | ||
| WTX deletion | 21 | 11 | 9 | 2 | ||
| WTX mutationc | 1 | 1 | 1 | 3 | ||
| R353X/wt ex | S210X | 1083delC/fs360X/mut ex | Q222X | |||
| R358X | ||||||
| T429I | ||||||
| P‐valued | 0.30 | 0.57 | 0.84 | 0.28 | ||
| Tumors with WTX and CTNNB1 alterations (ex 3 and ex 7/8) | 0 | 4 (1 and 3)e | 0 | 5 (2 and 3) | 0 | 6 (6 and unknown) |
Only tumors with WTX deletion are included.
P‐value indicates the difference in the incidence of tumors with WTX abnormality between the present and previous series of WT.
Of tumors with no corresponding normal tissues, those with nonsense mutation were included, but those with missense mutation were excluded.
P‐value indicates the difference in the incidence of tumors with WTX abnormality between the present and previous series of sporadic WTs with or without WT1 abnormality.
Four tumors had combined WTX and CTNNB1 alterations; one had mutation in exon 3 and 3 had mutation in exon 7 or 8 of CTNNB1. mut ex, mutant allele expression; wt ex, wild‐type allele expression.
Fluorescence in situ hybridization using DXZ1 and a BAC clone (RP11‐1105P13) containing WTX was performed in 17 of 25 tumors with WTX deletions identified by qPCR, and produced results consistent with the two methods with one exception.
A direct sequencing analysis performed in the remaining 89 tumors with no WTX deletion on the active X chromosome identified two single nucleotide changes in three tumors. A female tumor had a nonsense mutation (c. 1057C>T/R353X) that was previously reported in two WTs by other investigators (Table 2).7, 20 However, sequencing of cDNA from the same tumor revealed wild‐type WTX mRNA expression, but not mutant WTX mRNA expression. The other single nucleotide change (c. 85G>A/A29T) was found in tumors of one male and one female, and the same change was identified in peripheral blood cells of both patients, indicating that this nucleotide conversion was of germline origin.
WTX mRNA expression
We examined WTX mRNA expression in 49 WTs. WTX mRNA levels were similar between normal and fetal kidney tissues, and no WTX mRNA expression was found in five of nine WTs with WTX deletions (Fig. 2). In the remaining four tumors with WTX mRNA expression, the DNA ratios of WTX/MOCS2 were 0.52 in one tumor from a male, and 1.18, 1.37 and 1.45 in three tumors from females. Of 40 tumors with no WTX deletion, only one lacked WTX mRNA expression. In the other 39 WTs, WTX mRNA levels were equal to or greater than those in normal and fetal kidney tissues (Fig. 2).
Figure 2.
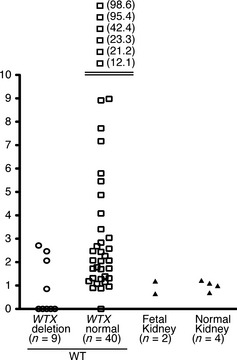
Results of quantitative polymerase chain reaction (qPCR) analysis of WTX mRNA. Expression levels of WTX mRNA in Wilms tumors (WTs) and fetal and infant kidneys were normalized to the average value of WTX mRNA in four infant kidneys. Each mark represents a WT or fetal and normal kidney. Numbers on the right side of each mark are the values of WTX mRNA level.
Loss of IGF2 imprinting, UPD, hemizygous loss, and retention of imprinting of IGF2
RNA was available for examining the allelic expression of IGF2 in 43 of 114 WTs, and the ApaI/AvaII polymorphism site was informative in 20 of the 43. Of 20 tumors, six (30.0%) showed biallelic expression indicating LOI, and 14 showed monoallelic expression indicating the retention of imprinting (ROI). Five and one tumor with biallelic IGF2 expression showed hypermethylated and hemimethylated CTCF6, a sixth CTCF (CCCTC‐binding factor)‐binding site, in the H19‐differentially methylated region (DMR), respectively, and we classified the hemimethylated tumor as having IGF2 LOI because the expression pattern is more important in determining IGF2 LOI than the methylation status of CTCF6 at H19‐DMR. All 14 tumors with monoallelic IGF2 expression showed hemimethylated CTCF6 at H19‐DMR as expected.
From the combined results of the IGF2 allelic expression pattern, the IGF2 SNP array pattern, and the methylation status of H19‐DMR, we identified IGF2 LOI in 24 (21.1%), IGF2 UPD in 36 (31.6%), hemizygous loss of the IGF2 region in seven (6.1%), a gain of the IGF2 region in three (2.6%), and IGF2 ROI in 44 (38.6%) of 114 WTs (Table S2). When we selected 101 tumors of sporadic origin, LOI, UPD, and ROI were found in 23 (22.8%), 34 (33.7%), and 44 (43.6%), respectively, in 101 sporadic tumors (Table 3).
Table 3.
Incidence rates of loss of imprinting (LOI), uniparental disomy (UPD) and retention of imprinting (ROI) of IGF2 in three series of Wilms tumor
| References | Total number | LOI of IGF2 | UPD of IGF2 | ROI of IGF2 |
|---|---|---|---|---|
| A | ||||
| Present study (sporadic tumors in Japan) (%) | 101 | 23 (22.8) | 34 (33.7)a | 44 (43.6)b |
| B | ||||
| Yuan et al.28 (sporadic tumors in USA) (%) | 58 | 22 (37.9) | 29 (50.0) | 7 (12.1) |
| C | ||||
| Fukuzawa et al.5 (unspecified tumors in New Zealand) (%) | 41 | 13 (31.7) | 17 (41.5) | 11 (26.8) |
Three tumors with 11p15.5 trisomy are included.
Seven tumors with hemizygous loss at 11p15.5 are included. LOI and UPD + ROI: A versus B P = 0.041, A versus C P = 0.267, A versus B + C P = 0.049.
Of 36 tumors with IGF2 UPD, 35 and one had a hypermethylated and hypomethylated CTCF6 at H19‐DMR, respectively, indicating that the IGF2 UPD in almost all tumors was of paternal origin. In addition, all of seven tumors with allelic loss of IGF2 and three tumors with a gain of IGF2 showed hypermethylated CTCF6 at H19‐DMR, indicating that the loss occurred in the maternal allele, and the gain in the paternal allele. The analysis with the SNP array showed that 68 tumors had a normal allelic balance with heterozygosity in the IGF2 region, and 23 (33.8%) of the 68 tumors had a hypermethylated CTCF6 at H19‐DMR, indicating IGF2 LOI. Thus, similar incidences of IGF2 LOI were obtained by the methylation‐based analysis of CTCF6 at H19‐DMR (33.8%) and by the allelic expression analysis of IGF2 at exon 9 (6/20, 30%).
Incidence of WT1,CTNNB1, or WTX abnormalities in Japanese children with Wilms tumor
WT1, CTNNB1 and WTX abnormalities were found in 36 (31.6%), 30 (26.3%) and 26 (22.8%) tumors, respectively, of the 114 tumors (Tables 1 and 2). None of the tumors had combined CTNNB1 and WTX abnormalities, suggesting mutual exclusion; this finding confirmed the previous one reported by other investigators.7 When tumors were classified into those with WT1 abnormalities and those without, CTNNB1 mutations were found to be more frequent in syndromic or sporadic tumors with WT1 abnormalities (P < 0.01 and P < 0.01). In contrast, WTX deletions/mutations were similarly frequent between sporadic tumors with and without WT1 abnormalities; the finding was previously reported by other investigators (Table 2).18
Discussion
As shown in Tables 1 and 2, we compared the incidence rate of WT1, CTNNB1 and WTX abnormalities in the present series with that in the former reports.7, 8, 18, 19, 20 The incidence of WT1 abnormalities in this cohort (31.6%) is much higher than that in two previous series, and comparable to that of two other series of WTs. When we included only sporadic WTs, the incidences of WT1 abnormality and CTNNB1 mutation were comparable between the present and three other series of WTs (Table 1).
We found an amino acid substitution (c. 85G>A/A29T) in WTX of germline origin in two tumors (Table S2), the same change was previously reported as a missense mutation in two WTs.7 Although the nucleotide change is difficult to assign as either a point mutation or rare SNP, we classified the change as a mutation if it proved to be of somatic origin. Thus, the present study found WTX deletions/mutations in 22.8% of 114 WTs, an incidence higher than that of two previous series, and comparable to that of three other series (Table 2). When we included only 101 sporadic WTs, the incidence of WTX deletions/mutations was also higher than or comparable to the previous series. In regard to various WTX mutations, half of them were of germinal origin, and somatic missense, nonsense and frameshift mutations were relatively rare (0.8–6.8%) (Table 2).
The present as well as previous studies showed that while CTNNB1 mutations were frequent in WTs with WT1 abnormalities, the incidence was much lower in WTs without WT1 abnormalities (Table 1). A lack of WT1 induces WT precursor cells to undergo apoptosis and mutant β‐catenin might rescue the cells by the activation of Wnt signaling.24 The present study showed the comparable frequencies of WTX abnormalities in sporadic WTs with and without WT1 abnormalities; the findings were previously reported by Ruteshouser et al. (Table 2).18 In addition to the inhibitory role in Wnt signaling pathway, WTX has another role in nuclear pathways implicated in the transcriptional regulation of cellular differentiation and cell death programs,24, 25 and the damage of this pathway by WTX alterations might involve in the tumorigenic process and explain why the alterations were found in both WT1‐wild‐type and WT1‐mutant type tumors.
While one study reported a lack of WTX mRNA expression in 11.5% of WTs without WTX deletions,20 the present study showed no WTX mRNA expression in only one (2.5%) of 40 WTs without WTX deletions (Fig. 2). In fetal and normal kidney tissues, levels of WTX mRNA were very low compared with the levels of GAPDH and ACTB. The sensitivity to detect WTX mRNA expression may be different between the two studies, and this might have caused the discrepancy.
There are two histological types of precancerous lesions for WT; perilobar nephrogenic rests (PLNRs) and intralobar nephrogenic rests (ILNRs). PLNRs are associated with IGF2 LOI, and ILNRs, with abnormalities of WT1.26, 27 While ILNRs and PLNRs were found in 20% and 17%, respectively, of Caucasians with WTs, the incidence rates were 25% and 2%, respectively, in Japanese with WTs.5 WTs in the present and previous series were classified into three types based on IGF2 status, and are summarized in Table 3. Yuan et al. identified IGF2 LOI in 37.9% of 58 sporadic WTs, and their frequency is lower than that of IGF2 LOI in the present series of 101 sporadic WTs (P = 0.04) (Table 3). The lower incidence of IGF2 LOI in Japanese than in Caucasians is consistent with the finding of a lower incidence of PLNRs in Japanese than in Caucasians. It is difficult to assess the difference in the incidence or WT with WT1 abnormality between two ethnic populations because of sampling bias for syndromic WT with WT1 abnormality. If we included only sporadic tumors, the frequencies of WT with WT1 or CTNNB1 abnormality were similar between the two populations. The incidence of WTX abnormality in Japanese WTs was also similar to those in three of the five Caucasian series of WT. Because the incidence of WT among Japanese is half of that in Caucasians, the population‐based rate of WT with IGF2 LOI may be much lower in Japanese than Caucasian children. In contrast, the population‐based incidences of WT1, CTNNB1, and WTX abnormality may be similar between the two populations (Fig. 3).
Figure 3.
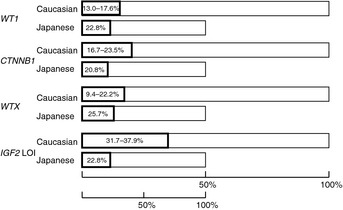
Abnormalities of the WT1,WTX and CTNNB1 genes, and loss of IGF2 imprinting (LOI) in sporadic Wilms tumors (WTs) in Japanese (the present series) and Caucasian children. The bar length for Japanese is half of that for Caucasians because the incidence rate is half that for Caucasians. The ranges of each abnormality in Caucasians indicate the lowest and highest values reported in the previous studies.5, 7, 8, 18, 28
Two groups of investigators reported the different incidences of IGF2 LOI in Japanese WTs.5, 6 To detect IGF2 LOI, we used allelic expression analysis and/or a combination of COBRA of H19 DMR and SNP arrays. Both methods detected similar incidence of IGF2 LOI. Yuan et al.28 used IGF2 LOI with methylation analysis of H19 DMR and SNP arrays. In contrast, Fukuzawa et al. used a methylation analysis of two imprinted regions of 11p15.5, H19 DMR and KvDMR1, to detect it.5 In addition, we and Yuan et al. showed the results of IGF2 LOI analysis in sporadic WTs; however, Fukuzawa et al. did not specify that the tumors were sporadic or not. Thus, we believe that the different incidences of IGF2 LOI between Japanese and Caucasians obtained by the present and Yuan et al. studies provided substantial evidence.
The present study raises the question of why the frequency of IGF2 LOI is lower in Japanese WTs. Beckwith‐Wiedemann syndrome (BWS) is an imprinting‐related growth disorder (OMIM #130650). Five to 10% of patients with BWS have methylation of H19 DMR on both parental chromosomes, resulting in IGF2 LOI.29 Interestingly, Japanese patients with BWS were shown to have a significantly lower frequency of H19‐DMR hypermethylation than North American and European patients, suggesting that susceptibility to epigenetic alterations differs between populations.30 The same decreased susceptibility to epigenetic change may have also caused the decreased incidence of WTs with IGF2 LOI.
A growing number of non‐genetic disorders of growth, such as intra‐uterine growth retardation, etc., have been associated with epigenetics since environmental signals perturb the embryonic hormonal/metabolic setting.31 Furthermore, a recent study indicated that post‐weaning diet affected genomic imprinting at the Igf2 locus in mice.32 It is important to clarify whether combinations of different susceptibilities to epigenetic alterations and different dietary habits or other environmental factors affect the imprinting status of IGF2.
Disclosure Statement
The authors have no conflicts of interest.
Supporting information
Table S1. Primers and TaqMan probes.
Table S2. Clinical, genetic, and epigenetic characteristics of 111 patients with 114 Wilms tumors.
Acknowledgments
We appreciate Drs V. Huff and E. C. Ruteshouser at M. D. Anderson Cancer Center, and Dr R. Fukuzawa at University of Otago for kindly providing some data on syndromic or sporadic origin of WT additional to the original ones reported in references 7 and 18. We also thank the parents and physicians who participated in the study of the Japan Wilms Tumor Study Group. This work was supported by Grants‐in‐Aid for scientific research (nos. 2039045, 2339045, and 21791021) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (Y. K.; M. H.) and the Kawano Masanori Memorial Foundation for the Promotion of Pediatrics (M. H.).
References
- 1. Kuroishi T, Takezaki T, Tominaga S, Tajima K. Cancer mortality in Japan (1950–2000). In: Tajima K, Kuroishi T, Ohshima A, eds. Cancer Mortality and Morbidity Statistics: Japan and the World‐2004. Tokyo: Japan Scientific Societies Press, 2004; 1–93. [Google Scholar]
- 2. Parkin DM, Stiller CA, Draper GJ, Bieber CA. The international incidence of childhood cancer. Int J Cancer 1988; 42: 511–20. [DOI] [PubMed] [Google Scholar]
- 3. Goodman MT, Yoshizawa CN, Kolonel LN. Ethnic patterns of childhood cancer in Hawaii between 1960 and 1984. Br J Cancer 1989; 60: 1758–63. [DOI] [PubMed] [Google Scholar]
- 4. Stiller CA, McKinney PA, Bunch KJ, Bailey CC, Lewis IJ. Childhood cancer and ethnic group in Britain: a United Kingdom children's Cancer Study Group (UKCCSG) study. Br J Cancer 1991; 64: 543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukuzawa R, Breslow NE, Morison IM et al Epigenetic differences between Wilms' tumours in white and east‐Asian children. Lancet 2004; 363: 446–51. [DOI] [PubMed] [Google Scholar]
- 6. Watanabe N, Nakadate H, Haruta M et al Association of 11q loss, trisomy 12, and possible 16q loss with loss of imprinting of insulin‐like growth factor‐II in Wilms tumor. Genes Chromosom Cancer 2006; 45: 592–601. [DOI] [PubMed] [Google Scholar]
- 7. Fukuzawa R, Anaka MR, Weeks RJ, Morison IM, Reeve AE. Canonical WNT signaling determines lineage specificity in Wilms tumour. Oncogene 2009; 28: 1063–75. [DOI] [PubMed] [Google Scholar]
- 8. Corbin M, de Reyniès A, Rickman DS et al WNT/beta‐catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries? Genes Chromosom Cancer 2009; 48: 816–27. [DOI] [PubMed] [Google Scholar]
- 9. Haruta M, Arai Y, Sugawara W et al Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chromosom Cancer 2008; 47: 712–27. [DOI] [PubMed] [Google Scholar]
- 10. Kreidberg JA, Sariola H, Loring JM et al WT‐1 is required for early kidney development. Cell 1993; 174: 679–91. [DOI] [PubMed] [Google Scholar]
- 11. Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 1995; 375: 34–9. [DOI] [PubMed] [Google Scholar]
- 12. Hu Q, Gao F, Tian W et al Wt1 ablation and Igf2 upregulation in mice result in Wilms tumors with elevated ERK1/2 phosphorylation. J Clin Invest 2010; 121: 174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koesters R, Ridder R, Kopp‐Schneider A et al Mutational activation of the β‐catenin proto‐oncogene is a common event in the development of Wilms tumors. Cancer Res 1999; 59: 3880–2. [PubMed] [Google Scholar]
- 14. Li CM, Kim CE, Margolin AA et al CTNNB1 mutations and overexpression of Wnt/beta‐catenin target genes in WT1‐mutant Wilms' tumors. Am J Pathol 2004; 165: 1943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maiti S, Alam R, Amos CI, Huff V. Frequent association of beta‐catenin and WT1 mutations in Wilms tumors. Cancer Res 2000; 60: 6288–92. [PubMed] [Google Scholar]
- 16. Rivera MN, Kim WJ, Wells J et al An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 2007; 315: 642–5. [DOI] [PubMed] [Google Scholar]
- 17. Major MB, Camp ND, Berndt JD et al Wilms tumor suppressor WTX negatively regulates WNT/beta‐catenin signaling. Science 2007; 316: 1043–6. [DOI] [PubMed] [Google Scholar]
- 18. Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one‐third of tumors. Genes Chromosom Cancer 2008; 47: 461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perotti D, Gamba B, Sardella M et al Functional inactivation of the WTX gene is not a frequent event in Wilms' tumors. Oncogene 2008; 27: 4625–32. [DOI] [PubMed] [Google Scholar]
- 20. Wegert J, Wittmann S, Leuschner I, Geissinger E, Graf N, Gessler M. WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosom Cancer 2009; 48: 1102–11. [DOI] [PubMed] [Google Scholar]
- 21. Nannya Y, Sanada M, Nakazaki K et al A robust algorithm for copy number detection using high‐density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 2005; 65: 6071–9. [DOI] [PubMed] [Google Scholar]
- 22. Yamamoto G, Nannya Y, Kato M et al Highly sensitive method for genome wide detection of allelic composition in nonpaired, primary specimens by use of Affymetrix single‐nucleotide‐polymorphism genotyping microarrays. Am J Hum Genet 2007; 81: 114–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Warburton PE, Greig GM, Haar T, Willard HF. PCR amplification of chromosome specific alpha satellite DNA: definition of centromeric STS markers and polymorphic analysis. Genomics 1991; 11: 324–33. [DOI] [PubMed] [Google Scholar]
- 24. Kim MK, Min DJ, Rabin M, Licht JD. Functional characterization of Wilms tumor‐suppressor WTX and tumor‐associated mutants. Oncogene 2010; 320: 832–42. [DOI] [PubMed] [Google Scholar]
- 25. Rivera MN, Kim WJ, Wells J et al The tumor suppressor WTX shuttles to the nucleus and modulates WT1 activity. Proc Natl Acad Sci USA 2009; 106: 8338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park S, Bernard A, Bove KE et al Inactivation of WT1 in nephrogenic rests, genetic precursors to Wilms tumour. Nat Genet 1993; 5: 363–7. [DOI] [PubMed] [Google Scholar]
- 27. Ohlsson R, Cui H, He L et al Mosaic allelic insulin‐like growth factor 2 expression patterns reveal a link between Wilms tumorigenesis and epigenetic heterogeneity. Cancer Res 1999; 59: 3889–92. [PubMed] [Google Scholar]
- 28. Yuan E, Li CM, Yamashiro DJ et al Genomic profiling maps loss of heterozygosity and defines the timing and stage dependence of epigenetic and genetic events in Wilms' tumors. Mol Cancer Res 2005; 3: 493–502. [DOI] [PubMed] [Google Scholar]
- 29. Cerrato F, Sparago A, Verde G et al Different mechanisms cause imprinting defects at the IGF2/H19 locus in Beckwith‐Wiedemann syndrome and Wilms' tumour. Hum Mol Genet 2008; 17: 1427–35. [DOI] [PubMed] [Google Scholar]
- 30. Sasaki K, Soejima H, Higashimoto K et al Japanese and North American/European patients with Beckwith‐Wiedemann syndrome have different frequencies of some epigenetic and genetic alterations. Eur J Hum Genet 2007; 15: 1205–10. [DOI] [PubMed] [Google Scholar]
- 31. Devaskar SU, Raychudhuri S. Epigenetics: a science of heritable biological adaptation. Pediatr Res 2007; 61: 1R–4R. [DOI] [PubMed] [Google Scholar]
- 32. Waterland RA, Lin JR, Smith CA, Jirtle RL. Post‐weaning diet affects genomic imprinting at the insulin‐like growth factor 2 (Igf2) locus. Hum Mol Genet 2006; 15: 705–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers and TaqMan probes.
Table S2. Clinical, genetic, and epigenetic characteristics of 111 patients with 114 Wilms tumors.