

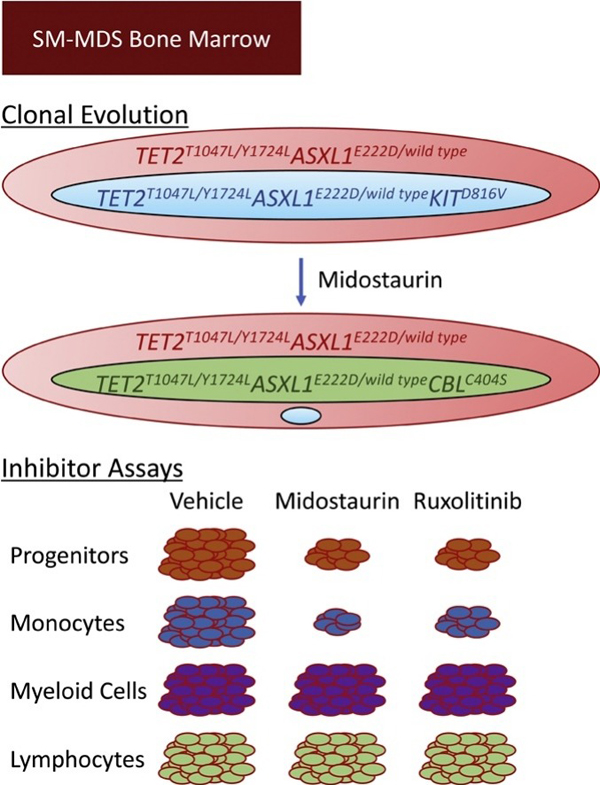
Graphical Abstract
Dear Editor,
Systemic mastocytosis (SM) is a heterogeneous malignancy with recurrent KIT mutations (frequently KITD816V) [1]. SM with an associated hematological neoplasm (SM-AHN), such as SM-myelodysplastic syndrome (SM-MDS), often harbors somatic mutations. In SM-AHN, TET2 and ASXL1 can precede KITD816V [2,3]. Little else is known about the temporal sequence of mutations and clonal dynamics in SM-AHN. RAS and STAT5-driven CD44 expression correlates with SM aggressiveness [4]. Otherwise, the molecular pathways governing malignant behavior in the SM and AHN components have not been well-defined. Understanding these pathways could identify therapeutic targets in this disease. We analyzed an SM-MDS patient sample harboring KITD816V, TET2T1047L, TET2Y1724L, ASXL1E222D, and CBLC404S mutations: we define the clonal architecture of this patient’s disease, the signaling activation status of this BM, and the differential effects of cytokines and signaling inhibitors on bone marrow (BM) subpopulations.
A 62-year-old female with progressive fatigue and maculopapular cutaneous mastocytosis (MPCM) concerning for systemic mastocytosis, presented for evaluation. Complete blood count (CBC) revealed bicytopenia: white blood cell (WBC) of 2.4 × 109/L, absolute neutrophil count (ANC) of 0.8 × 109/L, normal hemoglobin, and platelet count of 112 × 109/L. Serum tryptase was 162 μg/L (≤10.9 μg/L). A bone marrow (BM) biopsy (BMBx) showed trilineage hematopoiesis with increased cellularity (90 %) without increased blasts. The BM harbored abnormal clonal mast cells: several large aggregates of atypical CD117+trpytase+ mast cells by immunohistochemistry; CD2+CD25+ by flow cytometry, compatible with systemic mastocytosis [5]. Additionally, morphological examination revealed subtle dysgranulopoiesis characterized by rare neutrophils with slightly decreased cytoplasmic granules and irregular nuclei, concerning, but not sufficient, for a diagnosis of myelodysplasia. Cytogenetics were normal (Fig. 1A). Seven months later, fatigue, thrombocytopenia, and elevated tryptase progressed (platelets: 91 × 109/L; tryptase: 201 μg/L). BMBx was morphologically unchanged.
Fig. 1.
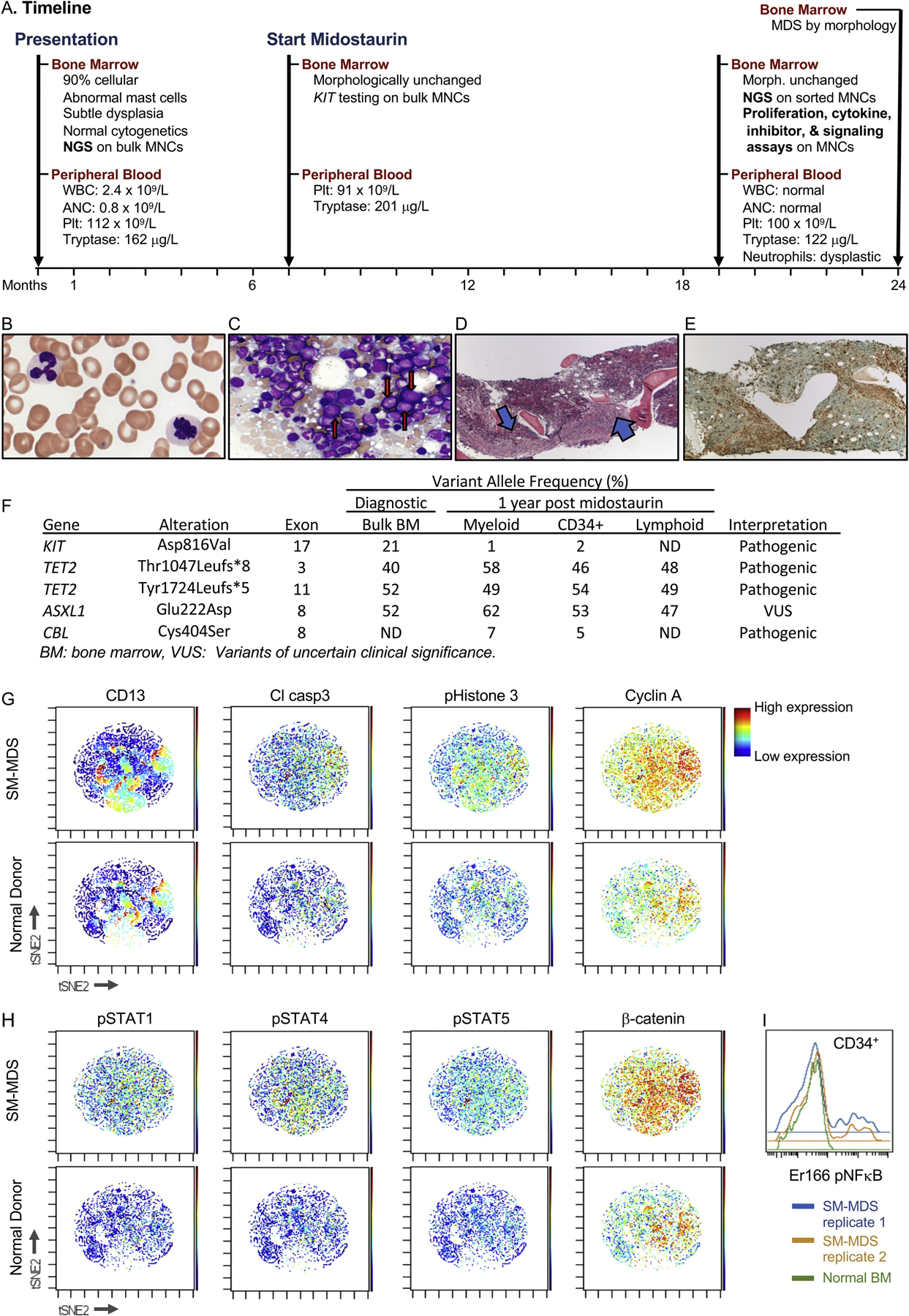
SM shares a common clone of origin with MDS in a patient with SM-MDS. A. Timeline describing the clinical course, bone marrow, and peripheral blood sampling. WBC: white blood cell count, ANC: absolute neutrophil count, Plt: platelets, NGS: next generation sequencing, MNC: mononuclear cells. B–E. Peripheral blood smear (B) and bone marrow biopsy (C–E) were obtained one year after midostaurin treatment was initiated and stained with Wright Giemsa (B–C), hematoxylin and eosin (H&E, D) or tryptase (E). B. Circulating dysplastic neutrophils with pale cytoplasm (100X magnification). C. Touch imprint. Blasts with increased M:E ratio are indicated with red arrows (50× magnification). D. Bone marrow core section with cellularity of approximately 95 %. Spindle-shaped mast cell aggregates are indicated by blue arrows (4× magnification). E. Spindle-shaped mast cell aggregates are confirmed by a tryptase IHC stain (4X magnification). F. Next generation sequencing results of bulk and sorted BM populations at diagnosis and 1 year post midostaurin. G–I. Mass cytometry (CyTOF) analysis reveals a unique signaling and proliferation pattern in SM-AHN bone marrow. Bone marrow mononuclear cells from SM-MDS (top panels) or a normal donor (bottom panels) were stained with metal-conjugated antibodies to cell surface and intracellular proteins and processed for mass cytometry (CyTOF). G–H. viSNE analysis represents each profiled cell. Each plot is colored as a heatmap according to levels of the epitope indicated. I. Histogram representing levels of pNFκB within the HSPC (CD34+) compartment.
She started midostaurin 100 mg twice/day and within 1 month, symptoms and laboratory parameters improved. WBC and ANC normalized to 3.6 × 109/L and 1.7 × 109/L, respectively, platelets increased to 114 × 109/L, and tryptase decreased to 75.9 μg/L. Despite excellent control of symptoms and tryptase levels, her thrombocytopenia persisted (47–121 × 109/L) and required midostaurin adjustments. One year after midostaurin initiation, WBC, ANC, and Hgb remained in the normal range, platelets remained low (100 × 109/L) and tryptase was 122 μg/L. BMBx showed atypical granulocyte precursors in addition to all previous findings (Fig. 1B–E). Concurrently, the peripheral blood demonstrated atypical neutrophils (decreased granulation and abnormal lobation): concerning but insufficient for diagnosing MDS. BMBx five months later revealed minimal changes in hypercellularity (75 %), blast percentage (4%), and mast cell aggregates (20 % of marrow cellularity) but increased hypogranular neutrophils and dyserythropoiesis (nuclear irregularity/budding and basophilic stippling) were diagnostic of MDS (SM-MDS).
SM-AHN BM aspirates were sorted and analyzed by next generation sequencing, intracellular signaling protein assessments by CyTOF, and proliferation and colony forming assays (see Supplementary Information).
We investigated the clonal dynamics and disease progression of this SM-AHN. Retrospective mutational profiling with next generation sequencing (NGS) was performed on the unsorted diagnostic BM cells and revealed a KITD816V mutation, as frequently reported in SM (Fig. 1F) [1]. This sample also harbored TET2T1047fs, TET2Y1724fs and ASXL1E222D, as frequently reported in MDS [6]. Notably, both the TET2 mutations and the ASXL1 mutation were present at near 50 % variant allele frequency (VAF) within the DNA sequencing data, suggesting that nearly all of the BM cells likely co-express these mutations in a heterozygous state. In contrast, KITD816V VAF was 21 %, indicating that a subclone of the predominant TET2T1047fs/Y1724fsASXL1E222D/wild type clone harbors KITD816V (TET2T1047fs/Y1724fsASXL1E222D/wild typeKITD816V). Since KIT mutations are known drivers of SM [1], these data suggest that SM developed from a subclone of a predominant clone (defined by TET2/ASXL1 mutations) comprising nearly all of the patient’s hematopoietic compartment. Next, we deciphered the contribution of each clone to hematopoiesis and disease evolution. BM mononuclear cells (BMNCs) from a subsequent BM aspirate (obtained 12 months after initiation of midostaurin) were sorted to isolate hematopoietic stem and progenitor cells (HSPCs, CD34+), myeloid cells (based upon CD45, SSC), and T cells (CD3+, Supplementary Figure) and submitted for NGS. As in the diagnostic specimen, TET2T1047L, TET2Y1724L, and ASXL1E222D were identified with a near 50 % VAF in all fractions. This finding indicates that this clone contributes to all or nearly all of this patient’s hematopoiesis and that these mutations likely co-occur in an early hematopoietic stem cell (HSC). Additionally, both the HSPC and myeloid fractions, but not the T cells, harbored a CBLC404S at low VAF, indicating that this newly detected subclone likely arose in the intervening time and contributes only to myelopoiesis. KITD816V was detected at VAF 1–2 % within the HSPCs and myeloid fractions, but was not detected in the T cells, indicating that this clone likely contributes to myelopoiesis. The significant reduction in KIT’s VAF after midostaurin treatment suggests that midostaurin selectively depletes this KITD816V subclone but does not impact the prevailing TET2T1047L/Y1724LASXL1E222D clone. Together, these findings demonstrate that the KITD816V clone likely shares a common origin with the MDS.
To assess the molecular mechanisms that might contribute to the malignant phenotype in this patient, we profiled signaling pathways and markers of proliferation and apoptosis in the patient’s BM and compared these to a normal donor BM using mass cytometry. The CD13+ maturing myeloid population was more prominent in SM-AHN BM than normal BM revealing expanded myeloid precursors (Fig. 1G), as seen in MDS. These BMNCs expressed increased cleaved caspase-3 (a marker of apoptosis) and phospho-histone 3 and cyclin A (markers of cell cycle progression, Fig. 1G); this pattern is consistent with increased proliferation and apoptosis previously documented in MDS [7]. Relative to normal BM, the patient’s BM displayed diffusely increased phosphorylated-STAT1, 4, and 5 and β-catenin in all compartments assayed, including HSPCs and myeloid progenitors (Fig. 1H); this finding is consistent with hyperactive signal transduction pathways as described in leukemogenesis [8]. Moreover, within HSPCs, a subset of cells expressed significantly higher levels of phospho-NFκB, a proleukemic mediator (Fig. 1I) [9]. Notably, NFκB and β-catenin have been implicated in increased self-renewal capacity of leukemia stem cells [10]. Hyperactivated STAT signaling has been well described in myeloid neoplasms, especially MPNs and SM.
Next, we assessed the responsiveness of hematopoietic lineages to physiologically- and clinically-relevant stimuli and drugs in BMNCs after 1 year of midostaurin treatment. This specimen harbored few mast cells (< 1%). We assessed the effect of in vitro treatment with thrombopoietin (TPO), stem cell factor (SCF), ruxolitinib (JAK inhibitor), or midostaurin (multi-kinase inhibitor) on the relative abundance of subpopulations. Both TPO and SCF increased CD34+ progenitor frequency, and both ruxolitinib and midostaurin reduced progenitor frequencies relative to vehicle (Fig. 2A–B). In contrast, monocyte frequencies were not affected by TPO or SCF. However, similar to the progenitors, both ruxolitinib and midostaurin reduced monocyte frequencies (Fig. 2A, C). Inhibitor treatment did not diminish the myeloid compartment as a whole. Within the myeloid compartment, however, midostaurin depleted the CD117−CD13- (immature myeloid precursors) subpopulation. In contrast, ruxolitinib depleted the more mature myeloid CD117−CD13+ subpopulation (Fig. 2D). CD13-selectivity was not observed in other subpopulations (CD34+, monocytes, or T cells). This finding suggests that midostaurin and ruxolitinib have distinct cell type-specific effects on myeloid cells and may be additive in controlling myeloid neoplasms.
Fig. 2.
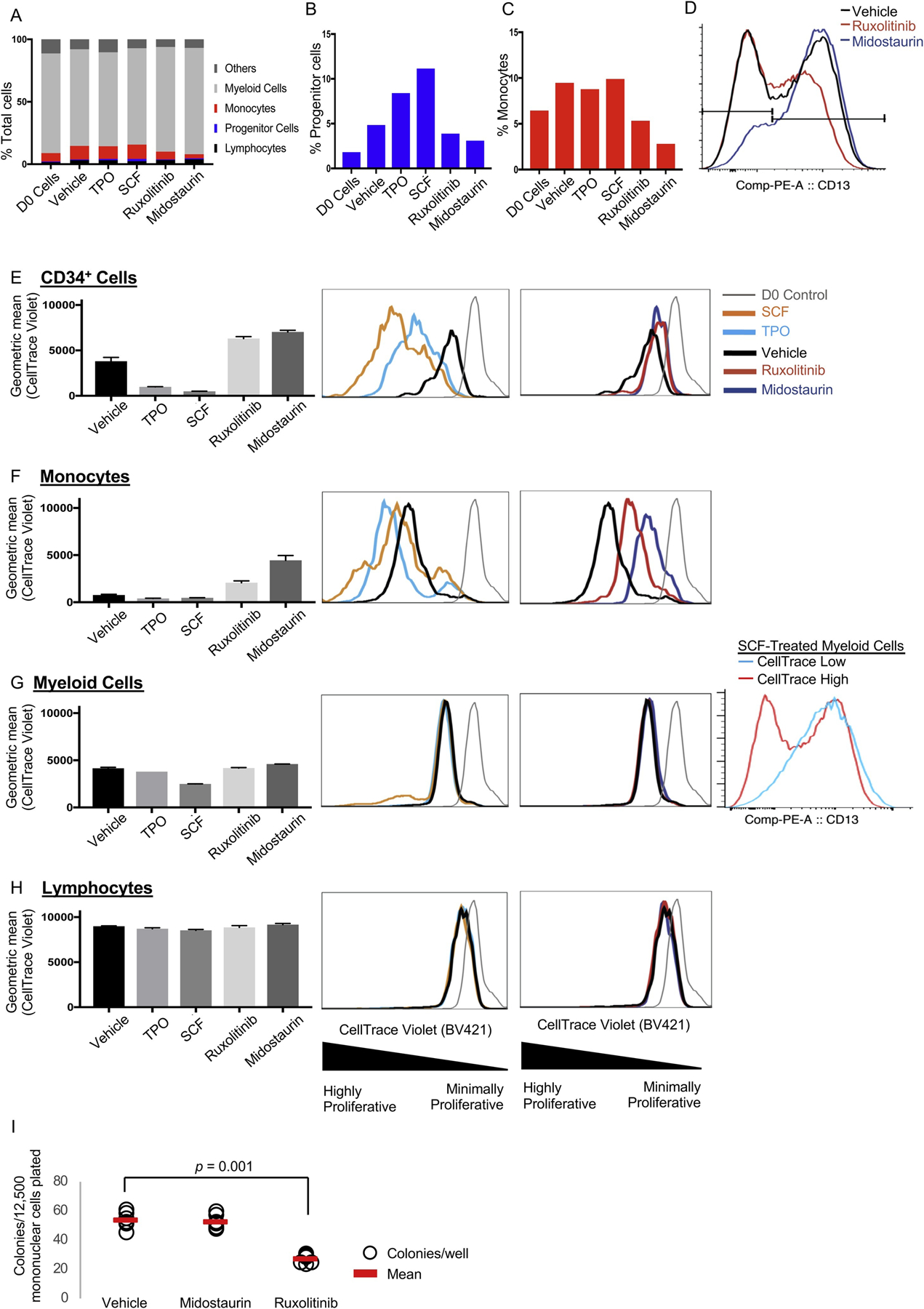
Bone marrow subpopulations display lineage-specific responsiveness to physiologically relevant stimuli and clinically relevant drugs. Bone marrow mononuclear cells were harvested one year after initiation of midostaurin treatment. A–H. Cells were plated in liquid culture and treated with cytokines, inhibitors, or vehicle. Each condition was plated in two technical replicates. The proportion of each population was assessed by flow cytometry after seven days of culture. A. Percentage of total or percentage of cells that are progenitors (CD34+, B) or monocytes (C). D. CD13 expression within the myeloid compartment. E–H. Cells were stained with CellTrace and plated in liquid culture for seven days in two technical replicates. D0 indicates CellTrace levels at the time of plating. I. Primary colony forming assay. Patient bone marrow mononuclear cells were plated in a semi-solid methylcellulose-based media (12,500 per well, six wells per condition). The colony number was scored after 7 days in culture. The colonies were of uniform size and appearance in all treatment groups. Treatment doses for all experiments: thrombopoietin (TPO) 10 ng/mL, stem cell factor (SCF) 10 ng/mL, ruxolitinib 400 nM, midostaurin 400 nM. Vehicle: DMSO. Error bars represent standard error of the mean.
Next, we assessed the proliferative responses of BM subpopulations to these cytokines and inhibitors in vitro. We used CellTrace, a fluorescent label diluted with each cell division to measure the proliferative history of cellular compartments. TPO or SCF treatment of HSPCs reduced CellTrace labeling (consistent with increased proliferation) relative to vehicle-treatment. In contrast, both midostaurin and ruxolitinib reduced proliferation of HSPCs (Fig. 2E). Like HSPCs, TPO or SCF treatment of monocytes increased proliferation while midostaurin or ruxolitinib reduced proliferation in this compartment (Fig. 2F). Vehicle-treated monocytes were highly proliferative, consistent with high monocyte counts in patients with ASXL1 [11] and TET2 [12] mutations. Notably, midostaurin normalizes monocyte counts in patients with SM-AHN [13]. The myeloid fraction was largely not impacted by these treatments, but a small subpopulation of myeloid cells, maturing myeloid precursors (CD117−CD13+), were highly proliferative in response to SCF (Fig. 2G, right-most panel). T cells were largely unaffected by treatment (Fig. 2H). These data indicate that this SM-AHN cell fractions have subpopulation-specific responses to treatments and suggest that ruxolitinib and midostaurin may have complimentary suppressive effects in this disease.
Colony forming assays (a surrogate for self-renewal capacity) of this patient’s BMNCs showed that ruxolitinib, but not midostaurin, inhibited colony formation by 50 % (Fig. 2I, p = 0.001); this finding suggests that ruxolitinib may target self-renewal more effectively than midostaurin, consistent with our mutational profiling data that showed that midostaurin reduced the frequency of the KIT mutant subclone but not the prevailing TET2T1047L/Y1724LASXL1E222D clone (Fig. 1F).
To our knowledge, this is the first report of mutational analysis correlated with intracellular signaling and proliferation of SM-AHN. We demonstrate that KITD816V SM likely shares a common clone of origin with the MDS. As has been shown in AHNs without an SM component, we found hyperactivation of intracellular signaling molecules and increased markers of cell cycle and apoptosis in this patient [14,15]. Our data also reveal cell type-specific effects of physiologically and clinically relevant cytokines and drugs and suggests that the therapeutic effects of ruxolitinib and midostaurin may be lineage-specific and may be utilized complementarily in SM-AHN treatment.
Supplementary Material
Acknowledgements
The authors would like to thank the staff of the M Health-Fairview Molecular Diagnostics Laboratory for processing NGS samples. This work utilized the resources of the Flow Cytometry Resource and other services of the Masonic Cancer Center (which is supported by NIHP30 CA77598) at the University of Minnesota. This work and Z.S. were supported by the American Cancer Society, Frederick A. DeLuca Foundation, Mentored Research Scholar Grant (MRSG-16-195-01-DDC); the Clinical and Translational Science Institute at the University of Minnesota KL2 Career Development Award NIH/NCATS ULI RR033183 & KL2 RR0333182; the University of Minnesota Department of Medicine Women’s Early Research Career Award; the division of Hematology, Oncology, and Transplantation, Department of Medicine; and the University of Minnesota Foundation donors. HDY is supported by Hematology T32 Research Training Grant (5T32 HL007062-42, NIH). ACN is supported by the American Cancer Society, Clinical Scientist Development Grant, CSDG-18-139-01-CSM.
Footnotes
Declaration of Competing Interest
The authors disclose no relevant conflicts of interest.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.leukres.2020.106404.
Contributor Information
Hyun Don Yun, Division of Hematology, Oncology and Transplantation, Department of Medicine, University of Minnesota, Minneapolis, MN, United States; Division of Hematology, Oncology and Cell Therapy, Department of Medicine, Rush University, Chicago, IL, United States.
Marie Lue Antony, Division of Hematology, Oncology and Transplantation, Department of Medicine, University of Minnesota, Minneapolis, MN, United States.
Michael A. Linden, Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN, United States
Klara E. Noble-Orcutt, Division of Hematology, Oncology and Transplantation, Department of Medicine, University of Minnesota, Minneapolis, MN, United States
Craig E. Eckfeldt, Division of Hematology, Oncology and Transplantation, Department of Medicine, University of Minnesota, Minneapolis, MN, United States; Masonic Cancer Center, University of Minnesota, Minneapolis, MN, United States
Celalettin Ustun, Division of Hematology, Oncology and Cell Therapy, Department of Medicine, Rush University, Chicago, IL, United States.
Andrew C. Nelson, Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN, United States.
Zohar Sachs, Division of Hematology, Oncology and Transplantation, Department of Medicine, University of Minnesota, Minneapolis, MN, United States; Masonic Cancer Center, University of Minnesota, Minneapolis, MN, United States.
References
- [1].Ustun C, et al. , Advanced systemic mastocytosis: from molecular and genetic progress to clinical practice, Haematologica 101 (10) (2016) 1133–1143. [DOI] [PubMed] [Google Scholar]
- [2].Jawhar M, et al. , Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis, Leukemia 30 (1) (2016) 136–143. [DOI] [PubMed] [Google Scholar]
- [3].Jawhar M, et al. , Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event, Leukemia 29 (5) (2015) 1115–1122. [DOI] [PubMed] [Google Scholar]
- [4].Mueller N, et al. , CD44 is a RAS/STAT5-regulated invasion receptor that triggers disease expansion in advanced mastocytosis, Blood 132 (18) (2018) 1936–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Valent P, Akin C, Metcalfe DD, Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts, Blood 129 (11) (2017) 1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bejar R, et al. , TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients, Blood 124 (17) (2014) 2705–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Behbehani GK, Bendall SC, Clutter MR, Fantl WJ, Nolan GP, Single-cell mass cytometry adapted to measurements of the cell cycle, Cytometry A 81 (7) (2012) 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dorritie KA, McCubrey JA, Johnson DE, STAT transcription factors in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention, Leukemia 28 (2) (2014) 248–257. [DOI] [PubMed] [Google Scholar]
- [9].Fisher DAC, et al. , Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia, Leukemia 31 (9) (2017) 1962–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang Y, et al. , The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML, Science 327 (5973) (2010) 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gelsi-Boyer V, et al. , ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia, Br. J. Haematol 151 (4) (2010) 365–375. [DOI] [PubMed] [Google Scholar]
- [12].Tefferi A, et al. , Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates, Leukemia 23 (5) (2009) 900–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gotlib J, et al. , Efficacy and safety of midostaurin in advanced systemic mastocytosis, N. Engl. J. Med 374 (26) (2016) 2530–2541. [DOI] [PubMed] [Google Scholar]
- [14].Sachs Z, et al. , Stat5 is critical for the development and maintenance of myeloproliferative neoplasm initiated by Nf1 deficiency, Haematologica 101 (10) (2016) 1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Economopoulou C, et al. , Cell cycle and apoptosis regulatory gene expression in the bone marrow of patients with de novo myelodysplastic syndromes (MDS), Ann. Hematol 89 (4) (2010) 349–358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.