

Abstract
Pharmacophore‐based techniques are nowadays an important part of many computer‐aided drug design workflows and have been successfully applied for tasks such as virtual screening, lead optimization and de novo design. Natural products, on the other hand, can serve as a valuable source for unconventional molecular scaffolds that stimulate ideas for novel lead compounds in a more diverse chemical space that does not follow the rules of traditional medicinal chemistry. The first part of this review provides a brief introduction to the pharmacophore concept, the methods for pharmacophore model generation, and their applications. The second, concluding part, presents examples for recent, pharmacophore method related research in the field of natural product chemistry. The selected examples show, that pharmacophore‐based methods which get mainly applied on synthetic drug‐like molecules work equally well in the realm of natural products and thus can serve as a valuable tool for researchers in the field of natural product inspired drug design.
Keywords: natural products, computer-aided drug design, pharmacophore, virtual screening, scaffold hopping
1. Introduction
Many modern blockbuster drugs originate from or were inspired by natural products (NP) and there is growing interest in employing computational approaches for the design of highly active compounds based on natural‐product derived fragments. [1] Chemotype‐specific synthetic compound libraries used to be the common approach in medicinal chemistry, in which compounds are designed according to a specific target profile and aim for a high degree of diversity. [2] As natural products can serve as source for novel molecular scaffolds and thus stimulate ideas for novel lead compounds, [3] a renewed and growing interest in these molecules can be observed which gets demonstrated by several recently published drug design projects. Natural products do not always follow the rules which are commonly applied in modern drug design (for instance Lipinski's “Rule of Five” [4] ). However, this can also be seen as a valuable chance to explore powerful molecules in a more diverse chemical space. Recently, anti‐cancer agents with scaffolds inspired by natural products were described. [5] Those compounds are synthetically accessible, and the achievement of linking the world of natural and synthetic compounds is a great benefit for medicinal chemistry. [6] Furthermore, software has been made available that enables an exploration of the NP‐derived chemical space in correlation with bioactivity profiles.[ 6 , 7 ]
As we are focusing on methods based on 3D pharmacophores and their application in natural product inspired drug design, we have reviewed some recently published drug design projects which relied on the application of pharmacophore‐based techniques. By employing screening databases, curated from structures of natural products, the in silico approach could be successfully linked to pharmacognostic profiles.
2. The Pharmacophore Concept
In medicinal chemistry, pharmacophore‐based methods can nowadays be considered as an indispensable component in any modern computer‐aided drug design (CADD) toolbox. Due to their abstract nature, pharmacophores are easy to comprehend and intuitive which renders them also useful as a tool for medicinal chemists to describe, explain and visualize ligand‐target binding modes.
Depending on background and context, the term pharmacophore was often attributed with different meanings. Historically, [8] the term “pharmacophore” was used to vaguely denote common structural or functional elements of a set of compounds essential for the activity towards a particular biological target. However, the official IUPAC definition for pharmacophores [9] is more specific and states:
‘A pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supra‐molecular interactions with a specific biological target structure and to trigger (or to block) its biological response’.
According to this definition, pharmacophores do not represent ensembles of particular functional groups (e. g. primary amines, thioamide, …) or characteristic structural fragments (e. g. a pyrrolidine ring), but are an abstract description of stereoelectronic molecular properties. Structurally different molecules possessing similar pharmacophoric patterns can therefore be assumed to be recognized by the same binding site of a biological target and thus also exhibit similar biological profiles. [8] The pharmacophore concept thus allows medicinal chemists to postulate pharmacophore models as the “essence” of the structure‐activity knowledge they have gained in an extensive structural study of a series of active and inactive molecules for a given drug target.
2.1. 3D Pharmacophore Representation
3D Pharmacophores represent the nature, as well as the location of the chemical features of a molecule involved in ligand‐target interactions as geometric entities like spheres, planes and vectors. Although simple, this kind of representation is usually sufficient to capture the active conformation of a molecule and all essential interactions contributing to its activity in 3D space. For pharmacophore matching the features of a query model only have to be met in their spatial arrangement and type but not in the underlying chemical structures. Therefore, pharmacophores have an inherent scaffold hopping ability which is reflected in a usually high structural diversity of the compounds in virtual screening hit‐lists (given that the screened compounds are structurally diverse).
Not surprisingly, the choice of the basic feature set for building pharmacophore models has an inevitable impact on model quality. While the choice of features used to be very specific at the early stage of pharmacophore modeling, [10] recent techniques build pharmacophore models in a more general way. [11] By using a very general feature set resulting models are interpretable and easy to comprehend, but might lack selectivity and have a lower discriminatory power by neglecting specific characteristics of functional groups. However, building a very restrictive model by employing a higher number of feature types can quickly lead to problems when it comes to the matching of structurally unrelated compounds that are active towards the same target. Thus, developing a feature set that represents a reasonable trade‐off between being too general and being too selective is one of the biggest challenges current software packages [12] for pharmacophore modeling are facing.
Table 1 provides an overview of the most important feature types and the non‐bonding interactions they may establish in presence of a complementary interaction partner. Vector and plane representations are used for feature types whose interactions are directed, i. e. a certain mutual orientation of complementary features is required. Spheres can be used for any feature type but are typically used only for features whose interactions are of undirected nature or where an orientation cannot be determined (e. g. a rotatable ‐OH group).
Table 1.
Summary of the most important pharmacophoric feature types and their interactions typically observed in biological systems.
|
Feature Type |
Geom. Representation |
Complementary Feature Type(s) |
Interaction Type(s) |
Structural Examples |
|---|---|---|---|---|
|
Hydrogen‐Bond Acceptor (HBA) |
Vector or Sphere |
HBD |
Hydrogen‐Bonding |
Amines, Carboxylates, Ketones, Alcoholes, Fluorine Substituents, … |
|
Hydrogen‐Bond Donor (HBD) |
Vector or Sphere |
HBA |
Hydrogen‐Bonding |
Amines, Amides, Alcoholes, … |
|
Aromatic (AR) |
Plane or Sphere |
AR, PI |
π‐Stacking, Cation‐π |
Any arom. Ring |
|
Positive Ionizable (PI) |
Sphere |
AR, NI |
Ionic, Cation‐π |
Ammonium Ion, Metal Cations, … |
|
Negative Ionizable (NI) |
Sphere |
PI |
Ionic |
Carboxylates |
|
Hydrophobic (H) |
Sphere |
H |
Hydrophobic Contact |
Halogen Substituents, Alkyl Groups, Alicycles, weakly or non‐polar arom. Rings, … |
Ligand Shape Constraints. A pharmacophore model representing just a set of key interactions usually does not describe all molecular characteristics, which are required for high affinity binding. Even if a molecule fits the pharmacophoric model well it can still fail to bind to the receptor due to steric clashes. Therefore, pharmacophore models must also account for spatial constraints imposed by the shape of the binding site. This can be achieved by the incorporation of so‐called exclusion volumes. Such volumes can be of different size since they are representing receptor areas where the ligand is not allowed to occupy space after an alignment [16] with the pharmacophore. The most reliable information about spatial restrictions can be extracted from X‐ray structures of ligand‐receptor complexes. [14] If such information is not available, the location and size of exclusion volumes has to be assigned either manually or by computer‐aided methods that distribute spheres based on the union of the molecular shapes of a set of aligned known actives. [17]
2.2. Generation of Pharmacophore Models
Pharmacophore models can be constructed manually, generated in an automated way starting from the structure of known active ligands (ligand‐based), or can be derived from the three‐dimensional structure of the target receptor (structure‐based). Which approach to choose mainly depends on data availability, data quality, computational resources and also the intended use of the generated pharmacophore models.
Manually created Pharmacophore Models. From an algorithmic point of view, this is the simplest way to obtain pharmacophore models. Manual construction usually requires a human user who needs considerable expert knowledge about the biological target and the key structural characteristics of a series of known active compounds. Due to the availability of computational methods which reliably analyze the common characteristics of a series of known actives or account for all possible ligand‐target interactions (see below), manual model construction largely lost its relevance and manual intervention has moved towards the refinement of automatically generated models.
Structure‐based Pharmacophore Models. Having access to the three‐dimensional structure of a ligand‐receptor complex [14] is a tremendous advantage when it comes to the generation of high quality pharmacophore models. If the bioactive conformation of the ligand is known, the atomic coordinates can be directly used to guide the correct placement of pharmacophoric features. Information about the receptor structure, on the other hand, enables the identification of relevant interactions with the ligand and the incorporation of binding site shape information (see section 2.1). There exists a variety of software tools [12] which are able to derive structure‐based pharmacophore models and shape constraints in a fully automated manner from the 3D structure of a ligand‐receptor complex. If only the unbound state structure of the target is accessible (as it is often the case), the generation of high quality models becomes much more challenging. For the generation of pharmacophores in such cases computational methods are available [18] but in general deliver models of poorer initial quality caused by the lack of complementary ligand information. Substantial validation and careful manual refinement of the initial models is usually required to work out a refined feature set that allows for a good discrimination between actual pocket binders and non‐binders.
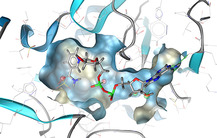
As an example, Figure 1 shows the structure‐based pharmacophore model of the natural product balanol which acts as a potent inhibitor of cAMP‐dependent protein kinase (cAPK). [13] The model contains only a subset of the features of balanol, those for which a suitable complementary interaction partner in the cAMP binding site environment could be identified.
Figure 1.
Structure‐based pharmacophore model of the NP inhibitor balanol in complex with the catalytic subunit of cAMP‐dependent protein kinase [13] (PDB [14] code: 1BX6) generated by LigandScout.[ 12d , 15 ] Red arrows represent H‐bond acceptors, green arrows H‐bond donors, yellow spheres hydrophobic features and the blue star a positively ionizable feature.
Ligand‐based Pharmacophore Models. Pharmaco‐phores can also be derived in a ligand‐based manner from a sufficient number of known actives. An important prerequisite for the success of this method is that the active ligands bind to the same receptor, at the same binding site and in the same orientation. Otherwise, the obtained pharmacophore models will not represent the correct mode of action and cannot be used for the identification of novel actives. For the generation of ligand‐based pharmacophore models several algorithms have been published which are described in detail elsewhere. [19] Since the bioactive conformation of the training set ligands is usually not known, a crucial processing step for all algorithms is the generation of ligand conformers such that at least one represents a good approximation of the bioactive conformation. In the next step, a chemical feature pattern has to be identified [20] that is common to all ligands. Usually, multiple pharmacophoric patterns can be extracted which are then ranked according to a suitable fitness function. Ligand‐based pharmacophore modeling is challenging because several variables have a significant impact on the outcome. It is therefore crucial to keep a critical view on the generated models and to validate them comprehensively. [21]
2.3. Pharmacophore‐based Virtual Screening
The most common application of pharmacophore models is their use as a query for the virtual screening of large compound libraries. The ultimate goal here is the discovery of novel compounds which exhibit a set of desired pharmacophoric features that are considered crucial for biological activity towards a particular target of interest. [22] Due to their abstract nature, pharmacophore screening usually delivers hit compounds with a high structural diversity. As an additional benefit, the simplicity of the pharmacophoric representation enables a fast in silico searching [23] of even large screening databases containing millions of compounds. Depending on the selectivity of the query pharmacophore, application specific match constraints and database size, tens to thousands of hit molecules are usually retrieved by a typical screening run. A portion of the hits will be false positives and show no significant activity at all, but in comparison with random sampling, hit rates obtained by pharmacophore searches are generally much higher and thus allow an enrichment of potentially active compounds in significantly smaller database subsets. This is especially important for academic research where the available resources are often quite limited and only a low‐throughput biological testing of relatively few compounds is possible. As already stated before, pharmacophore screening allows to identify novel and diverse chemotypes, which are for human eyes not obvious matches to the query pharmacophore. Hit‐lists made up of molecules belonging to different structural classes can serve as a valuable source of “ideas” for the development and optimization of novel lead compounds that might not have been discovered by a traditional rational drug design processes alone.
3. Applications of Pharmacophores in Natural Product inspired Drug Discovery
Natural products are often structurally complex and differ from synthetic drug‐like molecules in many ways. Studies [24] comparing these two compound classes have shown, that natural products contain a much larger fraction of sp 3‐hybridized atoms at bridgeheads and more chiral centres than synthetic small molecules. [24] On average, natural products also have a lower nitrogen but higher oxygen content and only 38% of the known natural products contain aromatic ring systems. [24a] Furthermore, around 50 % of the structurally resolved natural products in the Dictionary of Natural Products [25] do not have synthetic counterparts and only 20 % of the ring systems seen in natural products can also be found in drugs on the market. [24b] These findings clearly show, that a sophisticated use of the chemical space offered by natural products has the potential to drive innovation at nearly all stages of the drug design process. However, the higher structural complexity and conformational flexibility of natural products renders drug design also more challenging. Dedicated databases for pharmacophore‐based virtual screening usually store pre‐generated conformational ensembles of all the contained molecules in order to speed up the screening process. [23] To adequately account for a higher conformational flexibility, larger conformational ensembles need to be generated which increases both the demand for disk‐storage and required processing time. A further complication regarding pharmacophore modeling may arise from the often high polar atom content of natural products. This leads to a correspondingly higher number of H‐bond donor and acceptor features, many of which might be irrelevant for an energetically favorable interaction with the target receptor. Deriving a core set of essential features which is able to distinguish true actives from inactives with high confidence requires a considerable amount of information (like a series of known actives or co‐crystal X‐ray structures, see also section 2.2) which is often not available, especially in the context of natural products.
The presented “success stories” in the following sections will show, facing these challenges can be rewarding. All of the reviewed examples were taken from literature and apply pharmacophore‐based methods for the discovery of novel NP‐based drug candidates. An excellent and comprehensive review of similar drug discovery projects was already published by Schuster et al. in 2010. [26] This short review aims to follow up on this and will cover only more recent literature that was published after 2010.
3.1. Identification of Novel Natural Inhibitors of Trypanosoma brucei Glyceraldehyde‐3‐phosphate‐dehydrogenase
This work aimed at the identification of natural inhibitors of Trypanosoma brucei Glyceraldehyde‐3‐phosphate‐dehydrogenase (TbGAPDH). [27] Trypanosoma brucei is a protozoan parasite which can cause human African trypanosomiasis (HAT), also known as “sleeping sickness”. Being classified as one of the “Neglected Tropical Diseases” (NTDs) by the World Health Organization (WHO), this infectious disease is endangering more than 70 million sub‐Saharan African people. [28] Suggested as potential drug target to deprive the parasite of energy supply, [29] inhibitors of GAPDH represent promising trypanocidal agents.[ 29 , 30 ] Offering a vast structural diversity, natural products are known to exhibit high potential against protozoan infection diseases. [31]
For the virtual screening runs that were performed in this study, the natural product database MEGx [32] was selected. From the 4803 natural compounds contained in this database, 700 were kept after several filtering steps such as the Lipinski's rule of five [4] or limits on the number of stereocenters.
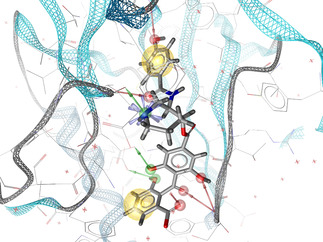
Three crystallographic structures (PDB‐IDs: 2X0N, 3IDS and 1GYP) of GAPDH from human pathogenic trypanosomatids were taken from the Protein Data Bank (PDB) [14] and used to generate four pharmacophore models by means of the software MOE. [12c] Three of them were structure‐based pharmacophore models established on co‐crystallized NAD+ (see Figure 2) and the last one was manually built by analyzing an electrostatic map of the glyceraldehyde‐3‐phosphate (G‐3‐P) binding site, due to the absence of the substrate.
Figure 2.
Structure‐based pharmacophore model of NAD+ in complex with GADPH (PDB [14] code: 2X0N) generated by LigandScout.[ 12d , 15 ] Red arrows represent H‐bond acceptors, green arrows H‐bond donors. The binding pocket surface is colored by lipophilicity where yellow corresponds to lipophilic areas and blue to polar areas.
A virtual screening run was performed for each pharmacophore model and the resulting hits were then docked in the respective binding site from which the initial pharmacophore model was derived. Based on their docking and virtual screening scores, 13 natural products were experimentally tested to validate the inhibition of GAPDH. Five compounds displaying an enzyme inhibition superior to 50 % at a concentration of 50 μM were eventually selected and their IC50 values determined.
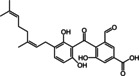
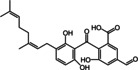
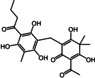
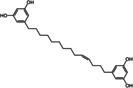
The structures of the selected natural products are shown in Table 2. Three of the compounds are geranylated benzophenone derivatives extracted from the fungus Geniculosporium sp., one is a flavaspidic acid AB extracted from the fern Dryopteris crassirhizoma, and the last one is a tetradecane derivative extracted from the tree Grevillea whiteana. All of the five compounds inhibited GAPDH and had IC50 values below 30 μM, with two of the geranylated benzophenone derivatives even showing IC50 values below 8 μM.
Table 2.
List of the five natural products exhibiting more than 50 % TbGAPDH inhibition at a concentration of 50 μM.
|
Compound Structure |
% of TbGAPDH inhibition at 50 μM |
IC50 (μM) |
Compound Class |
|---|---|---|---|
|
|
66 |
24.56±1.03 |
Geranylated benzophenone derivative |
|
|
98 |
4.73±1.03 |
Geranylated benzophenone derivative |
|
|
>90 |
6.68±1.04 |
Geranylated benzophenone derivative |
|
|
88 |
21.97±1.03 |
Flavaspidic acid AB |
|
|
>90 |
22.79±1.01 |
Tetradecane derivative |
3.2. Ginkgolic Acid as a Multi‐target Inhibitor of Key Enzymes in Pro‐inflammatory Lipid Mediator Biosynthesis
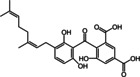
The primary goal of this project was the identification of novel NP‐based microsomal prostaglandin E2 synthase‐1 (mPGES‐1) inhibitors. [33] The enzyme mPGES‐1 has been found to be responsible for massive prostaglandin E2 (PGE2) biosynthesis at inflammatory sites and is thus considered as an attractive therapeutic target for the treatment of inflammation‐related disorders and also cancer. [34] Besides prostaglandins (PGs), leukotrienes (LTs) formed by 5‐lipoxygenase (5‐LO) are also heavily involved in the inflammatory response regulation network. Recent pharmacological strategies therefore focus on multiple target concepts, such as dual mPGES‐1/5‐LO inhibitors. [34] For this purpose, mPGES‐1 and 5‐LO pharmacophore models were established that in combination with virtual screening lead to the identification of various small synthetic molecules showing an inhibition of both PGE2 and LT synthesis. [35] The authors of this study used an analogous approach and screened an in‐house Chinese Herbal Medicine (CHM) database comprising 10,216 unique compounds with two earlier reported mPGES‐1 inhibitor pharmacophore models. [36] From the hit list of the less restrictive second model, ginkgolic acid (GA, shown in Figure 3), a 6‐alkenyl derivative of salicylic acid contained in Ginkgo biloba, was finally selected as the most promising candidate and subjected to extensive biological testing.
Figure 3.
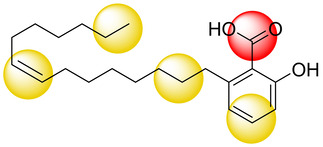
Structure of GA together with a schematic representation of the five pharmacophoric features that were matched by the mPGES‐1 inhibitor pharmacophore model used as a query for virtual screening. Yellow spheres correspond to lipophilic features and the red sphere specifies a negative ionizable feature.
In cell‐free assays, a fully reversible potent suppression of mPGES‐1 activity (IC50=0.7 μM) could be observed that was largely independent of substrate concentration. Cyclooxygenase (COX) 1 and thromboxane A2 synthase (TXAS) were identified as additional targets of GA with lower affinity (IC50=8.1 and 5.2 μM). 5‐LO was potently inhibited by GA with an IC50 value of 0.2 μM in a reversible and substrate‐independent manner. Docking studies that were performed in addition to the biological tests further substantiated a direct molecular interaction of GA with mPGES‐1 and 5‐LO binding sites. Analyses of lipid mediator (LM) profiles from bacteria‐stimulated human M1‐ and M2‐like macrophages finally confirmed the multi‐target features of GA and have shown a LM redirection towards the formation of 12‐/15‐LO products including specialized pro‐resolving mediators (SPMs).
Unfortunately, the allergic and genotoxic potential of GA renders its direct use as drug for humans highly unlikely. However, the revealed favorable multiple‐target‐inhibitor profile of GA makes it still perfectly suited to serve as a tool compound for the investigation of novel pharmacological strategies in the fight against diseases linked to inflammation‐related disorders.
3.3. Identification of Steroid Sulfatase Inhibiting Lanostane Triterpenes
This study aimed at the identification of potent steroid sulfatase (STS) inhibitors from natural sources. [37] STS is a membrane‐bound catabolic enzyme located in the rough endoplasmatic reticulum and responsible for the conversion of sulfated estrogenic steroids into free steroid hormones. The underlying mechanism is based on the hydrolysis of the sulfate ester moiety of the inactive precursor molecule releasing the active steroid hormone. [38] STS is a key enzyme involved in the increase of free steroid concentrations in tumors and thus has significant potential as a novel drug target in hormone‐dependent breast and prostate cancers and also in conditions like endometriosis. [39]
Although since the early 1990s reversible and irreversible STS inhibitors have been under heavy investigation, [40] the chemical space of natural products as a source of potential inhibitors remained largely unexplored. [41] For the identification of STS inhibiting natural compounds, the authors chose a pharmacophore‐based virtual screening approach employing ligand‐based pharmacophore models. In total three different models were generated:
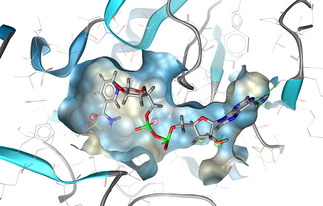
Model 1 was generated from two known steroidal STS inhibitors (CAS: 1360613‐74‐6, 1215117‐28‐4) and contained one HBA, one mixed HBA/HBD, and four hydrophobic features in addition to one optional hydrophobic feature. Model 2 was a variation of model 1 where the HBA feature in model 1 was converted to an HBD feature based on the observation that most of the known steroidal STS inhibitors contain a hydroxyl group instead of a carbonyl group in the concerned position. Model 3 was based on two non‐steroidal inhibitors (CAS: 634908‐04‐6, 512818‐32‐5) and contained one HBD, one HBA, one aromatic, and four hydrophobic features (see Figure 4).
Figure 4.
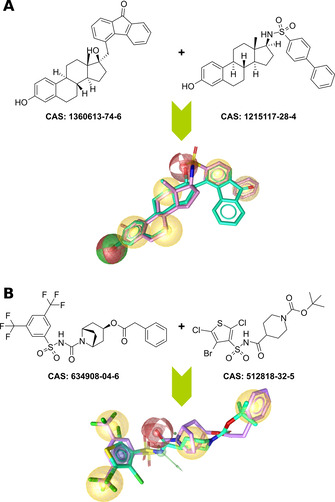
Ligand‐based pharmacophore modeling of STS inhibitors: A) Pharmacophore model based on two known steroidal STS inhibitors, B) model based on two potent non‐steroidal inhibitors. The models were generated by LigandScout[ 12d , 15 ] and are replicas of the models 1 (A) and 3 (B) described in section 3.3. Red spheres represent H‐bond acceptors, green spheres/arrows H‐bond donors, yellow spheres hydrophobic features, and the blue ring specifies an aromatic feature.
For model validation, the authors compiled a dataset including both steroidal and non‐steroidal scaffolds comprising 65 known STS inhibitors and 113 proven inactive compounds from literature sources. The steroid‐based models matched 58 (model 1) and 57 (model 2) of the known actives, but none of the inactive compounds. Pharmacophore model 3 matched 5 of the active molecules and also none of the inactive compounds. Taken together, the models were able to selectively retrieve 63 of the 65 active molecules without matching a single inactive molecule.
A prospective virtual screening of a database containing 32,119 secondary metabolites of natural products used in Traditional Chinese Medicine (TCM Database@Taiwan [42] ) returned a total of 21 hits. Model 1 retrieved 20 molecules, model 2 one molecule, and model 3 did not match any molecules. 20 % of the hits retrieved by model 1 solely belonged to the structure class of lanostane‐type triterpenes (LTTs) which are the main constituents of polypore fungi. Based on this observation, the authors focused their investigations on the general STS inhibition potential of LTTs and prepared extracts of the polypores Ganoderma lucidum Karst. (GL), Gloeophyllum odoratum Imazeki (GO), and Fomitopsis pinicola Karst. (FP) in order to have access to a higher number of LTT candidates for in vitro testing. In this way, 18 LTTs were finally obtained whose inhibitory activity towards STS was tested at 20 μM in JEG‐3 cells with the potent STS inhibitor STX64 (irosustat) serving as a positive control and reference for 100 % inhibition. Three of the tested compounds (Table 3), i. e. piptolinic acid D (1), pinicolic acid B (2), and ganoderol A (3), showed an STS inhibition and 74 % (IC50=10.5 μM), 72 % (IC50=12.4 μM), and 62 % (IC50=15.7 μM), respectively and represent the first non‐sulfated/non‐sulfamated STS inhibitors from natural sources reported so far. The remaining isolates showed only moderate, weak or no inhibition of STS.
Table 3.
List of the three most potent LTT based STS inhibitors isolated from two polypore species: Fomitopsis pinicola Karst. (FP), Ganoderma lucidum Karst. (GL).
|
Comp. |
Structure |
% of STS Inhibition at 20 μM |
IC50 (μM) |
Fungal Source |
|---|---|---|---|---|
|
1 |
|
74 |
10.5 |
FP |
|
2 |
|
72 |
12.4 |
FP |
|
3 |
|
62 |
15.7 |
GL |
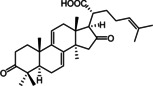
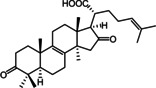
The identified STS inhibiting LTTs are by far not as potent as the irreversible inhibitors currently in clinical trials. However, they nevertheless provided valuable insight regarding the traditional use of TCM polypores in the field of cancer related conditions and might act as starting points for the design of alternative, but equally potent non‐sulfated/non‐sulfamated STS inhibitors.
3.4. Further Reading
For the interested reader, the following tabular overview provides references to further recently published projects and studies which successfully applied pharmacophore‐based methods in the field NP‐inspired drug discovery and design:
Table 4.
List of further notable research projects applying pharmacophore‐based methods in the context of NP‐inspired drug design.
|
Resarch Topic |
Appl. Pharm. Methods |
Year |
Ref. |
|---|---|---|---|
|
Synthesis, antimycobacterial evaluation and pharmacophore modeling of analogues of the natural product formononetin |
Ligand‐based pharm. modeling |
2015 |
|
|
Pharmacophore modeling and in silico toxicity assessment of potential anticancer agents from African medicinal plants |
Structure‐based pharm. modeling, virt. screening |
2016 |
|
|
Identification of hERG channel blocking Ipecac alkaloids by combined in silico – in vitro screening |
Virt. screening |
2016 |
|
|
Identification of natural products as novel PI3Kβ inhibitors |
Ligand‐based pharm. modeling, virt. screening |
2018 |
|
|
Pharmacophore mapping of natural products for pancreatic lipase inhibition |
Ligand‐based pharm. modeling, virt. screening |
2020 |
4. Summary
In this review we summarized selected published examples for natural product related drug design projects where the application of pharmacophore‐based techniques played an important role in achieving the set research goals. In modern computer‐aided drug design, medicinal chemists usually navigate solely in the less complex chemical space of molecules that strictly follow the traditional rules of drug‐likeness. [4] These rules have been derived from careful statistical analyses and have proven their validity in the development of many successful present day drugs. Natural products with their higher structural diversity and complexity often violate the established rules but have throughout human history shown, that they can act as equally powerful weapons in the treatment of a wide variety of diseases. For the present day medicinal chemist natural products can serve as a valuable source of ideas for the design of novel lead compounds outside the synthetic drug space and taking this chance may eventually lead to findings that were not accessible before. The reviewed examples have shown, that pharmacophore‐based methods which were primarily designed for the work with less complex synthetic drug‐like molecules can also be applied to the natural product space and thus represent a powerful addition to the toolbox of any researcher in field of natural product inspired drug design.
Conflict of interest
None declared.
Biographical Information
Thomas Seidel studied chemistry at the Vienna University of Technology where he received a PhD in synthetic organic chemistry in 2003. From 2004 until 2005 he worked as a postdoctoral research assistant in the group of Prof. Johann Gasteiger at the Computer Chemistry Center/University of Erlangen‐Nuremberg. After his postdoctoral stay he returned to Vienna for a position at the company InteLigand GmbH (Vienna) where he developed cheminformatics software for pharmacophore modeling and virtual screening. Since 2014 Thomas Seidel is a senior researcher at the Department of Pharmaceutical Chemistry/University of Vienna where he leads the Chemoinformatics Research Group.

Biographical Information
Oliver Wieder studied Biology, Sinology, Watermanagement and Environmental Engineering at the University of Vienna and the University of Natural Ressources and Life Sciences (Vienna). After finishing his studies in 2016 he started working as a software‐developer at InteLigand GmbH. In May 2018 he started his PhD in the Chemoinformatics Research Group at the Department of Pharmaceutical Chemistry in collaboration with Laboratoires Servier (Paris) and InteLigand GmbH. His research focuses on the development of AI‐guided applications for early drug discovery ‐ in particular deep‐learning related methods for lead optimization.

Biographical Information
Arthur Garon studied chemistry and biology at the University of Clermont‐Ferrand and obtained an MSc in chemoinformatics at the University of Strasbourg in 2016. Since 2017 he is a PhD student in the Chemoinformatics Research Group at the Department of Pharmaceutical Chemistry in collaboration with InteLigand GmbH and Laboratoires Servier. His research focuses on the development and implementation of new methods for pharmacophore‐based drug design where he is mainly working on the extraction and analysis of pharmacophore related information derived from crystallographic structures and molecular dynamics simulations.

Biographical Information
Thierry Langer holds an MSc degree in Pharmacy (1988) and a PhD in Pharmaceutical Chemistry (1991) from University of Vienna. He began his academic career at the Leopold‐Franzens‐University (Innsbruck) in 1992 after completing a postdoctoral fellowship with Prof. C.‐G. Wermuth in Strasbourg. In 2003, with colleagues, he founded the company Inte:Ligand GmbH which develops scientific software for computer aided molecular design, and served as the CEO until 2008. Then he was appointed as the CEO of Prestwick Chemical, Inc. in Strasbourg. Under his leadership, several successful drug discovery programs in different research target sectors were conducted. In 2013, he was nominated full professor for medicinal chemistry at the University of Vienna, where he currently heads the Department of Pharmaceutical Chemistry at the Faculty of Life Sciences.

T. Seidel, O. Wieder, A. Garon, T. Langer, Mol. Inf. 2020, 39, 2000059.
References
- 1. Rodrigues T., Reker D., Schneider P., Schneider G., Nat. Chem. 2016, 8, 531–541. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a.G. Schneider, K.-H. Baringhaus, Molecular design concepts and applications, Wiley-VCH, Weinheim, 2008;
- 2b. Schreiber S. L., Science 2000, 287, 1964–1969. [DOI] [PubMed] [Google Scholar]
- 3. Grabowski K., Baringhaus K. H., Schneider G., Nat. Prod. Rep. 2008, 25, 892–904. [DOI] [PubMed] [Google Scholar]
- 4. Lipinski C. A., Drug Discovery Dev. 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- 5. Elumalai N., Berg A., Natarajan K., Scharow A., Berg T., Angew. Chem. Int. Ed. 2015, 54, 4758–4763; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4840–4845. [Google Scholar]
- 6. Rodrigues T., Reker D., Schneider P., Schneider G., Nat. Chem. 2016, 8, 531–541. [DOI] [PubMed] [Google Scholar]
- 7. Larsson J., Gottfries J., Muresan S., Backlund A., J. Nat. Prod. 2007, 70, 789–794. [DOI] [PubMed] [Google Scholar]
- 8.C. G. Wermuth, Pharmacophores and pharmacophore searches, Wiley-VCH, Weinheim, 2006.
- 9. Wermuth C. G., Ganellin C. R., Lindberg P., Mitscher L. A., Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar]
- 10. Marshall G. R., Barry C. D., Bosshard H. E., Dammkoehler R. A., Dunn D. A., in Computer-Assisted Drug Design, Vol. 112, American Chemical Society, 1979, pp. 205–226. [Google Scholar]
- 11. Greene J., Kahn S., Savoj H., Sprague P., Teig S., J. Chem. Inf. Comput. Sci. 1994, 34, 1297–1308. [Google Scholar]
- 12.
- 12a.Discovery Studio, Dassault Systèmes BIOVIA, https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/;
- 12b.Phase, Schrödinger Inc., https://www.schrodinger.com/phase/;
- 12c.Molecular Operating Environment (MOE), Chemical Computing Group, https://www.chemcomp.com/Products.htm;
- 12d.LigandScout, Inte:Ligand GmbH, http://www.inteligand.com/ligandscout/.
- 13. Narayana N., Diller T. C., Koide K., Bunnage M. E., Nicolaou K. C., Brunton L. L., Xuong N.-H., Ten Eyck L. F., Taylor S. S., Biochemistry 1999, 38, 2367–2376. [DOI] [PubMed] [Google Scholar]
- 14. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wolber G., Langer T., J. Chem. Inf. Model. 2005, 45, 160–169. [DOI] [PubMed] [Google Scholar]
- 16. Wolber G., Dornhofer A. A., Langer T., J. Comput.-Aided Mol. Des. 2006, 20, 773–788. [DOI] [PubMed] [Google Scholar]
- 17. Dixon S. L., Smondyrev A. M., Knoll E. H., Rao S. N., Shaw D. E., Friesner R. A., J. Comput.-Aided Mol. Des. 2006, 20, 647–671. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Goodford P. J., J. Med. Chem. 1985, 28, 849–857; [DOI] [PubMed] [Google Scholar]
- 18b. Mortier J., Dhakal P., Volkamer A., Molecules 2018, 23, 1959; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18c. Ortuso F., Langer T., Alcaro S., Bioinformatics 2006, 22, 1449–1455; [DOI] [PubMed] [Google Scholar]
- 18d. Schuetz D. A., Seidel T., Garon A., Martini R., Körbel M., Ecker G. F., Langer T., J. Chem. Theory Comput. 2018, 14, 4958–4970; [DOI] [PubMed] [Google Scholar]
- 18e. Wade R. C., Goodford P. J., J. Med. Chem. 1993, 36, 148–156. [DOI] [PubMed] [Google Scholar]
- 19. Leach A. R., Gillet V. J., Lewis R. A., Taylor R., J. Med. Chem. 2010, 53, 539–558. [DOI] [PubMed] [Google Scholar]
- 20. Wolber G., Seidel T., Bendix F., Langer T., Drug Discovery Today 2008, 13, 23–29. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a.K. Poptodorov, T. Luu, R. D. Hoffmann, Pharmacophores and pharmacophore searches, Wiley-VCH, Weinheim, 2006;
- 21b.N. Triballeau, H.-O. Bertrand, F. Achner, Pharmacophores and pharmacophore searches, Wiley-VCH, Weinheim, 2006.
- 22.
- 22a. Clark D. E., Westhead D. R., Sykes R. A., Murray C. W., J. Comput.-Aided Mol. Des. 1996, 10, 397–416; [DOI] [PubMed] [Google Scholar]
- 22b. Manallack D. T., Drug Discovery Today 1996, 1, 231–238; [Google Scholar]
- 22c. Martin Y. C., J. Med. Chem. 1992, 35, 2145–2154. [DOI] [PubMed] [Google Scholar]
- 23. Seidel T., Ibis G., Bendix F., Wolber G., Drug Discovery Dev. 2010, 7, 203–270. [Google Scholar]
- 24.
- 24a. Henkel T., Brunne R. M., Müller H., Reichel F., Angew. Chem. Int. Ed. 1999, 38, 643–647; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 688–691; [Google Scholar]
- 24b. Lee M.-L., Schneider G., J. Comb. Chem. 2001, 3, 284–289. [DOI] [PubMed] [Google Scholar]
- 25.Dictionary of Natural Products (DNP), 28.2, CRC Press, Taylor&Francis Group, http://dnp.chemnetbase.com.
- 26. Schuster D., Wolber G., Curr. Pharm. Des. 2010, 16, 1666–1681. [DOI] [PubMed] [Google Scholar]
- 27. Herrmann F. C., Lenz M., Jose J., Kaiser M., Brun R., Schmidt T. J., Molecules 2015, 20, 16154–16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Third WHO report on neglected tropical diseases, World Health Organization, https://www.who.int/neglected_diseases/9789241564861/en/.
- 29.M. Gualdrón-López, P. A. M. Michels, W. Quiñones, A. J. Cáceres, L. Avilán, J.-L. Concepción, Trypanosomatid Diseases:Molecular Routes to Drug Discovery, Wiley-VCH, Weinheim, 2013.
- 30.
- 30a. Schuster R., Holzhutter H. G., Eur. J. Biochem. 1995, 229, 403–418; [PubMed] [Google Scholar]
- 30b. Caceres A. J., Michels P. A., Hannaert V., Mol. Biochem. Parasitol. 2010, 169, 50–54. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Schmidt T. J., Khalid S. A., Romanha A. J., Alves T. M., Biavatti M. W., Brun R., Da Costa F. B., de Castro S. L., Ferreira V. F., de Lacerda M. V., Lago J. H., Leon L. L., Lopes N. P., das Neves Amorim R. C., Niehues M., Ogungbe I. V., Pohlit A. M., Scotti M. T., Setzer W. N., de N. C. S. M., Steindel M., Tempone A. G., Curr. Med. Chem. 2012, 19, 2176–2228; [PubMed] [Google Scholar]
- 31b. Schmidt T. J., Khalid S. A., Romanha A. J., Alves T. M., Biavatti M. W., Brun R., Da Costa F. B., de Castro S. L., Ferreira V. F., de Lacerda M. V., Lago J. H., Leon L. L., Lopes N. P., das Neves Amorim R. C., Niehues M., Ogungbe I. V., Pohlit A. M., Scotti M. T., Setzer W. N., de N. C. S. M., Steindel M., Tempone A. G., Curr. Med. Chem. 2012, 19, 2128–2175. [DOI] [PubMed] [Google Scholar]
- 32.MEGx Database, AnalytiCon Discovery, https://ac-discovery.com/purified-natural-product-screening-compounds/.
- 33. Gerstmeier J., Seegers J., Witt F., Waltenberger B., Temml V., Rollinger J. M., Stuppner H., Koeberle A., Schuster D., Werz O., Front. Pharmacol. 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koeberle A., Werz O., Biochem. Pharmacol. 2015, 98, 1–15. [DOI] [PubMed] [Google Scholar]
- 35. Koeberle A., Werz O., Biotechnol. Adv. 2018, 36, 1709–1723. [DOI] [PubMed] [Google Scholar]
- 36. Waltenberger B., Wiechmann K., Bauer J., Markt P., Noha S. M., Wolber G., Rollinger J. M., Werz O., Schuster D., Stuppner H., J. Med. Chem. 2011, 54, 3163–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grienke U., Kaserer T., Kirchweger B., Lambrinidis G., Kandel R. T., Foster P. A., Schuster D., Mikros E., Rollinger J. M., Bioorg. Chem. 2020, 95, 103495. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Jameera Begam A., Jubie S., Nanjan M. J., Bioorg. Chem. 2017, 71, 257–274; [DOI] [PubMed] [Google Scholar]
- 38b. Barry V. L. P., J. Mol. Endocrinol. 2018, 61, T233-T252. [DOI] [PubMed] [Google Scholar]
- 39. Sang X., Han H., Poirier D., Lin S. X., J. Steroid Biochem. Mol. Biol. 2018, 183, 80–93. [DOI] [PubMed] [Google Scholar]
- 40. Williams S. J., Expert Opin. Ther. Pat. 2013, 23, 79–98. [DOI] [PubMed] [Google Scholar]
- 41. Grienke U., Foster P. A., Zwirchmayr J., Tahir A., Rollinger J. M., Mikros E., Sci. Rep. 2019, 9, 11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen C. Y.-C., PLoS One 2011, 6, e15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mutai P., Pavadai E., Wiid I., Ngwane A., Baker B., Chibale K., Bioorg. Med. Chem. Lett. 2015, 25, 2510–2513. [DOI] [PubMed] [Google Scholar]
- 44. Ntie-Kang F., Simoben C. V., Karaman B., Ngwa V. F., Judson P. N., Sippl W., Mbaze L. M., Drug Des. Dev. Ther. 2016, 10, 2137–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kratz J. M., Mair C. E., Oettl S. K., Saxena P., Scheel O., Schuster D., Hering S., Rollinger J. M., Planta Med. 2016, 82, 1009–1015. [DOI] [PubMed] [Google Scholar]
- 46. Jin X., Kwon W., Kim T. S., Heo J.-N., Chung H. C., Choi J., No K. T., Bull. Korean Chem. Soc. 2018, 39, 294–299. [Google Scholar]
- 47. de Oliveira M. G., de Souza W. R. N., Rodrigues R. P., Kawano D. F., Borges L. L., da Silva V. B., in Emerging Research in Science and Engineering Based on Advanced Experimental and Computational Strategies (Eds.: La Porta F. d. A., Taft C. A.), Springer International Publishing, Cham, 2020, pp. 305–338. [Google Scholar]