

Abstract
With an increase in sedentary lifestyle and dietary over nutrition, obesity has become one of the major public health problems worldwide and is a prevalent predisposing risk factor to non‐alcoholic fatty liver disease (NAFLD), the most common chronic liver disease in Western developed countries. NAFLD represents a series of diseased states ranging from non‐alcoholic fatty liver (NAFL) to steatohepatitis (NASH), which can lead to fibrosis and eventually to cirrhosis and hepatocellular carcinoma. Currently, the only effective treatment to cure end‐stage liver disease is liver transplantation. Macrophages have been reported to play a crucial role in the progression of NAFLD, thereby are a potential target for therapy. In this review, we discuss the current knowledge on the role of macrophages and inflammatory signalling pathways associated with obesity and chronic liver inflammation, and their contribution to NAFLD development and progression.
Keywords: lipid metabolism, macrophages, NAFLD
1. INTRODUCTION
Over the last decade, NAFLD—recently also defined as Metabolic Associated Fatty Liver Disease, MAFLD—has become the predominant cause of chronic liver disease worldwide. 1 In fact, the majority might be due to rapid increase of overweight and obesity. In 2016, the World Health Organization (WHO) estimated that close to 2 billion adults worldwide were obese or overweight and this number is continuing to rise as up to 38 million children under the age of 5 are overweight. 2 NAFLD constitutes a spectrum of liver diseases ranging from simple fat accumulation in the liver (NAFL, isolated steatosis) to inflammatory state, non‐alcoholic steatohepatitis (NASH), which can lead to fibrosis (scaring of tissue) and ultimately to cirrhosis (permanent tissue damage) and hepatocellular carcinoma (HCC). 3 The prevalence of NAFLD is estimated about 20%‐40% worldwide, and it is anticipated to increase exponentially due to high prevalence of obesity. 1 Moreover, the presence and severity of NAFLD are associated with the risks of serious extrahepatic diseases, such as hypertension, insulin resistance, dyslipidemia, colon cancer, chronic kidney diseases and type II diabetes. 4 , 5
The exact pathogenesis of NAFLD is not completely understood, although a ‘multi‐hit’ hypothesis of NALFD is generally accepted, where genetic, epigenetic and environmental factors influence the disease activity, making it a very dynamic disease. 6 Impaired function of other organs, such as disrupted gut barrier function or obese adipose tissue (AT), accelerates the progression of the disease (Figure 1A). 7 Chronic hepatic lipid accumulation causes oxidative stress and endoplasmic reticulum stress leading to induction of inflammatory pathways, in which macrophages play an essential role (Figure 1B). 8 However, the exact role of macrophages and the mechanisms by which metabolic stress leads to NASH with recruitment and activation of monocytes/macrophages are not fully understood. In this review, we focus on the contribution of macrophage functions to the progression of obesity‐induced NAFLD.
Figure 1.
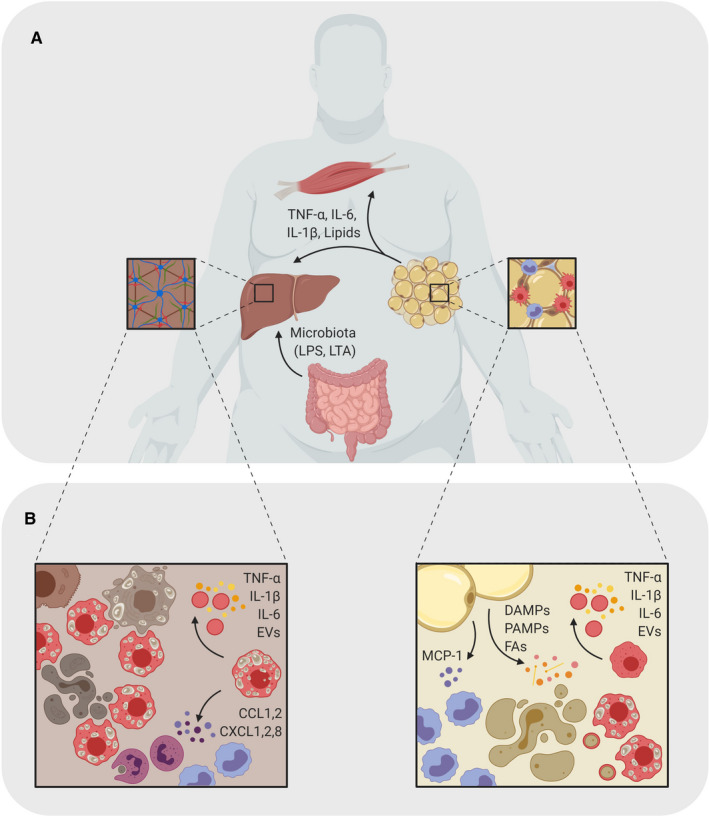
Role of macrophages in obesity‐driven NAFLD. A, Inflammatory macrophage activation (shown in red) promoting the progression of NAFLD involves a cross‐talk of AT, liver, gut and skeletal muscles. The inflammatory state in the AT leads to secretion of lipids and pro‐inflammatory cytokines into the blood stream where they travel to other metabolic organs, such as the skeletal muscles and the liver. Here, they induce an inflammatory state that can lead to metabolic diseases caused by chronic low‐grade inflammation. In obesity, the liver also receives LPS and lipoteichoic acid (LTA) from the gut, which also contribute to inflammation and tissue damage. B, During obesity, adipocytes stimulate monocyte infiltration by secreting MCP‐1. Adipocyte secretion of various DAMPs, PAMPs and FAs mediate a pro‐inflammatory polarization and differentiation of ATMs and monocytes. Within the liver, excess lipids, apoptitic bodies and AT‐derived cytokines induce an inflammatory environment with pro‐inflammatory hepatic macrophages and foam cells (shown in red). Their respond to the constant stimuli maintains the inflammatory state of the tissue by secreting pro‐inflammatory factors and neutrophil‐ and monocyte (shown in purple) attractants.
2. HEPATIC MACROPHAGES MEDIATE NAFLD‐NASH PROGRESSION
Non‐alcoholic fatty liver disease is the hepatic manifestation of metabolic syndrome and the most common chronic liver disease worldwide. It is a consequence of enhanced hepatic lipid accumulation. This excessive accumulation of hepatic fat leads to the deregulation of the immune system, activation of pro‐inflammatory signals, the breakdown of hepatic immune tolerance and development of NASH. Crucial to this transition process is hepatic macrophages. 9 , 10 , 11
Hepatic macrophages are a mixture of liver‐resident Kupffer cells and monocyte‐derived macrophages. Kupffer cells are the most abundant tissue‐resident macrophage cell type in mammalian bodies, accounting for 80%‐90% of total tissue‐resident macrophages. 12 They were first described by Karl Wilhelm von Kupffer as endothelial cells and components of liver vascular walls. They reside within the liver sinusoid where they receive portal venous blood from the gut and spleen as well as arterial blood from aorta. As such, Kupffer cells are the key sentinels at the interface of liver with other organs, such as the gut and AT. 13 They are the first responders to liver injury by sensing danger‐associated molecular patterns (DAMPs) including fatty acids, cholesterol from AT and pathogen‐associated molecular patterns (PAMPs), such as leaky gut‐derived endotoxins. 14
Recent data have demonstrated that Kupffer cells are predominantly from embryonic yolk sac‐derived macrophages and foetal liver monocytes with a minimal contribution from circulating blood monocytes at steady state. 15 In mice, Kupffer cells are characterized by the specific expression of C‐type lectin domain family 4 member F (CLEC‐4F) and TIM4+, 16 , 17 while a specific marker to identify human Kupffer cells remains to be elucidated. Kupffer cells have been described as important mediators of inflammation as for example Kupffer cell‐specific deletion through administration of gadolinium chloride in vivo attenuated NASH development. 10 However, under pathological condition, such as the progression from NAFL to NASH, there is massive recruitment of blood monocytes into the liver. 17 As such, monocyte‐derived hepatic macrophages might be the mediators and drivers in chronic low‐grade inflammation in NAFLD. Indeed, several studies demonstrated the contribution of both Kupffer cells and recruited monocytes in the development of NASH. 18 , 19 , 20 However, the underlying mechanisms and functional roles of different liver macrophage subsets are still not completely clear, given limitations due to the lack of tissue‐specific macrophage depletion models. It is still not well understood whether the microenvironment, due to excess of lipids, alters phenotype of resident Kupffer cells or provides the niche for infiltrating monocytes that differentiate into a new cell population. The recent identification of specific markers for Kupffer cells and new scientific methods (fate mapping and lineage tracing) makes it possible for a more targeted approach in the complex investigation of hepatic macrophage biology in homeostasis and disease. 21 Overall, the question is whether the origin of recruited monocytes determines their phenotype. To understand the impact and contribution of these different monocyte and macrophage subsets in the progression from NAFL to NASH, the population and repopulation of hepatic macrophages require further investigations.
Fibrosis is a hallmark in NASH and can lead to permanent scarring of the liver, which can further develop to HCC. Hepatic stellate cells are pericytes located in the perisinusoidal space. They produce components of the extracellular matrix and are the main contributors to fibrosis. 22 Recent studies have shown that macrophage populations in inflammatory tissue are highly heterogeneous and contain both pro‐ and anti‐inflammatory macrophages (Figure 2A). 23 During NASH progression, hepatic stellate cells are activated by apoptotic hepatocytes, oxidative stress and pro‐inflammatory as well as anti‐inflammatory immune cells. The secretion of pro‐inflammatory cytokines and pro‐fibrotic factors, such as TGF‐β, by hepatic macrophages and infiltrating monocytes enhances the pro‐fibrotic process of the hepatic stellate cells. 24 A potential biomarker for fibrosis is the scavenger receptor CD163. Soluble CD163 from adipose tissue macrophages (ATMs) and hepatic macrophages—which is increased in inflammatory macrophages—has been associated with fibrotic tissue. 25 The pro‐inflammatory and pro‐fibrotic processes amplify each other and might lead to cirrhosis.
Figure 2.
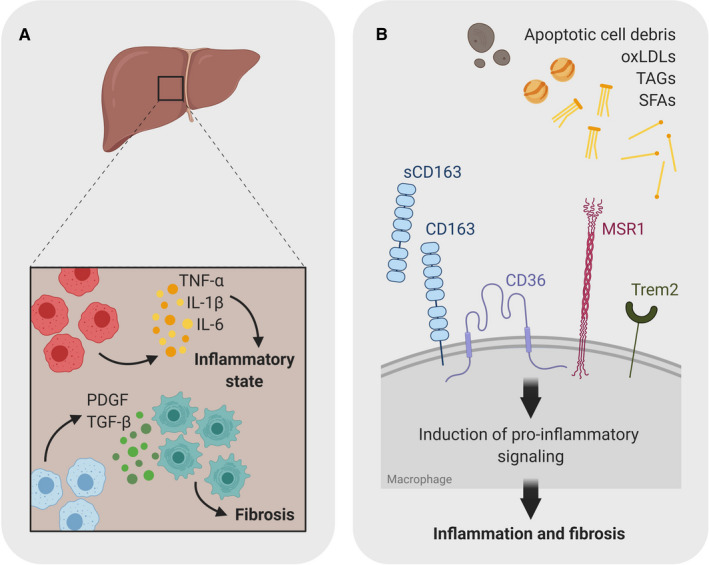
Scavenger receptors and Toll‐like receptors contribute to the inflammatory activation of macrophages. A, A variety of external cues can induce pro‐inflammatory macrophage polarization through the activation of inflammatory pathways. In obesity, these signals include a variety of lipids, cell debris, pathogen‐associated molecular patterns (PAMPs) that are recognized by scavenger receptors (SRs) and Toll‐like receptors (TLRs) on the cell surface of macrophages. Triggering these receptor contributes to the chronic low‐grade inflammation leading to tissue damage and ultimately fibrosis. B, Scavenger receptors expressed on myeloid cells are activated by apoptotic cell debris, TAGs, lipids etc Upon activation of these receptors, they induce pro‐inflammatory signalling in myeloid cells polarizing anti‐inflammatory tissue‐resident macrophages to pro‐inflammatory phenotype. Constant activation leads to a chronic low‐grade inflammation and ultimately to fibrosis
Cirrhosis is a major risk factor for the development of HCC, which is the most common type of liver cancer in adults. 26 Macrophages contribute to the development of HCC first by inducing a chronic low‐grade inflammatory environment with oxidative stress that induces DNA damage and hepatocyte death. 27 Infiltrating monocytes respond to the stimuli from the tumour microenvironment and differentiate into tumour‐associated macrophages (TAMs). TAMs are a heterogeneous population with a dominating anti‐inflammatory phenotype that is associated with a poor survival. 28
3. ADIPOCYTE‐MACROPHAGE CROSS‐TALK
Adipose tissue is not simply an organ for lipid storage and energy homeostasis, but in fact, accumulating evidence suggests that immune cells within AT contribute to the pathophysiology of obesity‐related diseases, such as NAFLD. 17 , 29 It is well accepted that prolonged and excess nutrient uptake induce chronic low‐grade inflammation within the AT, contributing to the manifestation of metabolic disorder, characterized by altered production of hormones, pro‐inflammatory cytokines and adipokines (Figure 1A). 30
Adipose tissue macrophages are believed to be one of the major players in regulating AT inflammation. 8 Healthy AT has a limited number of resident macrophages dominated by an anti‐inflammatory phenotype. In obesity, adipocytes start to enlarge and undergo cellular hypertrophy. As a consequence of adipocyte metabolic dysfunction, they increase production of the macrophage recruitment molecule, monocyte chemoattractant protein 1 (MCP‐1 or CC‐chemokine ligand 2; CCL2), which attracts monocytes to the AT. 31 , 32 In addition to recruitment, MCP‐1 promotes ATM proliferation within the tissue, significantly contributing to increased ATM numbers. 33 Further exposure of these ATMs to free fatty acids (FFAs) and triglycerides leads to their pro‐inflammatory activation and increased production of tumour necrosis factor‐alpha (TNF‐α), interleukin‐6 (IL‐6) and interleukin‐1β (IL‐1β) (Figure 1B), which has been shown to impair insulin sensitivity and glucose intolerance leading to insulin resistance, which is highly associated with other metabolic diseases as well as cancer. 34 , 35 , 36 , 37 Deletion of adipocyte MCP‐1 in vivo protected against obesity‐induced insulin and glucose resistance, indicating the importance of this pathway in the regulation of glucose metabolism. 31 However, genetic deletion of the murine MCP1 receptor, CCR2, showed inconsistent results in prevention of obesity, ATM recruitment and insulin/glucose insensitivity. 38 , 39 These data indicate that other factors might be involved in the activation of CCR2 and monocyte recruitment.
Adipose tissue macrophages activation status determines effector functions. Toll‐like receptor 4 (TLR4) is a well‐known pathogen recognition receptor (PRR) sensing lipopolysaccharide (LPS) from Gram‐negative bacteria. 40 In addition to LPS, TLR4 was also reported to be activated by FFAs; however, a recent in vitro study demonstrated that TLR4 is not a receptor for FFAs. 41 Our recent work indicates that the phagocytic receptor, macrophage scavenger receptor 1 (MSR1, SR‐A), is involved in FFA uptake followed by activation of JNK signalling independent of LPS, indicating an important role of scavenger receptors in lipid‐induced inflammation.
Activated ATMs have been linked to obesity and progression of NAFLD in mouse models and humans. 42 , 43 In unhealthy obesity, fat mobilization from adipocytes is impaired and insulin is unable to suppress lipolysis, which is the key mediator of increased lipid flux in ectopic organs such as liver and skeletal muscle. 44
4. MACROPHAGES AND LIPID SENSORS
Recently, it has been shown that different macrophage subsets display a distinct phenotype upon destabilisation of lipid homeostasis in the context of obesity. However, there is little known about the short‐ and long‐term effects on hepatic and extrahepatic complications. In recent years, single‐cell and bulk RNA sequencing have helped us enormously to better understand the pathophysiology of liver diseases. For instance, extensive single‐cell analysis of human NAFL and NASH livers defined a TREM2+, CD9+ macrophage subpopulation to be associated with fibrosis. 45 The scavenger receptor TREM2 (triggering receptor expressed on myeloid cells 2) has been found to be a major driver of tissue‐level immune cell remodelling. TREM2 regulates genes involved in phagocytosis, lipid catabolism and energy metabolism. 45 TREM2 has been recently linked to the lipid‐metabolizing lipid‐associated macrophages (LAMs) within AT that prevent the development of metabolic dysregulation in obesity. Additionally, role of TREM2 has been described in the development of disease‐associated microglia (DAMs) and their protective function in neurodegenerative diseases. 46 Notably, single‐cell RNA sequencing of AT from humans and mice showed a conserved TREM2 signature similar to the gene signature of DAMs. 46 , 47 Common signs of adiposity, neurodegeneration and atherosclerosis are the accumulation of extracellular lipids and inflammation. Therefore, TREM2 may function as a receptor for signals upon loss of tissue homeostasis by sensing extracellular lipids during metabolic dysfunction. 46
Another group of lipid sensors involved in lipid uptake is scavenger receptors. Scavenger receptors have been of major interest in the study of atherogenesis over the past decade and were first described in macrophages as alternative receptors to the low‐density lipoprotein (LDL) receptor in the uptake of cholesterol and lipids. 48 Interestingly, while most of scavenger receptors are important in the clearance of oxidized low‐/high‐density lipoproteins (LDL/HDL) or apolipoproteins, the exact underlying pathways are still unknown. We have recently shown that MSR1, which is highly expressed in hepatic macrophages, plays an important immunoregulatory role in the early development of obesity‐induced NAFLD and is responsible for foam cell formation—lipid laden macrophages associated with cardiovascular diseases, such as NAFLD (Figure 2B). Moreover, our data indicate that genetic variations in MSR1 could modify protein function, macrophage phenotype and thereby NAFLD development. These data support the notion of the role of genetic factors in susceptibility to NAFLD. 41 , 49 , 50 In conclusion, further characterization of scavenger receptors in the liver is thus of particular interest to better understand the pathophysiology of NAFLD and may potentially offer a powerful strategy for harnessing protective functions not only in the development and progression of NAFLD but also the metabolic syndrome in general.
5. CONCLUSIONS
Mounting evidence highlights the correlation of obesity with NAFLD, as the risk and severity factor for hepatic steatosis and steatohepatitis. However, there is still much to be understood regarding the mechanisms of the disease. Functional and phenotypic diversity of macrophages plays a key role in the spectrum of metabolic syndrome‐related diseases, such as NAFLD. Their function depends on their origin as well as activation, differentiation and polarization affected by both local and systemic signals in the progression of NAFLD. Moreover, there is also intertwined regulation of macrophage functions from different compartments such as circulation, bone marrow, AT, gut and liver. Mechanistically, it is of special interest to better understand the role of lipid metabolism in the regulation of macrophage function. More recently, scavenger receptors have come to focus as regulators of macrophage subsets, through which they affect inflammation and fibrosis. As such, additional studies are required to further investigate the functional role of specific macrophage subpopulations and their regulators in the form of scavenger receptors in NAFLD progression in order to target them for therapy.
AUTHOR CONTRIBUTIONS
SKP, OGB, OG and AH wrote the manuscript and designed the figures. All authors approved the manuscript and agreed to be accountable for the content of the work.
ACKNOWLEDGMENT
This study has been supported by the Knut och Alice Wallenberg Foundation Wallenberg Centre for molecular and translational medicine, University of Gothenburg, Sweden (to AH) and Åke Wirbergs Research funding #M18‐0121 (to AH) and Cancerfonden # 19 0352 Pj (to AH). Figures created with BioRender.com (licensed to AH). The authors would like to thank the members of AH's group and Patrycja Swacha for the critical reading of the manuscript. The authors disclose no conflicts.
Kragh Petersen S, Bilkei‐Gorzo O, Govaere O, Härtlova A. Macrophages and scavenger receptors in obesity‐associated non‐alcoholic liver fatty disease (NAFLD). Scand J Immunol. 2020;92:e12971 10.1111/sji.12971
Petersen and Bilkei‐Gorzo made equal contribution.
REFERENCES
- 1. Eslam M, Sanyal AJ, George J, International Consensus P . MAFLD: a consensus‐driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158(7):1999‐2014. [DOI] [PubMed] [Google Scholar]
- 2. WHOOaOW . 2020. [cited; Available from https://www.who.int/news‐room/fact‐sheets/detail/obesity‐and‐overweight. Accessed September 16, 2020.
- 3. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16:411‐428. [DOI] [PubMed] [Google Scholar]
- 4. Adams LA, Anstee QM, Tilg H, Targher G. Non‐alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. 2017;66:1138‐1153. [DOI] [PubMed] [Google Scholar]
- 5. Lefere S, Tacke F. Macrophages in obesity and non‐alcoholic fatty liver disease: crosstalk with metabolism. JHEP Rep. 2019;1:30‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anstee QM, Darlay R, Cockell S, et al. Genome‐wide association study of non‐alcoholic fatty liver and steatohepatitis in a histologically characterised cohort(). J Hepatol. 2020;73:505‐515. [DOI] [PubMed] [Google Scholar]
- 7. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple‐hit pathogenesis of non‐alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038‐1048. [DOI] [PubMed] [Google Scholar]
- 8. Lanthier N. Targeting Kupffer cells in non‐alcoholic fatty liver disease/non‐alcoholic steatohepatitis: why and how? World J Hepatol. 2015;8(7):2184‐2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stienstra R, Saudale F, Duval C, et al. Kupffer cells promote hepatic steatosis via interleukin‐1beta‐dependent suppression of peroxisome proliferator‐activated receptor alpha activity. Hepatology. 2010;51:511‐522. [DOI] [PubMed] [Google Scholar]
- 10. Huang W, Metlakunta A, Dedousis N, et al. Depletion of liver Kupffer cells prevents the development of diet‐induced hepatic steatosis and insulin resistance. Diabetes. 2010;59:347‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tosello‐Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet‐induced mouse model through tumor necrosis factor‐alpha production. J Biol Chem. 2012;23(287):40161‐40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bouwens L, Baekeland M, De Zanger R, Wisse E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology. 1986;6:718‐722. [DOI] [PubMed] [Google Scholar]
- 13. Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA, Eksteen B. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLoS One. 2016;11:e0159524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smedsrod B, De Bleser PJ, Braet F, et al. Cell biology of liver endothelial and Kupffer cells. Gut. 1994;35:1509‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;24(38):79‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scott CL, Zheng F, De Baetselier P, et al. Bone marrow‐derived monocytes give rise to self‐renewing and fully differentiated Kupffer cells. Nat Commun. 2016;27(7):10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Devisscher L, Scott CL, Lefere S, et al. Non‐alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol. 2017;322:74‐83. [DOI] [PubMed] [Google Scholar]
- 18. Xiong X, Kuang H, Ansari S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single‐cell secretome gene analysis. Mol Cell. 2019;8(75):644‐660 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krenkel O, Puengel T, Govaere O, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67:1270‐1283. [DOI] [PubMed] [Google Scholar]
- 20. Leroux A, Ferrere G, Godie V, et al. Toxic lipids stored by Kupffer cells correlates with their pro‐inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57:141‐149. [DOI] [PubMed] [Google Scholar]
- 21. Kebschull JM, Zador AM. Cellular barcoding: lineage tracing, screening and beyond. Nat Methods. 2018;15:871‐879. [DOI] [PubMed] [Google Scholar]
- 22. Sato M, Suzuki S, Senoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct. 2003;28:105‐112. [DOI] [PubMed] [Google Scholar]
- 23. Biswas SK, Gangi L, Paul S, et al. A distinct and unique transcriptional program expressed by tumor‐associated macrophages (defective NF‐kappaB and enhanced IRF‐3/STAT1 activation). Blood. 2006;1(107):2112‐2122. [DOI] [PubMed] [Google Scholar]
- 24. Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655‐663. [DOI] [PubMed] [Google Scholar]
- 25. Kazankov K, Barrera F, Møller HJ, et al. The macrophage activation marker sCD163 is associated with morphological disease stages in patients with non‐alcoholic fatty liver disease. Liver Int. 2016;36:1549‐1557. [DOI] [PubMed] [Google Scholar]
- 26. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35‐50. [DOI] [PubMed] [Google Scholar]
- 27. Lin Z, Xu SH, Wang HQ, et al. Prognostic value of DNA repair based stratification of hepatocellular carcinoma. Sci Rep. 2016;13(6):25999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Budhu A, Forgues M, Ye Q‐H, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99‐111. [DOI] [PubMed] [Google Scholar]
- 29. Rosso C, Kazankov K, Younes R, et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non‐alcoholic fatty liver disease. J Hepatol. 2019;71:1012‐1021. [DOI] [PubMed] [Google Scholar]
- 30. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanda H, Tateya S, Tamori Y, et al. MCP‐1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796‐1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Amano SU, Cohen JL, Vangala P, et al. Local proliferation of macrophages contributes to obesity‐associated adipose tissue inflammation. Cell Metab. 2014;7(19):162‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor‐alpha: direct role in obesity‐linked insulin resistance. Science. 1993;1(259):87‐91. [DOI] [PubMed] [Google Scholar]
- 35. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity‐induced insulin resistance in mice lacking TNF‐alpha function. Nature. 1997;9(389):610‐614. [DOI] [PubMed] [Google Scholar]
- 36. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity‐related insulin resistance. J Clin Invest. 2003;112:1821‐1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high‐fat feeding. J Clin Invest. 2006;116:115‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inouye KE, Shi H, Howard JK, et al. Absence of CC chemokine ligand 2 does not limit obesity‐associated infiltration of macrophages into adipose tissue. Diabetes. 2007;56:2242‐2250. [DOI] [PubMed] [Google Scholar]
- 40. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;20(86):973‐983. [DOI] [PubMed] [Google Scholar]
- 41. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid‐induced insulin resistance. J Clin Invest. 2006;116:3015‐3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cancello R, Tordjman J, Poitou C, et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes. 2006;55:1554‐1561. [DOI] [PubMed] [Google Scholar]
- 43. Govaere O, Martinez‐Lopez N, Petersen SK, et al. Macrophage Scavenger Receptor 1 mediates lipid‐induced inflammation in non‐alcoholic fatty liver disease. bioRxiv. 2020. 10.1101/2020.02.01.930115 [DOI] [PubMed] [Google Scholar]
- 44. Laurencikiene J, van Harmelen V, Arvidsson Nordstrom E, et al. NF‐kappaB is important for TNF‐alpha‐induced lipolysis in human adipocytes. J Lipid Res. 2007;48:1069‐1077. [DOI] [PubMed] [Google Scholar]
- 45. Ramachandran P, Dobie R, Wilson‐Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single‐cell level. Nature. 2019;575:512‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keren‐Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;15(169):1276‐1290 e17. [DOI] [PubMed] [Google Scholar]
- 47. Jaitin DA, Adlung L, Thaiss CA, et al. Lipid‐associated macrophages control metabolic homeostasis in a Trem2‐dependent manner. Cell. 2019;25(178):686‐698. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zani IA, Stephen SL, Mughal NA, et al. Scavenger receptor structure and function in health and disease. Cells. 2015;22(4):178‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eslam M, McLeod D, Kelaeng KS, et al. IFN‐lambda3, not IFN‐lambda4, likely mediates IFNL3‐IFNL4 haplotype‐dependent hepatic inflammation and fibrosis. Nat Genet. 2017;49:795‐800. [DOI] [PubMed] [Google Scholar]
- 50. Cai B, Dongiovanni P, Corey KE, et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2020;4(31):406‐421 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]