

Abstract
Approximately half of people infected with HIV (PWH) exhibit HIV-associated neuropathology (neuroHIV), even when receiving combined antiretroviral therapy. Opiate use is widespread in PWH and exacerbates neuroHIV. While neurons themselves are not infected, they incur sublethal damage and GABAergic disruption is selectively vulnerable to viral and inflammatory factors released by infected/affected glia. Here, we demonstrate diminished K+-Cl− cotransporter 2 (KCC2) levels in primary human neurons after exposure to HIV-1 or HIV-1 proteins ± morphine, resulting in disruption of GABAAR-mediated hyperpolarization/inhibition. We found that the HIV proteins Tat (acting through NMDA receptors), and R5-tropic gp120 (acting via CCR5), and morphine (acting through μ-opioid receptors) induce KCC2 loss. We demonstrate that modifying KCC2 levels or function, or antagonizing NMDAR, CCR5 or MOR rescues KCC2 and GABAAR-mediated hyperpolarization/inhibition in HIV, Tat, or gp120 ± morphine-exposed neurons. Using an inducible, Tat-transgenic mouse neuroHIV model, we found that chronic exposure to Tat also reduces KCC2. Our results identify KCC2 as a novel therapeutic target for ameliorating the pathobiology of neuroHIV, especially PWH exposed to opiates.
Keywords: neuroHIV, opioid abuse, C-C Motif Chemokine Receptor 5 (CCR5), intracellular chloride homeostasis, KCC2, GABAA hyperpolarization, Archon1, GCaMP6f
Introduction
Close to 38 million people worldwide are infected with HIV, with 50,000 new diagnoses of HIV-1 infection per year in the US alone (de Mendoza, 2019). Up to 50% of those infected with HIV-1 have CNS complications including HIV-associated neurocognitive disorders (HAND) (Antinori et al., 2007; Heaton et al., 2010). It is widely accepted that neurons are not infected by HIV-1. Neurological deficits in patients receiving combined antiretroviral therapy (cART) are likely due to sublethal neuronal stress and injury induced by viral proteins released from infected cells and persistent neuroinflammation, resulting in hyperexcitability (Anthony, Ramage, Carnie, Simmonds, & Bell, 2005; Everall et al., 2009; Nath & Steiner, 2014; Neuenburg et al., 2002). Exposure to HIV-1 transactivator of transcription (Tat) depolarizes neurons and leads to electrophysiological dysfunction and hyperexcitability, partly through interactions with N-methyl-D-aspartic acid (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Krogh, Wydeven, Wickman, & Thayer, 2014; Philippon et al., 1994). Another HIV-1 protein, gp120, binds co-receptors C-C chemokine receptor type 5 (CCR5) and/or C-X-C chemokine receptor type 4 (CXCR4) (Wang et al., 2003) (Kaul, Ma, Medders, Desai, & Lipton, 2007), leading to neurotoxic glial and inflammatory effects, as well as direct neurotoxicity. Opiate use is often comorbid with HIV-1 infection and has been implicated in increased severity of HAND (Bell, Arango, & Anthony, 2006; Byrd et al., 2011; Carrico, 2011; Smith, Simmonds, & Bell, 2014). This comorbidity is highlighted by recent HIV-1 outbreaks in communities experiencing a surge in opiate abuse (Conrad C, 2015). In vivo and in vitro models of HAND demonstrate that morphine, the primary bioactive metabolite of heroin in the CNS, exacerbates HIV-1-induced neuropathogenesis primarily through the activation of the μ-opioid receptor (MOR) on glia, and subsequent modulation of neuroinflammation and reduced glutamate buffering (Bokhari et al., 2009; Rodriguez et al., 2017; Shiping Zou et al., 2011).
GABAergic neurons seem to be selectively vulnerable to damage by Tat and HIV-1 infection in mouse models and post-mortem brain tissue from HIV-infected patients, highlighting disinhibition as a potential mechanism underlying hyperexcitability in the HIV-infected brain (Buzhdygan et al., 2016; Fitting et al., 2013; Gelman, Chen, et al., 2012; Marks et al., 2016). The deficits appear to result from a loss of GABAergic markers (including GAD1, GAD2, and GJD2) rather than the death of GABAergic interneurons in autopsy samples from PWH (Buzhdygan et al., 2016). Importantly, deficits in GABAergic markers differed significantly among brain regions and linked to worse cognitive performance, but these markers were unaffected by a history of drug abuse and differed minimally when comparing pre- and post-cART autopsy samples (Buzhdygan et al., 2016).
KCC2 extrudes Cl− to maintain a low intracellular Cl− concentration ([Cl−i in mature neurons (Blaesse, Airaksinen, Rivera, & Kaila, 2009). GABAA and glycine receptor-induced fast synaptic inhibition is predicated on the maintenance of low neuronal [Cl−]i provided by KCC2 activity. While upregulation and subsequent increases in KCC2 activity have been extensively studied in development, loss of KCC2 activity and/or expression have more recently been noted in a number of neurological disorders including epilepsy (M. Chen et al., 2017; Kahle et al., 2014; Puskarjov et al., 2014; Silayeva et al., 2015), traumatic brain injury (Lizhnyak, Muldoon, Pilaka, Povlishock, & Ottens, 2019), schizophrenia (Arion & Lewis, 2011; Hyde et al., 2011; Tao et al., 2012), Rett syndrome (Tang et al., 2016), Huntington’s disease (Dargaei et al., 2018), and morphine-induced hyperalgesia (Ferrini, Lorenzo, Godin, Quang, & De Koninck, 2017; Ferrini et al., 2013). The KCC2 activity enhancer, CLP257, and its prodrug, CLP290, have allowed manipulation of KCC2 activity and rescued deficits in KCC2-deficient states (Gagnon et al., 2013). While models of HAND have been shown to disrupt GABAergic functioning, loss of KCC2 activity has not yet been implicated in these deficits.
In addition to working with individual HIV-1 proteins, we exposed primary human neurons (hNeurons), derived and matured from neural progenitors (hNPC), to supernatant from HIV-1 infected monocytes (HIVsup). This supernatant reflects the complexity of an infective milieu in that it contains multiple inflammatory/reactive factors and virions, as well as viral proteins. The genetically encoded voltage (GEVI) and Ca2+ indicators (GECI), Archon1 (Piatkevich et al., 2018) and GCaMP6f (T. W. Chen et al., 2013), respectively, were used to examine the functional outcomes of interactions between an HIV+ environment and morphine on hNeurons. Specifically, we used Archon1 and GCaMP6f to examine changes in GABAAR-mediated hyperpolarization and inhibition, respectively. Importantly, this optical approach permitted us to manipulate and test functional outcomes absent the biohazards inherent with invasive electrophysiological measurements in the presence of infective HIV. Assessment of electrophysiological activity by Archon1 is equivalent to patch-clamp recording of neuronal spiking and subthreshold millivolt scale activity (Piatkevich et al., 2018). We demonstrate that HIVsup and morphine target KCC2 in hNeurons, resulting in loss of GABAAR mediated hyperpolarization (measured by Archon1) and inhibition (measured by GCaMP6f). This suggests that dysregulation of KCC2 may play a role in circuit hyperexcitability in the HIV-infected brain. Further, we identify Tat, acting via an NMDA receptor (NMDAR)-mediated mechanism, and gp120ADA, acting via a novel, CCR5-mediated mechanism, as viral proteins that can dysregulate KCC2 and GABAAR-mediated inhibition. We also examined the effects of Tat exposure on KCC2 in an in vivo model of HAND and found significantly diminished KCC2 in the striatum of these animals. Our results suggest KCC2 and its upstream regulators as promising therapeutic targets to alleviate the symptoms of HAND ± comorbid opiate use.
Materials and Methods
Primary hNPC culture.
Human neural progenitor cells (hNPCs) were derived from human CNS tissue at 15 - 17 weeks gestation (Advanced Biosciences Resources; Alameda, CA). Tissue was passed through 120 μM mesh, centrifuged and washed with medium twice, and dissociated cells were plated on poly-L-lysine (10 μg/mL) and laminin (2 μg/mL) coated flasks. hNPCs were maintained in NPC medium: DMEM F12 supplemented with 0.6% glucose, 10% B27 (without vitamin A; Thermo Fisher Scientific), 20 ng/mL fibroblast growth factor (FGF; R&D Systems, Minneapolis, MN), 20 ng/mL epidermal growth factor (EGF; EMD Millipore, Billerica, MA), 10 ng/mL leukemia inhibitory factor (LIF; EMD Millipore, Billerica, MA), 1% Pen/Strep, 5 mM HEPES (N-2-hydroxyethylpiperazine-N-3-ethane sulfonic acid; Thermo Fisher Scientific) with medium changed every 3 - 4 days.
Neuronal Differentiation.
Upon 80 - 90% confluency, hNPCs were detached using Accutase cell detachment solution (Millipore) and plated on poly-L-lysine (50 μg/mL) and laminin (100 μg/mL) coated 12-mm-diameter coverslips (immunostaining) or glass bottomed MatTek dishes (functional imaging) at 12,000 - 15,000 cells/coverslip in DMEM-F12 medium supplemented with 0.6% glucose, 10% B27 (without vitamin A; Thermo Fisher Scientific), 1% Pen/Strep, 5 mM HEPES (Thermo Fisher Scientific), and 10 ng/mL brain derived neurotrophic factor (BDNF; Sigma). After 7 DIV, medium was exchanged gradually (50% on days 7 and 9) and then fully (100% every 72 - 96 h) to BrainPhys™ (STEMCELL Technologies Inc, Vancouver, BC, CAN) supplemented with SM1 (STEMCELL).
HIV-1 propagation and HIVsup preparation.
HIV-1BaL was propagated using methods previously established (Balinang, Masvekar, Hauser, & Knapp, 2017). Briefly, isolated PBMCs from peripheral blood Leuko Paks (ZenBio, Research Triangle Park, NC) were activated with 1 mg/mL phytohemagglutinin (PHA) for 48 h, then infected with HIV-1BaL (NIH AIDS Reagent Program) at 1 ng/mL p24. After 72 h, infection was assayed using p24 antigen ELISA kit (Advanced Bioscience, Rockville, MD) and supernatant harvested and frozen at −80° C for HIVsup experiments. HIVsup used in all experiments was from the same source sample to reduce variability.
Treatments.
hNeuron cultures were treated with HIVsup at 125 - 500 pg/mL [p24] or HIV protein (10 - 100 nM HIV-1 Tat1-86 IIIB (clade B) or 250 pM - 1 nM R5-tropic gp120ADA, X4-tropic gp120IIIB, or dual-tropic gp120 MN; ImmunoDx, Woburn MA) ± 500 nM morphine sulfate for 6 or 24 h. Drug treatments: 10 μM CLP257 (Sigma), 50 μM AP5 (Alomone Labs, Jerusalem IL), 50 nM maraviroc (MVC; BOC Sciences, Shirley, NY) were applied 30 min prior to HIVsup, HIV protein, and morphine for 24 h experiments. 50 μM VU02440551 (Tocris, Bristol UK) was applied 2 h prior to GEVI/GECI imaging experiments. Supernatant from uninfected but activated monocytes (UNF) was used as a control for HIVsup. Vehicle controls were used for all other treatments.
Immunocytochemistry and Cell Counting.
Neuronal cultures were fixed for 15 min (4% paraformaldehyde), permeabilized for 15 min (0.1% Triton-X 100, 0.1% BSA) and blocked for 1 h at room temperature (0.1% BSA, 1% goat serum in PBS). Coverslips were then immunostained with rabbit anti-KCC2 (1:1000; Protein Tech, Rosemont, IL) and mouse anti-microtubule-associated protein (MAP2) (1:500; Millipore, Burlington, MA) for KCC2 immunoreactivity and differentiation studies. Rabbit anti-MAP2 (1:500; Abcam, Cambridge, UK), mouse anti-glial fibrillary acidic protein (GFAP) (1:1000; Millipore), rabbit anti-Na+-K+-Cl− cotransporter 1 (NKCC1) (1:100; Abcam), mouse anti-MAP2, mouse anti-glutamate decarboxylase (GAD) 67 (1:500; Abcam), rabbit anti-glutaminase (1:500; Abcam), and mouse anti-MOR (1:500; Antibodies Incorporated, Davis, CA). were used for differentiation studies. Alexa Fluor 488 and 594-conjugated secondary antibodies (1:1000; Thermo Fisher, Waltham, MA) were used for all studies as well as Hoechst 33342 (1:10000; Invitrogen, Carlsbad, CA) to detect nuclei. Cells were visualized and images obtained using a Zeiss LSM 700 confocal module configured to an Axio Observer Z.1 and Zen 2010 software (Zeiss Inc., Thornwood, NY). Cells were manually counted using the CellCounter plugin for Image J. At least 200 Hoechst+ cells were counted per sample.
GEVI/GECI Expression.
AAV serotype 1 containing GCaMP6f and Archon1 were acquired from AddGene (plasmid # 100837; Watertown, MA) and Vector Biolabs (Malvern, PA), respectively. hNeurons were treated with AAV1.hSyn.GCaMP6f.WPRE.SV40 or AAV1.hSyn.Archon1.WPRE.SV40 for 5 - 7 days prior to experiments. Human synapsin promoter (hSyn) was used to ensure neuronal specificity.
GEVI/GECI Imaging and Analysis.
Archon1 and GCaMP6f recordings were acquired at 2 or 1 KHz, respectively, with a Neuro-CCD camera (RedShirt Imaging, Decatur, GA) mounted on a Zeiss Axio Observer with 40× oil immersion objective. Archon1 and GCaMP6f illumination was achieved with an LED (UHP-T-LED; Prizmatix, Holon, IL) 640 nm excitation, 660LP emission for Archon1 (custom-made filter cube; Omega Optical, Brattleboro, VT) and 470/40 nm excitation, 495 dichroic, 525/50 nm emission for GCaMP6f (filter set 38; Zeiss). All recordings were performed in BrainPhys solution (STEMCELL) with 2 μM CGP55845 (GABABR antagonist; Abcam) or 2 μM CGP55845 and 100 μM picrotoxin (PTX) (GABAAR antagonist; Tocris) to validate Archon1 results (Fig. S3).
Soma ROI were used for all experiments. Archon1 and GCaMP6f traces were analyzed with a custom code in R. Both Archon1 and GCaMP6f traces were 5-point Savitzky-Golay smoothed and GCaMP6f traces were Loess subtracted, to remove slower transients. Fluorescence changes were calculated as ΔF/F = (FA - Fbl)/Fbl, where FA is the fluorescence level during perfusion and Fbl, is the average baseline fluorescence intensity 2.5 s prior to perfusion. Significant Ca2+ transients were counted when the trace exceeded the standard deviation (SD) of background activity (measured 2.5 s prior to perfusion) by 4-fold and returned to baseline levels (defined as within 0.5 × SD of mean activity).
Animals.
Male, doxycycline (DOX)-inducible, GFAP driven HIV-1 Tat-transgenic mice (10 – 12 weeks old) were used to explore the effects of Tat expression on KCC2. Mice expressing the tat and rtTA transgenes (Tat+) or mice lacking the tat transgene, but expressing the rtTA transgene (Tat−), were fed doxycycline-containing chow (6 mg/g, Harlan Indianapolis, IN) for 2 weeks prior to sacrifice. Animal procedures were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee and are in accordance with Association for Assessment and Accreditation of Laboratory Animal Care guidelines.
Immunoblot.
KCC2 presence was examined by immunoblotting striatal tissue from Tat+ and Tat− mice. Striata were freshly harvested and homogenized in RIPA lysis buffer (Sigma) and Halt™ Protease & Phosphatase Inhibitor Cocktail (Thermo Fisher). Lysates were then centrifuged and stored at −80° C. Protein concentration was measured using BCA protein assay (Pierce, Rockford, IL). 40 μg of lysates were loaded per well onto 4 – 20% Tris-HCl Ready Gels (Bio-Rad Laboratories, Hercules, CA). Proteins were transferred to PVDF membranes (Bio-Rad) and probed with antibodies against KCC2 (1:2000; Protein Tech) and GAPDH (1:2000; Abcam). Alexa Fluor 647 and 488-conjugated secondary antibodies (1:2000; Thermo Fisher) were used to detect proteins. A Bio-Rad ChemiDoc™ MP Imaging System and Image Lab were used to measure and analyze protein levels, respectively.
Statistics.
KCC2 immunocytochemistry studies were assessed using three or two-way analysis of variance (ANOVA; R) to determine the effects of Tat1-86/gp120/HIVsup, CLP257/AP5/MVC, and morphine on KCC2 expression at 24 h following viral/drug exposure. The 6 h studies were assessed using two-way ANOVA with HIV (i.e., Tat1-86, gp120, or HIVsup) and morphine treatments as factors. Interactions and main effects for both were examined via simple main effects using a Bonferroni post hoc test to determine individual group differences. Functional imaging studies were analyzed as above except the assessment of VU0240551 was determined separately using a two-tailed Student’s t test. A two-tailed Student’s t test was also used to examine differences between Tat+ and Tat− mice in western blots. Data were expressed as mean value ± standard error of the mean. Analyses were considered significant if p < 0.05. Cook’s distance was used to determine and remove outliers.
Results
hNPC differentiation and hNeuron characterization.
We developed a protocol to differentiate hNPCs into mature hNeurons. EGF, FGF, and LIF initially maintain hNPC pluripotency. These factors were replaced with BDNF to promote neuronal survival and differentiation, after which cells were gradually transitioned to BrainPhys medium, which promotes neural activity and more accurately mimics in vivo physiological conditions (Bardy et al., 2015). After 3 weeks in vitro, 53 ± 3.3% of cells are MAP2+ (GFAP−) neurons and 44 ± 3.5% GFAP+ astrocytes (Fig. 1). Neurons were found to be primarily glutaminase+ (~75%), and glutamate decarboxylase+ (~25%), with few tyrosine hydroxylase+ (~1%) cells (Fig. S1). hNeurons also expressed MOR (Fig. S1). NKCC1 is expressed on immature neurons and opposes KCC2 activity by increasing neuronal [Cl−]i. NKCC1 expression decreased to undetectable levels by 21 days in vitro (DIV) in our cultures, while KCC2 expression increased, as seen normally during neuronal maturation (Schulte, Wierenga, & Bruining, 2018) (Fig. 1C,D; Fig. S1). These characteristics suggested that hNeurons had matured enough to express KCC2 and to assess its role in GABAAR-induced hyperpolarization, as evident in experiments performed below.
Figure 1. Differentiation and characterization of hNeuron.

hNPCs differentiate to ~55% MAP2+/GFAP− (green) and 40% GFAP (red)+ by 14 DIV (A). Representative images from hNeuron cultures showing immunofluorescence of MAP2 and GFAP after 21 DIV (B). hNeurons lose NKCC1 (C) and gain KCC2 (D) immunoreactivity by 21 DIV.
HIV-1-infected PBMC supernatant reduces KCC2 immunoreactivity.
Previous studies have shown selective vulnerability of GABAergic neurons to HIV-1 and Tat (Gelman, Chen, et al., 2012; Marks et al., 2016). Here, we examined KCC2 immunoreactivity to determine if [Cl−]i dysregulation might relate to GABAergic deficits. To model the HIV-infected human brain, we exposed hNeurons to HIVsup. Experiments show ~55 % of Hoechst+ cells in control groups are immunoreactive for KCC2 (Fig. 2A), with significant reductions in co-expression when hNeurons were exposed for 6 or 24 h to HIVsup at 250 - 500 pg/mL [p24] (but not 125 pg/ml [p24]) or 500 nM morphine (p < 0.01; n = 6) (Fig. 2A,B). Loss of KCC2 was not due to cell death in the exposure groups examined as evidenced by continued MAP2 antigenicity and the LIVE/DEAD™ viability/cytotoxicity assay (Molecular Probes, Eugene, OR) (Fig. S2). Pretreatment (30 min) with the KCC2 enhancer, CLP257 (10 μM), restored KCC2 immunoreactivity (p < 0.05; n = 6) (Fig 2B). These results suggest that Cl− regulation and, therefore, GABA function may be altered in neurons of the HIV-1-exposed brain. HIVsup and morphine interactions were examined at 125 pg/mL p24, but not found). This interaction was not examined at 250-500 pg/mL p24 since cell death can occur with morphine co-exposure at the higher p24 levels (Masvekar, El-Hage, Hauser, & Knapp, 2015) and which might be confounding for KCC2 immunoreactivity.
Figure 2. HIVsup and morphine significantly reduce the percentage of cells immunoreactive for KCC2.

Treatment for 6 h (A) or 24 h (B) with 250 – 500 pg/mL [p24] HIVsup ± morphine significantly reduces the number of KCC2 immunoreactive cells (†p < 0.01 for respective controls; n = 6). CLP257 (blue) rescued this effect across all groups groups(*p < 0.05, n = 6). Naloxone and CTAP both antagonized the effects of morphine (C), suggesting the involvement of MOR (*p < 0.01; n = 6). AP5 (orange) restored KCC2 immunoreactivity after 250 – 500 pg/mL HIVsup exposure while MVC (maroon) significantly increased KCC2 in 500 pg/mL (*p > 0.05, n = 5 – 6). A trend towards rescue of KCC2 immunoreactivity was found at 250 pg/mL HIVsup (p = 0.061, n = 5 – 6). Co-exposure of HIVsup with both AP5 and MVC fully restored KCC2 levels, suggesting NMDAR and CCR5 as the primary pathways of HIVsup-mediated KCC2 reduction. Representative images for KCC2 immunofluorescence of vehicle, morphine, and morphine + CTAP-treated hNeurons. Scale bar = 50 μm (E).
The HIV proteins Tat and gp120 are both well-known to influence neuronal hyperexcitability and progression of HAND (Nath & Steiner, 2014), so we tested their contribution to the loss in KCC2 immunoreactivity induced by HIVsup. Since NMDAR and CCR5 are respective major targets of Tat and gp120, we pretreated cultures with the NMDAR antagonist AP5 (50 μM) and/or the CCR5 antagonist, MVC (50 nM). Independent exposures showed that both significantly increase KCC2 immunoreactivity at 500 pg/mL HIVsup and AP5 at 250 pg/mL HIVsup (p < 0.05, n = 5 – 6) with a trend towards rescue with MVC at 250 pg/mL HIVsup (p = 0.061, n = 5 – 6) (Fig 2D). When combined, MVC and AP5 are significantly different than either individually and completely restored KCC2 immunoreactivity (p < 0.01; n =5 - 6) (Fig 2D), suggesting NMDAR and CCR5 as the pathways mediating HIVsup-induced KCC2 reduction. Further examination revealed a significantly increased KCC2 after AP5 exposure compared to MVC exposure suggesting that while both CCR5 and NMDAR activation play a role, NMDAR is the dominant pathway towards diminished KCC2 in this context (p < 0.01; n = 5). Both the pan-opioid receptor antagonist, naloxone, and the selective MOR antagonist, CTAP, reversed KCC2 losses induced by morphine, suggesting MOR activation can suppress KCC2 activity (p < 0.01; n = 6) (Fig 2C).
HIVsup ± morphine diminishes GABAaR mediated hyperpolarization.
The GEVI, Archon1, is a far-red-shifted, membrane-bound, voltage-sensitive, opsin-based fluorescent protein that changes fluorescence intensity with membrane potential. This high speed, noninvasive measurement of electrophysiological activity was shown to be identical to patch-clamp recording of neuronal spiking (100 Hz) and subthreshold millivolt scale activity (Piatkevich et al., 2018). To determine HIVsup ± morphine effects on GABAAR activation, we measured Archon1 activity in cultures perfused with 100 μM GABA and GABABR antagonist, CGP55845 (2 μM) after 24 h exposure to 125 - 500 pg/mL [p24] HIVsup ± morphine or to UNF. Archon1-expressing hNeuron control groups showed robust hyperpolarization with peak ΔF/F deflection of −8.7 ± 0.5% during 115 s perfusion of 100 μM GABA, CGP55845 (Fig. 3). hNeurons exposed for 24 h to varying concentrations of HIVsup (125 - 500 pg/mL p24) or morphine (500 nM) showed 16 - 34% reductions of peak ΔF/F deflection during perfusion (p < 0.01; n = 15 - 24), suggesting reduced hyperpolarization in response to GABAAR activation (Fig. 3). Analysis revealed a main effect of CLP257 treatment in restoring GABAAR-mediated hyperpolarization. A comparison of individual groups revealed that CLP257 reversed the effects of 500 pg/mL [p24] HIVsup and morphine (p < 0.05; n = 15 - 24) (Fig. 3). Groups exposed to the KCC2 antagonist, VU0240551 (50 μM), recapitulated results of HIVsup and morphine groups (28% ΔF/F reduction; p < 0.05; n = 15) (Fig. 3). We confirmed that ΔF/F changes above were, in fact, due to GABAAR activation by perfusing GABA, CGP55845, and the noncompetitive GABAAR antagonist PTX (100 μM) and found no fluctuations in ΔF/F (Fig. S3). These results suggest that loss of KCC2 activity due to HIVsup ± morphine exposure results in dysregulated [Cl−]i and subsequent deficits in GABAAR activity, and that these deficits can be rescued by maintaining KCC2 expression with CLP257.
Figure 3. HIVsup and morphine decrease GABAAR-mediated hyperpolarization.

(A) Representative traces of 500 pg/mL [p24] HIVsup treated (red) and vehicle control (black) during GABA and CGP55845 perfusion (115 s). Arrow is representative of the diminished hyperpolarization quantified in the bar graph. (B) Bar graph shows peak ΔF/F deflection from each treatment groups normalized as a percentage of vehicle control corresponding to that sample during 115 s perfusion of 100 μM GABA and CGP55845. hNeurons expressing Archon1 exposed to 125 - 500 pg/mL [p24] HIV ± morphine shows reduced hyperpolarization (†p < 0.01, n = 15 - 21) and/or UNF (‡p < 0.01, n = 15 - 21). Analysis revealed a main effect of CLP257 and group comparisons showed that CLP257 reversed the effects of 500 pg/mL HIVsup and morphine groups (blue) (*p < 0.05, n = 15 - 21). Further, 2 h treatment with VU02440551 showed similar results (#p < 0.05, n = 15 - 21), suggesting that these effects are due to loss of KCC2 activity. Data were analyzed from 15 - 21 cells from 5 – 6 separate tissue samples per group.
HIV-1 proteins reduce KCC2 immunoreactivity.
Since HIVsup contains multiple inflammatory and viral factors found in the HIV-infected CNS, it served as a more translational model of neuronal injury. To determine the contribution of individual viral proteins to effects seen with HIVsup, we exposed hNeurons to Tat1-86 and gp120 (R5-tropic, X4-tropic, and dual-tropic), both of which are thought to be primary factors in neurodegenerative outcomes and HAND symptoms. We first examined the effects of Tat1-86 exposure in isolation because NMDAR is a primary target of Tat and its activation with regard to induction of KCC2 degradation is well-known (Medina et al., 2014). Both morphine (500 nM) and Tat1-86 (50 – 100 nM) alone significantly reduced percentages of Hoechst+ cells displaying KCC2 immunoreactivity, after both 6 and 24 h exposure, compared to vehicle groups (p < 0.01; n = 6) (Fig. 4B, C). Further, there was a significant interaction between Tat1-86 (50 nM) and morphine with 24 h co-exposed groups displaying exacerbated KCC2 loss (p < 0.05; n = 5 – 6), suggesting signaling convergence. Again, cell death was not responsible for Tat1-86 effects since the number of MAP2+ cells was stable (Fig. 4A) and no increases in dying/dead cells were evident using a cytotoxicity assay (Fig. S2). CLP257 significantly rescued KCC2 immunoreactivity in Tat1-86, morphine, and co-exposed groups (p < 0.01; n = 6) (Fig. 4C). Pharmacological blockade of NMDAR with AP5 rescued KCC2 immunoreactivity (Fig. 4C). These data suggest that Tat1-86 activation of NMDAR may suppress KCC2 levels sufficient to reduce the number of HIVsup-exposed KCC2 immunoreactive hNeurons without causing their death.
Figure 4. hNeurons lose KCC2 immunoreactivity after exposure to HIV-Tat ± morphine.

(A) Representative images of hNeuron cultures immunolabeled for MAP2 and KCC2. Loss of KCC2 immunoreactivity can be noted in cells treated with both 50 nM Tat1-86 and 50 nM Tat1-86 + morphine. Rescue is seen in 50 nM Tat1-86 + AP5 (scale bar = 50 μm). (B, C) The percentage of cells immunoreactive for KCC2. Both 6 h (B) and 24 h (C) exposure to 50 - 100 nM Tat1-86 ± morphine results in significant decrease in KCC2 immunoreactivity compared to their respective controls (†p < 0.01; n = 6 – 7). Further, a significant interaction between morphine and Tat1-86 was found at the 50 nM Tat1-86 level after 24 h exposure (#p < 0.05). Application of CLP257 (blue) and AP5 (white) showed significant restoration of KCC2 immunoreactivity across all groups and Tat1-86 exposed groups, respectively (*p < 0.01).
Effects on KCC2 vary with gp120 tropism.
We tested concentration -dependent effects of gp120 (250 pM-1 nM) from three different tropic strains of HIV. Exposure to gp120ADA (R5-tropic; p < 0.01; n = 6), but not gp120IIIB (X4-tropic) (p = 0.23; n = 5) or gp120MN (dual-tropic) (p = 0.078; n = 5) strains, significantly reduced KCC2 expression at both 6 h and 24 h compared to controls (Fig. 5A,B). Further, there was a trend towards interaction with morphine and gp120ADA at the 500 pM level (p = 0.068; n = 6). The reduction in KCC2 immunoreactivity was not due to cell death, although exposure to 1 nM gp120MN trended towards significant cell loss (p = 0.06; Fig. S2). While KCC2 is a well-studied target of NMDAR activation in other neuropathologic diseases, CCR5-mediated KCC2 regulation has not been described by others. To validate that CCR5 activation triggered KCC2 loss, we pretreated cultures with the CCR5 antagonist, MVC (50 nM), which maintained KCC2 expression (49.19 ± 1.2%; p < 0.01; n = 5). Interestingly, pretreatment with CLP257 did not rescue KCC2 immunoreactivity in these experiments. These results describe a novel mechanism of KCC2 regulation involving CCR5 and suggest that the activation of CCR5 by gp120ADA may contribute to KCC2 reductions after HIVsup exposure.
Figure 5. gp120 ADA reduces KCC2 immunoreactivity.

(A, B) Bar graphs quantifying percentage of cells immunoreactive to KCC2. Both 6 h (A) and 24 h (B) exposures to 500 pM - 1 nM gp120 ADA significantly reduced the percentage of KCC2 immunoreactive cells compared to their respective controls (†p < 0.01; n = 6), while gp120 IIIB or gp120 MN had no significant effect on KCC2 expression. Interestingly, CLP257 co-exposure failed to maintain KCC2 immunoreactivity in gp120 ADA exposed groups, but MVC showed significant rescue (*p < 0.05; n = 5 - 6), suggesting that CCR5 activation is responsible for gp120 loss.
HIV-1 proteins reduce inhibitory potential of GABAAR activation.
We next examined the functional effects of Tat1-86, gp120ADA, and morphine on GABAAR-induced inhibition by expressing the intracellular Ca2+ reporter GCaMP6f in hNeurons using AAV1.hSyn.GCaMP6f.WPRE. GABAAR-mediated inhibition was examined in these cells by recording neuronal Ca2+ activity during perfusion of BrainPhys solution containing 100 μM GABA, 25 μM glutamate, and 2 μM CGP55845 for 55 s and quantifying resultant neuronal spiking (the total number of significant Ca2+ transients during 55 s recording period). Analysis revealed a significant increase in 50 nM Tat1-86 and morphine groups, without interaction comparedto vehicle (p < 0.01; n = 15 – 21) (Fig. 6B). Thus, Tat1-86 and morphine independently reduced the inhibitory potential of GABAAR activation. Pretreatment with CLP257 ameliorated Tat1-86 and morphine effects (p < 0.01; n = 15 - 21), suggesting rescue of inhibitory activity in these groups, consistent with immunoreactivity results showing restored levels of KCC2. We also found disinhibition of hNeurons, as determined by significantly increased Ca2+ spiking in groups exposed to 500 pM gp120ADA (Fig. 6B). Three-way ANOVA revealed a main effect of gp120ADA regardless of CLP257 or morphine application (p < 0.05; n = 15 - 18). Pretreatment with MVC showed a strong tendency to prevent gp120-induced excitation presumably by retaining/restoring KCC2 function (p = 0.059). Consistent with immunoreactivity results, these results suggest a failure of CLP257 to maintain KCC2 expression following gp120ADA exposure and, thus a failure to restore the inhibitory effects of GABAAR activation. Selectively inhibiting KCC2 with VU0240551 recapitulated the effects of Tat1-86 or gp120ADA ± morphine effects (n = 15, p < 0.01) independently confirming the role of KCC2 in the overexcitation. These results suggest that hNeurons become disinhibited after Tat1-86 or gp120ADA ± morphine exposure due to a loss of KCC2 activity and that pharmacological maintenance of KCC2 can prevent or rescue the deleterious effects of Tat and/or morphine.
Figure 6. Tat1-86, gp120 ± morphine exposure decreased GABAAR mediated inhibition.

(A) Representative 30 s sample GCaMP6f traces of vehicle (top) and 50 nM Tat1-86 + morphine (bottom) exposed hNeurons during 25 μM glutamate, 100 μM GABA, and CGP55845 perfusion. (B) Exposure to 50 nM Tat1-86 ± morphine (500 nM) resulted in significantly increased Ca2+ transients measured by GCaMP6f activity during 55 s perfusion of 25 μM glutamate, 100 μM GABA, and CGP55845 compared to control (†; p < 0.05; n = 15 - 21). CLP257 (blue) rescued these effects (*; p < 0.01; n = 15 - 21). Exposure to 500 pM gp120 ADA showed significant increase in neuronal activity, regardless of CLP257 or morphine cotreatment (†; n = 15 - 21; p < 0.05). MVC exposure trended towards reversing gp120 effects (p = 0.059, n = 18). KCC2 antagonist, VU02440551 recapitulated results of Tat1-86, gp120, and morphine, suggesting that these effects are due to loss of KCC2 activity (#p < 0.01, n = 15). Ca2+ transients were considered significant and counted when the spike amplitude was > 4 SD above background activity (determined 2.5 s prior to perfusion; red line) and returned to within 0.5 x standard deviation of background activity (blue line).
Tat expression reduces KCC2 in the striatum of Tat-transgenic mice.
HIV-1 Tat transgenic mice show neuropathology and behavioral deficits similar to PWH (Fitting et al., 2013; Schier et al., 2017). To examine if KCC2 abnormalities found in culture could be seen in Tat-transgenic mice, we performed western blot analysis of striatal tissue after two weeks of Tat induction by DOX. Tat+ mice showed a significant decrease in total KCC2 levels compared to Tat− control mice (Fig. 7) (p < 0.05; n = 12). These results confirm that chronic Tat exposure induces KCC2 loss in vivo as well as in cultured hNeurons.
Figure 7. Tat+ mice show reduced striatal KCC2 compared to control Tat - mice.
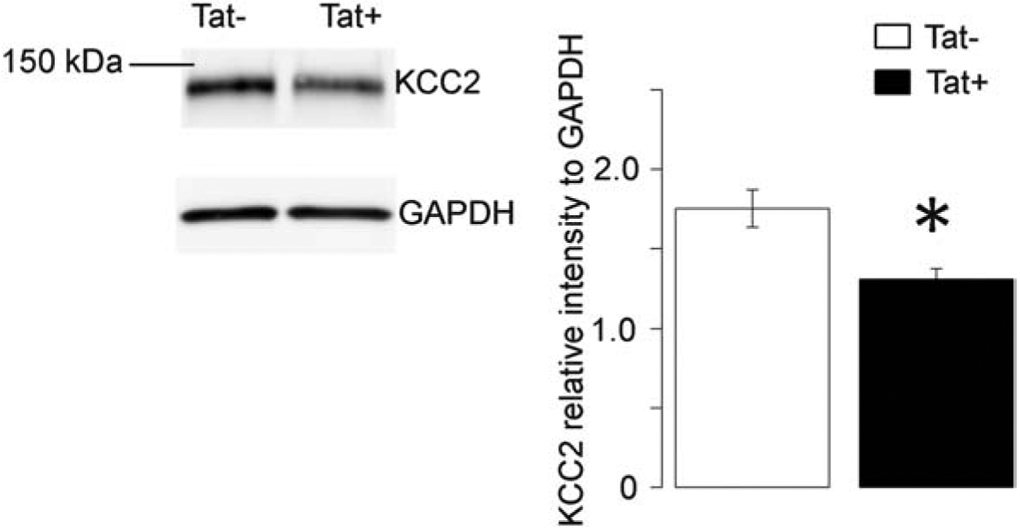
Two weeks of Tat expression significantly reduced the amount of KCC2 detected by western blot (n = 12; p < 0.05). Data are displayed as KCC2 intensity normalized to GAPDH.
Discussion
Previous studies including human post-mortem (Buzhdygan et al., 2016; Gelman, Chen, et al., 2012) and in vivo rodent studies (Fitting et al., 2013; Marks et al., 2016; Xu et al., 2016) have shown selective changes in GABAergic markers and function, driving an emerging concept that disrupted GABAergic transmission may be central to the development of HIV-related CNS dysfunction. Our data strengthen this perspective by demonstrating that KCC2 levels and subsequent GABAergic activity are diminished by an infective, HIV+ environment and by exposure to gp120ADA in vitro or HIV-1 Tat, both in vitro and in vivo. Importantly, HIV-infected individuals demonstrate a loss of GABAergic markers without the loss of GABAergic neurons (Buzhdygan et al., 2016) as seen in the present study. Taken together, our data along with findings from other investigators suggest that there may be an overall loss of inhibition via reduced inhibitory synapses and/or GABAergic function, and as we demonstrate here, diminished hyperpolarization at remaining inhibitory synapses caused by deficits in KCC2.
Endogenous levels of chemokine (C-C motif) ligand 5 (CCL5) are almost undetectable in the healthy CNS, but dramatic increases are found in several neuroinflammatory disorders (Louboutin & Strayer, 2013) including HAND (El-Hage et al., 2005; Letendre, Lanier, & McCutchan, 1999). In multiple sclerosis, CCL5 is elevated during early stages of the disease and is correlated with hyperexcitability (Mori et al., 2016; Sorensen et al., 1999). Our results demonstrate KCC2 as a target of CCR5 activation that may underlie this hyperexcitability. Targeting CCR5, perhaps with MVC, should be further investigated for therapeutic potential in multiple sclerosis and in other disorders in which elevated CCL5-CCR5 signaling may be operative, such as in neuroHIV (Kim et al., 2018).
Additionally, our results using the MOR-selective antagonist, CTAP (Fig. 2C) demonstrate that neuronal KCC2 expression is a target of MOR activation. It is important to note that we have used a mixed neuron-astrocyte culture model. Outcomes may thus be due to direct activation of neuronal MOR, or a secondary effect of MOR activation on astroglia. Our results only demonstrated an interaction between morphine and viral proteins at the 24 h timepoint. Our previous studies have shown neurotoxic interactions between morphine and HIV/viral proteins at 24 – 72h and also that neurotoxicity was largely if not completely driven by glia (S. Zou et al., 2011) [or similarly by CCR5 deletion from glia (Kim et al., 2018)]. Thus, astrocytes may be acting as the intermediary through which MOR activation leads to neuronal KCC2 loss in the data presented here. Overall, our implication of MOR in KCC2 loss is in line with previous findings that GABAAR activation on neurons in the ventral tegmental area of rats switches from an inhibitory to excitatory signal during the development of opiate dependency (Laviolette, Gallegos, Henriksen, & van der Kooy, 2004; Vargas-Perez et al., 2009), and that KCC2 loss contributes to states of opiate withdrawal and morphine-induced hyperalgesia via mechanisms involving microglia-mediated BDNF release and subsequent TrkB activation (Ferrini et al., 2017; Ferrini et al., 2013; Taylor et al., 2016). Further, we found an exacerbation of KCC2 losses at 50 nM Tat with co-exposure to morphine, suggesting that opiates and HIV protein interactions may similarly occur in the CNS and underlie the increased incidence/severity of HAND that has been noted in PWH who abuse opiates (Bell et al., 2006; Byrd et al., 2011; Carrico, 2011; Smith et al., 2014).
Interestingly, CLP257 was able to maintain KCC2 expression and functional response of hNeurons to Tat, but had no effect on gp120-exposed groups. While many factors regulate KCC2 activity/degradation, the mechanism by which CLP257 preserves KCC2 activity remains elusive. KCC2 stability is regulated by phosphorylation at multiple sites. For example, phosphorylation of S940 or T1007 by protein kinase C and WNK increases and decreases KCC2 membrane stability, respectively (Inoue et al., 2012; Lee, Deeb, Walker, Davies, & Moss, 2011). NMDAR activation can lead to protein phosphatase 1 activation and S940 dephosphorylation leading to decreased membrane localization and subsequent calpain-induced KCC2 degradation in the cytoplasm (Lee et al., 2011). Thus, there are several mechanisms that may underlie the effects of CLP257. gp120 and Tat may have differential effects on one or more of these pathways, resulting in these inconsistencies. MVC, an FDA-approved CCR5 antagonist commonly prescribed to HIV-infected individuals to prevent HIV from binding to this co-receptor, was able to rescue both KCC2 expression and functional responses to GABAAR activation in gp120ADA-exposed hNeurons. Thus, MVC treatment might prevent gp120ADA-induced KCC2 loss upstream of CLP257 and could be considered more widely for PWH with HAND symptomology.
Neuronal electrophysiological properties govern CNS function, and elucidating deficits at the cellular level can identify circuitry imbalances that underlie neurological disorders. To study electrophysiological responses in HIV-1 exposed neurons, we used the GEVI, Archon1. This noninvasive interrogation has the advantages of allowing ensemble activity to be studied both in vitro and in vivo, with the capability of tracking specific neurons long-term. This is a particularly useful alternative to traditional electrophysiology when biosafety is a concern. Most of the experiments here utilized acute exposure paradigms (6 and 24 h). While this approach allowed us to uncover novel targets and subsequent functional responses in primary human neurons, PWH are exposed to these factors over a much longer timeframe. Thus, we began to validate the observed changes using a chronic in vivo model and found reduced KCC2 in the striatum of Tat+ transgenic mice after 2 weeks of Tat exposure. Further studies are underway to test the effectiveness of a CLP257 prodrug (CLP290) to reverse KCC2 loss, the resultant electrophysiological and behavioral changes observed with Tat and/or CLP290 exposures, and any preferential vulnerability between the prominent neuronal subtypes in the striatum (dopamine D1 receptor-expressing vs. dopamine D2 receptor-expressing) to KCC2 loss. Interestingly, the loss of the expression of GABAergic markers is inversely correlated with increases in DRD2L in PWH (Buzhdygan et al., 2016) and increases in DRD2L in the prefrontal cortex positively correlated with the development of HAND (Gelman, Lisinicchia, et al., 2012). HIV-induced neurocognitive detriments vary among individuals, and length of exposure is likely a contributing factor along with age, genetics, comorbidities (including drug use), and the particular strains of HIV present. We used the HIVBaL strain to generate HIVsup as R5 tropism is prominent in the CNS (Schnell, Joseph, Spudich, Price, & Swanstrom, 2011). We also began to address viral heterogeneity by comparing effects of three different gp120 strains. Results here suggest that R5-tropic strains may be more likely to disrupt GABAergic circuitry. CSF viral titers and levels of HIV-1 Tat have been shown to vary among infected individuals whose blood titers are well-controlled (Henderson et al., 2019; Johnson et al., 2013). Thus, we expect that levels of viral proteins and inflammation within the brain parenchyma must also vary among PWH. As these levels are not easily measured, our experimental paradigm may over- or underestimate CNS exposure levels in HIV-infected individuals.
Similar situations of disinhibition through dysregulated [Cl−]i appear to contribute significantly to hyperexcitability in other neurological disorders, and restoration of KCC2 with CLP257 or its prodrug, CLP290, has shown promise in reversing the hyperexcitability in models of these disorders (B. Chen et al., 2018; Ferrini et al., 2017). Overall, our studies further implicate KCC2 in novel ways that could have broad impact for human health. We established KCC2 and upstream pathways as promising therapeutic targets to restore GABAergic function and to treat the symptoms of HAND, and this approach may be particularly relevant for PWH that use opiates. We also identified KCC2 as a novel link underlying hyperexcitability in conditions that involve elevated CCL5 and/or CCR5 activation in the CNS. Our results also highlight that opiate drugs of abuse may independently dysregulate KCC2 levels to cause GABAergic dysfunction. This finding has significance for the large population of opiate abusers who are not infected with HIV.
Supplementary Material
Highlights.
HIV-1 proteins, infective HIV, and morphine reduce KCC2 levels in primary human neurons
Viral factors reduced KCC2 via NMDAR and CCR5; morphine acted via μ-opioid receptors
The loss of KCC2 dysregulated GABAAR-mediated hyperpolarization and inhibition
KCC2 and neuronal dysfunction were rescued by manipulating KCC2 or upstream pathways
HIV-Tat expression in vivo diminished KCC2 levels in the striatum
Acknowledgments
Funding
This work was supported by the National Institutes of Health- National Institute on Drug Abuse Grants F31DA047195 (A.J.B.), R01DA034231 (K.F.H., P.E.K.), and R01DA018633 (K.F.H).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
No competing interests.
References
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, & Bell JE (2005). Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol, 64(6), 529–536. [DOI] [PubMed] [Google Scholar]
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, … Wojna VE (2007). Updated research nosology for HIV-associated neurocognitive disorders. Neurology, 69(18), 1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion D, & Lewis DA (2011). Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch Gen Psychiatry, 68(1), 21–31. doi: 10.1001/archgenpsychiatry.2010.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balinang JM, Masvekar RR, Hauser KF, & Knapp PE (2017). Productive infection of human neural progenitor cells by R5 tropic HIV-1: opiate co-exposure heightens infectivity and functional vulnerability. AIDS, 31(6), 753–764. doi: 10.1097/qad.0000000000001398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardy C, van den Hurk M, Eames T, Marchand C, Hernandez RV, Kellogg M, … Gage FH (2015). Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc Natl Acad Sci U S A, 112(20), E2725–2734. doi: 10.1073/pnas.1504393112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JE, Arango JC, & Anthony IC (2006). Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol, 1(2), 182–191. doi: 10.1007/s11481-006-9018-2 [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, & Kaila K (2009). Cation-chloride cotransporters and neuronal function. Neuron, 61(6), 820–838. doi: 10.1016/j.neuron.2009.03.003 [DOI] [PubMed] [Google Scholar]
- Bokhari SM, Yao H, Bethel-Brown C, Fuwang P, Williams R, Dhillon NK, … Buch SJ (2009). Morphine enhances Tat-induced activation in murine microglia. J Neurovirol, 15(3), 219–228. doi: 10.1080/13550280902913628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzhdygan T, Lisinicchia J, Patel V, Johnson K, Neugebauer V, Paessler S, … Gelman B (2016). Neuropsychological, Neurovirological and Neuroimmune Aspects of Abnormal GABAergic Transmission in HIV Infection. J Neuroimmune Pharmacol, 11(2), 279–293. doi: 10.1007/s11481-016-9652-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DA, Fellows RP, Morgello S, Franklin D, Heaton RK, Deutsch R, … Grant I (2011). Neurocognitive impact of substance use in HIV infection. J Acquir Immune Defic Syndr, 58(2), 154–162. doi: 10.1097/QAI.0b013e318229ba41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico AW (2011). Substance use and HIV disease progression in the HAART era: implications for the primary prevention of HIV. Life Sci, 88(21-22), 940–947. doi: 10.1016/j.lfs.2010.10.002 [DOI] [PubMed] [Google Scholar]
- Chen B, Li Y, Yu B, Zhang Z, Brommer B, Williams PR, … He Z (2018). Reactivation of Dormant Relay Pathways in Injured Spinal Cord by KCC2 Manipulations. Cell, 174(3), 521–535.e513. doi: 10.1016/j.cell.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Wang J, Jiang J, Zheng X, Justice NJ, Wang K, … Yang L (2017). APP modulates KCC2 expression and function in hippocampal GABAergic inhibition. eLife, 6, e20142. doi: 10.7554/eLife.20142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, … Kim DS (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature, 499(7458), 295–300. doi: 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, B. H., Broz D, Buddha S, Chapman EL, Galang RR, Hilman D, Hon J, Hoover KW, Patel MR, Perez A, Peters PJ, Pontones P, Roseberry JC, Sandoval M, Shielods J, Wlathall J, Waterhouse D, Widle PJ, Wu H, Duve JM; Centers for Disease Control and Prevention (CDC). (2015). Community outbreak of HIV infection linked to injection drug use of oxymorphone--Indiana, 2015. Morb Mortal Wkly Rep, 64(16), 443–444. [PMC free article] [PubMed] [Google Scholar]
- Dargaei Z, Bang JY, Mahadevan V, Khademullah CS, Bedard S, Parfitt GM, … Woodin MA (2018). Restoring GABAergic inhibition rescues memory deficits in a Huntington's disease mouse model. Proc Natl Acad Sci U S A, 115(7), E1618–e1626. doi: 10.1073/pnas.1716871115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mendoza C (2019). UNAIDS Update Global HIV Numbers. AIDS Rev, 21(3), 170–171. [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, & Hauser KF (2005). Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia, 50(2), 91–106. doi: 10.1002/glia.20148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall I, Vaida F, Khanlou N, Lazzaretto D, Achim C, Letendre S, … Masliah E (2009). Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol, 15(5-6), 360–370. doi: 10.3109/13550280903131915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, Lorenzo LE, Godin AG, Quang ML, & De Koninck Y (2017). Enhancing KCC2 function counteracts morphine-induced hyperalgesia. Sci Rep, 7(1), 3870. doi: 10.1038/s41598-017-04209-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'Guidice T, Lorenzo LE, … De Koninck Y (2013). Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(−) homeostasis. Nat Neurosci, 16(2), 183–192. doi: 10.1038/nn.3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, … Hauser KF (2013). Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry, 73(5), 443–453. doi: 10.1016/j.biopsych.2012.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon M, Bergeron MJ, Lavertu G, Castonguay A, Tripathy S, Bonin RP, … De Koninck Y (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med, 19(11), 1524–1528. doi: 10.1038/nm.3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman BB, Chen T, Lisinicchia JG, Soukup VM, Carmical JR, Starkey JM, … Susan Morgello12 for the National Neuro, A. T. C. (2012). The National NeuroAIDS Tissue Consortium Brain Gene Array: Two Types of HIV-Associated Neurocognitive Impairment. PLOS One, 7(9), e46178. doi: 10.1371/journal.pone.0046178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman BB, Lisinicchia JG, Chen T, Johnson KM, Jennings K, Freeman DH Jr., & Soukup VM (2012). Prefrontal dopaminergic and enkephalinergic synaptic accommodation in HIV-associated neurocognitive disorders and encephalitis. J Neuroimmune Pharmacol, 7(3), 686–700. doi: 10.1007/s11481-012-9345-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, Clifford DB, Franklin DR Jr., Woods SP, Ake C, Vaida F, … Grant I (2010). HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology, 75(23), 2087–2096. doi: 10.1212/WNL.0b013e318200d727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LJ, Johnson TP, Smith BR, Reoma LB, Santamaria UA, Bachani M, … Nath A (2019). Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS, 33 Suppl 2, S145–s157. doi: 10.1097/qad.0000000000002268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, … Kleinman JE (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci, 31(30), 11088–11095. doi: 10.1523/jneurosci.1234-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Furukawa T, Kumada T, Yamada J, Wang T, Inoue R, & Fukuda A (2012). Taurine inhibits K+-Cl− cotransporter KCC2 to regulate embryonic Cl− homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J Biol Chem, 287(25), 20839–20850. doi: 10.1074/jbc.M111.319418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, & Nath A (2013). Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A, 110(33), 13588–13593. doi: 10.1073/pnas.1308673110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, … Rouleau GA (2014). Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep, 15(7), 766–774. doi: 10.15252/embr.201438840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Ma Q, Medders KE, Desai MK, & Lipton SA (2007). HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ, 14(2), 296–305. doi: 10.1038/sj.cdd.4402006 [DOI] [PubMed] [Google Scholar]
- Kim S, Hahn YK, Podhaizer EM, McLane VD, Zou S, Hauser KF, & Knapp PE (2018). A central role for glial CCR5 in directing the neuropathological interactions of HIV-1 Tat and opiates. Journal of Neuroinflammation, 15(1), 285. doi: 10.1186/s12974-018-1320-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh KA, Wydeven N, Wickman K, & Thayer SA (2014). HIV-1 protein Tat produces biphasic changes in NMDA-evoked increases in intracellular Ca2+ concentration via activation of Src kinase and nitric oxide signaling pathways. J Neurochem, 130(5), 642–656. doi: 10.1111/jnc.12724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Gallegos RA, Henriksen SJ, & van der Kooy D (2004). Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat Neurosci, 7(2), 160–169. doi: 10.1038/nn1182 [DOI] [PubMed] [Google Scholar]
- Lee HH, Deeb TZ, Walker JA, Davies PA, & Moss SJ (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci, 14(6), 736–743. doi: 10.1038/nn.2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letendre SL, Lanier ER, & McCutchan JA (1999). Cerebrospinal fluid beta chemokine concentrations in neurocognitively impaired individuals infected with human immunodeficiency virus type 1. J Infect Dis, 180(2), 310–319. doi: 10.1086/314866 [DOI] [PubMed] [Google Scholar]
- Lizhnyak PN, Muldoon PP, Pilaka PP, Povlishock J, & Ottens AK (2019). Traumatic Brain Injury Temporal Proteome Guides KCC2-targeted Therapy. J Neurotrauma. doi: 10.1089/neu.2019.6415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louboutin JP, & Strayer DS (2013). Relationship between the chemokine receptor CCR5 and microglia in neurological disorders: consequences of targeting CCR5 on neuroinflammation, neuronal death and regeneration in a model of epilepsy. CNS Neurol Disord Drug Targets, 12(6), 815–829. [DOI] [PubMed] [Google Scholar]
- Marks WD, Paris JJ, Schier CJ, Denton MD, Fitting S, McQuiston AR, … Hauser KF (2016). HIV-1 Tat causes cognitive deficits and selective loss of parvalbumin, somatostatin, and neuronal nitric oxide synthase expressing hippocampal CA1 interneuron subpopulations. J Neurovirol, 22(6), 747–762. doi: 10.1007/s13365-016-0447-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masvekar RR, El-Hage N, Hauser KF, & Knapp PE (2015). GSK3beta-activation is a point of convergence for HIV-1 and opiate-mediated interactive neurotoxicity. Mol Cell Neurosci, 65, 11–20. doi: 10.1016/j.mcn.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P, & Pellegrino C (2014). Current view on the functional regulation of the neuronal K(+)-Cl(−) cotransporter KCC2. Front Cell Neurosci, 8, 27. doi: 10.3389/fncel.2014.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F, Nistico R, Nicoletti CG, Zagaglia S, Mandolesi G, Piccinin S, … Centonze D (2016). RANTES correlates with inflammatory activity and synaptic excitability in multiple sclerosis. Mult Scler, 22(11), 1405–1412. doi: 10.1177/1352458515621796 [DOI] [PubMed] [Google Scholar]
- Nath A, & Steiner J (2014). Synaptodendritic injury with HIV-Tat protein: What is the therapeutic target? Exp Neurol, 251, 112–114. doi: 10.1016/j.expneurol.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuenburg JK, Brodt HR, Herndier BG, Bickel M, Bacchetti P, Price RW, … Schlote W (2002). HIV-related neuropathology, 1985 to 1999: rising prevalence of HIV encephalopathy in the era of highly active antiretroviral therapy. J Acquir Immune Defic Syndr, 31(2), 171–177. doi: 10.1097/00126334-200210010-00007 [DOI] [PubMed] [Google Scholar]
- Philippon V, Vellutini C, Gambarelli D, Harkiss G, Arbuthnott G, Metzger D, … Filippi P (1994). The basic domain of the lentiviral Tat protein is responsible for damages in mouse brain: involvement of cytokines. Virology, 205(2), 519–529. doi: 10.1006/viro.1994.1673 [DOI] [PubMed] [Google Scholar]
- Piatkevich KD, Jung EE, Straub C, Linghu C, Park D, Suk HJ, … Boyden ES (2018). A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat Chem Biol, 14(4), 352–360. doi: 10.1038/s41589-018-0004-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, … Kaila K (2014). A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep, 15(6), 723–729. doi: 10.1002/embr.201438749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Lapierre J, Ojha CR, Estrada-Bueno H, Dever SM, Gewirtz DA, … El-Hage N (2017). Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes. Viruses, 9(8), 201. doi: 10.3390/v9080201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier CJ, Marks WD, Paris JJ, Barbour AJ, McLane VD, Maragos WF, … Hauser KF (2017). Selective Vulnerability of Striatal D2 versus D1 Dopamine Receptor-Expressing Medium Spiny Neurons in HIV-1 Tat Transgenic Male Mice. J Neurosci, 37(23), 5758–5769. doi: 10.1523/jneurosci.0622-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell G, Joseph S, Spudich S, Price RW, & Swanstrom R (2011). HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog, 7(10), e1002286. doi: 10.1371/journal.ppat.1002286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte JT, Wierenga CJ, & Bruining H (2018). Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci Biobehav Rev, 90, 260–271. doi: 10.1016/j.neubiorev.2018.05.001 [DOI] [PubMed] [Google Scholar]
- Silayeva L, Deeb TZ, Hines RM, Kelley MR, Munoz MB, Lee HH, … Moss SJ (2015). KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc Natl Acad Sci U S A, 112(11), 3523–3528. doi: 10.1073/pnas.1415126112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Simmonds P, & Bell JE (2014). Brain viral burden, neuroinflammation and neurodegeneration in HAART-treated HIV positive injecting drug users. J Neurovirol, 20(1), 28–38. doi: 10.1007/s13365-013-0225-3 [DOI] [PubMed] [Google Scholar]
- Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, … Ransohoff RM (1999). Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest, 103(6), 807–815. doi: 10.1172/jci5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, … Chen G (2016). KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci U S A, 113(3), 751–756. doi: 10.1073/pnas.1524013113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Li C, Newburn EN, Ye T, Lipska BK, Herman MM, … Hyde TM (2012). Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J Neurosci, 32(15), 5216–5222. doi: 10.1523/jneurosci.4626-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Castonguay A, Ghogha A, Vayssiere P, Pradhan AA, Xue L, … Cahill CM (2016). Neuroimmune Regulation of GABAergic Neurons Within the Ventral Tegmental Area During Withdrawal from Chronic Morphine. Neuropsychopharmacology, 41(4), 949–959. doi: 10.1038/npp.2015.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Perez H, Ting AKR, Walton CH, Hansen DM, Razavi R, Clarke L, … van der Kooy D (2009). Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science, 324(5935), 1732–1734. doi: 10.1126/science.1168501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, … Volsky DJ (2003). Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology, 312(1), 60–73. [DOI] [PubMed] [Google Scholar]
- Xu C, Hermes DJ, Mackie K, Lichtman AH, Ignatowska-Jankowska BM, & Fitting S (2016). Cannabinoids Occlude the HIV-1 Tat-Induced Decrease in GABAergic Neurotransmission in Prefrontal Cortex Slices. J Neuroimmune Pharmacol, 11(2), 316–331. doi: 10.1007/s11481-016-9664-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Fitting S, Hahn Y-K, Welch SP, El-Hage N, Hauser KF, & Knapp PE (2011). Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at μ-opioid receptor-expressing glia. Brain, 134(12), 3616–3631. doi: 10.1093/brain/awr281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, & Knapp PE (2011). Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain, 134(Pt 12), 3616–3631. doi: 10.1093/brain/awr281 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.