

Abstract
Purpose:
Metastasis to the brain from breast cancer remains a significant clinical challenge, and may be targeted with CAR-based immunotherapy. CAR design optimization for solid tumors is crucial due to the absence of truly restricted antigen expression and potential safety concerns with “on-target off-tumor” activity. Here, we have optimized HER2-CAR T cells for the treatment of breast to brain metastases, and determined optimal second-generation CAR design and route of administration for xenograft mouse models of breast metastatic brain tumors, including multifocal and leptomeningeal disease.
Experimental Design:
HER2-CAR constructs containing either CD28 or 4–1BB intracellular costimulatory signaling domains were compared for functional activity in vitro by measuring cytokine production, T-cell proliferation, and tumor killing capacity. We also evaluated HER2-CAR T cells delivered by intravenous, local intratumoral, or regional intraventricular routes of admin istration using in vivo human xenograft models of breast cancer that have metastasized to the brain.
Results:
Here, we have shown that HER2-CARs containing the 4–1BB costimulatory domain confer improved tumor targeting with reduced T-cell exhaustion phenotype and enhanced proliferative capacity compared with HER2-CARs containing the CD28 costimulatory domain. Local intracranial delivery of HER2-CARs showed potent in vivo antitumor activity in orthotopic xenograft models. Importantly, we demonstrated robust antitumor efficacy following regional intraventricular delivery of HER2-CAR T cells for the treatment of multifocal brain metastases and leptomeningeal disease.
Conclusions:
Our study shows the importance of CAR design in defining an optimized CAR T cell, and highlights intraventricular delivery of HER2-CAR T cells for treating multifocal brain metastases.
Introduction
Breast cancer is the most commonly diagnosed cancer in women, with over 40,000 expected to die from advanced metastatic disease in 2017 (1). Approximately 20% to 25% of breast cancers overexpress HER2 (2), which is an established therapeutic target of both mAbs and receptor tyrosine kinase inhibitors. With the advent of effective mAbs directed against HER2, the median overall survival of patients with metastatic HER2+ breast cancer has improved (3). However, management of metastatic disease in the brain and/or central nervous system (CNS), observed in up to 50% of HER2+ breast cancer patients, continues to be a clinical challenge in large part due to the inability of mAbs to sufficiently cross the blood–brain barrier. Although small-molecule inhibitors of HER2 exist and have been clinically approved, their single-agent efficacy in the context of metastatic disease to the brain has been limited (4, 5). While HER2-targeted therapy in combination with conventional agents has shown some promise for the treatment of patients with metastatic breast cancer, control of brain metastases remains a significant unmet clinical need, as most patients survive less than 2 years following CNS involvement. Recent advances in cellular immunotherapy approaches have underscored the potential for potent antitumor immune responses and clinical benefit against solid cancers, and may be effective in the treatment of HER2+ breast cancer that has metastasized to the brain.
Chimeric antigen receptor (CAR)-based T-cell immunotherapy is being actively investigated for the treatment of solid tumors (6, 7), including HER2+ cancers. Unfortunately, the first CAR T-cell clinical experience with targeting the HER2 antigen resulted in the death of a patient with HER2+ metastatic colon cancer due to “on-target, off-tumor” toxicities (8). Recent phase I clinical trials evaluating intravenous administration of HER2-CAR T cells have demonstrated safety and antitumor activity in patients with sarcoma (9) and recurrent glioblastoma (10). These three trials have highlighted multiple elements of CAR T-cell therapy that should be addressed, including CAR construct design and T-cell manufacturing considerations, preconditioning prior to CAR T-cell infusion, and CAR T-cell dose; all of which are critical factors in therapeutic outcome. In particular for treating primary and metastatic brain tumors, we also hypothesize that route of administration will be an important consideration for safety and efficacy. For example, introducing CAR T cells directly to the site of disease may potentially minimize systemic distribution of adoptively transferred cells, and resultant toxicities. Indeed, our most recent clinical experience with local and regional delivery of CAR T cells for patients with recurrent glioblastoma has demonstrated both safety and antitumor benefits (11, 12).
Here, we have developed a second-generation HER2-specific CAR T-cell for the treatment of breast cancer that has metastasized to the brain. Comparison of two intracellular costimulatory domains, namely 4–1BB and CD28 within the CAR construct, has revealed differences in HER2 specificity as well as CAR-dependent effector activities. Using orthotopic human tumor xenograft models of breast cancer metastasis to the brain, we also evaluated therapeutic efficacy of local intratumoral and regional intraventricular delivery of HER2-CAR T cells. Our findings provide rationale for clinically evaluating intraventricular delivery of 4–1BB–containing HER2-CAR T cells for the treatment of patients with breast cancer metastasis to the brain.
Materials and Methods
Cell lines
Human breast cancer cell lines MDA-MB-361 (ATCC HTB-27), MDA-MB-231 (ATCC CRM-HTB-26), SKBR3 (ATCC HTB-30), and BT474 (ATCC HTB-20) were cultured in DMEM/Ham F-12 (F12; 1:1) containing 10% FBS (Hyclone), and 1× antibiotic–antimycotic (AA, Gibco; complete DMEM/F12). MDA-MB-468 breast cancer cells (ATCC HTB-132) were cultured in DMEM containing 10% FBS and 1× AA (complete DMEM). The low-passage patient-derived tumor line (BBM1) generated at City of Hope was cultured as described previously (13). Briefly, cells were maintained in complete DMEM/F12 on 300 μg/mL collagen-coated plates, and kept at low-passage in culture prior to use in vitro and in animal studies. The brain-seeking MDA-MB-231 (231BR) cells (a kind gift from Dr. Patricia S. Steeg, NIH, Bethesda, MD; ref. 14) were cultured in complete DMEM/F12. The human fibrosarcoma cell line HT1080 (ATCC CCL-121) and the human embryonic kidney cell line 293T (ATCC CRL-3216), were cultured in complete DMEM supplemented with 25 mmol/L HEPES (Irvine Scientific), and 2 mmol/L L-glutamine (Thermo Fisher Scientific). An EBV-transformed lymphoblastoid cell line (LCL) and LCL cells containing a membrane-tethered CD3 epsilon specific scFv agonist OKT3 (LCL-OKT3; ref. 15) were cultured in RPMI1640 containing 10% FBS and 1× AA. All cells were cultured at 37°C with 5% CO2.
DNA constructs and lentivirus production
The HER2-targeted scFv sequence was derived from the humanized mAb trastuzumab (4D5) and cloned into the antigen-binding domain of the HER2-CAR. The extracellular spacer domain included the 229-amino acid IgG4 Fc spacer with a double mutation (L235E;N297Q, abbreviated EQ; ref. 16). The intracellular costimulatory signaling domain contained either CD28 with a CD28 transmembrane domain, or 4–1BB with a CD8 transmembrane domain. The CD3ζ cytolytic domain was described previously (17). The CAR construct sequence was separated from a truncated CD19 gene (CD19t) using a T2A ribosomal skip sequence, and cloned in an epHIV7 lentiviral backbone under the control of the EF1α promoter.
Lentivirus was generated by plating 293T cells in T-225 tissue culture flasks 1-day prior to transfection with packaging plasmids and desired CAR lentiviral backbone plasmid. Supernatants were collected after 3 to 4 days, filtered, and centrifuged to remove cell debris, and incubated with 2 mmol/L magnesium and 25 U/mL Benzonase endonuclease (EMD Millipore) to remove contaminating nucleic acids. Supernatants were combined and concentrated via high-speed centrifugation (6,080× g) overnight at 4°C. Lentiviral pellets were then resuspended in PBS-lactose solution (4 g lactose per 100 mL PBS), aliquoted and stored at −80°C for later use. Lentiviral titers, as determined by CD19t expression, were quantified using HT1080 cells.
T-cell isolation, lentiviral transduction, and ex vivo expansion
Leukapheresis products were obtained from consented research participants (healthy donors) under protocols approved by the City of Hope Internal Review Board (IRB). On the day of leukapheresis, peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation over Ficoll-Paque (GE Healthcare) followed by multiple washes in PBS/EDTA (Miltenyi Biotec). Cells were rested overnight at room temperature on a rotator, and subsequently washed and resuspended in complete X-VIVO. Up to 5 × 109 PBMC were incubated with anti-CD14, anti-CD25, and anti-CD45RA microbeads (Miltenyi Biotec) for 30 minutes at room temperature and magnetically depleted using the CliniMACS system (Miltenyi Biotec) according to the manufacturer’s protocol. Depleted PBMCs were then enriched for central memory T cells (TCM) by incubating with biotinylated anti-CD62L antibody (produced by the Center for Biomedicine and Genetics at City of Hope) for 30 minutes at room temperature, and then with anti-Biotin microbeads (Miltenyi Biotec) for an additional 30 minutes at room temperature. TCM cells were then magnetically enriched using the autoMACS system (Miltenyi Biotec) according to the manufacturer’s protocol. For studies utilizing TCM cells, cells were immediately frozen in CryoStor (CS 5) (BioLife Solutions). Purity and phenotype of TCM cells were verified by flow cytometry.
Freshly thawed TCM cells were washed once and cultured in complete X-VIVO containing 50 U/mL recombinant human IL2 (rhIL2, Novartis Oncology) and 0.5 ng/mL recombinant human IL15 (rhIL15, CellGenix). For HER2-CAR lentiviral transduction, T cells were cultured with CD3/CD28 Dynabeads (Life Technologies), protamine sulfate (APP Pharmaceuticals), cytokine mixture (as stated above), and desired lentivirus at varying MOI either the day of, or the day following, bead stimulation. Spinoculation was performed by centrifugation at 2,000 rpm for 30 minutes at 32°C with no brake. Cells were then cultured in and replenished with fresh complete X-VIVO containing cytokines every 2–3 days. After 7–9 days, beads were magnetically removed, and cells were further expanded in complete X-VIVO containing cytokines to achieve desired cell yield. HER2-CAR–expressing T cells were then positively selected for CD19t using the EasySep CD19 Positive Enrichment Kit I or II (StemCell Technologies) according to the manufacturer’s protocol. Following further expansion, cells were frozen prior to in vitro functional assays and in vivo tumor models. Purity and phenotype of CAR T cells were verified by flow cytometry.
Intracellular/extracellular staining and flow cytometry
For flow cytometric analysis, cells were resuspended in FACS buffer (Hank balanced salt solution without Ca2+, Mg2+, or phenol red (HBSS−/−, Life Technologies) containing 2% FBS and 1× AA). For detecting CAR scFv, biotinylated Protein-L (GenScript USA) was used as described previously (18). Cells were incubated with primary antibodies for 30 minutes at 4°C in the dark before proceeding to secondary staining. For extracellular and secondary staining, cells were washed twice prior to 30-minute incubation at 4°C in the dark with fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll protein complex (PerCP), PerCP-Cy5.5, PE-Cy7, allophycocyanin (APC), and APC-Cy7 (or APC-eFluor780)-conjugated antibodies (CD3, CD4, CD8, CD19, CD45, CD45RA, CD45RO, CD62L, CD95, CD107a, HER2, LAG3 (CD223), PD-1 (CD279), TIM3 (CD366), CCR7, IFNγ) purchased from BioLegend, eBioscience, BD Biosciences or Thermo Fisher Scientific. Recombinant HER2-Fc chimera protein (R&D Systems) was labeled with an Alexa Fluor 647 antibody labeling kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Cell viability was determined using 4′, 6-diamidino-2-phenylindole (DAPI, Sigma). For intracellular staining, cells were fixed, permeabilized, and processed according to manufacturer’s protocol (BD Biosciences). Cells were then incubated with fluorophore-conjugated antibodies for 30 minutes at 4°C in the dark, and washed twice prior to resuspension in FACS buffer and acquisition on a MACSQuant Analyzer 10 (Miltenyi Biotec). Data were analyzed with FlowJo software (v10, TreeStar).
In vitro T-cell functional assays
For degranulation and intracellular cytokine assays, CAR T cells and tumor targets were cocultured at varying effector:target (E:T) ratios in complete X-VIVO in the absence of exogenous cytokines in round-bottom 96-well tissue culture-treated plates (Corning). FITC-CD107a was added to cultures and after incubating for 4–6 hours at 37°C, the cells were fixed and permeabilized before analysis by flow cytometry as described above. For tumor killing assays, CAR T cells and tumor targets were cocultured at varying E: T ratios in complete X-VIVO in the absence of exogenous cytokines in 96-well plates for 1–5 days and analyzed by flow cytometry as described above. Tumor killing by CAR T cells was calculated by comparing CD45-negative cell counts relative to those observed when targets were cocultured with Mock (untransduced) T cells, to account for any potential non-CAR–dependent killing activity. For T-cell activation assays, CAR T cells and tumor targets were cocultured at an E:T ratio of 1:1 in complete X-VIVO in the absence of exogenous cytokines in 96-well plates for the indicated time points and analyzed by flow cytometry for specific markers of T-cell activation. For T-cell proliferation studies, mock and HER2-CAR T cells were incubated with CFDA-SE (1–10 μmol/L, Thermo Fisher Scientific) prior to coculture with tumor targets at an E:T ratio of 1:1 in complete X-VIVO in the absence of exogenous cytokines in 96-well plates for 3–5 days and analyzed by flow cytometry.
ELISA cytokine assays
Varying concentrations of recombinant human ErbB2/HER2 Fc chimera protein (R&D Systems) were coated overnight in 1× PBS at 4°C on high-affinity 96-well flat bottom plates (Corning). Wells were washed twice with 1× PBS, blocked with 10% FBS for 1 hour, and washed again. CAR T cells (5 × 103) were added to protein-coated wells. Where specified, tumor targets (5 × 103) were incubated with T cells in noncoated wells (final volume of 200 μL). Following an overnight incubation at 37°C, supernatants were harvested and processed according to the Human IFNγ ELISA Ready-SET-GO! (eBioscience) manufacturer’s protocol. Plates were read at 450 nm using the Wallac Victor3 1420 Multilabel Counter (Perkin-Elmer) and Wallac 1420 Workstation software.
In vivo tumor studies
All animal experiments were performed under protocols approved by the City of Hope Institutional Animal Care and Use Committee. Tumor cells (BBM1, BT474, MDA-MB-468) were transduced with lentivirus carrying enhanced GFP (eGFP)/firefly luciferase (ffluc) under the control of the EF1a promoter to allow for noninvasive optical imaging (Xenogen, LagoX) once implanted into mice. BBM1 cells were ZsGreen positive, and thus were transduced with lentivirus carrying ffluc only.
For tumor models, mice were anesthetized by intraperitoneal (i.p.) injection of ketamine/xylazine and gaseous isoflurane prior to tumor injection. BBM1 (2 × 105), BT474 (1.5 × 105), or MDA-MB-468 (2 × 105) were prepared in HBSS−/− (2 μL per mouse) and injected orthotopically in the brain parenchyma of female NSG mice via stereotactic injection as described previously (19). Tumor growth was monitored at least once a week via optical imaging (Xenogen, LagoX) and flux signals were analyzed with Living Image software (Xenogen). For imaging, mice were injected intraperitoneally with 150 μL d-luciferin potassium salt (Perkin Elmer) suspended in PBS at 4.29 mg/mouse. Once flux signals reached desired levels, CAR T cells were prepared in PBS and mice were treated either by intratumoral/intracranial (i.c.) or intraventricular (intracerebroventricular, i.c.v.) injection in 3 μL final volume. At desired time points or at moribund status, mice were euthanized and tissues were processed for IHC as described below.
IHC
Brain tissue was fixed for up to 3 days in 4% paraformaldehyde (Boston BioProducts) and stored in 70% ethanol until further processing. Histology was performed by the Pathology Core at City of Hope. Briefly, paraffin-embedded sections (10-μm) were stained with mouse anti-human CD3 (DAKO), mouse anti-human CD4 (DAKO), mouse anti-human CD8 (DAKO), rat anti-human HER2 (DAKO), rat anti-human Granzyme-B (eBioscience), and anti-human nuclei (Millipore) antibodies. Images were obtained using the Nanozoomer 2.0HT digital slide scanner and the associated NDP.view2 software (Hamamatzu).
Statistical analysis
Data are presented as mean ± SEM, unless otherwise stated. Statistical comparisons between groups were performed using the unpaired two-tailed Student t test to calculate P values. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Results
HER2-CAR T cells containing a 4–1BB intracellular costimulatory domain show reduced cytokine production with similar cytolytic activity compared with CD28
We first investigated the impact of different intracellular costimulatory domains in HER2-specific CAR T cells by constructing CD28-containing (HER2–28z) and 4–1BB–containing (HER2-BBz) CARs. Both HER2-CAR constructs contained the humanized HER2-targeted scFv derived from trastuzumab (humanized 4D5 clone), the IgG4 Fc extracellular spacer with a double mutation (EQ) to reduce Fc receptor recognition (16), and the CD3z cytolytic domain followed by a truncated CD19 (CD19t) for cell tracking (Fig. 1A). HER2–28ζ and HER2-BBζ CARs were stably expressed (Fig. 1B) and exhibited comparable ex vivo T-cell expansion kinetics (Fig. 1C). Cell-surface T-cell phenotypes were also comparable with respect to the expression of CD8, CD4, CD45RO, CD45RA, CD62L, CD95, and CCR7 (Fig. 1D). For the following in vitro and in vivo studies, we utilized HER2-CARs engineered in TCM cells, as described previously (20, 21).
Figure 1.
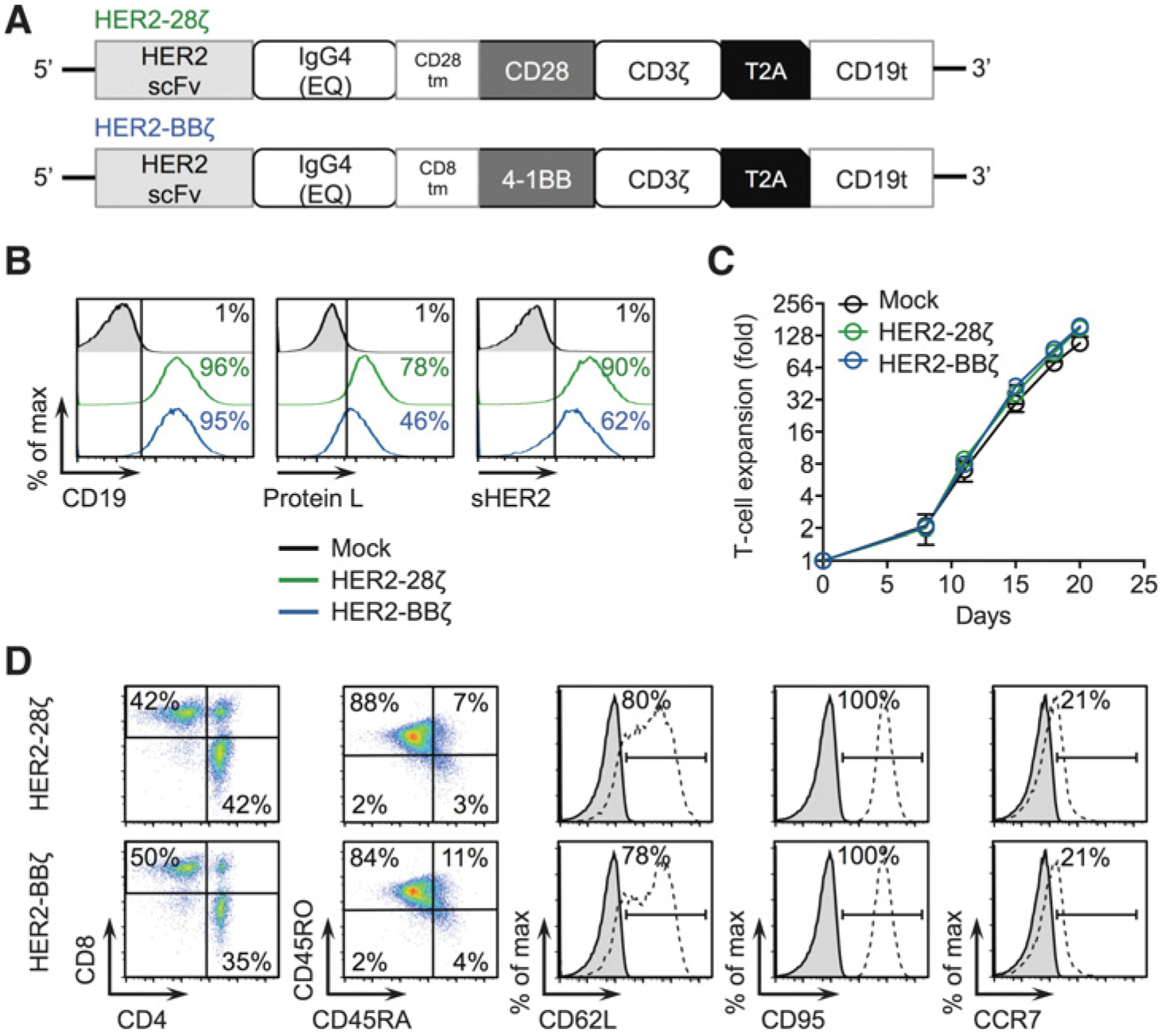
CAR design and T-cell engineering of HER2-CARs containing CD28 or 4–1BB intracellular costimulatory domains. A, Diagram of the lentiviral expression cassette with HER2-CARs containing the scFv from trastuzumab targeting HER2, with a 229–amino acid human IgG4 Fc linker containing a double mutation in the CH2 domain (EQ), a transmembrane domain (either CD28 or CD8), a cytoplasmic CD28 or 4–1BB costimulatory domain, and a cytolytic CD3z domain. A truncated nonsignaling CD19 (CD19t) is separated from the CAR with a T2A ribosomal skip sequence for tracking CAR-expressing cells. B, Mock (untransduced), HER2–28ζ, or HER2-BBζ CAR T cells expressing CD19 to detect lentiviral transduction of CARs (left histogram), Protein L to detect the scFv (middle histogram), or binding to Alexa Fluor-647–labeled soluble HER2 (right histogram) as determined by flow cytometry. C, Ex vivo expansion kinetics for Mock and HER2-CAR T cells over3 weeks in culture. D, Cell-surface expression of indicated markers on HER2-CAR T cells as determined by flow cytometry. All data are representative of at least two independent experiments.
Next, we assessed the tumor targeting abilities of HER2–28ζ and HER2-BBζ CAR T cells by evaluating antigen-specific T-cell activation. For these studies, we used several human cancer cell lines with varying HER2 cell surface expression (Fig. 2A). The human lymphoblastoid cell line, LCL, and the human breast cancer cell line, MDA-MB-468, are negative for HER2 expression as determined by flow cytometry. The human breast cancer cell lines MDA-MB-361 and MDA-MB-231BR (“brain-seeking” version; ref. 14) express low levels of HER2. SKBR3, BT474, and BBM1 (a patient-derived tumor line derived from the surgical resection of a brain metastatic lesion from a patient with breast cancer; ref. 13) express high levels of HER2. HER2-CAR T-cell function was first evaluated by measuring intracellular IFNγ levels and cell surface CD107a as an early measure of effector activity following a 5-hour incubation with MDA-MB-468 (HER2-negative) and BBM1 (HER2-expressing). As shown in Fig. 2B, while Mock (untransduced) T cells lacked activity against either tumor target, both HER2–28ζ and HER2-BBζ CAR T cells showed robust activity when cocultured with BBM1 cells and minimal activity when cocultured with MDA-MB-468 cells. Interestingly, HER2–28ζ CAR T cells showed higher intracellular IFNγ expression compared with HER2-BBζ CAR T cells against BBM1 cells. Both versions of HER2-CAR T cells were activated when cocultured with a panel of HER2-expressing tumor cells or with plate-bound recombinant HER2 protein, but HER2-BBζ CAR T cells produced significantly less IFNγ than HER2–28ζ CAR T cells (Fig. 2C–E).
Figure 2.
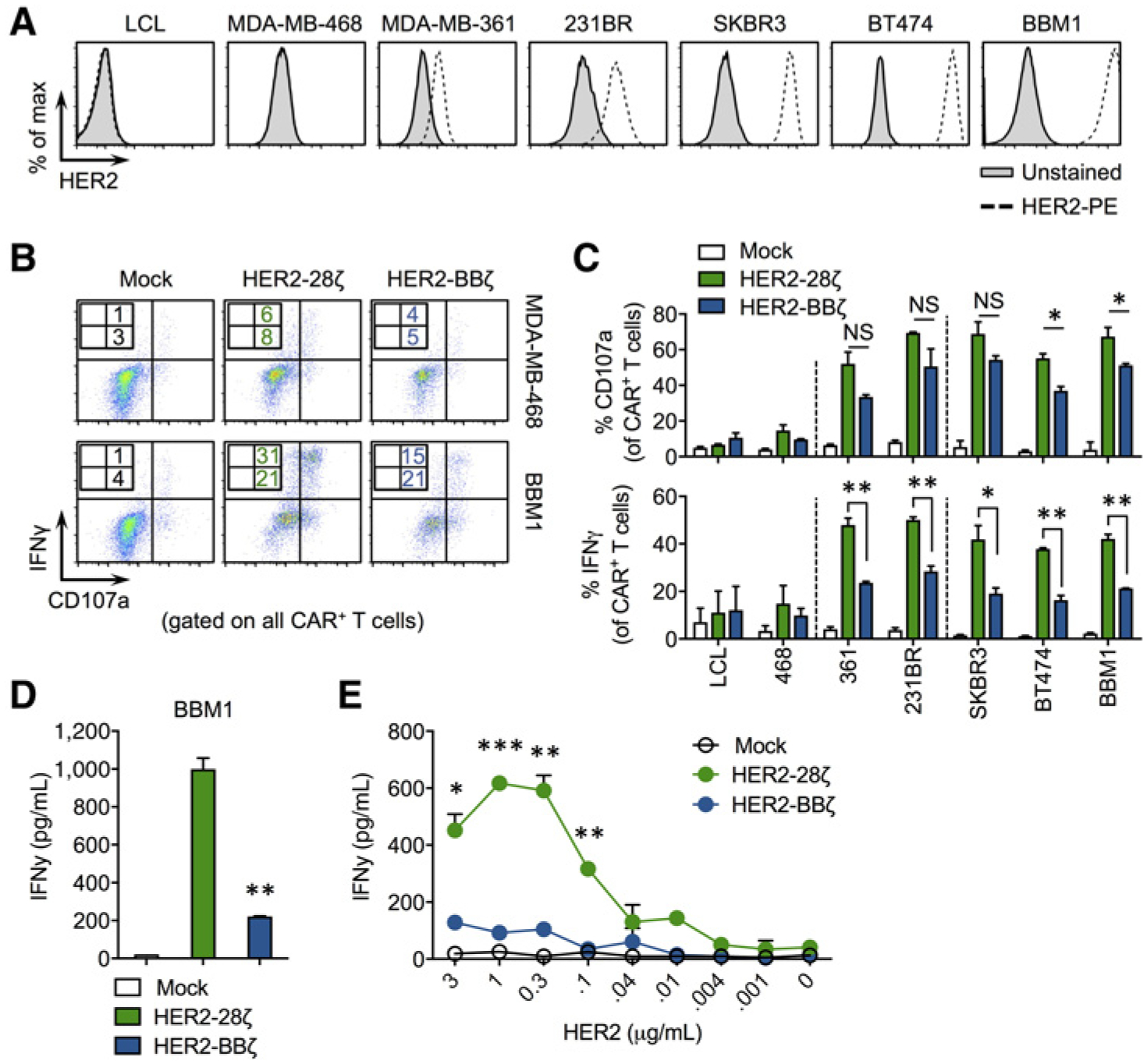
HER2-BBζ CAR T cells show reduced but antigen-specific cytokine production compared with HER2–28ζ CAR T cells. A, Histograms of HER2 expression in human cancer cell lines. LCL is an EBV-transformed human lymphoblastoid cell line. MDA-MB-468, MDA-MB-361, MDA-MB-231BR (231BR), SKBR3, and BT474 are breast cancer cell lines with varying HER2 expression. BBM1 is a patient-derived tumor line generated from the brain metastasis of a breast cancer patient. B, Representative zebra plots showing cell surface CD107a and intracellular IFNγ expression by Mock, HER2–28ζ, or HER2-BBζ CAR T cells following a 5-hour coculture with indicated tumor targets at an E:T ratio of 1:1. C, Quantification of CD107a degranulation and IFNγ production by HER2-CAR T cells using data generated from histograms as depicted in B. D, IFNγ production quantified by ELISA in supernatants from HER2-CAR T cells cultured overnight with BBM1 tumor cells at a 1:1 E:T ratio. E, IFNγ production quantified by ELISA in supernatants from Mock and HER2-CAR T cells cultured on plate-bound recombinant human HER2 at the indicated protein concentrations. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
4–1BB–containing HER2-CARs demonstrate improved tumor killing, with reduced T-cell exhaustion and greater proliferative capacity compared with CD28-containing HER2-CARs
Current findings suggest that costimulation with 4–1BB yields a more durable therapy compared to CD28 in some CAR settings (22, 23), although this has not yet been demonstrated with HER2-CAR T cells. To date, only HER2–28ζ and HER2–28BBζ CAR T cells have been evaluated preclinically and clinically as second-generation and third-generation CARs. To further explore functional differences between CD28- and 4–1BB–containing HER2-CARs, we performed in vitro tumor killing assays. HER2–28ζ or HER2-BBζ CAR T cells were cocultured with various tumor targets for 72 hours and flow cytometry was used to quantify tumor cell killing. While HER2–28ζ and HER2-BBζ CAR T cells killed low and high HER2-expressing tumor cells with similar efficiency, HER2–28ζ showed reproducible targeting of HER2-negative MDA-MB-468 cells to a greater extent than did HER2-BBζ (Fig. 3A). Similarly potent killing ability was observed with both CARs when the E:T ratio was titrated down to one CAR T-cell per eight tumor cells (Fig. 3B). In addition to higher selectivity, HER2-BBζ CAR T cells exhibited reduced expression of exhaustion markers compared to HER2–28ζ CAR T cells, including programmed death-1 (PD-1) lymphocyte-activation gene-3 (LAG3) and T-cell immunoglobulin and mucin-domain-3 (TIM3; Fig. 3C and D), Following 3 days of coculture, HER2–28ζ CAR T cells showed comparable proliferation compared with HER2-BBζ CAR T cells against BBM1 cells (Fig. 3E). By day 5 of coculture, however, HER2-BBζ CAR T cells proliferated to a greater extent, with approximately 50% of cells reaching more than 4 divisions compared with less than 20% for HER2–28ζ CAR T cells. Taken together, these data suggest that, in contrast to CD28 costimulation, 4–1BB costimulation selectively kills HER2-expressing tumor cells, with lower levels of T-cell exhaustion and superior in vitro antigen-dependent proliferation.
Figure 3.
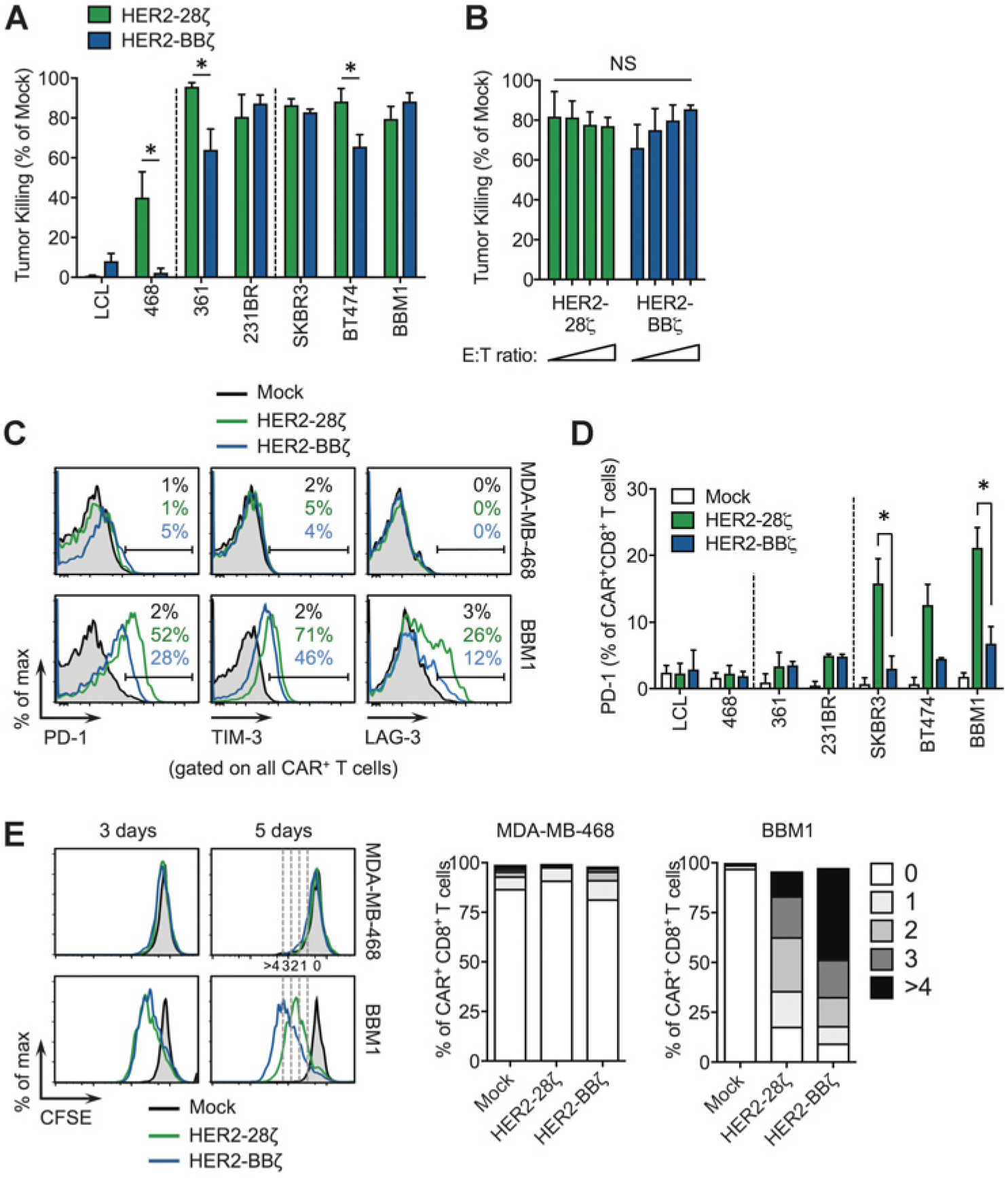
HER2-BBζ CAR T cells show similar killing ability with reduced exhaustion phenotype compared with HER2–28ζ CAR T cells. A, Tumor killing assay comparing HER2–28ζ and HER2-BBζ CAR T cells following a 3-day coculture with the indicated tumor targets, assessed by flow cytometry. B, Tumor killing assay against BBM1 with varying E:T ratios: 1:1, 1:2, 1:4, 1:8 (bars left to right). Data are pooled from two independent studies. C, Representative histograms of PD-1, TIM-3, and LAG-3 expression in Mock and HER2-CAR T cells following a 3-day coculture with HER2-negative MDA-MB-468 or HER2+ BBM1 tumor targets. D, Quantification of PD-1 expression in CD8+ CAR+ T cells following a 3-day coculture with indicated tumor targets at a 1:1 E:T ratio. Data are representativeof at least two independent studies. E, CFSE-based T-cell proliferation assay following a 3- to 5-day coculture with MDA-MB-468 or BBM1 tumor targets at a 1:1 E:T ratio. Representative histograms of CFSE intensity in CD8+ HER2-CAR T cells (left) and quantification of proliferation at 5 days (right). *, P < 0.05 (0 to >4 CFSE dilutions) in T cells (right) at 5 days.
Regression of breast cancer brain metastasis following local delivery of HER2-CAR T cells
To evaluate our HER2-CAR T cells in an orthotopic tumor xenograft model of breast cancer brain metastasis, we engrafted female NSG mice with BBM1 cells by stereotactic injection in the brain parenchyma. BBM1 tumors developed locally and progressively in the brain, maintaining high levels of HER2 expression as determined by IHC (Supplementary Fig. S1). At day 8 after tumor engraftment, we locally delivered0.5 × 106 HER2-CAR T cells intracranially (i.c.) into the tumor bed (Fig. 4A) and observed complete tumor regression, with antitumor responses within the first week in the majority of mice (Fig. 4B and C). Similar responses were observed with HER2–28ζ and HER2-BBζ CAR T cells, resulting in extended survival of CAR T-cell–treated mice compared with untreated (tumor only) or Mock T-cell–treated mice (Fig. 4D). These data support local intracranially delivery of HER2-CAR T cells as an effective method of targeting breast cancer brain metastasis in mice.
Figure 4.
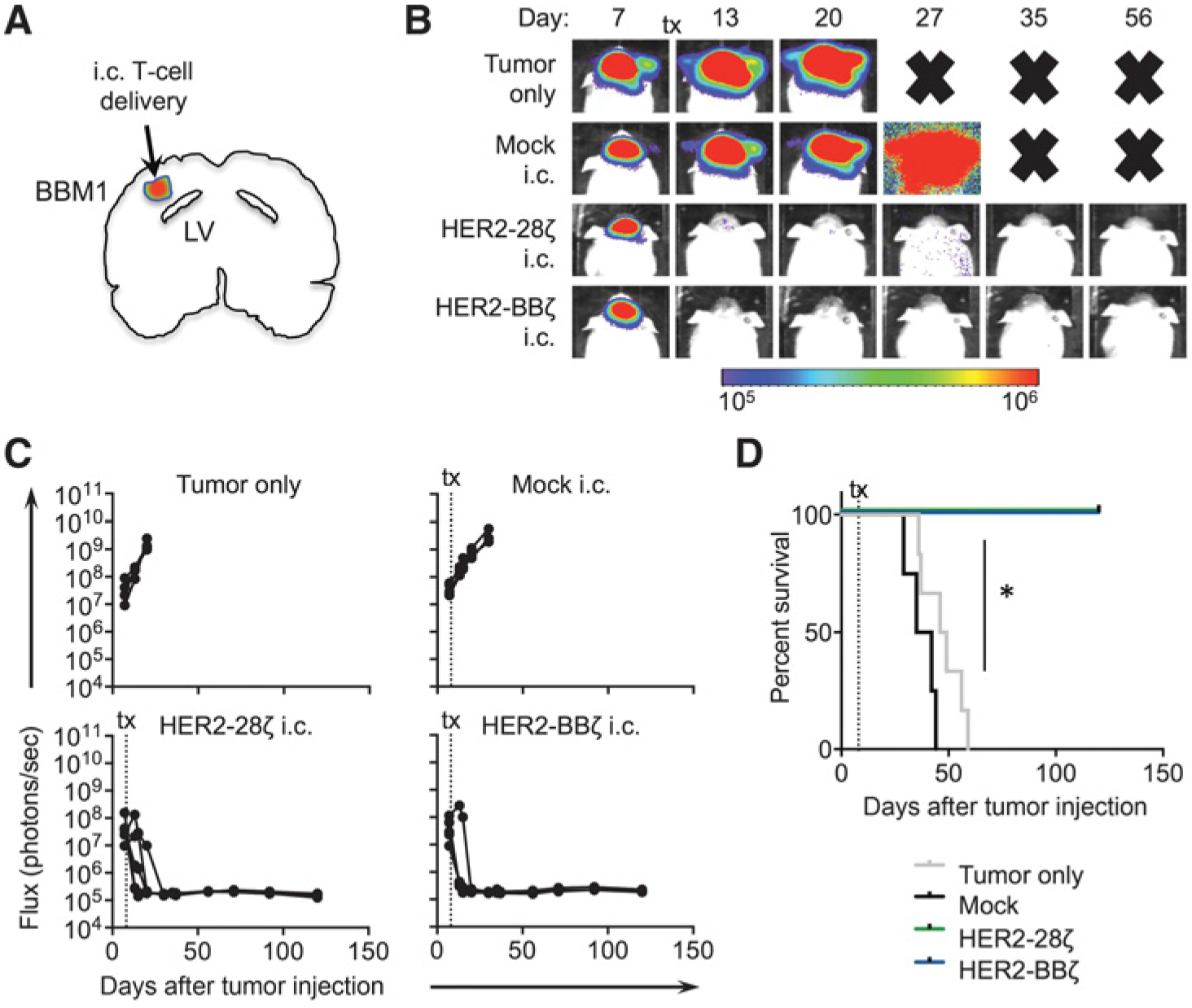
Local intratumoral delivery of HER2-CAR T cells eradicates xenograft models of breast cancer brain metastasis. A, Illustration of a mouse brain orthotopically engrafted with BBM1-eGFP/ffluc cells and treated by intratumoral (intracranial, i.c.) delivery of CAR T cells. B, BBM1 tumor-bearing mice (0.2 × 106 cells) were treated 8 days after tumor injection with Mock, HER2–28ζ, or HER2-BBζ CAR T cells (0.5 × 106 cells). The “Tumor only” group was either left untreated or treated with PBS. Representative flux images of BBM1-tumor–bearing mice at indicated time points after T-cell treatment. Bold “X” indicates removal of mice from study because of morbidity (see Materials and Methods). C, Quantification of flux signal from individual mice in each group at indicated time points after tumor injection. D, Kaplan–Meier survival for Mock, HER2–28ζ, or HER2-BBζ CAR T cells, and a “Tumor only” group. N > 4 mice per group. Data are representative of at least two independent experiments. *, P < 0.05.
Regional intraventricular delivery of HER2-BBζ CAR T cells eradicates breast cancer brain metastasis
Brain metastases are often multifocal and may therefore benefit from regional, in contrast to strictly local, delivery of CAR T cells. A recent case report by our group using IL13Rα2-specific CAR T cells in a patient with recurrent glioblastoma was the first to demonstrate the utility of intraventricular (i.c.v.) CAR T-cell delivery in treating multifocal disease (11). Thus, we compared regional i.c.v. to local i.c. HER2-CAR T-cell delivery in BBM1 tumor-bearing mice (Fig. 5A). We observed equivalent antitumor responses (Fig. 5B and C) and extended survival of mice (Fig. 5D) after i.c. and i.c.v. delivery using stereotactic infusion of 0.5 × 106 HER2-BBζ CAR T cells. Notably, i.c.v. delivery of HER2-CAR T cells exhibited delayed responses in some mice compared with local intratumoral delivery, likely owing to the required trafficking of these cells from the ventricle to the tumor site. Intraventricularly delivered HER2-BBζ CAR T-cell–mediated antitumor activity was also observed with larger tumor burdens, in BBM1-bearing mice treated at day 14 after tumor engraftment (Supplementary Fig. S2). We further validated i.c.v. delivery of HER2-BBζ CAR T cells in a second HER2-amplified xenograft model, BT474 (Supplementary Fig. S3A and S3B). Against the triple-negative xenograft model, MDA-MB-468, no antitumor responses were observed (Supplementary Fig. S3C and S3D). Taken together, these preclinical studies demonstrate the efficacy of intraventricular HER2-CAR T-cell therapy in treating HER2-positive breast cancer brain metastasis.
Figure 5.
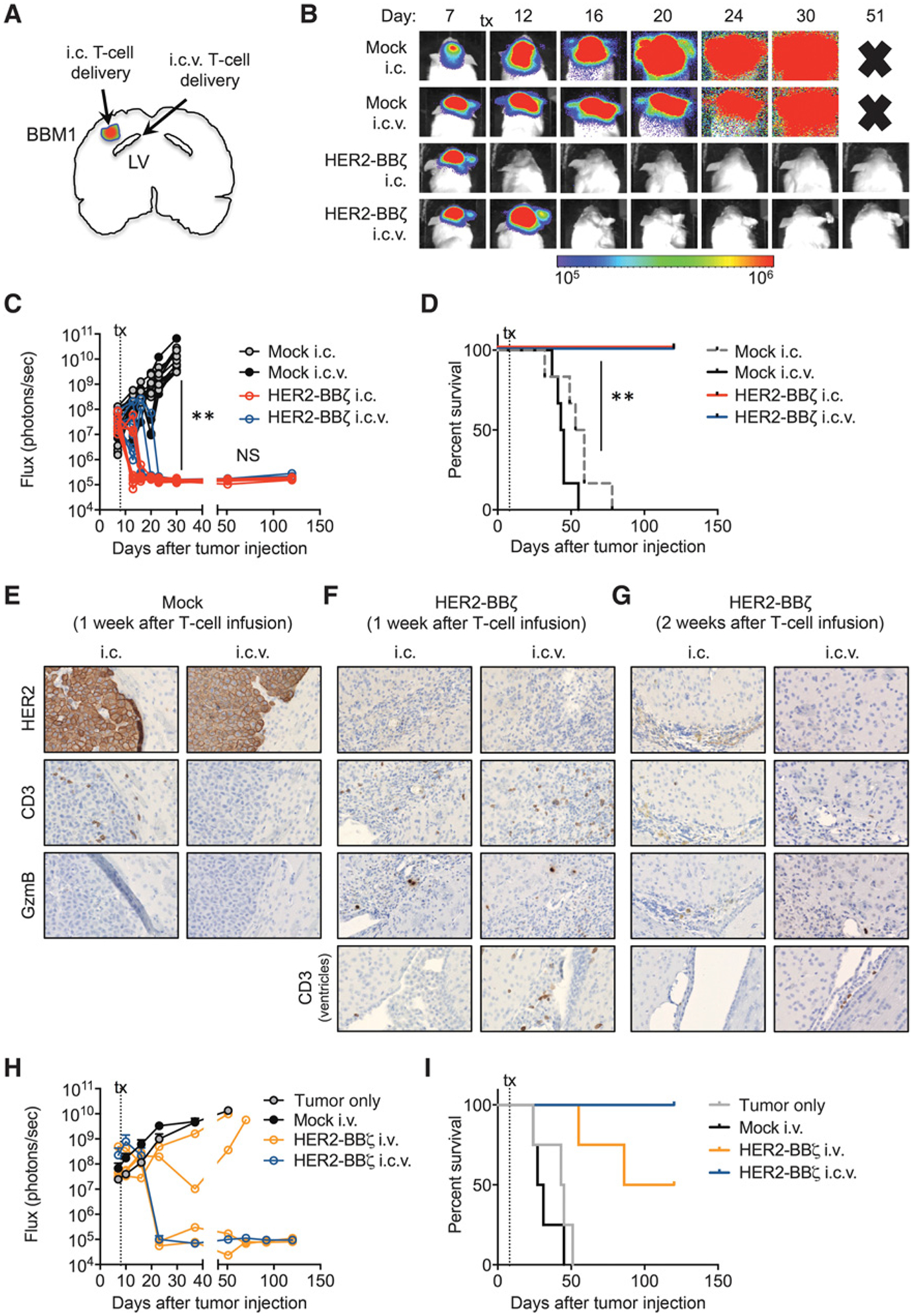
Regional intraventricular HER2-BBζ CAR T-cell delivery is an effective administration route for xenograft models of breast cancer brain metastasistherapy. A, Illustration of a mouse brain orthotopically engrafted with BBM1-eGFP/ffluc cells and treated with T cells by intracranial (i.c.) or intraventricular (i.c.v.) injection. B, BBM1 tumor–bearing mice were treated 8 days after tumor injection with i.c.- or i.c.v.-delivered Mock or HER2-BBζ CAR T cells (0.5 × 106 cells). Representative flux images of BBM1 tumor–bearing mice at indicated time points after T-cell treatment. Time points with “X” refer to removal of mice from study due to morbidity (see Materials and Methods). C, Quantification of flux signal from individual mice in each group at indicated time points after tumor injection. N ≥ 5 mice per group. Data are representative of at least two independent experiments. D, Kaplan–Meier survival curve for i.c.- or i.c.v.-delivered Mock and HER2-BBζ CAR T cells. E, Representative histology of HER2, CD3, and Granzyme B (GzmB) expression in BBM1 tumors, 1 week after i.c.and i.c.v. Mock T-cell infusion. F, Representative histology of HER2, CD3, and Granzyme B (GzmB) expression in BBM1 tumors, 1 week after i.c. and i.c.v. delivery of HER2-BBζ CAR T cells. Bottom, CD3 expression near or in ventricles. G, Representative histology of HER2, CD3, and Granzyme B (GzmB) expression in BBM1 tumors, 2 weeks after i.c. and i.c.v. HER2-BBζ T-cell infusion. Bottom, CD3 near or in ventricles. H, Quantification of flux signal from mice treatedby i.c.v or intravenous (i.v.) delivery of HER2-BBζ CAR T cells at indicated time points after tumor injection. Individual mice are shown for the “HER2-BBζ (i.v.)” group. I, Kaplan–Meier survival for Mock T cells (i.v.), HER2-BBζ CAR T cells (i.v.), HER2-BBζ CAR T cells (i.c.v.), and a “Tumor only” group. N ≥ 4 mice per group. Data are representative of at least two independent experiments. **, P < 0.01.
To evaluate T-cell persistence and functional activity, we compared BBM1 tumors by histology at one or two weeks post T-cell infusion. Mock T cells were found in tumors one week following local i.c. therapy, but were undetectable following i.c.v. delivery (Fig. 5E). However, HER2-BBζ CAR T cells were detected in at the tumor site at one-week post i.c. or i.c.v. delivery, with evidence of functional activity via Granzyme B staining (Fig. 5F). Tumor-infiltrating CAR T cells were comprised of both CD4 and CD8 T cells (Supplementary Fig. S4). These findings suggest antigen-dependent trafficking of HER2-CAR T cells following i.c.v. delivery. Importantly, T cells were also observed near the ventricle one week following i.c.v. but not i.c. delivery. At two weeks after T-cell infusion, rare T cells were observed in BBM1 tumors, although we detected a few T cells near the ventricles following i.c.v. therapy (Fig. 5G).
Two recent phase I clinical trials have demonstrated transient antitumor responses following systemic delivery of HER2-CAR T cells (with a CD28 intracellular costimulatory domain) in patients with HER2-positive sarcoma (9) and progressive glioblastoma (10), the latter suggesting trafficking of CAR T cells across the blood–brain barrier to target brain tumors. We compared regional (i.c.v.) and systemic (intravenous, i.v.) delivery of HER2-BBζ CAR T cells in our preclinical BBM1 model. HER2-CAR T cells delivered i.c.v. (0.5 × 106) showed complete regression of tumors, whereas i.v. delivery of a 10-fold greater dose (5 × 106) showed only partial tumor regression (Fig. 5H and I). Interestingly, we did not observe HER2-CAR T cells by flow cytometry in the peripheral blood of i.c.v. HER2-CAR T-cell–treated mice, suggesting limited systemic trafficking of i.c.v.-delivered T cells in this tumor model (data not shown). Overall, these studies demonstrate potent antitumor efficacy with HER2-CAR T cells following intraventricular delivery in preclinical models of HER2-positive breast cancer brain metastasis.
Regression of multifocal CNS disease following intraventricular HER2-BBζ CAR T-cell delivery
Our recent experience with i.c.v. delivery of CAR T cells for glioblastoma suggests the potential for broad central nervous system (CNS) distribution of T cells for simultaneous targeting of multiple brain tumors (11). We evaluated whether i.c.v. delivery of HER2-CAR T cells would target multifocal CNS disease. Interestingly, BBM1 tumors injected locally in the brain parenchyma trafficked down the spine with leptomeningeal disease developing in approximately 40% of mice. This disseminated disease was potently inhibited by i.c.v. delivery of HER2-BBζ CAR T cells (Fig. 6A and B), and showed similar extension of survival in mice as in Fig. 5D. Histology of the spinal tissue confirmed human HER2-positive diseases in the Mock-treated mice, but not in the HER2-BBζ CAR T-cell–treated mice (Fig. 6C). To model multifocal brain metastasis, we next established BBM1 tumors in both brain hemispheres in mice, and treated with i.c.v.-delivered HER2-CAR T cells. Interestingly, HER2-BBζ CAR T cells preferentially targeted BBM1 tumors at the side of i.c.v delivery at one-week post T-cell infusion, but eradicated both tumors by two weeks (Fig. 6D and E). Finally, as our data suggested that regionally delivered CAR T cells persisted for at least two weeks at the tumor site and in the ventricular space, we performed a tumor rechallenge experiment at two weeks following CAR T-cell delivery, and found effective control of rechallenge disease (Fig. 6F) and 100% survival of rechallenged mice (data not shown) without additional infusion of CAR T cells. Collectively, our preclinical models suggest that HER2-BBζ CAR T cells are potent in eradicating brain metastatic breast cancer, including multifocal and leptomeningeal HER2+ CNS disease.
Figure 6.
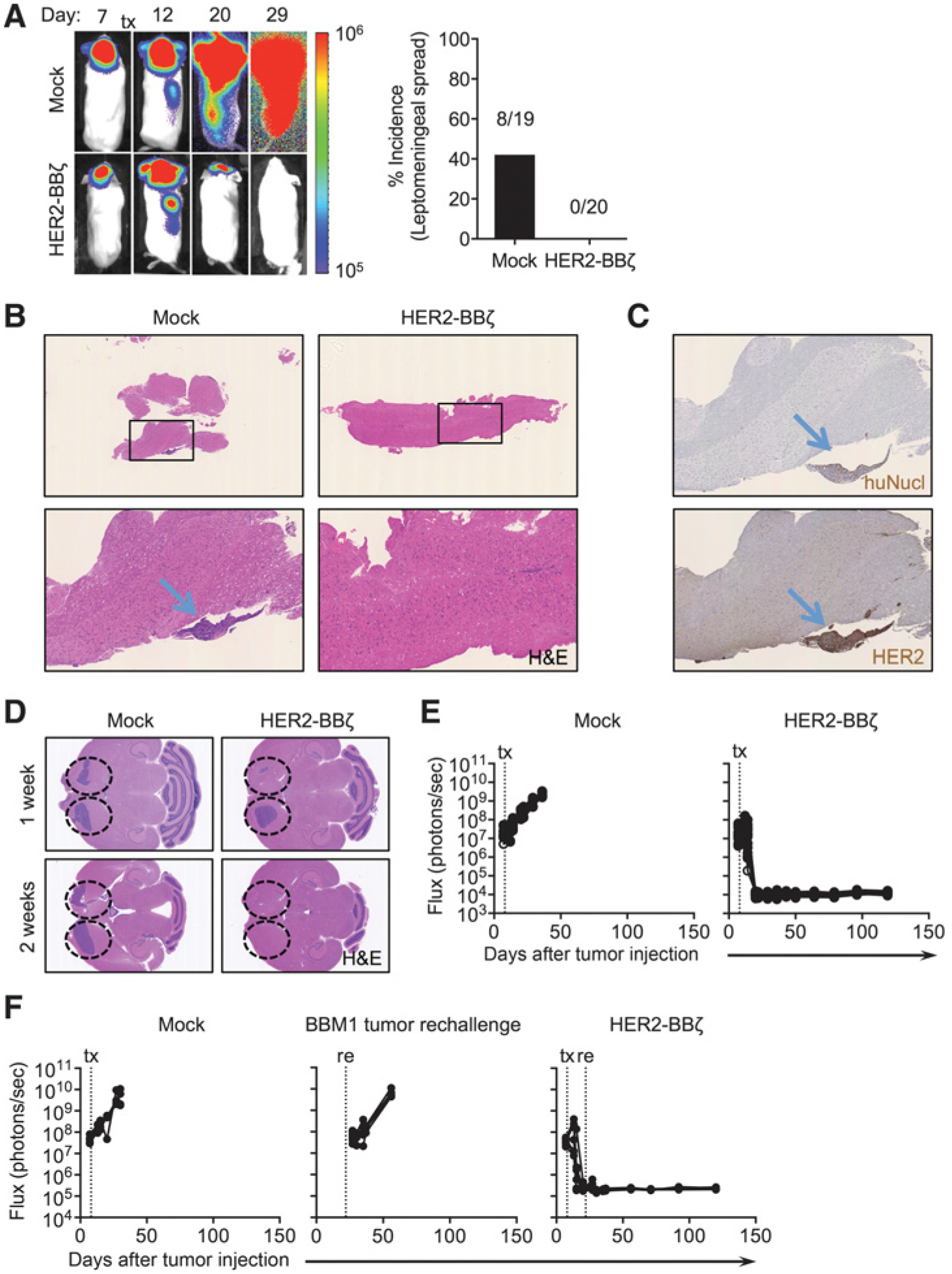
Intraventricular HER2-BBζ CAR T-cell therapy is effective against leptomeningeal and multifocal disease. A, Representative whole-body flux images of leptomeningeal tumor spread from i.c.v. delivery of Mock- and HER2-BBζ CAR T cells at 7 weeks after BBM1 tumor injection (left). Quantification of incidence of leptomeningeal spread (%) from two independent studies is shown (right). B, Representative H&E stains of spines at week 6 after tumor injection, from Mock- and HER2-BBζ CAR T-cell–treated mice. C, Representative histology of human nuclei (huNucl) for detection of human cells and HER2 in Mock T-cell–treated mice at 6 weeks after tumor injection. D, Mice were injected in left and right hemispheres with BBM1 cells at day 0 to mimic multifocal disease, and i.c. or i.c.v. delivery of Mock or HER2-BBζ CAR T cells (0.5 × 106 cells) at day 8 after tumor injection. Representative H&E stains of brains from Mock (i.c.), and i.c. and i.c.v. HER2-CAR T-cell–treated mice at 1 or 2 weeks after T-cell infusion. E, Quantification of flux signal from individual mice in each group at indicated time points after tumor injection. N ≥ 5 mice per group. F, Mice were injected with BBM1 cells at day 0 and treated with Mock- or HER2-BBζ CAR T cells (i.c.v., 0.5 × 106 cells) at day 8 after tumor injection. At day 22(14 days after T-cell infusion), mice were rechallenged with BBM1 tumor cells in the opposite hemisphere. The “tumor rechallenge” group comprised naïve non–tumor-bearing mice that received BBM1 tumor cells on day 22 of the study. Quantification of flux signal from individual mice at indicated time points after tumor injection. N ≥ 4 mice per group.
Discussion
This study has demonstrated robust antitumor responses in human xenograft models of HER2+ breast cancer metastasis to the brain after local intratumoral or regional intraventricular delivery of HER2-BBζ CAR T cells. In contrast, intravenous delivery of HER2-CAR T cells achieved only partial antitumor responses in mice even at 10-fold higher doses compared with local or regional delivery to the brain. Our investigation of the route of administration is timely given the recent clinical trial of HER2-CAR virus–specific T cells for the treatment of recurrent glioblastoma (10). In that study, clinical benefits of intravenously administrated HER2-CAR T cells were observed in approximately 50% of patients. While tumor antigen heterogeneity and low T-cell persistence may have contributed to the moderate response rate in that trial, it is also possible that local or regional delivery of HER2-CAR T cells may have improved the efficacy. In our models, i.c.v. delivery of HER2-BBz CAR T cells demonstrated robust antitumor activity against multifocal disease and leptomeningeal spread, which is clinically relevant to CNS-metastatic breast cancer where leptomeningeal disease is associated with shorter survival (24). Our findings will ultimately inform the design of our upcoming phase I trial in which HER2-BBζ CAR T cells will be delivered i.c.v. to patients with HER2+ brain metastases and/or leptomeningeal carcinomatosis.
Solid tumor-targeted CAR T-cell therapy faces concern of “on-target off-tumor” toxicity by virtue of the low but appreciable normal tissue expression of most overexpressed cell-surface tumor antigens. We propose a HER2-CAR T-cell therapeutic strategy that may obviate this concern in two ways. First, local or regional delivery of CAR T cells for primary brain and metastatic brain tumors may circumvent systemic targeting of less restricted tumor antigens, like HER2. Our group was the first to clinically evaluate both local intratumoral and regional intraventricular delivery of adoptive T cells for the treatment of brain tumors, demonstrating patient safety and clinical benefit of CAR T cells targeting IL13Rα2 (11, 12). Indeed, the recent case report describing a patient with recurrent glioblastoma demonstrated the potential of regional i.c.v. delivery of CAR T cells targeting IL13Rα2 to mediate a complete response against multifocal disease, including lesions in the spine. This clinical response was associated with local CAR T-cell persistence and increased inflammatory cytokines in the cerebrospinal fluid, but not in the peripheral blood. While we did not directly test intrathecal delivery of HER2-CAR T cells in our preclinical models, we believe this route of administration may also hold promise for more regional distribution of CAR T cells for the treatment of multifocal brain metastases. Although i.c. and i.c.v. infusion of HER2-CAR T cells in our preclinical models showed comparable antitumor efficacy, the regional distribution of CAR T cells in the brain via i.c.v. delivery might hold promise for the treatment of multifocal brain metastases, while potentially limiting systemic T-cell distribution.
In addition to the regional delivery route, our proposed HER2-CAR T-cell immunotherapy also addresses potential toxicity issues by the choice of intracellular costimulatory signaling domain (8–10, 25–27). Our data show significant dampening of cytokine production, along with a tendency to require higher HER2 expression for cytokine production by HER2-BBζ compared with HER2–28ζ CAR T cells. Our work also confirms prior studies showing the benefits of 4–1BB costimulation compared with CD28, which include increased T-cell proliferation and reduced expression of exhaustion markers (23, 28). Thus, we propose that 4–1BB costimulation, along with our novel route of HER2-CAR T-cell administration, may provide for a safe and effective CAR T-cell platform for patients with HER2+ breast cancer brain metastases.
The death of a HER2+ metastatic colon cancer patient following HER2-CAR T-cell infusion prompted much debate in the immunotherapy field regarding HER2 as a CAR target and the design of CAR T-cell clinical trials. The case report described that following a lymphodepleting preconditioning regimen, third-generation HER2-CAR T cells (containing both CD28 and 4–1BB costimulatory signaling domains) were infused intravenously at a dose of approximately 8 billion CAR+ T cells with concurrent administration of high-dose IL2 (8). More recent trials for sarcoma and glioblastoma used systemic delivery of second-generation HER2-CAR T cells (containing a CD28 costimulatory signaling domain) without prior lymphodepleting chemotherapy. These trials demonstrated safety and antitumor activity, suggesting that the toxicity in the aforementioned case report might have been associated with the third-generation CAR construct, preconditioning, and/or high CAR T-cell dose. In addition to the novel strategy of regional i.c.v. delivery of HER2-CAR T cells for brain metastasis, our second-generation HER2-BBζ CAR construct may improve safety. The specific scFv antigen-binding domain has also been suggested to contribute to differences in safety observed in the HER2-CAR trials thus far. The recent sarcoma and recurrent glioblastoma trials with HER2-CAR T cells were designed with the murine FRP5-based scFv clone, whereas the first case report (and our CAR described here) was designed with the a trastuzumab-based scFv (humanized derivative of the murine 4D5 antibody). The baseline affinities of 4D5 and FRP5 are 0.15 nmol/L (29) and 6.5 nmol/L (30) respectively. However, the potential loss of affinity by humanization of 4D5 (31) and the scFv conversion of both full antibodies (32) may have created more comparable affinities of these two scFvs. Trastuzumab-based CARs may also demonstrate improved T-cell persistence compared with the murine FRP5-based CARs by virtue of reduced human anti-CAR (and in particular anti-mouse) antibodies. Together, these observations support that the HER2-CAR design, infused T-cell dose, and preconditioning regimens are critical factors in the resulting safety demonstrated in the sarcoma and glioblastoma phase I HER2-CAR T-cell studies.
We are, to our knowledge, the first to provide preclinical evidence for effectively targeting breast cancer metastasis to the brain with regional intraventricular delivery of HER2-CAR T cells. We further directly compare second-generation HER2-CAR T cells with either CD28 or 4–1BB costimulation, demonstrating selective targeting of HER2+ breast tumors with HER2-BBζ CAR T cells. However, we also acknowledge the limitations of these preclinical studies. The lack of an intact immune system in NSG mice precludes a detailed understanding of the complex tumor micro-environment in influencing overall CAR T-cell efficacy. We additionally did not assess the safety of targeting HER2 with our CAR T cells in the current work, but the recent safety of HER2-CAR T cells in two clinical trials support the clinical evaluation of HER2-CAR T cells for breast cancer patients with brain metastases. Collectively, our preclinical data demonstrate potent and selective targeting of HER2+ brain metastases from breast cancer with regional intraventricular delivery of HER2-BBζ CAR T cells, and have directed our strategic platform for translating HER2-CAR T-cell therapy to the clinic.
Supplementary Material
Translational Relevance.
Metastatic disease in the brain and/or central nervous system continues to be a major clinical challenge for HER2+ breast cancer patients. CAR-based T-cell immunotherapy is being actively investigated for the treatment of solid tumors, including HER2+ cancers. Here, we evaluate CAR design as well as the route of CAR T-cell administration for the treatment of HER2+ breast cancer that has metastasized to the brain. Our preclinical data demonstrate effective targeting of breast cancer brain metastases with intraventricular delivery of HER2-BBζ CAR T cells, and are directing our translation of HER2-CAR T-cell therapy to the clinic. The widespread application of these findings may also affect the development of CAR T cells for other HER2+ solid tumors and their brain metastases.
Acknowledgments
The authors thank staff members of the Flow Cytometry Core, the Animal Facility Core, and the Pathology Core in the Beckman Research Institute at the City of Hope Comprehensive Cancer Center (supported by the NCI, P30CA033572) for excellent technical assistance. They also thank Dr. Kurt Jenkins at the City of Hope for assistance with labeling soluble HER2-Fc. Research reported in this publication was supported by the Norris Foundation and the Gateway Foundation.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
S.J. Priceman, C.E. Brown, and S.J. Forman are listed as coinventors on a patent on the development of HER2-CAR T cells and intraventricular delivery of adoptive cellular immunotherapy, which is owned by City of Hope and licensed to Mustang Therapeutics. No other potential conflicts of interest were disclosed.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Eroglu Z, Tagawa T, Somlo G. Human epidermal growth factor receptor family-targeted therapies in the treatment of HER2-overexpressing breast cancer. Oncologist 2014;19:135–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azim HA, Azim HA Jr. Systemic treatment of brain metastases in HER2-positive breast cancer: current status and future directions. Future Oncol 2012;8:135–44. [DOI] [PubMed] [Google Scholar]
- 5.Leone JP, Leone BA. Breast cancer brain metastases: the last frontier. Exp Hematol Oncol 2015;4:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol 2015;27:466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res 2016;22:1875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18:843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol 2015;33:1688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol 2017;3:1094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015;21:4062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A 2014;111:984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmieri D, Bronder JL, Herring JM, Yoneda T, Weil RJ, Stark AM, et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res 2007;67:4190–8. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood 2011;117:1888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther 2015;23:757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood 2003;101:1637–44. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Z, Chinnasamy N, Morgan RA. Protein L: a novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl Med 2012;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res 2012;18:2199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang WC, et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother 2012;35:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 2016;127:2980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015;21:581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott BJ, Oberheim-Bush NA, Kesari S. Leptomeningeal metastasis in breast cancer - a systematic review. Oncotarget 2016;7:3740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Textor A, Listopad JJ, Wuhrmann LL, Perez C, Kruschinski A, Chmielewski M, et al. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res 2014;74:6796–805. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther 2009;17: 1779–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun M, Shi H, Liu C, Liu J, Liu X, Sun Y. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res 2014;16:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 2016;126:3130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 1992;89:4285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wels W, Harwerth IM, Mueller M, Groner B, Hynes NE. Selective inhibition of tumor cell growth by a recombinant single-chain antibody-toxin specific for the erbB-2 receptor. Cancer Res 1992;52:6310–7. [PubMed] [Google Scholar]
- 31.Makabe K, Nakanishi T, Tsumoto K, Tanaka Y, Kondo H, Umetsu M, et al. Thermodynamic consequences of mutations in vernier zone residues of a humanized anti-human epidermal growth factor receptor murine antibody, 528. J Biol Chem 2008;283:1156–66. [DOI] [PubMed] [Google Scholar]
- 32.Menzel C, Schirrmann T, Konthur Z, Jostock T, Dubel S. Human antibody RNase fusion protein targeting CD30+ lymphomas. Blood 2008;111: 3830–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.