

Abstract
Fetal hemoglobin (HbF) can blunt the pathophysiology, temper the clinical course, and offer prospects for curative therapy of sickle cell disease. This review focuses on (1) HbF quantitative trait loci and the geography of β-globin gene haplotypes, especially those found in the Middle East; (2) how HbF might differentially impact the pathophysiology and many subphenotypes of sickle cell disease; (3) clinical implications of person-to-person variation in the distribution of HbF among HbF-containing erythrocytes; and (4) reactivation of HbF gene expression using both pharmacologic and cell-based therapeutic approaches. A confluence of detailed understanding of the molecular basis of HbF gene expression, coupled with the ability to precisely target by genomic editing most areas of the genome, is producing important preliminary therapeutic results that could provide new options for cell-based therapeutics with curative intent.
Visual Abstract

Introduction
Fetal hemoglobin (HbF; α2γ2), a minor hemoglobin of normal adults, has major clinical significance for sickle cell disease. γ-Globin is encoded in HBG2 (Gγ) and HBG1 (Aγ), nearly identical genes found in a developmentally regulated gene cluster on chromosome 11p15 (5′—ϵ—Gγ—Aγ—δ—β—3′). Sickle cell disease is caused by homozygosity for the sickle hemoglobin (HbS) gene (β7glu-val, GAG-GTG) or compound heterozygosity of the HbS gene and other HBB disorders.1 DeoxyHbS polymerization drives the disease pathophysiology. The rate and extent of polymerization is modified by the erythrocyte HbF concentration.2 Abnormal sickle erythrocytes transit small vessels slowly providing time for deoxyHbS fiber formation with resulting cell injury. HbF prolongs the time needed for HbS polymerization (delay time), allowing the escape of sickle erythrocytes from the microcirculation into larger veins and hence the lungs, where on reoxygenation, HbS polymer melts.3 HbF is variably increased in nearly all HbS homozygotes.4 The most efficient means of treatment is interrupting disease pathophysiology by preventing deoxyHbS polymerization. This can be accomplished by inducing sufficiently high levels of HbF in most sickle erythrocytes.5
Ten years have elapsed since we reviewed HbF as a modulator of sickle cell disease.4 Important developments since then include the following: (1) detailed understanding of the switch from fetal to adult hemoglobin gene expression; (2) therapeutic exploitation of this knowledge; (3) how HBB haplotypes influence HbF; (4) understanding the importance of heterogeneity of the distribution of HbF concentrations among HbF-containing erythrocytes; (5) expanded use of hydroxyurea; and (6) new targets for HbF induction. This review focuses on advances since 2011.
Quantitative trait loci associated with HbF
Inactivation and activation of genes within the β-globin gene cluster holds the keys to reactivating for therapeutic gain the usually dormant HbF genes.6-12 Transcription factor complexes interact with the locus control region (LCR) enhancers upstream of the β-globin gene cluster and with globin gene promoters, silencing embryonic and fetal genes while activating adult genes. In addition to BCL11A and ZBTB7A, the predominant HbF repressors discussed below, developmental factors including the LIN28B and IGF2BP1 RNA-binding factors and the let-7 family of microRNAs also modulate HbF gene expression. Reduction of let-7 or overexpression of its regulator LIN28B is associated with increased HBG expression.13
Three quantitative trait loci (QTL), BCL11A, MYB, and a locus in the promotor of HBG2 modify HbF gene expression. BCL11A (chr2p16), a zinc finger transcription factor, was first identified by genome-wide association studies.4,14-16 BCL11A is a repressor of HbF gene expression. Reducing bcl11a expression in transgenic sickle mice derepressed γ-globin genes.17,18 Functional variants of BCL11A are marked by single nucleotide polymorphisms (SNPs) in intron 2 that tag an erythroid-specific enhancer.19 The sentinel SNP marking this QTL is rs1427407. This enhancer consists of 3 DNase hypersensitive sites +62, +58, and +55 kb from the transcription initiation site.19 Variants associated with increased HbF are present in ∼20% of sickle cell anemia patients. BCL11A binds TGACCA motifs in the HBB gene cluster, preferentially binding to position −115 in the HBG2 and HBG1 promoters.20,21 BCL11A expression is controlled by transcriptional and/or translational mechanisms.22-24 ZBTB7A (LRF) is another zinc finger HbF gene silencer. Its inactivation in HUDEP-2 cells led to ∼50% HbF; BCL11A plus ZBTB7A knockout caused > 90% HbF.25 The relative increase in γ globin is magnified by the decrease in β globin. ZBTB7A is not polymorphic and does not account for HbF variation in patients with sickle cell anemia.26
Natural mutations and clustered regulary interspaced short palindromic repeat (CRISPR)-Cas9 disruption of the BCL11A and ZBTB7A binding sites are associated with a large effect on expression of the γ-globin genes.12,21,27 Krüppel-like factor 1 (KLF1) binds to the promoter of ZBTB7A increasing its expression; KLF1 also activates the expression of BCL11A.28,29 Reduced KLF1 expression or activity is associated with HbF derepression.
The C-T polymorphism 158 bp 5′ of HBG2, a cleavage site for Xmn1, is associated with increased HbF in the Senegal and Arab-Indian HbS gene haplotypes. CRISPR-Cas9 disruption of this site in sickle cell CD34+ cells caused ∼25% γ globin and 55 ± 5% F-cells. Disruption of BCL11A and ZBTB7A binding sites led to 30% to 40% γ globin and 75% to 80% F-cells.27 Only HBG2 expression is affected by the −158 polymorphism; both HbF genes are affected by BCL11A polymorphisms.30 HBG2 levels fall from ∼70% at birth to ∼40% 6 months postnatally, making it unsurprising that targeting the HBG2 site had a smaller effect on HbF. The transcription factors binding to the −158 HBG2 motif are uncharacterized.
Four markers representing the 3 HbF QTL, rs654815 and rs1427407 in BCL11A, rs66650371, the 3-bp deletion in MYB, and rs7482144 in the HBG2 promoter, accounted for 21.8% of HbF variability in 581 HbS homozygotes and patients with HbS-β0 thalassemia. Similar results were found in 186 patients with compound heterozygosity for HbS and HbC genes (HbSC) disease and 994 Tanzanian HbS homozygotes.31
HbF, geography, and HBB haplotypes
The HbS gene had a single origin in West Central Africa or in the Sahara during the Holocene Wet Phase between 3400 and 11 100 years ago or in agriculturists ∼22 000 years ago, which was introduced into rainforest dwellers within the last 6000 years.32,33 The earlier origin in agriculturists might have resulted from destruction of the rain forest canopy that created conditions favorable for malaria. Malaria, in turn, provided the selective pressure driving the rise of the HbS gene. From the primordial haplotype the HbS gene subsequently became associated with 5 common HBB haplotypes: Bantu (CAR or Central African Republic), Cameroon, Arab-Indian (AI), Senegal, and Benin.34 AI, Senegal, and Benin haplotypes are derived from the CAR and Cameroon haplotypes.32 Each haplotype is associated with a distinctive mean HbF level, albeit with wide dispersion, making ascertainment of haplotype in individuals of little prognostic value.
Adult HbS homozygotes with the AI haplotype have a mean baseline HbF of 17%35; Senegal haplotype patients have HbF of ∼10%. Among Benin, Bantu, and Cameroon haplotype patients, the HbF level of individuals of African and Arab descent appear to differ. Benin haplotype patients of African descent have HbF of ∼6%; Saudi Arab patients with the Benin haplotype have HbF of ∼11%.36 Saudi patients with Bantu and Cameroon haplotypes have HbF of ∼8% compared with ∼5% in their African counterparts.35 Children with the AI haplotype have HbF of ∼30%.37 As HbF levels decline, AI haplotype adults have more numerous symptoms, and their disease begins to resemble that of African patients, but with a lower incidence of stroke, no leg ulcers, and >70% prevalence of splenomegaly.37,38 HbF differences among haplotypes were not associated with BCL11A polymorphisms.39 With the possible exception of the AI and Senegal haplotypes, the genetic basis of the variance of HbF levels among HbS gene haplotypes remains undefined.
The AI haplotype in Saudi patients is associated with a unique pattern of SNPs in putative cis-acting globin regulatory elements and is linked to a novel variant in ANTXR1.40,41
A 3-SNP subhaplotype of the AI haplotype defined by 2 polymorphisms in the LCR and rs7482144 was unique to Saudi Arabs. Homozygosity for minor alleles at rs16912979, rs7119428, and rs7482144 (T/A/T) were hypothesized, but not mechanistically verified, to represent a cis-acting domain modulating HBG2 expression by facilitating LCR contacts with the HBG2 promoter.40 The Senegal haplotype, with intermediate HbF levels, contains rs7482144; Benin, Bantu, and Cameroon haplotypes lacking rs7482144 have the lowest HbF.
BCL11A and MYB accounted for only 8.8% of HbF variance in the AI haplotype population, suggesting that other trans-acting elements might influence their high HbF. An intronic SNP (rs35685045) in ANTXR1 (TEM8; chr2p13) was associated with HbF in 4 cohorts of adult AI haplotype patients.41,42 This variant explained ∼10% of HbF variability compared with ∼8% for BCL11A. Like BCL11A and MYB, ANTXR1 appeared to be a repressor of HbF. ANTXR1, a type 1 transmembrane protein, is expressed at high levels in bone marrow. It remains possible that ANTXR1 could be acting through BCL11A despite their additive and independent effects on HbF and absence of linkage disequilibrium between ANTXR1 and BCL11A SNPs.
F-cells: heterogeneity of HbF concentrations
Erythrocytes with detectable HbF are called F-cells. These cells arise stochastically from erythroid precursors that can give rise to both HbF-containing and non–HbF-containing erythrocytes. BCL11A and ZBTB7A are not differentially expressed in F-erythroblasts compared with non–HbF-expressing erythroid precursors. F-cells do not resemble fetal erythrocytes save for their high levels of HbF.43 Patients with sickle cell anemia have 2% to 80% F-cells, which survive longer than non-HbF cells. Improved F-cell survival is likely a result of HbF protecting the cell from HbS polymer-induced damage.44-47 This protection provided by HbF can be nearly absolute or only partial. Treatment with hydroxyurea increases the number of F-cells and the concentration of HbF/F-cell.
Each erythrocyte contains ∼30 pg hemoglobin. The threshold for F-cell detection by fluorescent activated cell sorting (FACS) using an antibody reactive with HbF is ∼6 pg of HbF/F-cell. When HbF/F-cell is 9 to 12 pg, calculations suggested that deoxyHbS polymerization should be prevented at physiologic venous and capillary O2 saturations of 40% to 70%, affording the sickle erythrocyte nearly total protection from HbS polymer-induced damage, although recent studies call into question this observation.3,48,49 In compound heterozygotes for HbS and the common gene deletions HPFH-1 and HPFH-2 that cause hereditary persistence of HbF (HPFH), total HbF level is ∼30%. HbF is distributed nearly evenly, or pancellularly, in HbS-HPFH erythrocytes, endowing each cell with ∼10 pg HbF; HbS polymer is not present in these erythrocytes. Individuals with this genotype do not have hemolytic anemia and are usually asymptomatic.50 In sickle cell anemia, HbS polymer is present at 70% O2 saturation, and the polymer fraction rises steeply as O2 saturation falls.51 Patients with untreated sickle cell anemia, sickle cell anemia treated with hydroxyurea or HbS-δβ thalassemia, who can have total HbF levels similar to those found in HbS-HPFH, are usually symptomatic; all have hemolytic anemia. The contrast between the clinical and hematologic features of HbS-HPFH, and these conditions suggest that HbF level or F-cell percentage are poor predictors of disease severity in an individual because neither accounts for the variable distribution of HbF levels among F-cells. We mathematically modeled possible distributions of HbF/F-cell in patients with HbF levels of 5%, 10%, 20%, and 30%, reflecting, respectively, the mean levels of HbF in adult African-origin patients with the Benin/Bantu/Cameroon and Senegal haplotypes and adult and pediatric patients with the AI haplotype.52 It is nearly impossible to have a clinically important number of fully protected F-cells when HbF is 5%; no more than 15% of cells can be protected when HbF is 10%.52 This is consistent with disease severity in most patients with these HbF levels and individuals with Bantu/Benin/Cameroon haplotypes. Consistent with the phenotype of most patients with natural or therapy-induced HbF levels of 20%, it is possible for nearly 25% of F-cells to have polymer-inhibiting HbF levels. When HbF is near 30%, disease can be relatively benign and 70% of cells can be protected.52-54 (Figure 1). The number of F-cells with polymer-inhibiting concentrations of HbF is likely to be a more important determinant of the features of sickle cell anemia than the total number of F-cells or the concentration of HbF in the hemolysate. A prospective study where the distribution of HbF concentrations in F-cells can be rapidly and repeatedly measured and that controls for other hematologic variables like α thalassemia is needed to confirm this hypothesis.
Figure 1.
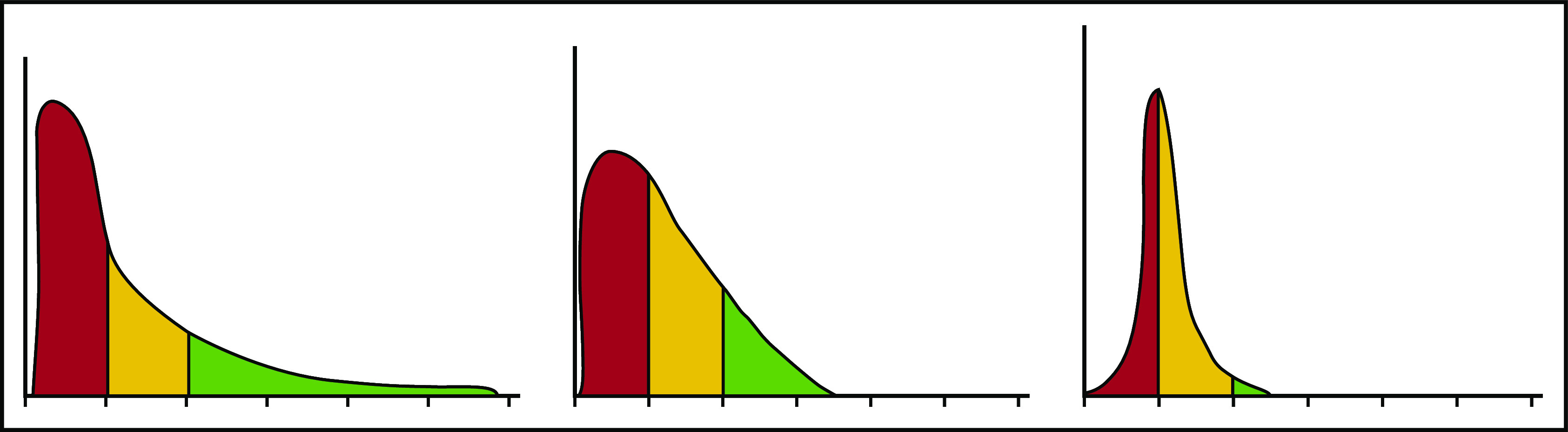
HbF distribution in sickle erythrocytes from 3 patients with 20% HbF. An example of 3 possible distributions of HbF/F-cell in 1000 cells from 3 individuals with mean HbF levels of 20%. The y axis represents numbers of cells, and the x axis represents HbF concentration in 5-pg increments. In red are cells likely to be least protected from HbS polymer damage. These cells have HbF < 6 pg and are not visible by FACS. In yellow are cells visible by FACS but not fully protected from HbS polymerization. They have HbF concentrations of 6 to 10 pg. In green, are cells with HbF concentrations >10 pg. These cells should be fully protected from HbS polymerization. Many other distributions are possible with the same mean HbF accounting for the varied phenotypes associated with the same HbF concentration. Data are derived from Steinberg et al.52
HbF effects on clinical and hematologic phenotypes
High HbF is strongly associated with a reduced rate of acute painful episodes, fewer leg ulcers, and longevity.55 Any protection afforded by HbF against the development of priapism, renal functional impairment, cerebrovascular disease, and perhaps sickle vasculopathy is questionable. HbF is also associated with higher O2 saturations.56 Unequal protection by HbF across the gamut of sickle cell disease complications might be partially explained by differences in the pathophysiologic roots of these complications.57,58 Acute painful episodes and acute chest syndrome are a result of sickle vasoocclusion; HbF has the most noticeable beneficial effect on these subphenotypes. Priapism, nephropathy, and sickle vasculopathy are associated with hemolysis. In these complications HbF has a less obvious beneficial effect, perhaps because only a small number of lysed sickle erythrocytes are needed to trigger these subphenotypes.59
HbF and survival
In 3764 patients in the pre-hydroxyurea era, HbF > 8.6% was associated with improved survival.60 Recent studies have documented the importance of hydroxyurea-induced increases in HbF. A meta-analysis of 7 studies with 2477 hydroxyurea era cases showed that HbF was associated with reduced mortality (hazard ratio [HR]: 0.97; 95% confidence interval [CI]: 0.94-1.0).61 Increased HbF associated with taking 15 to 35 mg/kg hydroxyurea improved survival in 393 HbS homozygotes (HR: 0.36; 95% CI: 0.17-0.73).62 Using the PedsQL Sickle Cell Disease Module, health-related quality of life in 123 Omani children with sickle cell disease was predicted by HbF independent of other measured elements (95% CI: 0.15-1.12).63
To identify prognostic factors for clinical complications, 57 infants were studied at 4.4 ± 1 months of age and followed for ∼19 months. HbF, which fell from 42% to 21% during the observation period, was the strongest prognostic factor for disease complications.64 Of 230 children treated with hydroxyurea with 510 patient-years of follow-up, patients with HbF less than 20% had twice the risk of hospitalization for pain and acute chest syndrome and 2 times the risk of fever as patients with >20% HbF.65
HbF and cerebrovascular disease
Pre-hydroxyurea era studies of patients with a mean age of 14.1 ± 12.7 years suggested that neither stroke nor silent cerebrovascular disease was associated with HbF.66,67 More recent work suggested a protective effect of HbF. Children aged 12.1 ± 2.6 years with the highest HbF were protected from silent infarctions.68 Of 375 children with sickle cell anemia or HbS-β0 thalassemia followed in a longitudinal prospective study, 59 had cerebrovascular disease or a stroke at a mean age of 4.4 years; HbF was protective (HR: 0.68; 95% CI: 0.48-0.96).69 Eighty-three neurologically intact adults with sickle cell anemia had silent white matter changes that were associated with low HbF.70 In early childhood, high HbF is likely to be associated with more F-cells containing protective HbF levels. As HbF falls and fewer F-cells are protected from HbS polymer damage, the propensity for vascular occlusion and intravascular hemolysis increases with the release of heme products into the circulation that scavenge nitric oxide causing endothelial damage and vasculopathy.57-59
HbF and malaria
HbF did not provide protection from malaria in African (HbF, 5.6%) or Indian (HbF, 18.2%) sickle cell anemia patients.71,72 Nevertheless, an open-label trial in 606 African children aged 1 to 10 years treated with hydroxyurea found that HbF increased from 10.9% to 23.4%. This was associated with a decrease in the rate of malaria infection by more than 60%.73 Malarial parasites grow poorly in HbF-containing erythrocytes. The stability of the HbF compared with the HbA tetramer might account for part of this effect.74
Therapeutic induction of HbF
Hydroxyurea
Hydroxyurea is recommended for nearly all patients with sickle cell anemia or HbS-β0 thalassemia starting in the first year of life; it is also is used in symptomatic HbSC disease and HbS-β+ thalassemia. Fourteen children with HbSC disease, mean age 11 years, were given hydroxyurea at the maximum tolerated dose. HbF increased from 1.7 ± 1.9% to 6.3 ± 6.7%.75 After 12 months, HbF increased from ∼2.8% to 5.5% in 133 hydroxyurea-treated HbSC disease patients.76 In 39 Italian patients with HbS-β+ thalassemia receiving more than 15 mg/kg of hydroxyurea, HbF increased from 4.7% to 10.9%.77 Greek adults with HbS-β+ thalassemia treated with hydroxyurea showed a HbF increase from a median of ∼6.8% to a median of ∼20%.78 Hydroxyurea is also used effectively in low-resource countries.73,79,80
By inducing increased levels of HbF through mechanisms that include suppressing erythropoiesis, hydroxyurea was first associated with reduction of morbidity and mortality in adults.81,82 Hydroxyurea treatment was associated with progressive increases in the polymerization delay time, coincident with the increase of F-cells.83 Some benefits of hydroxyurea might also be unrelated to HbF induction.83
HbF induction by hydroxyurea depends in part on a proliferating erythroid bone marrow, which is most vigorous in childhood. With time, intramedullary sickling and marrow infarction reduce the erythroid capacity of the marrow.84,85 Following 2 years of hydroxyurea in adults with baseline HbF of ∼5%, the top quartile of HbF responders had ∼12% HbF.86 In contrast, all children seem to have a robust HbF response to hydroxyurea that retards the natural age-related fall in HbF.87 At a fixed dose of 20 mg/kg, after at least 18 months of observation, the percent decrease in HbF in hydroxyurea-treated patients was 13% compared with 37% in placebo-treated controls. When hydroxyurea was started at 9 to 18 months of age, HbF remained at its baseline level of 25%. Nonetheless, even in treated patients, HbF levels might decline with age. Following 2 years of treating children <2 years old at a dose of 20 mg/kg, HbF was 19.7%; after 15 years, HbF fell to 15.1%. Therefore, even when started early in life, the HbF response to hydroxyurea might wane.88 Hydroxyurea started at ∼11 months at a dose of ∼27 mg/kg was associated with HbF levels of 33.3 ± 9.1%; 33% of patients had HbF > 40% with a hemoglobin concentration of 10.1 ± 1.3 g/dL; acute events were markedly reduced; and toxicity was minimal.89 This dosing approach is setting a new standard for using hydroxyurea with a profound effect on the disease in childhood. Dose escalation was also superior to fixed dosing when studied in Ugandan children.80 The ultimate safety and efficacy of this approach will require long-term follow-up.
Hydroxyurea was noninferior to transfusion for reducing transcranial Doppler flow velocities in selected children at high risk for stroke. Whether this observation was a direct benefit of HbF, a secondary benefit of the higher hemoglobin concentration caused by increased HbF, or a combination of these is uncertain.90
Novel agents and targets
Other drugs that act epigenetically or with novel mechanisms of action have increased HbF levels in preclinical and early-phase clinical trials; none are approved for treatment. The current status of these agents and new targets for HbF induction is summarized in Table 1.
Table 1.
Agents and targets for inducing HbF expression
| Agent/target | Mechanism | Status |
|---|---|---|
| Metformin | Enhanced FOXO3 expression. | Two- to threefold increase in F-cells in vitro91; small increment in HbF in vivo in 3 of 6 patients92; in an administrative database, patients with diabetes treated with metformin had fewer medical encounters; no HbF data.93 |
| Pomalidomide | Alters erythroid maturation.94 | HbF increased in 2 of 4 patients in phase 1 study. Myeloma patients treated with pomalidomide had increased γ-globin in RBC precursors95 and increased F-cells in vitro.43 |
| Tranylcypromine, RN-1, other LSD1 inhibitors | Inhibition of lysine-specific demethylase 1 via demethylation of H3K4.96 | Mouse, primate, and in vitro studies.96-99 |
| Decitabine/other demethylating agents | DNA methyltransferase depletion, histone methyltransferase inhibition decreases H3K9Me2 and increases H3K9Ac at the γ-globin gene locus.100 | Phase 1 trial of oral tetrahydrouridine + decitabine, HbF increased 4-9%, F-cells 40-80%, hemoglobin 1-2 g/dL in 3 patients with highest baseline HbF at highest drug dose.101 Methyltransferase (EHMT1/2) inhibition studied only in vitro. |
| PDE9 inhibitors | Inhibition of PDE9, increases cGMP and protein kinase G that increased HbF. | HbF increased in CD34+ cells. Sickle mice had greater than threefold increase in HbF and decrease in sickle cells and hemolysis.102 In phase 1B trial (NCT02114203), no effect on HbF or hemoglobin; decreased soluble E-selectin and WBC-platelet aggregates.103 |
| IGF2BP1 | mRNA-binding protein. Lentivirus delivery of IGF2BP1 to CD34+ cells increased HbF, fetal γ-globin mRNA and decreased β-globin mRNA. Contacts between the LCR and γ-globin genes increased. | Cell-based studies only.104 |
| LCR-promoter facilitation | Zinc finger-ldb1-facilitated looping of LCR to γ-globin gene promoters; increased γ-globin mRNA 40-fold in HUDEP-2 and CD34+ cells. | Cell-based studies only.105,106 |
| Dimethyl fumarate (DMF) | Through a Nrf2-mediated pathway, increased HbF in sickle CD34+ cells, sickle mice and primates. | Preclinical studies only.107 |
| BCL2LI | Overexpression in CD34+ cells increased HBG expression 11-fold. | Cell-based studies only.108 |
| SIRT1 agonists | Ectopic expression and SIRT1 agonists promote LCR-HBG interaction and increased HBG expression. | Cell-based studies only.109,110 |
| EIF2AK1 | EIF2AK1, the heme-regulated inhibitor, promotes the translation of ATF4, activating BCL11A transcription and silencing HBG. Inhibition of EIF2AK1 reduces BCL11A increasing HbF.111 | Cell-based studies only.112 |
| MBD2-NuRD | Knockdown of methyl-CpG binding domain protein 2 (MBD2) increased γ/γ+β mRNA > 30-fold and γ globin 10-fold. | Cell-based studies only.113 |
Agents and pathways studied in preclinical and early-phase clinical studies to stimulate increased HbF.
Cell-based HbF-inducing therapeutics
Cure of sickle cell anemias is possible if enough HbF can be induced in enough sickle erythrocytes to thwart their premature destruction and inhibit sickle vasoocclusion. This has not yet been achieved with drug therapy but might be possible with cell-based therapeutics. Cell-mediated therapy either adds a β-globin gene with a HbF-like substitution or reverses the silencing of the HbF genes by curbing HbF repressors or disrupting their binding motifs in γ-globin gene promoters.
Lentiviral-mediated gene addition.
More than 40 years ago, Nagel et al114 showed that the glutamine (Q) at position 87 in the γ-globin chain was 1 of 2 residues accounting for the inhibitory effect of HbF on HbS polymerization. Replacing β87 threonine (T) with Q (HbAT87Q) and incorporating this gene into a lentiviral vector that was transferred into hematopoietic stem cells corrected the main features of sickle cell disease in transgenic mice.115 In a phase 1/2 study, a similar HbAT87Q-lentiviral construct was introduced into autologous CD34+ stem and progenitor cells that were infused into myeloablated patients with severe forms β thalassemia. The requirement for chronic transfusion was reduced or eliminated in all patients.116 This same approach was used in a patient with sickle cell anemia. About 1 copy of this vector was present in each cell. After 15 months, total hemoglobin level was 11 to 12 g/dL; HbAT87Q formed about half of the total hemoglobin; and the patient was asymptomatic.117 Using a modified manufacturing process, 13 patients had plerixafor mobilized CD34+ cells transfected with the HbAT87Q-containing vector and were followed for 3 to 15 months. The vector copy number was 3.8, HbAT87Q was nearly 50% of total hemoglobin, and hemoglobin levels were 10.2 to 15 g/dL; patients became asymptomatic.118
Lentiviral-mediated short hairpin RNA knockdown of BCL11A.
A lentiviral vector containing a short hairpin RNA (shRNA) embedded in a microRNA scaffold was used to allow erythroid-specific knockdown of BCL11A (shRNAmiR) after insertion into autologous CD34+ cells.119 Preclinical testing in sickle CD34+ cells transduced with this shRNAmiR achieved a vector copy > 5 and gene marking of > 80%, resulting in a three- to fivefold induction of HbF. Growth, differentiation, and engraftment of gene-modified cells in vitro or in vivo were unaffected. Three patients treated with this vector and followed for 7 to 15 months had HbF of 23.7% to 40.7% and F-cells of 67.7% to 76.4%; total hemoglobin was >11 g/dL; and acute vasoocclusive events stopped (NCT03282656).120
Targeting HbF repressors by genomic editing.
The toolkit for genome editing is rapidly expanding. CRISPR-Cas dominates the field because of the ease of using guide RNAs directing the CRISPR complex to specific DNA sequences and the increasing ability to target most sites in the genome.121,122 In addition to the ability to create small deletions and insertions using traditional CRISPR-Cas9 technology, it is also possible to achieve some single base edits by nicking but not cleaving DNA. The favored repair mechanism for double-strand DNA breaks in human cells is nonhomologous end joining that leads to disruption of normal sequence in the targeted area, introducing small insertions or deletions. In the context of sickle cell disease treatment discussed below, CRISPR-Cas9 disrupts specific enhancer motifs or binding sites for HbF repressor proteins. Correcting the HbS mutation itself requires a slightly different approach relying on homology-directed repair, which as currently used, is a less efficient reparative pathway.
Several approaches to reducing the expression of HbF suppressors in CD34+ cells have entered clinical trials, including the first use of CRISPR-Cas9 in humans (Figure 2).123-125 They include disrupting the erythroid enhancer of BCL11A or the binding sites for BCL11A in the γ-globin gene promoters.126 Clinical trials are targeting BCL11A with CRISPR/Cas and zinc finger nucleases (NCT03745287, NCT03655678, and NCT03653247). In a phase 1/2 study, after stem cell mobilization with plerixafor and myeloablative conditioning with busulfan, a single infusion of autologous CD34+ cells that were edited with CRISPR-Cas9 using a guide RNA specific for the erythroid enhancer region of BCL11A was given to a patient with sickle cell anemia. Engraftment occurred after 30 days. The baseline HbF of 9.1% increased to 25.9% after 2 months and appeared to stabilize between 46% and 48% when tested at 4 through 9 months; F-cells increased from 33.9% to 99.7%; total hemoglobin stabilized at 11 to 12 g/dL; and vaso-occlusive episodes stopped.125 Two patients with β thalassemia treated similarly achieved HbF >97% of the hemolysate with a hemoglobin concentration at 15 months of 14 g/dL.125
Figure 2.

Genome editing to increase HbF production. Only HBG2 and its upstream binding sites for BCL11A and ZBTB7A are depicted. The expression of BCL11A, 1 of 2 major repressors of HbF gene expression, is controlled by an erythroid-specific enhancer. BCL11A binds to a TGACCA motif centered at position −115 in the promoters of both HBG2 and HBG1. ZBTB7A, the other major HbF gene repressor, not shown, binds at positions −195 to −197 and −201 to −202 upstream of both γ-globin genes. A still unknown transcription factor(s) is likely to bind the −158 site. The −158 polymorphism is found only in HBG2. CRISPR-Cas9 editing of either the BCL11A erythroid-specific enhancer, shown as a double-strand break, or its binding sites in the HbF gene promoters, shown before editing, reverses the repression of these genes increasing HbF.
All cell-based modalities had similar results with close to 50% HbF or HbAT87Q. Many of these studies had follow-up of more than 1 year, with near normalization of the hematologic findings. This suggests that stress erythropoiesis contributes minimally, if at all, to high HbF. Genomic editing and shRNA-mediated knockdown of BCL11A should erase lingering doubts that the clinical and hematologic phenotypes of sickle cell anemia can be reversed by high HbF levels in most sickle erythrocytes. If these genetic modalities are safe, effective, and similarly priced, how might one choose between them and allogeneic stem cell transplantation? Gene therapy does not require the immunosuppression. Nevertheless, experience with allogeneic transplantation is far greater, and matched-sibling transplants, especially in young children, provide excellent results.127 Gene therapy will also have to compete with the improving prospects for haploidentical allogeneic transplantation that could extend the proven effectiveness of allogeneic transplantation.128 Myeloablative conditioning is required for all cell-based therapeutics but has been associated with myelodysplasia; other forms of conditioning might be available in the future.129,130 Genomic editing does not use lentiviral transduction. Off-target consequences of genome editing, adverse effects of semirandom integration of lentivirus, and the sustainability of therapeutic effects need long-term study.
Conclusion
Cell-based therapeutics are developing rapidly. Perhaps in 10 years, when HbF is next reviewed, the current elegant efforts will be seen as crude approaches displaced by in vivo gene therapies with intravenous introduction of a therapeutic vector that homes to the targeted stem cell. Even better would be an affordable oral drug, or combination of drugs, that pancellularly raises HbF levels or otherwise prevents HbS polymerization.
Acknowledgments
This study was supported in part by the National Heart, Lung, and Blood Institute, National Institutes of Health grants R01 HL068970 and R01 HL133350 and the Office of Collaboration and Knowledge Exchange, University of Dammam, Saudi Arabia grant SP 11/2011.
Authorship
Contribution: M.H.S. wrote the paper.
Conflict-of-interest disclosure: M.H.S. sits on the advisor board for Vertex/CRISPR and Fulcrum Therapeutics and on the data monitoring committee for Imara.
Correspondence: Martin H Steinberg, Boston Medical Center, Room 211, Evans Building, 72 E Concord St, Boston, MA 02118; e-mail: mhsteinb@bu.edu.
REFERENCES
- 1.Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561-1573. [DOI] [PubMed] [Google Scholar]
- 2.Eaton WA, Hofrichter J. Hemoglobin S gelation and sickle cell disease. Blood. 1987;70(5):1245-1266. [PubMed] [Google Scholar]
- 3.Henry ER, Cellmer T, Dunkelberger EB, et al. Allosteric control of hemoglobin S fiber formation by oxygen and its relation to the pathophysiology of sickle cell disease. Proc Natl Acad Sci USA. 2020;117(26):15018-15027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017;129(20):2719-2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikawa Y, Miccio A, Magrin E, Kwiatkowski JL, Rivella S, Cavazzana M. Gene therapy of hemoglobinopathies: progress and future challenges. Hum Mol Genet. 2019;28(R1):R24-R30. [DOI] [PubMed] [Google Scholar]
- 7.Vinjamur DS, Bauer DE, Orkin SH. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br J Haematol. 2018;180(5):630-643. [DOI] [PubMed] [Google Scholar]
- 8.Blobel GA, Bodine D, Brand M, et al. An international effort to cure a global health problem: A report on the 19th Hemoglobin Switching Conference. Exp Hematol. 2015;43(10):821-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menzel S, Thein SL. Genetic modifiers of fetal haemoglobin in sickle cell disease. Mol Diagn Ther. 2019;23(2):235-244. [DOI] [PubMed] [Google Scholar]
- 10.Orkin SH, Bauer DE. Emerging genetic therapy for sickle cell disease. Annu Rev Med. 2019;70(1):257-271. [DOI] [PubMed] [Google Scholar]
- 11.Lettre G, Bauer DE. Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet. 2016;387(10037):2554-2564. [DOI] [PubMed] [Google Scholar]
- 12.Wienert B, Martyn GE, Funnell APW, Quinlan KGR, Crossley M. Wake-up sleepy gene: reactivating fetal globin for beta-hemoglobinopathies. Trends Genet. 2018;34(12):927-940. [DOI] [PubMed] [Google Scholar]
- 13.Lee YT, de Vasconcellos JF, Yuan J, et al. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood. 2013;122(6):1034-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197-1199. [DOI] [PubMed] [Google Scholar]
- 15.Sedgewick AE, Timofeev N, Sebastiani P, et al. BCL11A is a major HbF quantitative trait locus in three different populations with β-hemoglobinopathies. Blood Cells Mol Dis. 2008;41(3):255-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA. 2008;105(5):1620-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839-1842. [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Peng C, Sankaran VG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334(6058):993-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342(6155):253-257.24115442 [Google Scholar]
- 20.Liu N, Hargreaves VV, Zhu Q, et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173(2):430-442.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martyn GE, Wienert B, Yang L, et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet. 2018;50(4):498-503. [DOI] [PubMed] [Google Scholar]
- 22.Basak A, Munschauer M, Lareau CA, et al. Control of human hemoglobin switching by LIN28B-mediated regulation of BCL11A translation. Nat Genet. 2020;52(2):138-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lessard S, Beaudoin M, Orkin SH, Bauer DE, Lettre G. 14q32 and let-7 microRNAs regulate transcriptional networks in fetal and adult human erythroblasts. Hum Mol Genet. 2018;27(8):1411-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang P, Keller CA, Giardine B, et al. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 2017;31(16):1704-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masuda T, Wang X, Maeda M, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351(6270):285-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaikho EM, Habara AH, Alsultan A, et al. Variants of ZBTB7A (LRF) and its β-globin gene cluster binding motifs in sickle cell anemia. Blood Cells Mol Dis. 2016;59:49-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber L, Frati G, Felix T, et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. 2020;6:eaay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norton LJ, Funnell APW, Burdach J, et al. KLF1 directly activates expression of the novel fetal globin repressor ZBTB7A/LRF in erythroid cells. Blood Adv. 2017;1(11):685-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bieker JJ. Putting a finger on the switch. Nat Genet. 2010;42(9):733-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaikho EM, Farrell JJ, Chui DHK, Sebastiani P, Steinberg MH. Cis- and trans-acting expression quantitative trait loci differentially regulate gamma-globin gene expression. bioRxiv. 2018. 10.1101/304899 [Google Scholar]
- 31.Gardner K, Fulford T, Silver N, et al. g(HbF): a genetic model of fetal hemoglobin in sickle cell disease. Blood Adv. 2018;2(3):235-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shriner D, Rotimi CN. Whole-genome-sequence-based haplotypes reveal single origin of the sickle allele during the Holocene wet phase. Am J Hum Genet. 2018;102(4):547-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laval G, Peyrégne S, Zidane N, et al. Recent adaptive acquisition by African rainforest hunter-gatherers of the late Pleistocene sickle-cell mutation suggests past differences in malaria exposure. Am J Hum Genet. 2019;104(3):553-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagel RL, Steinberg MH. Genetics of the βs gene: origins, genetic epidemiology, and epistasis in sickle cell anemia In: Steinberg M, Forget BG, Higgs DR, Nagel RL, eds.. Disorders of Hemoglobin: Genetics, Pathophysiology, Clinical Management, Cambridge, United Kingdom: Cambridge University Press; 2001:711-755. [Google Scholar]
- 35.Al-Ali AK, Alsulaiman A, Alzahrani AJ, et al. Prevalence and diversity of haplotypes of sickle cell disease in the Eastern Province of Saudi Arabia. Hemoglobin. 2020;44(2):78-81. [DOI] [PubMed] [Google Scholar]
- 36.Alsultan A, Solovieff N, Aleem A, et al. Fetal hemoglobin in sickle cell anemia: Saudi patients from the Southwestern province have similar HBB haplotypes but higher HbF levels than African Americans. Am J Hematol. 2011;86(7):612-614. [DOI] [PubMed] [Google Scholar]
- 37.Perrine RP, Brown MJ, Clegg JB, Weatherall DJ, May A. Benign sickle-cell anaemia. Lancet. 1972;2(7788):1163-1167. [DOI] [PubMed] [Google Scholar]
- 38.Alsultan A, Alabdulaali MK, Griffin PJ, et al. Sickle cell disease in Saudi Arabia: the phenotype in adults with the Arab-Indian haplotype is not benign. Br J Haematol. 2014;164(4):597-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sebastiani P, Farrell JJ, Alsultan A, et al. BCL11A enhancer haplotypes and fetal hemoglobin in sickle cell anemia. Blood Cells Mol Dis. 2015;54(3):224-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vathipadiekal V, Alsultan A, Baltrusaitis K, et al. Homozygosity for a haplotype in the HBG2-OR51B4 region is exclusive to Arab-Indian haplotype sickle cell anemia. Am J Hematol. 2016;91(6):E308-E311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vathipadiekal V, Farrell JJ, Wang S, et al. A candidate transacting modulator of fetal hemoglobin gene expression in the Arab-Indian haplotype of sickle cell anemia. Am J Hematol. 2016;91(11):1118-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Ali ZA, Fallatah RK, Aljaffer EA, et al. ANTXR1 intronic variants are associated with fetal hemoglobin in the Arab-Indian haplotype of sickle cell disease. Acta Haematol. 2018;140(1):55-59. [DOI] [PubMed] [Google Scholar]
- 43.Khandros E, Huang P, Peslak SA, et al. Understanding heterogeneity of fetal hemoglobin induction through comparative analysis of F and A-erythroblasts. Blood. 2020;135(22):1957-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bertles JF, Milner PF. Irreversibly sickled erythrocytes: a consequence of the heterogeneous distribution of hemoglobin types in sickle-cell anemia. J Clin Invest. 1968;47(8):1731-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dover GJ, Boyer SH, Charache S, Heintzelman K. Individual variation in the production and survival of F cells in sickle-cell disease. N Engl J Med. 1978;299(26):1428-1435. [DOI] [PubMed] [Google Scholar]
- 46.Horiuchi K, Osterhout ML, Ohene-Frempong K. Survival of F-reticulocytes in sickle cell disease. Biochem Biophys Res Commun. 1995;217(3):924-930. [DOI] [PubMed] [Google Scholar]
- 47.Franco RS, Yasin Z, Palascak MB, Ciraolo P, Joiner CH, Rucknagel DL. The effect of fetal hemoglobin on the survival characteristics of sickle cells. Blood. 2006;108(3):1073-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maier-Redelsperger M, Noguchi CT, de Montalembert M, et al. Variation in fetal hemoglobin parameters and predicted hemoglobin S polymerization in sickle cell children in the first two years of life: Parisian Prospective Study on Sickle Cell Disease. Blood. 1994;84(9):3182-3188. [PubMed] [Google Scholar]
- 49.Horiuchi K, Osterhout ML, Kamma H, Bekoe NA, Hirokawa KJ. Estimation of fetal hemoglobin levels in individual red cells via fluorescence image cytometry. Cytometry. 1995;20(3):261-267. [DOI] [PubMed] [Google Scholar]
- 50.Ngo DA, Aygun B, Akinsheye I, et al. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol. 2012;156(2):259-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood. 1985;65(1):183-189. [PubMed] [Google Scholar]
- 52.Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123(4):481-485. [DOI] [PubMed] [Google Scholar]
- 53.Perrine RP, Pembrey ME, John P, Perrine S, Shoup F. Natural history of sickle cell anemia in Saudi Arabs. A study of 270 subjects. Ann Intern Med. 1978;88(1):1-6. [DOI] [PubMed] [Google Scholar]
- 54.Perrine RP, John P, Pembrey M, Perrine S. Sickle cell disease in Saudi Arabs in early childhood. Arch Dis Child. 1981;56(3):187-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87(8):795-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nkya S, Mgaya J, Urio F, et al. Fetal hemoglobin is associated with peripheral oxygen saturation in sickle cell disease in Tanzania. EBioMedicine. 2017;23:146-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383-1389. [DOI] [PubMed] [Google Scholar]
- 60.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644. [DOI] [PubMed] [Google Scholar]
- 61.Maitra P, Caughey M, Robinson L, et al. Risk factors for mortality in adult patients with sickle cell disease: a meta-analysis of studies in North America and Europe. Haematologica. 2017;102(4):626-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fitzhugh CD, Hsieh MM, Allen D, et al. Hydroxyurea-Increased fetal hemoglobin Is associated with less organ damage and longer survival in adults with sickle cell anemia. PLoS One. 2015;10(11):e0141706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boulassel M-R, Al-Badi A, Elshinawy M, et al. Hemoglobin F as a predictor of health-related quality of life in children with sickle cell anemia. Qual Life Res. 2019;28(2):473-479. [DOI] [PubMed] [Google Scholar]
- 64.Brousse V, El Hoss S, Bouazza N, et al. Prognostic factors of disease severity in infants with sickle cell anemia: A comprehensive longitudinal cohort study. Am J Hematol. 2018;93(11):1411-1419. [DOI] [PubMed] [Google Scholar]
- 65.Estepp JH, Smeltzer MP, Kang G, et al. A clinically meaningful fetal hemoglobin threshold for children with sickle cell anemia during hydroxyurea therapy. Am J Hematol. 2017;92(12):1333-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288-294. [PubMed] [Google Scholar]
- 67.Kinney TR, Sleeper LA, Wang WC, et al. ; The Cooperative Study of Sickle Cell Disease . Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. Pediatrics. 1999;103(3):640-645. [DOI] [PubMed] [Google Scholar]
- 68.van der Land V, Mutsaerts HJ, Engelen M, et al. Risk factor analysis of cerebral white matter hyperintensities in children with sickle cell disease. Br J Haematol. 2016;172(2):274-284. [DOI] [PubMed] [Google Scholar]
- 69.Sommet J, Alberti C, Couque N, et al. Clinical and haematological risk factors for cerebral macrovasculopathy in a sickle cell disease newborn cohort: a prospective study. Br J Haematol. 2016;172(6):966-977. [DOI] [PubMed] [Google Scholar]
- 70.Calvet D, Tuilier T, Mélé N, et al. Low fetal hemoglobin percentage is associated with silent brain lesions in adults with homozygous sickle cell disease. Blood Adv. 2017;1(26):2503-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mmbando BP, Mgaya J, Cox SE, et al. Negative epistasis between sickle and foetal haemoglobin suggests a reduction in protection against malaria. PLoS One. 2015;10(5):e0125929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Purohit P, Patel S, Mohanty PK, Das P, Panigrahi J. Fetal hemoglobin modifies the disease manifestation of severe Plasmodium Falciparum malaria in adult patients with sickle cell anemia. Mediterr J Hematol Infect Dis. 2016;8(1):e2016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tshilolo L, Tomlinson G, Williams TN, et al. ; REACH Investigators . Hydroxyurea for children with sickle cell anemia in Sub-Saharan Africa. N Engl J Med. 2019;380(2):121-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manning JM, Manning LR, Dumoulin A, Padovan JC, Chait B. Embryonic and fetal human hemoglobins: structures, oxygen binding, and physiological roles. Subcell Biochem. 2020;94:275-296. [DOI] [PubMed] [Google Scholar]
- 75.Summarell CC, Sheehan VA. Original Research: Use of hydroxyurea and phlebotomy in pediatric patients with hemoglobin SC disease. Exp Biol Med (Maywood). 2016;241(7):737-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luchtman-Jones L, Pressel S, Hilliard L, et al. Effects of hydroxyurea treatment for patients with hemoglobin SC disease. Am J Hematol. 2016;91(2):238-242. [DOI] [PubMed] [Google Scholar]
- 77.Di Maggio R, Hsieh MM, Zhao X, et al. Chronic administration of hydroxyurea (HU) benefits caucasian patients with sickle-beta thalassemia. Int J Mol Sci. 2018;19(3):E681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood. 2010;115(12):2354-2363. [DOI] [PubMed] [Google Scholar]
- 79.Deshpande SV, Bhatwadekar SS, Desai P, et al. Hydroxyurea in sickle cell disease: our experience in Western India. Indian J Hematol Blood Transfus. 2016;32(2):215-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.John CC, Opoka RO, Latham TS, et al. Hydroxyurea dose escalation for sickle cell anemia in Sub-Saharan Africa. N Engl J Med. 2020;382(26):2524-2533. [DOI] [PubMed] [Google Scholar]
- 81.Charache S, Terrin ML, Moore RD, et al. ; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia . Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332(20):1317-1322. [DOI] [PubMed] [Google Scholar]
- 82.Steinberg MH, McCarthy WF, Castro O, et al. ; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia and MSH Patients’ Follow-Up . The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. 2010;85(6):403-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood. 1996;88(12):4701-4710. [PubMed] [Google Scholar]
- 84.Hayes RJ, Beckford M, Grandison Y, Mason K, Serjeant BE, Serjeant GR. The haematology of steady state homozygous sickle cell disease: frequency distributions, variation with age and sex, longitudinal observations. Br J Haematol. 1985;59(2):369-382. [DOI] [PubMed] [Google Scholar]
- 85.West MS, Wethers D, Smith J, Steinberg M; The Cooperative Study of Sickle Cell Disease . Laboratory profile of sickle cell disease: a cross-sectional analysis. J Clin Epidemiol. 1992;45(8):893-909. [DOI] [PubMed] [Google Scholar]
- 86.Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ; Multicenter Study of Hydroxyurea . Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Blood. 1997;89(3):1078-1088. [PubMed] [Google Scholar]
- 87.Wang WC, Ware RE, Miller ST, et al. ; BABY HUG investigators . Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hankins JS, Aygun B, Nottage K, et al. From infancy to adolescence: fifteen years of continuous treatment with hydroxyurea in sickle cell anemia. Medicine (Baltimore). 2014;93(28):e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McGann PT, Niss O, Dong M, et al. Robust clinical and laboratory response to hydroxyurea using pharmacokinetically guided dosing for young children with sickle cell anemia. Am J Hematol. 2019;94(8):871-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ware RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387(10019):661-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Paikari A, Sumazin P, et al. Metformin induces FOXO3-dependent fetal hemoglobin production in human primary erythroid cells. Blood. 2018;132(3):321-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han J, Saraf SL, Molokie RE, Gordeuk VR. Use of metformin in patients with sickle cell disease. Am J Hematol. 2018;94(1):E15-E17. [DOI] [PubMed] [Google Scholar]
- 93.Badawy SM, Payne AB. Association between clinical outcomes and metformin use in adults with sickle cell disease and diabetes mellitus. Blood Adv. 2019;3(21):3297-3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moutouh-de Parseval LA, Verhelle D, Glezer E, et al. Pomalidomide and lenalidomide regulate erythropoiesis and fetal hemoglobin production in human CD34+ cells. J Clin Invest. 2008;118(1):248-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dulmovits BM, Appiah-Kubi AO, Papoin J, et al. Pomalidomide reverses γ-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood. 2016;127(11):1481-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shi L, Cui S, Engel JD, Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med. 2013;19(3):291-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cui S, Lim KC, Shi L, et al. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood. 2015;126(3):386-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Le CQ, Myers G, Habara A, et al. Inhibition of LSD1 by small molecule inhibitors stimulates fetal hemoglobin synthesis. Blood. 2019;133(22):2455-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rivers A, Vaitkus K, Ibanez V, et al. The LSD1 inhibitor RN-1 recapitulates the fetal pattern of hemoglobin synthesis in baboons (P. anubis). Haematologica. 2016;101(6):688-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Renneville A, Van Galen P, Canver MC, et al. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood. 2015;126(16):1930-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Molokie R, Lavelle D, Gowhari M, et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Med. 2017;14(9):e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McArthur JG, Svenstrup N, Chen C, et al. A novel, highly potent and selective phosphodiesterase-9 inhibitor for the treatment of sickle cell disease. Haematologica. 2020;105(3):623-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Charnigo RJ, Beidler D, Rybin D, et al. PF‐04447943, a phosphodiesterase 9A inhibitor, in stable sickle cell disease patients: A phase Ib randomized, placebo‐controlled study. Clin Transl Sci. 2019;12(2):180-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chambers CB, Gross J, Pratt K, et al. The mRNA-binding protein IGF2BP1 restores fetal hemoglobin in cultured erythroid cells from patients with β-hemoglobin disorders. Mol Ther Methods Clin Dev. 2020;17:429-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Breda L, Motta I, Lourenco S, et al. Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood. 2016;128(8):1139-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Deng W, Rupon JW, Krivega I, et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158(4):849-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krishnamoorthy S, Pace B, Gupta D, et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI Insight. 2017;2(20):96409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dai Y, Shaikho EM, Perez J, et al. BCL2L1 is associated with γ-globin gene expression. Blood Adv. 2019;3(20):2995-3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dai Y, Chen T, Ijaz H, Cho EH, Steinberg MH. SIRT1 activates the expression of fetal hemoglobin genes. Am J Hematol. 2017;92(11):1177-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bosquesi PL, Melchior ACB, Pavan AR, et al. Synthesis and evaluation of resveratrol derivatives as fetal hemoglobin inducers. Bioorg Chem. 2020;100:103948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang P, Peslak SA, Lan X, et al. The HRI-regulated transcription factor ATF4 activates BCL11A transcription to silence fetal hemoglobin expression. Blood. 2020;135(24):2121-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grevet JD, Lan X, Hamagami N, et al. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science. 2018;361(6399):285-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu X, Azzo A, Bilinovich SM, et al. Disruption of the MBD2-NuRD complex but not MBD3-NuRD induces high level HbF expression in human adult erythroid cells. Haematologica. 2019;104(12):2361-2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci USA. 1979;76(2):670-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pawliuk R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294(5550):2368-2371. [DOI] [PubMed] [Google Scholar]
- 116.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479-1493. [DOI] [PubMed] [Google Scholar]
- 117.Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848-855. [DOI] [PubMed] [Google Scholar]
- 118.Kanter J, Tisdale JF, Mapare M, et al. Resolution of sickle cell disease manifestations in patients treated with lentiglobin gene therapy: updates results from the phase 1/2 HGB-206 group C study. Blood. 2019;134(suppl 1):990. [Google Scholar]
- 119.Brendel C, Negre O, Rothe M, et al. Preclinical evaluation of a novel lentiviral vector driving lineage-specific BCL11A knockdown for sickle cell gene therapy. Mol Ther Methods Clin Dev. 2020;17:589-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Esrick EB, Achebe M, Armant M, et al. Validation of BCL11A as therapeutic target in sickle cell disease: results from the adult cohort of a pilot/feasibility gene therapy trial inducing sustained expression of fetal hemoglobin using post-transcriptional gene silencing. Blood. 2019;134(suppl 2):LBA-5. [Google Scholar]
- 121.Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578(7794):229-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368(6488):290-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Porteus MH. Genome Editing for the β-Hemoglobinopathies. Adv Exp Med Biol. 2017;1013:203-217. [DOI] [PubMed] [Google Scholar]
- 124.Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood. 2016;127(7):839-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Corbacioglu S, Cappellini MD, Chapin J, et al. Initial safety and efficacy results with a single dose of autologous crispr-cas9 modified CD34+ hematopoietic stem and progenitor cells in transfusion-dependent β-thalassemia and sickle cell disease. Eur Soc Haematol. 2020;EHA25:S280. [Google Scholar]
- 126.Métais JY, Doerfler PA, Mayuranathan T, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3(21):3379-3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Krishnamurti L. Should young children with sickle cell disease and an available human leukocyte antigen identical sibling donor be offered hematopoietic cell transplantation? Hematol Oncol Stem Cell Ther. 2020;13(2):53-57. [DOI] [PubMed] [Google Scholar]
- 128.Leonard A, Tisdale J, Abraham A. Curative options for sickle cell disease: haploidentical stem cell transplantation or gene therapy? Br J Haematol. 2020;189(3):408-423. [DOI] [PubMed] [Google Scholar]
- 129.Hsieh MM, Bonner M, Pierciey FJ, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4(9):2058-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gao C, Schroeder JA, Xue F, et al. Nongenotoxic antibody-drug conjugate conditioning enables safe and effective platelet gene therapy of hemophilia A mice. Blood Adv. 2019;3(18):2700-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]