

Abstract
The domestic Nili-Ravi water buffalo (Bubalus bubalis) is the best dairy animal contributing 68% to total milk production in Pakistan. In this study, we identified genome-wide single nucleotide polymorphisms (SNPs) to estimate various population genetic parameters such as diversity, pairwise population differentiation, linkage disequilibrium (LD) distribution and for genome-wide association study for milk yield and body weight traits in the Nili-Ravi dairy bulls that they may pass on to their daughters who are retained for milking purposes. The genotyping by sequencing approach revealed 13,039 reference genome-anchored SNPs with minor allele frequency of 0.05 among 167 buffalos. Population structure analysis revealed that the bulls were grouped into two clusters (K = 2), which indicates the presence of two different lineages in the Pakistani Nili-Ravi water buffalo population, and we showed the extent of admixture of these two lineages in our bull collection. LD analysis revealed 4169 significant SNP associations, with an average LD decay of 90 kb for these buffalo genome. Genome-wide association study involved a multi-locus mixed linear model for milk yield and body weight to identify genome-wide male effects. Our study further illustrates the utility of the genotyping by sequencing approach for identifying genomic regions to uncover additional demographic complexity and to improve the complex dairy traits of the Pakistani Nili-Ravi water buffalo population that would provide the lot of economic benefits to dairy industry.
Introduction
The domestic Nili-Ravi water buffalo (Bubalus bubalis) is a well-used dairy animal in livestock in Pakistan because it contributes about 68% of the total milk production in Pakistan [1]. Besides this, it is more adaptive to environment such as heat, disease and environmental stress and a better converter of poor quality roughages into nutritious and quality food [1, 2]. There is a high variation in phenotypic attributes, which indicates that further enhancement might be possible. Milk yield of the Nili-Ravi breed is commonly associated with many influential factors; however, the breed has high milk potential as compared with other buffalo species in Asia [2]. This variability indicates the possibility of using genomic selection to improve milk traits in future breeding programs. Some bulls excel in certain economic traits but not all. All the selected bulls for this study have been made available for artificial insemination, and so these bulls were genetically tested for various traits deemed important in milk production and body weight [3].
Several genetic studies for milk yield exist and a meta-analysis suggested that a majority of female traits that contributed to milk yield in dairy cattle tend to be lowly heritable (0.02 to 0.04) when compared to the studies involving male animals with mean heritability estimates between 0.05 and 0.22 [4]. Though superior genetic effects in both males and females are fundamental to breed for high milk production, male genetics is more informative as breeding values of AI bulls can be estimated with lesser bias [5].
Advances in high-throughput sequencing technologies provide the ability to identify genome-wide diversity and bottlenecks in domestication. Superior gene pools can be enriched to reproduce buffalos efficiently with the existing genomic resources and plan future conservation strategies [2, 6]. Genetic control of any trait specifies the interaction of environmental and genetic variance [7]. Inbreeding could cause further loss of genetic diversity and could accumulate deleterious alleles in the genomes, thereby resulting in deterioration of the breed, reduced milk yield, calving performance, udder health, fertility and survivability [8, 9]. Inbreeding gives rise to continuous segments of homozygous genotypes that are linked to inbreeding depression [10]. For example, one percent increase in FROH (association between increased inbreeding based on ROH) was associated with 20 kg decrease in 205-days milk yield [11], and also with 0.4-days extended insemination interval in heifers [12]. Informed decisions based on molecular knowledge could result in selecting diverse and superior animals for sustaining milk production.
With next generation sequencing (NGS), genome-wide single nucleotide polymorphisms (SNPs) can be developed affordably and can be effectively used for genome analysis of various traits in unexplored livestock such as water buffalo of Pakistan. A normal dam typically conceives on average up to 5 or 6 times in whole life-span but once an elite bull is effectively identified, its ability to produce a large number of offspring in life-time can be maximized by using artificial insemination. The detection and development of genome-wide SNP markers are essential tools for mapping and analyses of quantitative traits for superior bull selection [13–15].
Recently, many genotyping by sequencing (GBS) studies have been conducted on dairy breeds because it is a highly multiplexed and simple approach to construct NGS libraries [16–23]. De Donato et al. [24] used the GBS approach for genotyping 47 cattle breeds from Africa and the United States, producing 1.4 million unique reads per animal, and identified 63,697 SNPs. The authors concluded that the GBS approach is flexible, novel, sufficiently high-throughput and capable of providing acceptable genetic markers for genomic selection and genome-wide association study (GWAS). Ibeagha et al. [25] used the GBS approach to identify population-specific SNPs for use in improving complex dairy traits. Akanno et al. [26] reported seven SNPs with significant dominance associations for birth, weaning and yearling weight. Surya et al. [27] annotated the genome-wide SNPs of B. bubalis and reported 436 SNPs in 38 genes affecting fertility and 483 SNPs in 66 genes affecting milk production. Gao et al. [20] used SNP genotyping and identified two novel SNPs associated with body measurement traits of cattle.
The present study aimed to identify the chromosome-based distribution of SNPs, resolve the genetic diversity, and examine linkage disequilibrium (LD) and population structure for use in GWAS of body weight and milk yield traits in Nili-Ravi buffalo bulls from Pakistan to determine the quantity of specific traits that they may pass to their daughters. In addition, we resolved chromosome-based population demography and bottlenecks.
Materials and methods
The animal study was reviewed and approved by institutional guidelines of ethical review committee, University of Veterinary and Animal Sciences, Lahore, Pakistan.
Population and phenotypes
In total, 167 bulls were randomly selected for this study comprising single generation. Out of 167 breeding bulls, 155 bulls were from Semen Production Unit, Qadirabad, District Sahiwal, Punjab, Pakistan and other animals were from various other private dairy farms of Punjab in Pakistan. Animals selected for the study had no history of genital infections [28] and had diverse physical parameters with reference to the length from shoulder to pin-bone, wither height, height at sacrum, width between angles of the hip, and girth and width of the face above the eyes. In addition, bulls were selected based on reproductive parameters (scrotal circumference and scrotal length). All animals were dewormed and vaccinated regularly for foot-and-mouth disease, black quarter and haemorrhagic septicaemia. Blood samples were collected by a competent veterinary surgeon after the regular quarantine of animals. Source and phenotypic details of bulls in the study are in S1 Table.
Genotyping by sequencing
Genomic DNA for genotyping assay was isolated from blood samples of Nili-Ravi breeding bulls by using Qiagen columns (QIAamp DNA minikit, Qiagen, Hilden, Germany). The quantity of extracted DNA was measured by using the Qubit 3 Fluorometer with a double-stranded DNA HS assay kit (Invitrogen by Thermo Fischer Scientific, USA). GBS was performed as described [29, 30]. Briefly, total DNA from all samples was digested with ApeKI to reduce genome complexity. Digested fragments were ligated to common and barcode adapters with appropriate overhangs. Ligated products were then further pooled and amplified with compatible primers. Amplified sample pools constituted a sequencing “library”. After PCR reaction, pooled samples were gel-purified; a GBS “library” distribution of fragment sizes was checked by using BioAnalyzer (Agilent Technologies, USA). The GBS library was considered appropriate for sequencing reaction if the number of adapter dimers (approximately 128 bp in length) was minimal and the length of most of the genomic DNA fragment lengths was 170 to 350 bp. Templates were then quantified and diluted perfectly for sequencing reactions by using NextSeq 500 (Illumina, San Diego, CA).
Raw sequence reads were trimmed with use of Sickle (https://github.com/najoshi/sickle), adopting a minimum PHRED quality threshold of 20 in a sliding window. Then reads were demultiplexed and barcodes were removed. Trimmed reads were aligned to the B. bubalis reference sequences (www.ncbi.nlm.nih.gov/biosample/SAMN08640746) with Burrows-Wheeler Aligner [29]. Aligned reads were processed to identify SNPs by using SAMtools [31, 32] and barcode reads were handled and interpreted into a distinct sequence tag set, with a single TagCounts file generated per input file of FASTQ. We used TASSEL-GBS, an efficient bioinformatics pipeline considered for processing raw sequence dataset into a variant genotype file [33].
Genetic diversity and population structure analysis
Expected nucleotide diversity (θ per bp) and Tajima’s D for various chromosomes were estimated with sliding-window analysis by using TASSEL v5.0 as described [34]. Estimation of fixation index (FST) was based on Wright’s F statistic [35] with use of SNP and Variation Suite (SVS) v8.1.5. For quantitative assessment of the number of groups in the panel, we used a Bayesian clustering analysis with a model-based approach implemented in STRUCTURE v2.2 [36]. This approach involves use of multi-locus genotypic data to assign individuals to clusters or groups (K) without prior knowledge of their population affinities. The program was run with SNP markers for K values 1 to 9 (hypothetical number of subgroups), with 100,000 burn-in iterations, followed by 500,000 Markov Chain Monte Carlo iterations for accurate parameter estimates with a high-performance cluster. To verify the consistency of the results, we used 3 independent runs for each K. An admixture model with correlated allele frequencies was used. The optimal K value was determined by use of an ad-hoc statistic, ΔK [37]. The number of Ks in each dataset was evaluated by ΔK values estimated with the software Structure Harvester, a website and program for visualizing STRUCTURE output and implementing the Evanno method. In a second approach, we used principal component analysis (PCA) with SVS v7.7.6 (Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com). Female genetics was not included in the present study, which is in similar lines with several reports [3, 38–42].
Linkage disequilibrium (LD) analysis
For GBS study, we considered only SNPs that were successfully mapped to the B. bubalis whole genome sequence (WGS) draft to avoid spurious LD and incorrect association mapping. Before analyzing LD distribution, a block of the haplotypes was called for all adjacent SNPs by using SVS v8.1.5. Adjacent and pairwise measurements of LD were calculated separately for each chromosome. For computing LD, we used an expectation-maximization (EM) algorithm, formalized by Dempster et al. [43], an iterative technique for obtaining maximum likelihood estimates of sample haplotype frequencies.
Identification of runs of homozygosity (ROH)
ROHs were estimated using SNP and Variation Suite (SVS) that uses a sliding window to identify both ROHs and consecutive stretches that contain a minimum number of homozygous SNPs, at a minimum pre-specified distance. With this method, the software carries out basic detections of homozygous stretches identified by the sliding window, and the user only needs to define the parameter “minimum size” of segments to be identified [44].
GWAS mapping
GWAS comprises a multiple-locus mixed linear model that utilizes a backward and forward stepwise manner to identify genetic markers in the model as fixed-effects covariates and applied in SVS v8.1.5 [45]. Manhattan plots were evaluated in Genome Browse v1.0 (Golden Helix) for significantly associated SNPs. The SNP P-values from GWAS underwent sequential Bonferroni adjustments [46] and false discovery rate (FDR) study [47, 48]. Gene ontology and annotation terms for the SNP-containing sequences for each trait were identified with the WGS draft of the B. bubalis genome assembly.
Results
Genotyping of water buffalo collection and variation identification
We genotyped 167 water buffalo samples by GBS, which generated approximately 3.71 billion reads of 75 bp long. The median number of reads for each sample was 1.5 million (range ~100K to 11.0 million). Good barcoded tags with at least 10 read counts, corresponding to ~1.75 billion reads, were obtained and used for SNP calling. The 1.75-billion GBS reads were aligned to the B. bubalis reference genome (version 1; https://www.ncbi.nlm.nih.gov/genome/791?genomeassemblyid=374666), with 98% of the unique tags mapped to the genome. From the alignments, we identified 114,760 SNPs. After retaining SNPs with ≤5 0% missing data (representing at least 617 accessions) and minor allele frequency (MAF) ≥ 0.01 (i.e., SNPs present in at least 7 accessions), we obtained 24,319 SNPs distributed across the B. bubalis reference genome, with an average of one SNP per 10.6 kb. Only eight regions >500 kb in the reference genome were not covered by SNPs, because these eight regions were centromeric or pericentromeric. The MAF distribution of these SNPs is shown in S1 Fig. Among the 24,319 SNPs with ≤ 50% missing data and MAF ≥ 0.01, only biallelic SNPs were retained in the final SNP dataset.
Chromosome-wise SNPs
When screening for 70% call rate with 0.05 MAF, we isolated 13,039 SNPs for the 167 bulls studied; 659, 482, 507, 417, 847, 254, 317, 270, 285, 4566, 231, 230, 279, 694, 166, 236, 293, 149, 220, 145, 124, 158, 189, 89 and 1229 were mapped to the WGS draft and located on chromosomes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, X and Y, respectively.
Genetic diversity
Using the final SNP dataset, we performed PCA, which illustrated two clusters and some outliers (Fig 1 and S2 Table). Our results are consistent with those from Lu et al. [49], who also classified water buffalos into two primary groups, with some outliers. To validate the results of PCA, we analyzed the population structure of the water buffalo with the Bayesian clustering algorithm implemented in the STUCTURE program.
Fig 1. Principal component analysis (PCA) of 13,039 single nucleotide polymorphisms (SNPs) in 167 Pakistani Nili-Ravi breeding bulls.
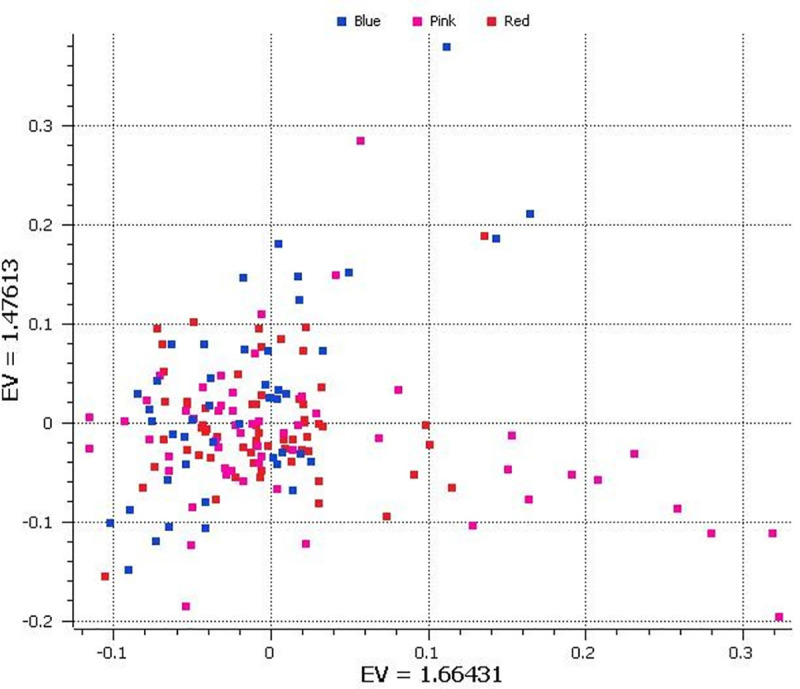
Colors are representative of high (blue), medium (pink) and low (red) milk yield bulls, majorly (93%) based on Semen Production Unit, Qadirabad, District Sahiwal, Pakistan while others are collected from different dairy farms of Punjab in Pakistan. Each point represents an individual animal and position of the points can be identified using Eigen values in S2 Table.
ΔK analysis showed that two populations (K = 2) represented the best number of clusters for these 167 bulls (Fig 2 and S1 Fig). As shown in Fig 2, at K = 2, bulls were grouped into two groups, which indicated the presence of two lineages in Pakistan and the extent of admixture of these two lineages (S2 Fig). The population structure at this optimal K was consistent with the PCA results; all suggested two primary clusters in the Pakistan water buffalo bull collection. These two lineages indicated that Nili-Ravi is crossbred buffalo originated from the Nili and River-type buffalos.
Fig 2. Two clusters inferred from the Pakistani Nili-Ravi population by using STRUCTURE analysis.

Nucleotide diversity
Because of the strong population structure, we assessed patterns of expected nucleotide diversity (θ) and Tajima’s D separately for each chromosome to understand genome-wide bottleneck effects. Such information will be of immense use when inferring the evolutionary dynamics of domestication across the water buffalo genome. Water buffalo domestication is often associated with “population bottlenecks” because of the limited number of founding individuals experiencing domestication events. The frequency of segregating SNPs, as reflected by various chromosomal measures of mean θ and Tajima’s D, is presented in Fig 3. For water buffalo, chromosomes 5, 10 and 14 showed reduced nucleotide diversity as compared with the remaining chromosomes. For chromosome 10, Tajima’s D was also reduced along with mean θ, which indicates purifying selection as compared with the other chromosomes. Chromosome 21 showed increased nucleotide diversity and Tajima’s D, which indicates the accumulation of rapid mutations on this chromosome.
Fig 3. Frequency spectrum for chromosomal means for expected nucleotide diversity (θ) and Tajima’s D for Nili-Ravi breeding bulls to understand genome-wide bottleneck effects.
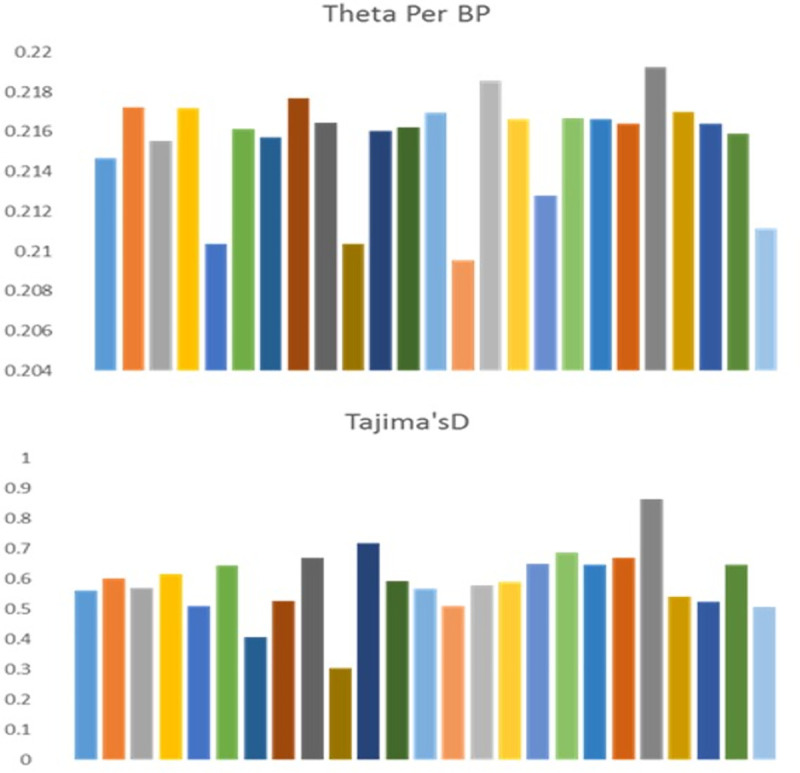
It shows highly variable nucleotide diversity and Tajima’s D across various chromosomes.
LD and recombination rate analysis across the chromosomes
Distribution of LD is essential to understand patterns of historical recombination and the non-random association of loci. We performed LD analysis for the whole dataset of water buffalo breeding bulls, involving all the adjacent pairs of SNPs mapped to various chromosomes (Fig 4). For computing LD, we used the EM algorithm [43] as an iterative technique for obtaining maximum likelihood estimates of sample haplotype frequencies. The number of associations mean LD block and maximum LD block sizes for various chromosomes are presented in Table 1. As expected, the largest LD block was in chromosome Y, because the Y chromosome in mammals is fixed and will not undergo recombination. In this study, we identified 4169 adjacent SNP pairs in LD, with an average LD decay at 90 kb for the Pakistan water buffalo genome.
Fig 4. Linkage disequilibrium decay as measured by r2 averaged in distance intervals across the 25 Nili-Ravi bull chromosomes.
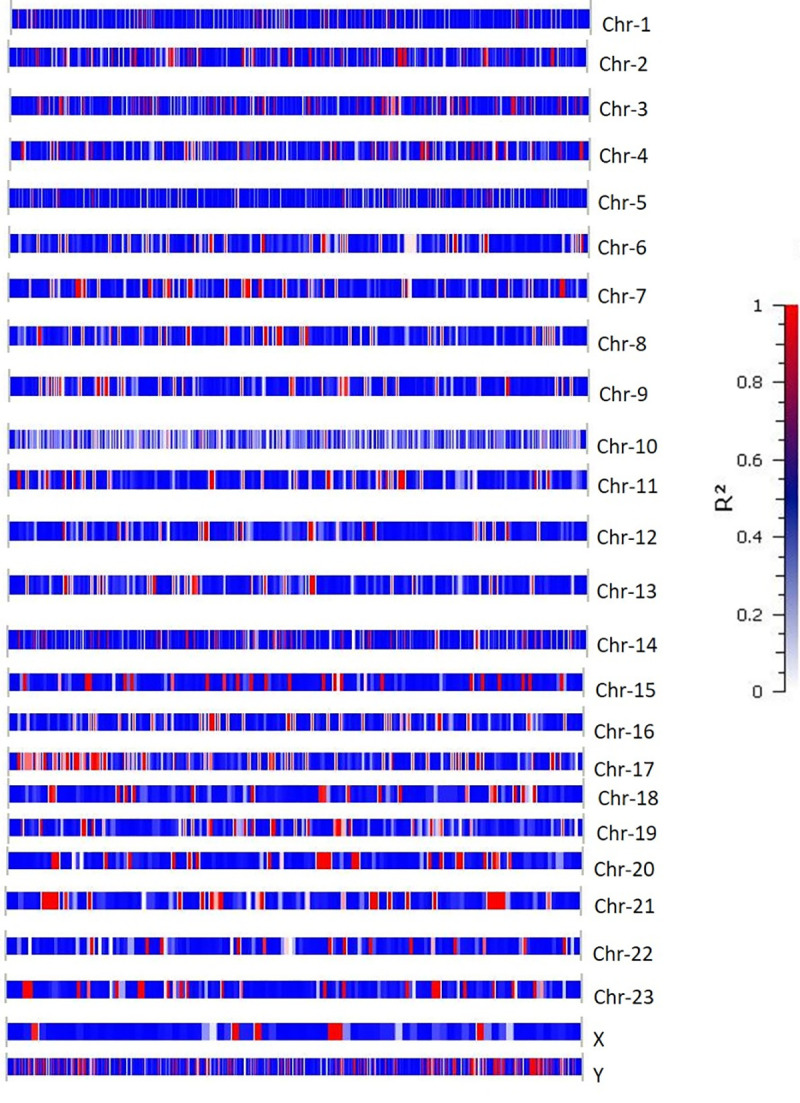
Each LD bar represents a chromosome, and individual LD blocks are colored based on the scale presented.
Table 1. Chromosome-wise distribution of linkage disequilibrium (LD) blocks for Nili-Ravi.
| Chromosome number | No. of Associations | Mean LD Block size (Kb) | Maximum LD Block size (Kb) |
|---|---|---|---|
| 1 | 243 | 65.45 | 2068.56 |
| 2 | 179 | 100.46 | 3047.04 |
| 3 | 169 | 63.43 | 3715.18 |
| 4 | 156 | 109 | 1598.18 |
| 5 | 221 | 47.47 | 2895.55 |
| 6 | 100 | 110.17 | 1793.58 |
| 7 | 101 | 102.76 | 3821.12 |
| 8 | 94 | 109.24 | 2068.86 |
| 9 | 93 | 122.87 | 2611.62 |
| 10 | 1231 | 13.53 | 3259.68 |
| 11 | 84 | 103.8 | 3043.47 |
| 12 | 63 | 116.018 | 2032.92 |
| 13 | 98 | 53.43 | 1425.26 |
| 14 | 204 | 27.52 | 1445.27 |
| 15 | 50 | 117.41 | 2297.35 |
| 16 | 85 | 81.23 | 2490.50 |
| 17 | 132 | 51.64 | 1773.93 |
| 18 | 51 | 86.14 | 2075.73 |
| 19 | 91 | 56.24 | 2481.07 |
| 20 | 47 | 170.13 | 4782.65 |
| 21 | 52 | 159.45 | 2232.41 |
| 22 | 51 | 67.072 | 1182.09 |
| 23 | 65 | 150.68 | 3362.06 |
| X | 27 | 117.03 | 1098.19 |
| Y | 482 | 51.73 | 16734.94 |
| Overall | 4169 | 90.156 |
Distribution of ROH and haplotype blocks
The genome-wide pattern of ROH was determined to examine local homozygosity in Pakistan water buffalo bulls (Fig 5 and S3 Table). ROH regions can be critically analyzed to identify susceptibility/deleterious loci across the genome where there has been significant evolutionary pressure. ROH regions form as a result of selective sweeps around the gene candidates that have evolutionary significance. Large regions of homozygous SNPs can be common between subpopulations without a direct common lineage, and these regions can be areas of functional significance [50].https://doc.goldenhelix.com/SVS/latest/svsmanual/references.html#lencz2007 In this study, we resolved ROH regions on chromosomes 1, 3, 5, 10, 16 and Y of varied lengths. The Y chromosome showed the highest ROH islands, which is expected, as in other mammals [51].
Fig 5. Incidence of each single nucleotide polymorphism (SNP) in a run of homozygosity (ROH) across the Nili-Ravi water buffalo chromosomes.
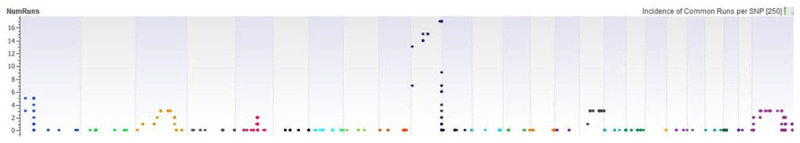
The X-axis shows the distribution of ROH across the genome and Y-axis represents the frequency (%) of overlapping ROH shared among samples. See S3 Table for a list of ROH across all the chromosomes.
Characterization of selection sweeps and domestication signature
A highly significant pairwise FST (P<0.001) distribution with low and high milk yield and body weight for the buffalo bulls is presented in Fig 6 and S4 Table. These sliding windows allowed for identifying differential evolutionary signatures across the genome for milk yield and body weight of the bulls. Furthermore, the patterns of FST variation indicated genomic areas with the signatures of positive selection and sweeps across various chromosomes. The pairwise analysis involving low and high bull groups showed higher genetic differentiation relative to low and high milk yield rather than body weight. Selection signatures detected loci with larger effects under strong selection on chromosomes 10, 18 and 20 for milk yield with 0.4 cut-off value (Fig 6A). The sliding window of pairwise FST for low and high body-weight bulls showed higher positive selection signals on chromosomes 7 and 18 with 0.35 cut-off value (Fig 6B).
Fig 6. Highly significant pairwise FST (P<0.001) distribution (low vs. high) for a) milk yield and b) body weight across the Nili-Ravi buffalo chromosomes.

SNPs were plotted relative to their physical positions within each autosome. Individual FST values are in S4 Table.
GWAS for milk yield and body weight
A Multi-Locus Mixed Linear model was implemented by using the EMMAX algorithm that involves stepwise regression and Byes Information Criteria (BIC) [45] to identify significantly associated SNPs for milk yield and body weight. Variance plots (Figs 7A and 8A) include partitioned error, genetic variance, variance explained in the current model and variance explained by covariates. Partition of variance explained in current study for milk yield and body weight was in the range of 70% to 80%. The–log10 P-values for the tested SNPs from GWAS analyses for both traits are shown as Manhattan plots (Figs 7B and 8B). All significant SNPs identified by GWAS showed cumulative phenotypic variance of 14% for milk yield and 20% for body weight. On chromosome 10, 14 SNPs were strongly associated with both milk yield and body weight. These 14 SNPs formed a tag, which is a representative in a region of the genome with high linkage disequilibrium. Such tag SNPs would reduce time and expense of mapping the genome areas associated with milk yield and body weight, because they eliminate the need to study each SNP [52]. The tag we identified on chromosome 10 explained the phenotypic variance of 14% and 22% for milk yield and body weight, respectively.
Fig 7. Genome-wide association results for dam's yield (liters) in Nili-Ravi buffalo.

a) Variance plots partitioned error, genetic variance, variance explained in current model and variance explained by covariates. b) Manhattan plot of the chromosome coordinates on the X-axis, with the negative log-10 of the association P-value for each SNP on the Y-axis. High negative log-10 indicates strong association with milk yield.
Fig 8. Genome-wide association results for body weight (Kg) by mixed linear model analysis.

a) Variance plots partitioned error, genetic variance, variance explained in current model and variance explained by covariates. b) Manhattan plot: the black line represents the threshold at −log 10 P-value and plots above this line are significantly associated with body weight.
P-values, regression beta and minor and major allelic effects are presented for milk yield and body weight in Tables 2 and 3. Level of significance, allelic effects and phenotypic variance explained for the other milk yield-associated SNPs on chromosomes 1, 3, 5, 8, 9, 12, 13, 16, 18, X and Y are also presented. A total of 14% and 85% of the significant SNPs exhibited positive contribution for milk yield and body weight, respectively. For milk yield, significant SNPs present on chromosomes 3 and 8 participated in positive contribution. For body weight, chromosomes 1, 6, 7, 9, 10, 14 and Y positively contributed to the phenotypic variance. For body weight, each significant SNP accounted for about 16% for chromosome 1, 18% for chromosomes 6 and 9, 21% for chromosome 7, 22% for chromosome 10, and 19% for chromosome Y. Independently, all SNPs for milk yield showed a phenotypic variance of 12% for chromosomes 1, 5 and 8; 13% for chromosomes 3, 12, 18 and Y; 16% for chromosomes 9 and 16; 14% for chromosome 10 and X; and 15% for chromosome 13.
Table 2. Major SNPs pertaining to milk yield in Nili-Ravi.
| SNP | Chr | P-Value | -LOG10 (P-Value) | Regression Beta | Beta Standard Error | Prop. Var. Explained | Minor Allele (Test Allele)→Major Allele | Minor Allele D Frequency |
|---|---|---|---|---|---|---|---|---|
| S1_158054912 | 1 | 0.00083 | 3.08 | -1627.16 | 468.80 | 0.13 | T→A | 0.06 |
| S3_172820617 | 3 | 0.000322 | 3.49 | -1768.04 | 470.74 | 0.15 | T→C | 0.06 |
| S3_55703175 | 3 | 0.000724 | 3.14 | 791.81 | 225.39 | 0.13 | G→A | 0.39 |
| S5_76106561 | 5 | 0.000757 | 3.12 | -1092.39 | 312.16 | 0.13 | T→C | 0.09 |
| S8_2495291 | 8 | 0.000884 | 3.05 | 829.70 | 240.40 | 0.13 | T→A | 0.49 |
| S8_2495296 | 8 | 0.000884 | 3.05 | 829.70 | 240.40 | 0.13 | G→C | 0.49 |
| S8_2495349 | 8 | 0.000884 | 3.05 | 829.70 | 240.40 | 0.13 | C→T | 0.49 |
| S9_4493004 | 9 | 0.000118 | 3.93 | -1888.13 | 466.93 | 0.17 | C→G | 0.07 |
| S9_4493022 | 9 | 0.000118 | 3.93 | -1888.13 | 466.93 | 0.17 | T→C | 0.07 |
| S10_104237148 | 10 | 0.000654 | 3.18 | -1216.83 | 343.36 | 0.13 | T→G | 0.06 |
| S10_104295834 | 10 | 0.000627 | 3.20 | -1031.78 | 290.09 | 0.13 | A→T | 0.15 |
| S10_104295869 | 10 | 0.000627 | 3.20 | -1031.78 | 290.09 | 0.13 | T→G | 0.15 |
| S10_104332442 | 10 | 9.22E-05 | 4.04 | -1521.84 | 369.98 | 0.17 | C→G | 0.07 |
| S10_104379763 | 10 | 0.000744 | 3.13 | -930.90 | 265.64 | 0.13 | G→A | 0.16 |
| S10_104401718 | 10 | 0.000302 | 3.52 | -1037.95 | 274.97 | 0.15 | T→A | 0.15 |
| S10_104401719 | 10 | 0.000302 | 3.52 | -1037.95 | 274.97 | 0.15 | A→T | 0.15 |
| S10_104401762 | 10 | 0.000302 | 3.52 | -1037.95 | 274.97 | 0.15 | C→G | 0.15 |
| S10_104410613 | 10 | 0.000207 | 3.68 | -1377.10 | 354.59 | 0.16 | C→G | 0.07 |
| S10_104422767 | 10 | 0.000481 | 3.32 | -1731.56 | 476.12 | 0.14 | A→T | 0.08 |
| S10_104434319 | 10 | 0.000632 | 3.20 | -948.36 | 266.81 | 0.13 | A→G | 0.23 |
| S10_104440997 | 10 | 0.000264 | 3.58 | -1297.72 | 340.24 | 0.15 | T→C | 0.08 |
| S10_104484172 | 10 | 0.000111 | 3.95 | -1278.47 | 314.77 | 0.17 | C→A | 0.11 |
| S10_104514430 | 10 | 0.000726 | 3.14 | -1077.40 | 306.76 | 0.13 | A→G | 0.11 |
| S12_75087502 | 12 | 0.000166 | 3.78 | -1688.66 | 427.71 | 0.16 | T→G | 0.07 |
| S13_18088155 | 13 | 0.00021 | 3.68 | -1449.74 | 373.59 | 0.16 | T→C | 0.05 |
| S16_3831231 | 16 | 0.00015 | 3.83 | -1866.75 | 469.36 | 0.16 | G→A | 0.05 |
| S18_39186829 | 18 | 0.000708 | 3.15 | -1045.17 | 296.96 | 0.13 | T→C | 0.09 |
| S24_1645 | 24 | 0.000372 | 3.43 | -1418.84 | 382.08 | 0.14 | C→T | 0.06 |
| S25_42968291 | 25 | 0.000649 | 3.19 | -1235.26 | 348.32 | 0.13 | G→T | 0.08 |
Table 3. Association results pertaining to body weight in Nili-Ravi.
| SNP | Chr | P-Value | -LOG10 | Regression Beta | Beta Standard Error | Prop. Var. Explained | SNP allele | Minor Allele Frequency |
|---|---|---|---|---|---|---|---|---|
| (P-Value) | ||||||||
| S1_38497677 | 1 | 0.000926 | 3.03 | 151.94 | 43.60 | 0.17 | G→T | 0.06 |
| S6_67621520 | 6 | 0.000469 | 3.33 | 78.10 | 21.10 | 0.19 | A→T | 0.26 |
| S7_114945813 | 7 | 0.000128 | 3.89 | 136.36 | 33.29 | 0.22 | G→A | 0.10 |
| S9_17523778 | 9 | 0.000528 | 3.28 | 96.66 | 26.38 | 0.18 | C→T | 0.15 |
| S10_104244729 | 10 | 8.13E-05 | 4.09 | 119.56 | 28.27 | 0.23 | G→C | 0.15 |
| S10_104244743 | 10 | 8.13E-05 | 4.09 | 119.56 | 28.27 | 0.23 | C→T | 0.15 |
| S10_104244754 | 10 | 8.13E-05 | 4.09 | 119.56 | 28.27 | 0.23 | A→T | 0.15 |
| S10_104303781 | 10 | 5.74E-05 | 4.24 | -146.72 | 33.88 | 0.24 | T→G | 0.11 |
| S10_104392764 | 10 | 0.000151 | 3.82 | 95.22 | 23.53 | 0.21 | C→A | 0.35 |
| S10_26780436 | 10 | 0.00033 | 3.48 | 83.16 | 21.83 | 0.19 | T→A | 0.19 |
| S14_83340556 | 14 | 0.000112 | 3.95 | 140.95 | 34.09 | 0.22 | G→T | 0.13 |
| S25_110079376 | 25 | 0.000134 | 3.87 | 141.97 | 34.77 | 0.22 | A→T | 0.41 |
| S25_115449301 | 25 | 0.000756 | 3.12 | -106.37 | 29.96 | 0.17 | C→A | 0.18 |
Some of the important gene candidates associated with milk yield were gamma-secretase subunit APH-1B pseudogene/Rho guanine nucleotide exchange factor 26 (S1_158054912), 26S proteasome non-ATPase regulatory subunit 5 (S3_172820617), Coilin (S3_55703175), uncharacterized LOC112586670/3-beta-hydroxysteroid-Delta(8), Delta(7)-isomerase pseudogene (S8_2495291, S8_2495296 and S8_2495349), kelch-like family member 29 (S12_75087502), multidrug resistance-associated protein 4-like (S13_18088155), fatty acid desaturase 2-like protein FADS2P1/olfactory receptor 5G3-like (S16_3831231), MARVEL domain-containing 3/U6 spliceosomal RNA (S18_39186829), MAPK-interacting and spindle-stabilizing protein-like (S24_1645) and EF-hand domain-containing 2 (S25_42968291). Apart from the strongly associated tag on chromosome 10, other SNPs associated with body weight were on chromosomes 1, 6, 7, 9, 14, X and Y. Levels of significance, allelic effects and phenotypic variance for the body weight-associated SNPs are also presented in Table 3. Notable candidate genes in the genomic regions associated with body weight were alpha-N-acetylgalactosaminide alpha-2,6-sialyltransferase 3 (S6_67621520), U6 spliceosomal RNA/Bardet-Biedl syndrome 7 (S7_114945813), FAM172A family with sequence similarity 172 member A (S9_17523778), glutaredoxin-related protein 5, mitochondrial (S14_83340556), spermatid nuclear transition protein 3-like (S25_110079376) and Bombesin receptor subtype 3/Protein CXorf40A-like (S25_115449301).
Discussion
Nili-Ravi bulls are currently classified as a half herd and are an important source of milk in Pakistan [2, 53]. Because of the narrow gene pool among this herd, transferring superior alleles to progeny is important. Previous genotyping methods using low density marker systems had limitations in understanding admixture and genome-wide diversity [54]. This is the first report of the use of genome-wide SNPs generated using GBS to estimate population structure and demographic history across various chromosomes in Nili-Ravi bulls. In addition, this study is important to breeders because we identified several genomic regions associated with milk yield and body weight that could help in genetic improvement of these important economic traits. Boison et al. [55] summarized various advantages of using genomic methods for breeding buffalo bulls.
Among the 167 animals included in this study, we noted two different lineages based on admixture analysis. Similar findings were also found by Lu et al. [49] for Chinese indigenous buffalo breeds. We further noted highly variable nucleotide diversity and Tajima’s D across various chromosomes, which also agrees with previous studies of Asian buffalo breeds that used microsatellite [56–58] and mitochondrial markers [59, 60]. For example, chromosome 10 showed reduced nucleotide diversity and Tajima’s D, and chromosome 21 indicated higher nucleotide diversity and Tajima’s D.
Parameters such as diversity, Tajima’s D pattern, LD decay and ROH address various aspects of population demography and help in understanding the dynamics of genome-wide bottleneck effects for predicting genetic gain for commercial traits. LD is a random association of alleles in subpopulations and is an important parameter for LD mapping or GWAS [61–64]. One description for such varying LD would be the “Bulmer effect,” whereby eminent LD-carrying regions are mostly clustered with unique sweeps harboring crucial genes underlying domestication [65, 66]. In this study, LD analysis revealed 4169 significant SNP associations, with an average LD decay of 90 kb for the Pakistani Nili-Ravi population. We observed the maximum LD score for chromosome 10 and largest LD block for chromosome Y. Similar results observed the LD decline at approximately 50 kb, with an average r2 ≥ 0.2 in Chinese crossbred buffalo groups [Jianghan × Nili-Ravi × Mediterranean and Nili-Ravi × Murrah × local (Fuzhong or Xilin)] [67] and an r2 value from 0.412 to 0.139 in Indian Nili-Ravi buffalo [68], which is much lower than that found in our study. However, comparing our findings directly with previous reports is difficult because of differences in the scale of sampling, given that the level of LD decreases with increasing sample size [69]. Recently, the pattern of LD decay was reported in various cattle and buffalo breeds across the world, including an Iranian water buffalo population at an average of 57 kb decay [70], Chinese buffalo breeds at 50 kb for the river group and 15 kb for the swamp group [49], Tunisian cattle at 73.4 kb [71], Korean cattle breeds from 26 to 32 kb [72] and Colombian Creole cattle breeds from 70 to 100 kb [73].
We performed GWAS using genome-wide SNPs for several novel candidate genes related to milk yield and body weight traits that are significant breeding goals for dairy industry. Gamma-secretase subunit APH-1B pseudogene/Rho guanine nucleotide exchange factor 26 gene (Gene ID: S1_158054912) present on chromosome 1 encodes a large protein that functions as a GDP-to-GTP exchange factor and is reported to be a significant candidate gene for the maternal effect on weaning weight and milk yield traits in cattle [74–76]. In this study, chromosome 3 harbored at least two genes that affect milk yield. The first is 26S proteasome non-ATPase regulatory subunit 5 (Gene ID: S3_172820617). The second is Coilin (Gene ID: S3_55703175) and is one of the main molecular components of Cajal bodies. Both of these gene members are involved in protein-mediated degradation found in mammary glands of cows, and this mechanism is involved in milk production of dairy cows [77, 78]. The formation of various cellular bodies and small nuclear ribonucleoproteins depends on specific structural proteins, such as Coilin in Cajal bodies [79, 80]. A subset of nuclear bodies uses specific long noncoding RNA (lncRNA) as their scaffolding molecule and is required for mammary gland development and lactation [81, 82].
Uncharacterized LOC112586670/3-beta-hydroxysteroid-Delta(8) is one of the major enzymes that participates early in lactation in dairy cattle. An lncRNA (LOC112586670) fragment is a candidate gene associated with buffalo milk production [49, 74, 83–86]. From functional analysis of liver microarray data from mid-lactating Holstein cows, delta(7)-isomerase pseudogene (Gene ID: S8_2495291, S8_2495296, S8_2495349) is involved in the cholesterol biosynthesis pathway [87, 88]. This finding suggests that the candidate gene may contribute to milk metabolic pathways. Many studies have reported the kelch-like family member 29 (Gene ID: S12_75087502) involved in growth [89] and milk production in dairy cattle and buffalos [78, 90, 91]. Recently, it was also identified as the closest gene mapped by novel QTL-lead SNP for milking speed in French Holstein cattle, and T was an effect allele with a −0.08 allelic substitution effect of the SNP [92]. Multidrug resistance-associated protein 4-like (Gene ID: S13_18088155) is a member of C subfamily of ATP-binding cassette (ABC) transporters. ABCC4 acts in basic metabolic pathways and is highly polymorphic, with potential effects in a variety of phenotypes, and has also been detected in milk during early lactation [74, 91, 93–95]. The genetic effects of ABCG2 polymorphism have been observed for milk yield traits in Chinese Holstein cattle [96, 97]. Alim et al. [98] reported that the A allele was the more frequent in Chinese Holstein cattle (0.53), followed by the C allele (0.47). The authors identified that the CC genotype was associated with increased milk yield and protein percentage during the first and second lactation, whereas the C-A genotype decreased protein yield, so the C allele is associated with superior milk performance [97, 98], which is in partial agreement with our findings. Multidrug resistance-associated protein 4-like may be a novel candidate gene associated with milk yield.
In the bovine, fatty acid desaturase 2-like protein FADS2P1/olfactory receptor 5G3-like (Gene ID: S16_3831231) is a potential genetic marker for fatty acid composition in cattle milk [99–101]. In Jersey cattle, the major allele was A, and significant associations were recorded for the milk lauric, behenic, lignoceric, oleic, eicosatrienoic, and docosadienoic fatty acids. In Polish Holstein-Friesian cows, the G allele was prevalent, and significant associations were observed for erucic and docosahexaenoic acids [99]. MARVEL domain-containing 3/U6 spliceosomal RNA (Gene ID: S18_39186829) could be a novel marker related to milk trait because on relative expression analysis, its closest members (RNU6B, RNU5A and RNU1A) were used to assess the suitability of a combination of small nuclear RNAs as a reference RNA in milk somatic cells of lactating yaks [91, 102, 103]. MAPK-interacting and spindle-stabilizing protein-like (Gene ID: S24_1645) is involved in regulating milk synthesis [91, 104–106]. EF-hand domain-containing 2 (Gene ID: S25_42968291) is one of the most common calcium-binding structural motifs, and a well-defined helix-loop-helix structural domain, present in many calcium-binding proteins, has been identified in milk [107–110]. Our results support these important insights into the synthesis of milk proteins and potential targets for the future improvement of milk quality.
We identified alpha-N-acetylgalactosaminide alpha-2,6-sialyltransferase 3 (Gene ID: S6_67621520) as a novel marker for body weight, and it also contributes to protein glycosylation for milk synthesis [111–113]. U6 spliceosomal RNA/Bardet-Biedl syndrome 7 (Gene ID: S7_114945813) is associated with a rare hereditary disorder that has several phenotypic features including retinopathy, obesity, polydactyly and hypogenitalism. Among them, obesity is the major clinical finding and the incidence is reported to be 72% to 86% in the Bardet-Biedl syndrome population [114, 115]. From previous findings, Bardet-Biedl syndrome 7 gene could be considered a good marker for body weight. FAM172A family with sequence similarity 172 member A (Gene ID: S9_17523778) plays an important role in cell proliferation and facilitates S-phase entry. Thus, this gene is important as a cell growth regulator [116, 117]. FAM172A is also reported as a candidate gene for CHARGE syndrome (heart defects, atresia of choanae, retardation of growth and genital abnormalities) [118]. Glutaredoxin-related protein 5, mitochondrial (Gene ID: S14_83340556) is required for normal cell growth [119, 120]. Loss of glutaredoxin 3 impedes mammary lobuloalveolar development during pregnancy and lactation [121]. Our study adds further support to the importance of this gene in relation to the body weight trait. Spermatid nuclear transition protein 3-like (Gene ID: S25_110079376) was found a new marker for body mass. Large testes in relation to body mass (relative testes mass) is a strong predictor of high sperm competition levels in many taxa [122]. This gene family member is essential in chromatin condensation during spermiogenesis and hence, the family members are candidate genes for identifying sperm motility markers [123] and for semen quality traits [124]. Bombesin receptor subtype 3/Protein CXorf40A-like (Gene ID: S25_115449301) has been studied in animals as an important regulator of body weight, energy expenditure and glucose homeostasis [125–127].
Several female fertility traits in dairy cattle showed reduced genetic correlations among milk production traits because of low or moderate heritabilities [4, 128]. The male lines received the greatest attention in selection because of accurate prediction of sire's heritability [129]. Estimated breeding values of AI bulls are considered to be more reliable for various predictions [130]. For this reason, many GWAS studies in dairy cattle has been carried out based on bulls’ performance for use in genomic selection programs. The use of female data in GS programs has raised some debatable issues such as readjustment of phenotypes for comparison with the bulls [131]. A study that identified 105 genome-wide significant SNPs for milk production traits was through a paternal transmission disequilibrium test that explored associations from the sire families [132]. In similar lines to the current study, Nayeri and Stothard [86] utilized 3,729 North American Holstein bulls for GWAS and a genome scan with 4280 Nordic Red Cattle bulls was used to identify loci affecting milk yields Touru et al. [133].
We also performed pairwise population FST (P<0.001) differentiation for low and high bulls for milk yield and body weight to understand pattern of selection [134]. All significant SNPs present on chromosomes 10 and 18, including MARVEL domain-containing 3/U6 spliceosomal RNA (S18_39186829), had high FST values indicating positive selection at these regions. Flori et al. [135] indicated reduced FST values for GHR gene in milk yield of cattle and also identified 13 highly significant genomic regions with varied pairwise FST subjected to recent and/or strong positive selection. For body weight in our study, chromosomes 7 and 18 had high positive selection signals.
Defining ROH in a complex pedigree can be a way to examine homozygosity levels across the genome that has resulted by inbreeding and selection [136]. In this study, we observed ROH regions across the buffalo genome on chromosomes 1, 3, 5, 10, 16 and Y, with varied lengths. Chromosome 10 showed the longest ROH indicating functional significance of several important milk yield associated loci. Similar results were reported in several cattle breeds: shorter ROH results from the crosses with distant haplotype relatedness, and recombination has had more time to trim down ROH that is the target of selection sweeps, whereas greater ROH length is mostly caused by recent inbreeding [137, 138]. We identified two significant SNPs (S5_76106561; S10_104237148) in ROH that were negatively associated with the milk yield trait, so those could be deleterious alleles and fixed in the population. Three other body weight associated SNPs (S10_104303781; S10_104392764; S25_110079376) are in the ROH region, S10_104303781 was negatively associated with the weight. Longer ROH had stronger linear relationship with the number of individuals that carry deleterious homozygous mutations reducing the biological fitness [51]. For example, a large number of genomic regions that contain long ROH manifested unfavorable associations because of deleterious mutations with milk yield in Holstein cattle [139].
Conclusion
Our study demonstrates the utility of the GBS approach for genetic analysis of complex dairy traits and for understanding the population structure of Pakistani Nili-Ravi buffalos. Our SNP analysis revealed that Nili-Ravi bulls are admixed with two lineages. Our GWAS for milk yield and body weight identified SNPs located in candidate genes previously identified in other dairy breeds. Results of this study suggested that male effects contribute strong genetic effects for milk yield. In future, favorable allele combinations from this study could be used to generate Nili-Ravi breeding bulls with high milk yield and body weight by selecting the bull with higher genetic merit is tantamount that will produce a daughter, who in-turn produces the greatest net revenue for dairy industry in terms of improved or superior commercial traits. Performance of their daughters will later be used to update these measures. Since these dairy bull data were publicly available, individual bull efficiency results can be disseminated to potential semen buyers.
Supporting information
Each individual is represented by a single vertical line divided into K colored segments, where K is the number of ancestral populations.
(TIF)
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability
The raw Illumina reads generated and/or analyzed during the current study are available in the Sequence Read Archive repository (NCBI-SRA) under the following accession number PRJNA604217.
Funding Statement
This research was funded by the Punjab Agricultural Research Board, Lahore [Project No-492], and Higher Education Commission Pakistan.
References
- 1.GOP. Agricultural census organization, statistics division, government of Pakistan In: Ali S, editor. Livestock census. Pakistan: Pakistan Bureau of Statistics; 2015. pp. 1–73. [Google Scholar]
- 2.Khan MS, Ahmad N, Khan MA. Genetic resources and diversity in dairy buffaloes of Pakistan. Pak Vet J. 2007; 27(4):201–7. [Google Scholar]
- 3.Whitt CE, Tauer LW, Huson H. Bull efficiency using dairy genetic traits. PLoS One. 2019; 14(11):e0223436 10.1371/journal.pone.0223436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berry D, Wall E, Pryce J. Genetics and genomics of reproductive performance in dairy and beef cattle. Animal 2014; 8(S1):105–21. 10.1017/S1751731114000743 [DOI] [PubMed] [Google Scholar]
- 5.Spelman R, Hayes BJ, Berry DP. Use of molecular technologies for the advancement of animal breeding: genomic selection in dairy cattle populations in Australia, Ireland and New Zealand. Animal Production Science 2013; 53:869–75. [Google Scholar]
- 6.Warriach HM, McGill DM, Bush RD, Wynn PC, Chohan KR. A review of recent developments in buffalo reproduction. Asian-Australas J Anim Sci. 2015; 28(3):451–5. 10.5713/ajas.14.0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan MS. Animal recording for improved breeding and management strategies of buffalo in Pakistan. Intern Comm Anim Record Tech Series. 2000; 4:21–3. [Google Scholar]
- 8.Parland SM, Kearney JF, Rath M, Berry DP. Inbreeding effects on milk production, calving performance, fertility, and conformation in Irish Holstein-Friesians. J Dairy Sci. 2007; 90(9):4411–9. 10.3168/jds.2007-0227 [DOI] [PubMed] [Google Scholar]
- 9.Santana ML Jr, Aspilcueta-Borquis RR, Bignardi AB, Albuquerque LG, Tonhati H. Population structure and effects of inbreeding on milk yield and quality of Murrah buffaloes. J Dairy Sci. 2011; 94(10):5204–11. 10.3168/jds.2011-4377 . [DOI] [PubMed] [Google Scholar]
- 10.Martikainen K, Koivula M, Uimari P. Identification of runs of homozygosity affecting female fertility and milk production traits in Finnish Ayrshire cattle. Sci Rep. 2020; 10(1):3804 10.1038/s41598-020-60830-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjelland D, Weigel K, Vukasinovic N, Nkrumah J. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J Dairy Sci. 2013; 96(7):4697–706. 10.3168/jds.2012-6435 [DOI] [PubMed] [Google Scholar]
- 12.Martikainen K, Tyrisevä A-M, Matilainen K, Pösö J, Uimari P. Estimation of inbreeding depression on female fertility in the Finnish Ayrshire population. J Anim Breed Genet. 2017;134(5):383–92. 10.1111/jbg.12285 [DOI] [PubMed] [Google Scholar]
- 13.Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009; 4(4):e5350 10.1371/journal.pone.0005350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snelling WM, Allan MF, Keele JW, Kuehn LA, McDaneld T, Smith TP, et al. Genome-wide association study of growth in crossbred beef cattle. J Anim Sci. 2010; 88(3):837–48. 10.2527/jas.2009-2257 . [DOI] [PubMed] [Google Scholar]
- 15.Peters SO, Kizilkaya K, Garrick DJ, Fernando RL, Reecy JM, Weaber RL, et al. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J Anim Sci. 2012; 90(10):3398–409. 10.2527/jas.2012-4507 . [DOI] [PubMed] [Google Scholar]
- 16.Abed A, Legare G, Pomerleau S, St-Cyr J, Boyle B, Belzile FJ. Genotyping-by-sequencing on the ion torrent platform in barley. Methods Mol Biol. 2019; 1900: 233–52. 10.1007/978-1-4939-8944-7_15 . [DOI] [PubMed] [Google Scholar]
- 17.Sasaki S, Muraki E, Inoue Y, Suezawa R, Nikadori H, Yoshida Y, et al. Genotypes and allele frequencies of buried SNPs in a bovine single-nucleotide polymorphism array in Japanese Black cattle. Anim Sci J. 2019; 90(12):1503–9. 10.1111/asj.13293 [DOI] [PubMed] [Google Scholar]
- 18.Nguyen Q, Tellam RL, Kijas J, Barendse W, Dalrymple BP. P1037 Predicting regulatory SNPs within enhancers and promoters in cattle. J Anim Sci. 2016; 94(suppl_4):32–3. 10.2527/jas2016.94supplement432a [DOI] [Google Scholar]
- 19.Upadhyay M, Bortoluzzi C, Barbato M, Ajmone-Marsan P, Colli L, Ginja C, et al. Deciphering the patterns of genetic admixture and diversity in southern European cattle using genome-wide SNPs. Evol Appl. 2019; 12(5):951–63. Epub 2019/05/14. 10.1111/eva.12770 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y, Huang B, Bai F, Wu F, Zhou Z, Lai Z, et al. Two novel SNPs in RET gene are associated with cattle body measurement traits. Animals 2019; 9(10):836 10.3390/ani9100836 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi JW, Choi BH, Lee SH, Lee SS, Kim HC, Yu D, et al. Whole-genome resequencing analysis of Hanwoo and Yanbian cattle to identify genome-wide SNPs and signatures of selection. Molecules and Cells 2015; 38(5):466–73. Epub 2015/05/29. 10.14348/molcells.2015.0019 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothammer S, Kunz E, Seichter D, Krebs S, Wassertheurer M, Fries R, et al. Detection of two non-synonymous SNPs in SLC45A2 on BTA20 as candidate causal mutations for oculocutaneous albinism in Braunvieh cattle. Genet Sel Evol. 2017; 49(1):73 Epub 2017/10/07. 10.1186/s12711-017-0349-7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang F, Qu K, Chen N, Hanif Q, Jia Y, Huang Y, et al. Genome-wide SNPs and indels characteristics of three Chinese cattle breeds. Animals 2019; 9(9):596 10.3390/ani9090596 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Donato M, Peters SO, Mitchell SE, Hussain T, Imumorin IG. Genotyping-by-sequencing (GBS): a novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS One 2013; 8(5):e62137 10.1371/journal.pone.0062137 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibeagha-Awemu EM, Peters SO, Akwanji KA, Imumorin IG, Zhao X. High density genome wide genotyping-by-sequencing and association identifies common and low frequency SNPs, and novel candidate genes influencing cow milk traits. Sci Rep. 2016; 6:31109 10.1038/srep31109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akanno EC, Chen L, Abo-Ismail MK, Crowley JJ, Wang Z, Li C, et al. Genome-wide association scan for heterotic quantitative trait loci in multi-breed and crossbred beef cattle. Genet Sel Evol. 2018; 50(1):48 10.1186/s12711-018-0405-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surya T, Vineeth MR, Sivalingam J, Tantia MS, Dixit SP, Niranjan SK, et al. Genomewide identification and annotation of SNPs in Bubalus bubalis. Genomics 2019; 111(6):1695–8. 10.1016/j.ygeno.2018.11.021 [DOI] [PubMed] [Google Scholar]
- 28.Islam S, Shehzad W, Bajwa AA, Imran M, Zahoor MY, Abdullah M, et al. Molecular detection of brucellosis, leptospirosis and campylobacteriosis by multiplex PCR and screening by ELISA assays in buffalo breeding bulls. Pak Vet J. 2020; 40(1):81–7. 10.29261/pakvetj/2019.092. [DOI] [Google Scholar]
- 29.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011; 6(5):e19379 10.1371/journal.pone.0019379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy UK, Nimmakayala P, Levi A, Abburi VL, Saminathan T, Tomason YR, et al. High-resolution genetic map for understanding the effect of genome-wide recombination rate on nucleotide diversity in watermelon. G3-Genes Genom Genet. 2014; 4(11):2219–30. 10.1534/g3.114.012815 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25(14):1754–60. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9. 10.1093/bioinformatics/btp352 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, et al. Tassel-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 2014; 9(2):e90346 10.1371/journal.pone.0090346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korneliussen TS, Moltke I, Albrechtsen A, Nielsen R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics 2013; 14(1):289 10.1186/1471-2105-14-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution 1984:1358–70. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- 36.Pritchard J, Stephens M, Rosenberg N, Donnelly P. Association mapping in structured populations. Am J HumGenet. 2000; 67(1):170–81. 10.1086/302959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005; 14(8):2611–20. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- 38.Gautier M, Faraut T, Moazami-Goudarzi K, Navratil V, Foglio M, Grohs C, et al. Genetic and haplotypic structure in 14 European and African cattle breeds. Genetics 2007; 177:1059–70. 10.1534/genetics.107.075804 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makina SO, Muchadeyi FC, van Marle-Köster E, MacNeil MD, Maiwashe A. Genetic diversity and population structure among six cattle breeds in South Africa using a whole genome SNP panel. Frontiers in Genetics 2014; 5:333 10.3389/fgene.2014.00333 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung N, Szyda J, Frąszczak M, Fries HR, Lund MS, Guldbrandtsen B, et al. Population structure analysis of Bull genomes of European and Western Ancestry. Sci Rep. 2017; 7:40688 10.1038/srep40688 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sermyagin AA, Dotsev AV, Gladyr EA, Traspov AA, Deniskova TE, Kostyunina OV, et al. Whole-genome SNP analysis elucidates the genetic structure of Russian cattle and its relationship with Eurasian taurine breeds. Genet Sel Evol. 2018; 50:37 10.1186/s12711-018-0408-8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M‐ H, Tapio I, Vilkki J, Ivanova Z, Kiselyova T, Marzanov, et al. The genetic structure of cattle populations (Bos taurus) in northern Eurasia and the neighbouring Near Eastern regions: implications for breeding strategies and conservation. Mol Ecol. 2007; 16:3839–53. 10.1111/j.1365-294X.2007.03437.x . [DOI] [PubMed] [Google Scholar]
- 43.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Series B Stat Methodol. 1977; 39(1):1–38. [Google Scholar]
- 44.Curik I, Ferenčaković M, Sölkner J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest Sci. 2014; 166:26–34. 10.1016/j.livsci.2014.05.034. [DOI] [Google Scholar]
- 45.Segura V, Vilhjalmsson BJ, Platt A, Korte A, Seren U, Long Q, et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet. 2012; 44(7):825–30. 10.1038/ng.2314 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holm S. A simple sequentially rejective multiple test procedure. ScandJ Stat. 1979; 6(2):65–70. [Google Scholar]
- 47.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995; 57(1):289–300. [Google Scholar]
- 48.Storey JD. A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodol. 2002; 64(3):479–98. 10.1111/1467-9868.00346 [DOI] [Google Scholar]
- 49.Lu X-R, Duan A-Q, Liang S-S, Ma X-Y, Liang X-W, Deng T-X. Genome-wide analysis reveals genetic diversity, linkage disequilibrium, and selection for milk traits in Chinese buffalo breeds. bioRxiv. 2019:701045 10.1101/701045 [DOI] [PubMed] [Google Scholar]
- 50.Lencz T, Lambert C, DeRosse P, Burdick KE, Morgan TV, Kane JM, et al. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Nat Acad Sci USA. 2007; 104(50):19942–7. 10.1073/pnas.0710021104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim ES, Cole JB, Huson H, Wiggans GR, Van Tassell CP, Crooker BA, et al. Effect of artificial selection on runs of homozygosity in US Holstein cattle. PLoS One 2013; 8(11):e80813 10.1371/journal.pone.0080813 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42(Database issue):D1001–6. 10.1093/nar/gkt1229 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bashir MK, Khan MS, Bhatti SA, Iqbal A. Lifetime performance of Nili-ravi buffaloes in Pakistan. Asian-Aust J Anim Sci. 2007; 20(5):661–8. [Google Scholar]
- 54.Cleveland MA, Hickey JM. Practical implementation of cost-effective genomic selection in commercial pig breeding using imputation. J Anim Sci. 2013; 91(8):3583–92. 10.2527/jas.2013-6270 . [DOI] [PubMed] [Google Scholar]
- 55.Boison SA, Santos DJA, Utsunomiya AHT, Carvalheiro R, Neves HHR, O’Brien AMP, et al. Strategies for single nucleotide polymorphism (SNP) genotyping to enhance genotype imputation in Gyr (Bos indicus) dairy cattle: comparison of commercially available SNP chips. J Dairy Sci. 2015; 98(7):4969–89. 10.3168/jds.2014-9213 [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Sun D, Yu Y, Zhang Y. Genetic variation and divergence among swamp buffalo, river buffalo and cattle: a microsatellite survey on five populations in China. Asian-Australas J Anim Sci. 2008; 21(9):1238–43. 10.5713/ajas.2008.70746 [DOI] [Google Scholar]
- 57.Barker JS, Moore SS, Hetzel DJ, Evans D, Tan SG, Byrne K. Genetic diversity of Asian water buffalo (Bubalus bubalis): microsatellite variation and a comparison with protein-coding loci. AnimGenet. 1997; 28(2):103–15. Epub 1997/04/01. 10.1111/j.1365-2052.1997.00085.x . [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Sun D, Yu Y, Zhang Y. Genetic diversity and differentiation of Chinese domestic buffalo based on 30 microsatellite markers. Anim Genet. 2007; 38(6):569–75. 10.1111/j.1365-2052.2007.01648.x [DOI] [PubMed] [Google Scholar]
- 59.Kathiravan P, Kataria RS, Mishra BP, Dubey PK, Sadana DK, Joshi BK. Population structure and phylogeography of Toda buffalo in Nilgiris throw light on possible origin of aboriginal Toda tribe of South India. J Anim Breed Genet. 2011; 128(4):295–304. 10.1111/j.1439-0388.2011.00921.x [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Lu Y, Yindee M, Li K-Y, Kuo H-Y, Ju Y-T, et al. Strong and stable geographic differentiation of swamp buffalo maternal and paternal lineages indicates domestication in the China/Indochina border region. Mol Ecol. 2016;25(7):1530–50. 10.1111/mec.13518 [DOI] [PubMed] [Google Scholar]
- 61.Flint-Garcia SA, Thornsberry JM, IV ESB. Structure of linkage disequilibrium in plants. Ann Rev Plant Biol. 2003; 54(1):357–74. 10.1146/annurev.arplant.54.031902.134907 . [DOI] [PubMed] [Google Scholar]
- 62.Nicolas SD, Péros J-P, Lacombe T, Launay A, Le Paslier M-C, Bérard A, et al. Genetic diversity, linkage disequilibrium and power of a large grapevine (Vitis vinifera L) diversity panel newly designed for association studies. BMC Plant Biol. 2016; 16(1):74 10.1186/s12870-016-0754-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bush WS, Moore JH. Genome-wide association studies. PLoS Comput Biol. 2012; 8(12):e1002822 10.1371/journal.pcbi.1002822 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin T, König S. Genome-wide associations and detection of potential candidate genes for direct genetic and maternal genetic effects influencing dairy cattle body weight at different ages. Genet Sel Evol. 2019; 51(1):4 10.1186/s12711-018-0444-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bulmer MG. The effect of selection on genetic variability. Am Nat. 1971; 105(943):201–11. 10.1086/282718 [DOI] [Google Scholar]
- 66.Kovi MR, Fjellheim S, Sandve SR, Larsen A, Rudi H, Asp T, et al. Population structure, genetic variation, and linkage disequilibrium in perennial ryegrass populations divergently selected for freezing tolerance. Front Plant Sci. 2015; 6(929). 10.3389/fpls.2015.00929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deng T, Liang A, Liu J, Hua G, Ye T, Liu S, et al. Genome-wide SNP data revealed the extent of linkage disequilibrium, persistence of phase and effective population size in purebred and crossbred buffalo populations. Front Genet. 2019; 9(688). 10.3389/fgene.2018.00688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thakor PB, Hinsu AT, Bhatiya DR, Shah TM, Nayee N, Sudhakar A, et al. High-throughput genotype based population structure analysis of selected buffalo breeds. bioRxiv. 2018:395681 10.1101/395681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968; 38(6):226–31. 10.1007/BF01245622 . [DOI] [PubMed] [Google Scholar]
- 70.Mokhber M, Shahrbabak MM, Sadeghi M, Shahrbabak HM, Stella A, Nicolzzi E, et al. Study of whole genome linkage disequilibrium patterns of Iranian water buffalo breeds using the Axiom Buffalo Genotyping 90K Array. PLoS One 2019; 14(5):e0217687 10.1371/journal.pone.0217687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jemaa SB, Thamri N, Mnara S, Rebours E, Rocha D, Boussaha M. Linkage disequilibrium and past effective population size in native Tunisian cattle. Genet Mol Biol. 2019; 42(1):52–61. 10.1590/1678-4685-GMB-2017-0342 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim S, Cheong HS, Shin HD, Lee SS, Roh HJ, Jeon DY, et al. Genetic diversity and divergence among Korean cattle breeds assessed using a BovineHD single-nucleotide polymorphism chip. Asian-Australas J Anim Sci. 2018; 31(11):1691–9. 10.5713/ajas.17.0419 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bejarano D, Martinez R, Manrique C, Parra LM, Rocha JF, Gomez Y, et al. Linkage disequilibrium levels and allele frequency distribution in Blanco Orejinegro and Romosinuano Creole cattle using medium density SNP chip data. Genet Mol Biol. 2018; 41(2):426–33. 10.1590/1678-4685-GMB-2016-0310 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reinhardt TA, Lippolis JD. Developmental changes in the milk fat globule membrane proteome during the transition from colostrum to milk. J Dairy Sci. 2008; 91(6):2307–18. 10.3168/jds.2007-0952 [DOI] [PubMed] [Google Scholar]
- 75.Michenet A, Barbat M, Saintilan R, Venot E, Phocas F. Detection of quantitative trait loci for maternal traits using high-density genotypes of Blonde d’Aquitaine beef cattle. BMC Genet. 2016; 17(1):88 10.1186/s12863-016-0397-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turner MD, Wilde CJ, Burgoyne RD. Exocytosis from permeabilized lactating mouse mammary epithelial cells. Stimulation by Ca2+ and phorbol ester, but inhibition of regulated exocytosis by guanosine 5'-[gamma-thio]triphosphate. Biochem J. 1992; 286(Pt 1):13–5. 10.1042/bj2860013 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pawlowski K, Pires JAA, Faulconnier Y, Chambon C, Germon P, Boby C, et al. Mammary gland transcriptome and proteome modifications by nutrient restriction in early lactation holstein cows challenged with intra-mammary lipopolysaccharide. Int J Mol Sci. 2019; 20(5). 10.3390/ijms20051156 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dai W, Wang Q, Zhao F, Liu J, Liu H. Understanding the regulatory mechanisms of milk production using integrative transcriptomic and proteomic analyses: improving inefficient utilization of crop by-products as forage in dairy industry. BMC Genomics 2018; 19(1):403 10.1186/s12864-018-4808-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tucker KE, Berciano MT, Jacobs EY, LePage DF, Shpargel KB, Rossire JJ, et al. Residual Cajal bodies in coilin knockout mice fail to recruit Sm snRNPs and SMN, the spinal muscular atrophy gene product. J Cell Biol. 2001; 154(2):293–307. 10.1083/jcb.200104083 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nizami ZF, Deryusheva S, Gall JG. Cajal bodies and histone locus bodies in Drosophila and Xenopus. Cold Spring Harb Symp Quant Biol. 2010; 75:313–20. 10.1101/sqb.2010.75.005 . [DOI] [PubMed] [Google Scholar]
- 81.Yamazaki T, Hirose T. The building process of the functional paraspeckle with long non-coding RNAs. Front Biosci. 2015; 7:1–41. 10.2741/715 . [DOI] [PubMed] [Google Scholar]
- 82.Standaert L, Adriaens C, Radaelli E, Van Keymeulen A, Blanpain C, Hirose T, et al. The long noncoding RNA Neat1 is required for mammary gland development and lactation. RNA 2014; 20(12):1844–9. 10.1261/rna.047332.114 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai W, Li C, Liu S, Zhou C, Yin H, Song J, et al. Genome wide identification of novel long non-coding RNAs and their potential associations with milk proteins in Chinese holstein cows. Front Genet. 2018; 9:281 10.3389/fgene.2018.00281 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hu J, Zhang Z, Shen WJ, Azhar S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr Metab. 2010; 7:47 10.1186/1743-7075-7-47 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004; 25(6):947–70. 10.1210/er.2003-0030 [DOI] [PubMed] [Google Scholar]
- 86.Nayeri S, Stothard P. Tissues, metabolic pathways and genes of key importance in lactating dairy cattle. Springer Sci Rev. 2016; 4(2):49–77. 10.1007/s40362-016-0040-3 [DOI] [Google Scholar]
- 87.Silvente-Poirot S, Dalenc F, Poirot M. The effects of cholesterol-derived oncometabolites on nuclear receptor function in cancer. Cancer Res. 2018; 78(17):4803–8. 10.1158/0008-5472.CAN-18-1487 . [DOI] [PubMed] [Google Scholar]
- 88.Akbar H, Bionaz M, Carlson DB, Rodriguez-Zas SL, Everts RE, Lewin HA, et al. Feed restriction, but not l-carnitine infusion, alters the liver transcriptome by inhibiting sterol synthesis and mitochondrial oxidative phosphorylation and increasing gluconeogenesis in mid-lactation dairy cows. J Dairy Sci. 2013; 96(4):2201–13. 10.3168/jds.2012-6036 [DOI] [PubMed] [Google Scholar]
- 89.Xu L, Yang L, Zhu B, Zhang W, Wang Z, Chen Y, et al. Genome-wide scan reveals genetic divergence and diverse adaptive selection in Chinese local cattle. BMC Genomics 2019; 20(1):494 10.1186/s12864-019-5822-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Iamartino D, Williams JL, Sonstegard TS, Reecy JM, Tassell CP, Nicolazzi EL, et al. The buffalo genome and the application of genomics in animal management and improvement. Buffalo Bull. 2013; 32(1):151–8. [Google Scholar]
- 91.Du C, Deng T, Zhou Y, Ye T, Zhou Z, Zhang S, et al. Systematic analyses for candidate genes of milk production traits in water buffalo (Bubalus bubalis). Anim Genet. 2019; 50(3):207–16. 10.1111/age.12739 . [DOI] [PubMed] [Google Scholar]
- 92.Marete A, Sahana G, Fritz S, Lefebvre R, Barbat A, Lund MS, et al. Genome-wide association study for milking speed in French Holstein cows. J Dairy Sci. 2018; 101(7):6205–19. 10.3168/jds.2017-14067 [DOI] [PubMed] [Google Scholar]
- 93.de Camargo GM, Aspilcueta-Borquis RR, Fortes MR, Porto-Neto R, Cardoso DF, Santos DJ, et al. Prospecting major genes in dairy buffaloes. BMC Genomics. 2015; 16:872 10.1186/s12864-015-1986-2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Blazquez AMG, Macias RIR, Cives-Losada C, de la Iglesia A, Marin JJG, Monte MJ. Lactation during cholestasis: role of ABC proteins in bile acid traffic across the mammary gland. Sci Rep. 2017; 7(1):7475 10.1038/s41598-017-06315-8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Delosière M, Pires J, Bernard L, Cassar-Malek I, Bonnet M. Milk proteome from in silico data aggregation allows the identification of putative biomarkers of negative energy balance in dairy cows. Sci Rep. 2019; 9(1):9718 10.1038/s41598-019-46142-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li C, Sun D, Zhang S, Yang S, Alim MA, Zhang Q, et al. Genetic effects of FASN, PPARGC1A, ABCG2 and IGF1 revealing the association with milk fatty acids in a Chinese Holstein cattle population based on a post genome-wide association study. BMC Genetics 2016; 17:110 10.1186/s12863-016-0418-x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yue W, Fang X, Zhang C, Pang Y, Xu H, Gu C, et al. Two novel SNPs of the ABCG2 gene and its associations with milk traits in Chinese Holsteins. Mol Biol Rep. 2011; 38(5):2927–32. 10.1007/s11033-010-9955-y . [DOI] [PubMed] [Google Scholar]
- 98.Alim MA, Xie Y, Fan Y, Wu X, Sun D, Zhang Y, et al. Genetic effects of ABCG2 polymorphism on milk production traits in the Chinese Holstein cattle. J Appl Anim Res. 2013; 41(3):333–8. 10.1080/09712119.2013.782873 [DOI] [Google Scholar]
- 99.Proskura WS, Liput M, Zaborski D, Sobek Z, Yu YH, Cheng YH, et al. The effect of polymorphism in the FADS2 gene on the fatty acid composition of bovine milk. Arch Anim Breed. 2019; 62(2):547–55. 10.5194/aab-62-547-2019 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rudolph MC, McManaman JL, Phang T, Russell T, Kominsky DJ, Serkova NJ, et al. Metabolic regulation in the lactating mammary gland: a lipid synthesizing machine. Physiol Genomics. 2007; 28(3):323–36. 10.1152/physiolgenomics.00020.2006 . [DOI] [PubMed] [Google Scholar]
- 101.Ibeagha-Awemu EM, Akwanji KA, Beaudoin F, Zhao X. Associations between variants of FADS genes and omega-3 and omega-6 milk fatty acids of Canadian Holstein cows. BMC Genetics 2014; 15:25 10.1186/1471-2156-15-25 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bai WL, Yin RH, Yang RJ, Khan WA, Ma ZJ, Zhao SJ, et al. Identification of suitable normalizers for microRNA expression analysis in milk somatic cells of the yak (Bos grunniens). J Dairy Sci. 2013; 96(7):4529–34. 10.3168/jds.2013-6603 [DOI] [PubMed] [Google Scholar]
- 103.Wang M, Moisa S, Khan MJ, Wang J, Bu D, Loor JJ. MicroRNA expression patterns in the bovine mammary gland are affected by stage of lactation. J Dairy Sci. 2012; 95(11):6529–35. 10.3168/jds.2012-5748 . [DOI] [PubMed] [Google Scholar]
- 104.Yu TC, Chen SE, Ho TH, Peh HC, Liu WB, Tiantong A, et al. Involvement of TNF-alpha and MAPK pathway in the intramammary MMP-9 release via degranulation of cow neutrophils during acute mammary gland involution. Vet Immunol Immunop. 2012; 147(3–4):161–9. 10.1016/j.vetimm.2012.04.011 . [DOI] [PubMed] [Google Scholar]
- 105.Lu LM, Li QZ, Huang JG, Gao XJ. Proteomic and functional analyses reveal MAPK1 regulates milk protein synthesis. Molecules 2012; 18(1):263–75. 10.3390/molecules18010263 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bhat SA, Ahmad SM, Ibeagha-Awemu EM, Bhat BA, Dar MA, Mumtaz PT, et al. Comparative transcriptome analysis of mammary epithelial cells at different stages of lactation reveals wide differences in gene expression and pathways regulating milk synthesis between Jersey and Kashmiri cattle. PLoS One 2019; 14(2):e0211773 10.1371/journal.pone.0211773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ha M, Sabherwal M, Duncan E, Stevens S, Stockwell P, McConnell M, et al. In-depth characterization of sheep (Ovis aries) milk whey proteome and comparison with cow (Bos taurus). PLoS One 2015; 10(10):e0139774 10.1371/journal.pone.0139774 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Makina SO, Muchadeyi FC, van Marle-Koster E, Taylor JF, Makgahlela ML, Maiwashe A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet Sel Evol. 2015; 47:92 10.1186/s12711-015-0173-x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pisanu S, Cacciotto C, Pagnozzi D, Puggioni GMG, Uzzau S, Ciaramella P, et al. Proteomic changes in the milk of water buffaloes (Bubalus bubalis) with subclinical mastitis due to intramammary infection by Staphylococcus aureus and by non-aureus staphylococci. Sci Rep. 2019; 9(1):15850 10.1038/s41598-019-52063-2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Grzybowska EA. Calcium-binding proteins with disordered structure and their role in secretion, storage, and cellular signaling. Biomolecules 2018; 8(2). 10.3390/biom8020042 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maksimovic J, Sharp JA, Nicholas KR, Cocks BG, Savin K. Conservation of the ST6Gal I gene and its expression in the mammary gland. Glycobiol. 2011; 21(4):467–81. 10.1093/glycob/cwq185 . [DOI] [PubMed] [Google Scholar]
- 112.Boehm G, Stahl B. Oligosaccharides from milk. J Nutr. 2007; 137(3 Suppl 2):847S–9S. 10.1093/jn/137.3.847S . [DOI] [PubMed] [Google Scholar]
- 113.Poulsen NA, Robinson RC, Barile D, Larsen LB, Buitenhuis B. A genome-wide association study reveals specific transferases as candidate loci for bovine milk oligosaccharides synthesis. BMC Genomics 2019; 20(1):404 10.1186/s12864-019-5786-y . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guo DF, Rahmouni K. Molecular basis of the obesity associated with Bardet-Biedl syndrome. Trends Endocrin Met. 2011; 22(7):286–93. 10.1016/j.tem.2011.02.009 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J HumGenet.2013; 21(1):8–13. 10.1038/ejhg.2012.115 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feng Z, Li H, Liu S, Cheng J, Xiang G, Zhang J. FAM172A induces S phase arrest of HepG2 cells via Notch 3. Oncol Rep. 2013; 29(3):1154–60. 10.3892/or.2013.2235 . [DOI] [PubMed] [Google Scholar]
- 117.Li LX, Zhou WB, Tao Z, Deng WJ, Liang WC, Yang ZH, et al. Effect of FAM172A protein on apoptosis and proliferation in HEK293 cells. Zhonghua Yi Xue Za Zhi. 2010; 90(34):2424–7. . [PubMed] [Google Scholar]
- 118.Belanger C, Berube-Simard FA, Leduc E, Bernas G, Campeau PM, Lalani SR, et al. Dysregulation of cotranscriptional alternative splicing underlies CHARGE syndrome. Proc Natl Acad Sci USA. 2018; 115(4):E620–E9. 10.1073/pnas.1715378115 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brautigam L, Schutte LD, Godoy JR, Prozorovski T, Gellert M, Hauptmann G, et al. Vertebrate-specific glutaredoxin is essential for brain development. Proc Natl Acad Sci USA. 2011; 108(51):20532–7. 10.1073/pnas.1110085108 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Inves. 2010; 120(5):1749–61. 10.1172/JCI40372 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pham K, Dong J, Jiang X, Qu Y, Yu H, Yang Y, et al. Loss of glutaredoxin 3 impedes mammary lobuloalveolar development during pregnancy and lactation. Am. J. Physiol. Endocrinol. Metab. 2017; 312(3):E136–E49. 10.1152/ajpendo.00150.2016 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kenagy GJ, Trombulak SC. Size and function of mammalian testes in relation to body size. J Mammal. 1986; 67(1):1–22. 10.2307/1380997 [DOI] [Google Scholar]
- 123.Hirenallur Maheshwarappa Y, Kumar S, Chaudhary R, Mishra C, Ayyar S, Kumar A, et al. Identification of sperm motility markers in bovine transition protein genes. Zuchthygiene 2019; 54(2):365–72. 10.1111/rda.13369 . [DOI] [PubMed] [Google Scholar]
- 124.Gao Q, Ju Z, Zhang Y, Huang J, Zhang X, Qi C, et al. Association of TNP2 gene polymorphisms of the bta-miR-154 target site with the semen quality traits of Chinese Holstein bulls. PLoS One 2014; 9(1):e84355 10.1371/journal.pone.0084355 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gonzalez N, Moreno P, Jensen RT. Bombesin receptor subtype 3 as a potential target for obesity and diabetes. Expert Opin Ther Tar. 2015; 19(9):1153–70. 10.1517/14728222.2015.1056154 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Majumdar ID, Weber HC. Biology and pharmacology of bombesin receptor subtype-3. Curr Opin Endocrinol Diabetes Obes. 2012; 19(1):3–7. 10.1097/MED.0b013e32834ec77d 01266029-201202000-00003. [DOI] [PubMed] [Google Scholar]
- 127.Ramos-Alvarez I, Martin-Duce A, Moreno-Villegas Z, Sanz R, Aparicio C, Portal-Nunez S, et al. Bombesin receptor subtype-3 (BRS-3), a novel candidate as therapeutic molecular target in obesity and diabetes. Mol CellEndocrin. 2013; 367(1–2):109–15. 10.1016/j.mce.2012.12.025 . [DOI] [PubMed] [Google Scholar]
- 128.Berry DP, Friggens NC, Lucy M, Roche JR. Milk production and fertility in cattle. Annu Rev Anim Biosci. 2016; 4:269–90. 10.1146/annurev-animal-021815-111406 [DOI] [PubMed] [Google Scholar]
- 129.Graves RR. Improving dairy cattle by the continuous use of the proved sire. J. Dairy Sci. 1925; 8:391–404. [Google Scholar]
- 130.Kolbehdari D, Wang Z, Grant JR, et al. A whole genome scan to map QTL for milk production traits and somatic cell score in Canadian Holstein bulls. J Anim Breed Genet. 2009; 126(3):216–27. 10.1111/j.1439-0388.2008.00793.x . [DOI] [PubMed] [Google Scholar]
- 131.Wiggans GR, Cooper TA, VanRaden PM, Cole JB. Technical note: Adjustment of traditional cow evaluations to improve accuracy of genomic predictions. J. Dairy Sci. 2011; 94:6188–93. 10.3168/jds.2011-4481 [DOI] [PubMed] [Google Scholar]
- 132.Jiang L, Liu J, Sun D, et al. Genome wide association studies for milk production traits in Chinese Holstein population. PLoS One 2010; 5(10):e13661 10.1371/journal.pone.0013661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Touru T, Sahana G, Guldbrandtsen B, et al. Genome-wide association analysis of milk yield traits in Nordic Red Cattle using imputed whole genome sequence variants. BMC Genet. 2016; 17:55 10.1186/s12863-016-0363-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Akanno EC, Abo-Ismail MK, Chen L, Crowley JJ, Wang Z, Li C, et al. Modeling heterotic effects in beef cattle using genome-wide SNP-marker genotypes. J Anim Sci. 2018; 96(3):830–45. 10.1093/jas/skx002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Flori L, Fritz S, Jaffrezic F, Boussaha M, Gut I, Heath S, et al. The genome response to artificial selection: a case study in dairy cattle. PLoS One 2009; 4(8):e6595 10.1371/journal.pone.0006595 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008; 83(3):359–72. 10.1016/j.ajhg.2008.08.007 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mukherjee A, Mukherjee S, Dhakal R, Mech M, Longkumer I, Haque N, et al. High-density genotyping reveals genomic characterization, population structure and genetic diversity of Indian Mithun (Bos frontalis). Sci Rep. 2018; 8(1):10316 10.1038/s41598-018-28718-x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Szmatoła T, Gurgul A, Ropka-Molik K, Jasielczuk I, Ząbek T, Bugno-Poniewierska M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest Sci. 2016; 188:72–80. 10.1016/j.livsci.2016.04.006. [DOI] [Google Scholar]
- 139.Pryce JE, Haile-Mariam M, Goddard ME, Hayes BJ. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle. Genet Sel Evol. 2014; 46:71 10.1186/s12711-014-0071-7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Each individual is represented by a single vertical line divided into K colored segments, where K is the number of ancestral populations.
(TIF)
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
The raw Illumina reads generated and/or analyzed during the current study are available in the Sequence Read Archive repository (NCBI-SRA) under the following accession number PRJNA604217.