

Abstract
BACKGROUND
Identifying mechanisms of diseases with complex inheritance patterns, such as macular telangiectasia type 2, is challenging. A link between macular telangiectasia type 2 and altered serine metabolism has been established previously.
METHODS
Through exome sequence analysis of a patient with macular telangiectasia type 2 and his family members, we identified a variant in SPTLC1 encoding a subunit of serine palmitoyltransferase (SPT). Because mutations affecting SPT are known to cause hereditary sensory and autonomic neuropathy type 1 (HSAN1), we examined 10 additional persons with HSAN1 for ophthalmologic disease. We assayed serum amino acid and sphingoid base levels, including levels of deoxysphingolipids, in patients who had macular telangiectasia type 2 but did not have HSAN1 or pathogenic variants affecting SPT. We characterized mice with low serine levels and tested the effects of deoxysphingolipids on human retinal organoids.
RESULTS
Two variants known to cause HSAN1 were identified as causal for macular telangiectasia type 2: of 11 patients with HSAN1, 9 also had macular telangiectasia type 2. Circulating deoxysphingolipid levels were 84.2% higher among 125 patients with macular telangiectasia type 2 who did not have pathogenic variants affecting SPT than among 94 unaffected controls. Deoxysphingolipid levels were negatively correlated with serine levels, which were 20.6% lower than among controls. Reduction of serine levels in mice led to increases in levels of retinal deoxysphingolipids and compromised visual function. Deoxysphingolipids caused photoreceptor-cell death in retinal organoids, but not in the presence of regulators of lipid metabolism.
CONCLUSIONS
Elevated levels of atypical deoxysphingolipids, caused by variant SPTLC1 or SPTLC2 or by low serine levels, were risk factors for macular telangiectasia type 2, as well as for peripheral neuropathy. (Funded by the Lowy Medical Research Institute and others.)
UNDERSTANDING THE PATHOPHYSIOLOGY of diseases with complex genetic inheritance patterns is challenging. Macular telangiectasia type 2, which has a prevalence of 0.0045% to 0.06%, is a rare macular disease that leads to loss of central vision, with the onset of symptoms occurring in the fifth or sixth decade of life.1–4 The condition has a strong genetic component, as evidenced by extended families having multiple affected family members.5 However, the inheritance pattern is not clear because of the variable penetrance and expressivity of the disease.6,7 Recent insights into macular telangiectasia type 2 have come from genomewide association and metabolomic studies, which have suggested that it is associated with low serine levels in blood.8 Serine is a substrate in numerous metabolic pathways, including protein, nucleotide, and lipid synthesis. Whether and how low levels of serine affect macular health is unknown.
Serine palmitoyltransferase (SPT) condenses serine and palmitoyl-CoA and is the rate-limiting enzyme in sphingolipid synthesis.9 Variants in the genes SPTLC1 and SPTLC2, encoding two SPT subunits, result in the increased synthesis of atypical deoxysphingolipids and are causative for the rare disorder hereditary sensory and autonomic neuropathy type 1 (HSAN1).10–12 Deoxysphingolipids are toxic to multiple cell types, especially neurons.10,13–17 In vitro studies have shown that deoxysphingolipids can also accumulate in the absence of mutations in SPTLC1 or SPTLC2 when levels of serine are low.18 In this study, we investigated the role of deoxysphingolipids in macular telangiectasia type 2 and examined how a metabolic defect might lead to disease in both the eye and the peripheral nervous system.
METHODS
STUDY PARTICIPANTS AND GENETIC ANALYSES
We enrolled patients with macular telangiectasia type 2, HSAN1, or both conditions, as well as available family members and unaffected controls; all participants provided written informed consent, and the study was approved by local institutional review boards. Clinical diagnoses were confirmed by experienced retina specialists at the Moorfields Eye Hospital Reading Centre who were not aware of previous diagnoses. Genomic DNA from patients with macular telangiectasia type 2 and their family members was analyzed by exome sequencing. Exome sequence was captured with SeqCap EZ Exome, version 3. Raw sequence data were quality filtered with CASAVA (Illumina) and aligned to the Human Reference Genome, build hg19. All potentially disease-associated variants that were detected were confirmed by Sanger sequencing.
CLINICAL EXAMINATIONS
All participants underwent dilated retinal examination and standard ophthalmic imaging, conducted with the use of a Heidelberg Spectralis confocal imaging system. Dual-wavelength auto-fluorescence macular pigment imaging and fluorescence lifetime imaging ophthalmoscopy (FLIO) were performed with prototype instruments provided by Heidelberg Engineering.19
STATISTICAL ANALYSIS
Linear mixed models controlling for covariates with a significant effect on metabolite levels in plasma were used to assess differences in these levels between unaffected persons and persons with macular telangiectasia type 2. For all experiments, P values from tests performed on the same data set were corrected for multiple testing by the Benjamini–Hochberg procedure. The regression coefficient b represents the magnitude of the effect. (Additional details of the statistical analysis, as well as details of the experiments conducted in mice and cell culture, are provided in Supplementary Appendix 1, available with the full text of this article at NEJM.org.)
RESULTS
HSAN1 AND SUSCEPTIBILITY TO MACULAR TELANGIECTASIA TYPE 2
A 21-year-old man (Family Member III-3 [Family 1] in Fig. 1 and Table 1) presented with bull’s-eye maculopathy in both eyes. Clinical examination revealed findings diagnostic for macular telangiectasia type 2, including parafoveal telangiectatic retinal vessels, “right angle” venules, retinal opacification, pigment clumping, low levels of macular carotenoid pigment, late and mild leakage on fluorescein angiography, blue light–reflectance abnormalities, and intraretinal cysts and ellipsoid zone defects on optical coherence tomography (OCT). His parents and both siblings were then examined, which led to a diagnosis of macular telangiectasia type 2 in the father (II-2). One sister (III-2) was described as “possibly affected” because of reduced macular pigment and subtle parafoveal leakage as revealed by late-phase fluorescein angiography. The other sister (III-1) was not affected. The proband and his father and possibly affected sister reported having a familial peripheral neuropathy that had been diagnosed as Charcot–Marie–Tooth disease.
Figure 1 (facing page). Macular Telangiectasia Type 2 in Patients with Hereditary Sensory and Autonomic Neuropathy Type 1A (HSAN1A) and the SPTLC1 p.Cys133Tyr Variant.
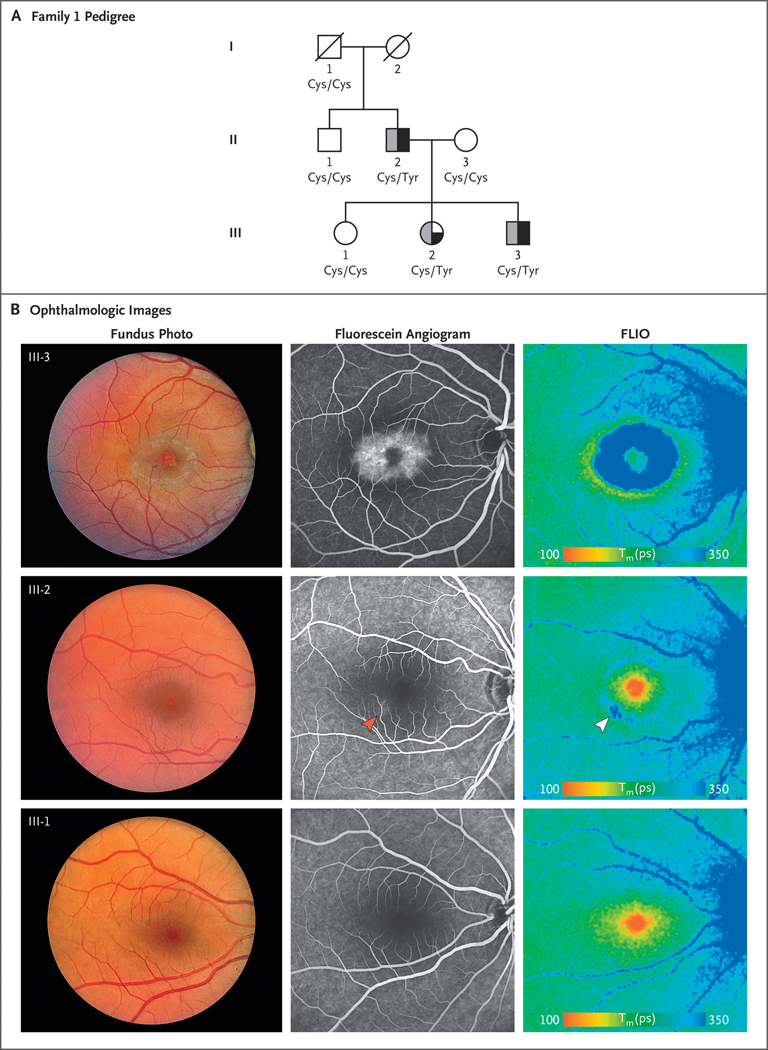
Panel A shows a pedigree of Family 1. Squares indicate male family members, and circles female family members; a slash through a symbol indicates that the family member is deceased. A half-solid symbol indicates macular telangiectasia type 2, and a half-gray symbol indicates HSAN1A. The quarter-solid symbol representing Family Member III-2 indicates possibly affected status. The amino acid status of SPTLC1 at position 133, as predicted on the basis of the genetic variants, is shown. Panel B shows ophthalmologic images of Family Members III-3 (affected), III-2 (possibly affected), and III-1 (unaffected), highlighting features of macular telangiectasia type 2, including temporal right-angle vessels, parafoveal opacification, and pigment clumping. Late-fluorescein angiographic images show strong parafoveal hyperfluorescence in III-3, subtle inferotemporal hyperfluorescence in III-2 (red arrowhead), and no hyperfluorescence in the unaffected family member, III-1. Fluorescence lifetime imaging ophthalmoscopy (FLIO) images of Family Member III-3 show a characteristic parafoveal blue ring of long FLIO lifetimes and redistribution of the foveal macular pigment in an orange ring of short FLIO lifetimes around the macular telangiectasia type 2 area. Family Member III-2 has a less intense blue parafoveal ring that is most pronounced at the same site where fluorescein staining was observed (white arrowhead). The unaffected sister (III-1) has a normal FLIO with normal macular pigment (orange spot centered on the fovea) and no parafoveal blue ring. Tm(ps) denotes mean fluorescence lifetime in picoseconds.
Table 1.
Genetic and Clinical Features of Patients with Hereditary Sensory and Autonomic Neuropathy Type 1.*
| Family and Patient No. | Variant Affecting SPT | Sex | Birth Year (Age in Years) | Macular Telangiectasia Type 2 Status | FLIO | Serine Supplementation† |
|---|---|---|---|---|---|---|
| Family 1 | ||||||
| III-3 | SPTLC1 p.Cys133Tyr | M | 1994 (24) | Affected | Positive | No |
| II-2 | SPTLC1 p.Cys133Tyr | M | 1965 (53) | Affected | Positive | No |
| III-2 | SPTLC1 p.Cys133Tyr | F | 1992 (26) | Possibly affected | Positive | No |
| Family 2 | ||||||
| III-2 | SPTLC1 p.Cys133Tyr | F | 1980 (38) | Unaffected | Positive | Yes |
| II-2 | SPTLC1 p.Cys133Tyr | F | 1951 (67) | Affected | Positive | Yes |
| III-3 | SPTLC1 p.Cys133Tyr | M | 1953 (65) | Affected | NT | Yes |
| III-1 | SPTLC1 p.Cys133Tyr | M | 1974 (44) | Affected | Positive | Yes |
| IV-1 | SPTLC1 p.Cys133Tyr | F | 1972 (46) | Affected | NT | Yes |
| Patient 1 | SPTLC1 p.Cys133Tyr | F | 1963 (55) | Affected | NT | Yes |
| Family 3 | ||||||
| II-1 | SPTLC2 p.Ser384Phe | M | 1974 (44) | Affected | NT | No |
| I-2 | SPTLC2 p.Ser384Phe | M | 1953 (65) | Affected | NT | No |
| Patient 2 | SPTLC1 p.Cys133Trp | F | 1980 (38) | Unaffected | NT | Yes |
| Patient 3 | SPTLC1 p.Cys133Trp | M | 1973 (45) | Unaffected | NT | Yes |
FLIO denotes fluorescence lifetime imaging ophthalmoscopy, NT not tested, and SPT serine palmitoyltransferase.
The patients who received serine supplementation had been taking high-dose serine supplements for at least 1 year before examination.
Exome sequencing revealed that these family members had the p.Cys133Tyr mutation in SPTLC1 (variant NM_006415.3 [SPTLC1]: c.398G→A; genomic location, 9:92080045 [GRCh38]; ClinVar accession number, VCV000004800.1),20 which is diagnostic of the rare disorder hereditary sensory and autonomic neuropathy type 1A (HSAN1A).20 The variant is pathogenic according to American College of Medical Genetics criteria for causality21 and cosegregated with both macular telangiectasia type 2 and HSAN1A in this family (Fig. 1B).
To determine whether HSAN1-associated mutations are causal for macular telangiectasia type 2, we examined 10 consecutive patients with HSAN1 (including members of two additional affected families) who had no known relationship to the index family (Table 1, and Fig. S1 in Supplementary Appendix 1). Six patients carried the p.Cys133Tyr mutation, two carried the p.Cys133Trp mutation in SPTLC1, and two carried the p.Ser384Phe mutation in SPTLC2, a variant previously shown to result in increased deoxysphingolipid levels and to cause HSAN1C, a disease closely related to HSAN1A (Table 1).22 Macular telangiectasia type 2 is an orphan disease, affecting approximately 1 in 5000 persons.3,4 We observed macular telangiectasia type 2 in 9 of the 13 patients with HSAN1 we examined (P = 2.2×10−16 by one-sided binomial test). Of the remaining 4 patients with HSAN1, 2 had abnormal macular features (III-2 in Family 1) or macular telangiectasia type 2–associated changes (III-2 in Family 1 and III-2 in Family 2). All 4 patients were younger than 50 years of age and were too young for a definitive diagnosis; they were also taking serine supplements, which may delay disease onset. The macular telangiectasia type 2 phenotype observed in the patients with HSAN1 was typical of that observed in the patients without HSAN1. We further characterized 6 patients with HSAN1 (including those graded as unaffected or possibly affected) with the use of FLIO. Each patient had changes characteristic of macular telangiectasia type 2 (parafoveal shifts to longer fluorescence lifetimes),19 whereas family members without HSAN1A-associated mutations had normal FLIO images (Fig. 1B, and Fig. S1B in Supplementary Appendix 1).
Family Members I-2 and II-1 in Family 3, who were carrying the p.Ser384Phe mutation in SPTLC2 (causing HSAN1C22), also had a diagnosis of macular telangiectasia type 2 (Fig. S1 in Supplementary Appendix 1). The two patients with HSAN1A who had the SPTLC1 p.Cys133Trp variant (Patient 2 and Patient 3), which is also associated with high deoxysphingolipid levels,18 were designated as not affected with macular telangiectasia type 2. Both patients were younger than 50 years of age.
DEOXYSPHINGOLIPID LEVELS IN PATIENTS WITH MACULAR TELANGIECTASIA TYPE 2
Although the majority of patients with macular telangiectasia type 2 do not have a pathogenic variant affecting either subunit of SPT, they have lower levels of serine in serum than do unaffected controls.8 Under low-serine conditions, alanine incorporation by SPT is increased, which results in the generation of deoxysphingolipids that lack the hydroxyl moiety critical for synthesis and degradation of more complex sphingolipids (Fig. 2A).9,23 To determine whether deoxysphingolipid levels are elevated in patients with macular telangiectasia type 2, we assayed amino acids and total hydrolyzed sphingoid bases in serum specimens from 125 nonconsanguineous patients with the condition and 94 unaffected controls (Table S1 in Supplementary Appendix 1). We had previously sequenced the exomes of these patients and found no pathogenic variants in SPTLC1 or SPTLC2.
Figure 2. Deoxysphinganine and Serine Levels in Patients with Macular Telangiectasia Type 2.

Panel A shows the generation of deoxysphingolipids under various conditions. Double arrows denote two sequential reactions in the biosynthetic pathway. SPT denotes serine palmitoyltransferase, and SPTvar variant SPT. Panels B through D show the quantitative levels of the metabolites sphinganine (Panel B), deoxysphinganine (Panel C), and serine (Panel D) in serum from 125 patients with macular telangiectasia type 2 (MacTel) and 94 controls; patients with known SPTLC1 or SPTLC2 variants were excluded. Data represented by the box-and-whisker plots are the median, first and third quartiles, and 5th and 95th percentiles. Linear mixed modeling was performed for statistical comparison. Panels E and F show correlation plots of serine and alanine levels, respectively, as compared with deoxysphinganine levels. The term rs denotes the Spearman rank correlation coefficient; the P values are from linear mixed models.
We used linear mixed models to assess whether there were differences in metabolite levels between patients with macular telangiectasia type 2 and unaffected controls. The effect of available covariates (ethnic group, sex at birth, age, collection site, and diabetes status) was estimated, and only significant covariates were included in the final model. We detected no significant differences in levels of sphinganine (b = 0.043, P = 0.476) but found that levels of serum deoxysphinganine were, on average, 84.2% higher in the cohort with macular telangiectasia type 2 (b = 0.611, P = 1.04×10−10), indicating that levels of these toxic lipids are elevated in the broader (non-HSAN1) population of patients with the condition (Fig. 2B and 2C; the data set for the graphs shown in Fig. 2 are provided in Supplementary Appendix 2, available at NEJM.org). To address whether deoxysphingolipid levels correlated with disease severity, we analyzed the relationship between deoxysphingolipid levels and a quantifiable anatomical abnormality, loss of the ellipsoid zone (representing a break in the OCT reflectance pattern). The extent of loss is known to correlate with function as assessed by microperimetry24–27 and hence can be used as a measure of disease progression. We found a positive relationship between ellipsoid-zone loss and deoxysphinganine levels in the 125 patients with macular telangiectasia type 2 (b = 0.148; 95% confidence interval, 0.004 to 0.299). (Additional details of these analyses are provided in Tables S2 through S6, S9, and S10 in Supplementary Appendix 1.)
Elevated deoxysphinganine levels have been reported in patients with type 2 diabetes,13,28 and patients with macular telangiectasia type 2 have a higher prevalence of type 2 diabetes than the general population.29 We therefore evaluated the relationship among these two conditions and deoxysphinganine levels. We found that deoxysphinganine levels were not significantly higher in patients with macular telangiectasia type 2 who had type 2 diabetes than in those who did not have type 2 diabetes (b = 0.328, P = 0.07) (Table S5 in Supplementary Appendix 1). Among persons who did not have macular telangiectasia type 2, levels did not differ significantly between those who had type 2 diabetes and those who did not (b = −0.116, P = 0.46). When patients with type 2 diabetes were excluded from the entire group, levels of deoxysphinganine were higher among patients with macular telangiectasia type 2 than among unaffected controls (b = 0.516, P = 9.3×10−7), which suggests that the elevation in deoxysphinganine levels in patients with macular telangiectasia type 2 is not driven by the greater number of patients with type 2 diabetes in this group (Table S1 in Supplementary Appendix 1).
Consistent with our previous findings,8 serine levels were significantly lower in serum specimens from patients with macular telangiectasia type 2 than among controls, by 20.6% on average (b = −0.231, P = 6.63×10−10) (Fig. 2D). In addition, levels of deoxysphinganine showed the strongest correlations with levels of plasma serine (b = −1.27, P = 5.55×10−18) and alanine (b = 0.63, P = 7.59×10−5) (Fig. 2E and 2F), associations that remained significant when we controlled for other covariates and macular telangiectasia type 2 status. Patients with macular telangiectasia type 2 had total deoxysphinganine levels that were similar to, and in some cases higher than, those of patients with HSAN1, which suggested that reduced serine levels are sufficient to increase deoxysphinganine to disease-associated levels. These results show that reduced levels of circulating serine, regardless of the cause, result in accumulation of deoxysphinganine in serum in patients. (Additional details of these analyses are provided in Tables S6 through S10 and Figure S2A in Supplementary Appendix 1.)
We next determined the specific sphingolipid species that were represented in the total hydrolyzed deoxysphinganine pool and the way in which they were transported systemically (Fig. S2B and S2C in Supplementary Appendix 1). By comparing levels of free deoxysphinganine with levels of hydrolyzed deoxysphinganine (the sum of free deoxysphinganine plus deoxydihydroceramide), we found that nearly 90% of the pool of deoxysphinganines was derived from deoxydihydroceramide. When we measured the deoxysphinganine levels in fractionated serum we found that, similar to other sphingolipids,30 deoxysphinganines were complexed with the low- and verylow-density lipoproteins, which suggests that the liver is the primary source of circulating deoxysphingolipids.
LOW SERINE LEVELS IN A MOUSE MODEL
We next tested whether low levels of circulating serine (macular telangiectasia type 2 phenotype) would be sufficient to increase tissue deoxysphingolipid levels. We fed 8-week-old male mice an isocaloric and isonitrogenous control diet or a diet that was depleted of serine and glycine (depletion of glycine prevents its compensatory conversion to serine31). We found no appreciable difference in food consumption, body weight, or body composition between the two groups of mice (Fig. S3A through S3D in Supplementary Appendix 1). Mice that were fed a serine- and glycine-free diet had lower serine levels throughout the day than did those that were fed the control diet (P = 9.3×10−14) (Fig. 3A).
Figure 3. Serine and Deoxysphinganine Levels and Retinal and Peripheral Neural Defects in Wild-Type Mice.

Panel A shows serine levels in plasma measured in mice that were fed a control diet or serine- and glycine-free diet for 2 weeks (5 mice in each group). Panels B through E show the quantification of total hydrolyzed deoxysphinganine in plasma and in the indicated tissues in mice that were fed a control diet or a serine- and glycine-free diet for 3 months (5 mice in each group). Deoxysphinganine levels are normalized to the milligrams of tissue protein. RPE denotes retinal pigment epithelium. Panels F and G show electroretinographic (ERG) measurements of photopic responses to a flash in light-adapted mice that were fed a control or a serine- and glycine-free diet for 10 months (8 to 10 eyes in each group). Panel H shows representative photopic flicker responses from mice that were fed a control or a serine- and glycine-free diet. Panel I shows peripheral neural function evaluated by hot-plate assay indicating “time to flick” (i.e., time to a visible reaction to a thermal stimulus) in mice that were fed a control or a serine- and glycine-free diet for 10 months (8 mice in each group). Data represented by the box-and-whisker plots are the median, first and third quartile, and minimum and maximum values. P values were determined with Student’s t-test and corrected for multiple testing by the Benjamini–Hochberg procedure (Tables S14 and S15 in Supplementary Appendix 1).
The mice with low circulating serine levels had deoxysphinganine levels that were 3 times as high as those in the mice that were fed the control diet (P = 0.002) and had no change in sphinganine levels (Fig. 3B). We found a negative correlation between serine levels and deoxysphinganine levels (r = −0.803). The retina and retinal pigment epithelium from mice that were fed the serine- and glycine-free diet also had elevated deoxysphinganine levels (P<0.001 and P = 0.006, respectively) (Fig. 3C and 3D). We examined the sciatic nerve and found a similar elevation in deoxysphinganine relative to the nerve in control mice (P = 0.04) (Fig. 3E). The ratio of deoxysphinganine to sphinganine was also elevated in the plasma and tissues. (Additional details of these analyses are provided in Figure S3 in Supplementary Appendix 1.)
Retinal and peripheral sensory deficits developed in mice that were fed the serine- and glycine-free diet for 10 months. Electroretinographic measurements showed significant deficits in the photopic and flicker responses (P = 0.002 and P = 0.005, respectively) (Fig. 3F, 3G, and 3H, and Fig. S4 in Supplementary Appendix 1). Similar to a mouse model of HSAN1 in which mutant SPTLC1 was overexpressed,12 mice that were fed the depleted diet also had an elevated thermal latency threshold (P = 0.005) (Fig. 3I). A reduction in serine and glycine levels in plasma is sufficient to increase plasma and tissue levels of deoxysphingolipids and causes functional retinal and peripheral sensory deficits.
RETINAL TOXICITY OF DEOXYDIHYDROCERAMIDE
A characteristic late-stage manifestation of macular telangiectasia type 2 is the loss of photoreceptors in the macula32; this process can be modeled in vitro with human induced pluripotent stem cell–derived retinal organoids, which are stratified retinal tissues containing most of the major neural retinal cell types, including functional photoreceptors and Müller glia (Fig. 4A).33 Treatment of retinal organoids with deoxysphinganine induced photoreceptor apoptosis in a dosedependent manner (Fig. 4B), with toxic effects at 50 nmol per liter (P = 0.03), a concentration detected in patients with macular telangiectasia type 2 and HSAN1 (Fig. S2A in Supplementary Appendix 1). In contrast, toxic effects were not observed when organoids were treated with two distinct deoxysphingosine species.34 Inhibition of ceramide synthase with fumonisin B1 prevented deoxysphinganine-induced cell death (P = 6×10−16) (Fig. 4C and 4D), indicating that the toxic effects were due to one or both of its downstream products, deoxydihydroceramide and deoxyceramide (Fig. 4D). To distinguish between these, we quantified ceramide species after treatment with deoxysphinganine, sphinganine, sphingosine, or either species of deoxysphingosine and found that only the levels of deoxydihydroceramide correlated with toxicity. (Additional details of these analyses are provided in Figures S5 through S7 in Supplementary Appendix 1.)
Figure 4. Effect of PPAR-α Agonist Fenofibrate on Deoxysphingolipid Toxicity in Human Photoreceptors.

Panel A, subpanel a shows live bright-field image of human retinal organoid tissue showing outer segments (OS) projecting from the outer nuclear layer (ONL); subpanels b through d show representative confocal images of cell death (indicated by terminal deoxynucleotidyl transferase dUTP nick end labeling [TUNEL] staining) within the photoreceptors (indicated by the presence of α-recoverin) of the retinal organoid ONL after 4 days of treatment with control media (subpanel b), deoxysphinganine (1 μmol per liter) (subpanel c), or deoxysphinganine (1 μmol per liter) plus fenofibrate (20 μmol per liter) (subpanel d). Blue staining with 4′,6-diamidino-2-phenylindole (DAPI) indicates the presence of cell nuclei. Panel B shows the toxicity dose response of deoxysphinganine in retinal organoids. Cell death was quantified in human photoreceptors cultured in varying concentrations of deoxysphinganine for 8 days (9 per group) (Table S15 in Supplementary Appendix 1). Panel C shows pharmacologic rescue of deoxysphinganine toxicity. Cell death was quantified in human photoreceptors after treatment with control media (6 organoids), deoxysphinganine (1 μmol per liter) (22 organoids), deoxysphinganine (1 μmol per liter) plus fumonisin B1 (35 μmol per liter) (12 organoids), or deoxysphinganine (1 μmol per liter) plus fenofibrate (20 μmol per liter) (21 organoids). Bar graphs with T bars indicate the means and standard errors. P values were determined with the Wald test on a linear regression analysis. Cell death (the number of dead cells within the defined area) was normalized by a square-root transformation. Model assumptions were assessed with model diagnostics plots (Tables S16 and S17 in Supplementary Appendix 1). Panel D shows the steps in the ceramide–deoxyceramide pathway, including the interaction of fumonisin B1 and fenofibrate. Both pathways share the enzymes shown in gray. Deoxysphingosine is degraded through ω-hydroxylation; it cannot be degraded through the sphingosine-1-phosphate (S1P) pathway. Double arrows denote two sequential reactions in the biosynthetic pathway. CerS denotes ceramide synthase, and SPT serine palmitoyltransferase.
FENOFIBRATE AND DEOXYSPHINGOLIPID-INDUCED RETINAL TOXICITY
We sought to ameliorate the toxic effects of deoxydihydroceramide with the use of the peroxi-some proliferator-activated receptor α (PPAR-α) agonist fenofibrate (Fig. 4D). Fenofibrate has been shown to stimulate degradation of deoxysphingolipids in a human embryonic kidney cell line and has been used to reduce deoxysphinganine levels in patients with dyslipidemia.35,36 We found that concurrent treatment of retinal organoids with fenofibrate at a concentration of 20 μmol per liter decreased deoxysphinganine-induced cell death in photoreceptors by approximately 80% (P = 1.85×10−13) (Fig. 4A and 4C).
DISCUSSION
In addition to finding that altered amino acid and sphingolipid metabolism were the basis of the photoreceptor loss in patients with macular telangiectasia type 2, we have expanded the definition of diseases caused by variants in SPTLC1 or SPTLC2 and have shown a link between abnormal systemic serine metabolism and tissue-specific disease. HSAN1-associated variants in these genes lead to pathologic accumulation of deoxysphinganine; our data show that these mutations also cause macular telangiectasia type 2. The vast majority of patients with macular telangiectasia type 2 do not have variants in SPTLC1 or SPTLC2, but these patients do have low serine levels and elevated deoxysphingolipid levels. Reducing circulating serine in mice is sufficient to increase deoxysphingolipid levels and cause functional defects in the retina and peripheral nerves. We therefore conclude that elevated deoxysphingolipid levels can cause macular disease in the broader population of patients with macular telangiectasia type 2, as well as in patients with HSAN1.
We do not know why serine levels are systemically low in patients with macular telangiectasia type 2. Given that serine is central to numerous metabolic pathways, the genetic regulators are probably diverse. A genomewide association study identified loci associated with the condition within or near two of the three genes involved in de novo serine synthesis — PHGDH and PSPH8 — which supports speculation that perhaps serine synthesis is impaired in some patients. Perhaps, as well, some patients have elevated levels of deoxysphingolipids because of high alanine levels or impaired lipid degradation. The multiple means of regulating serine and deoxysphingolipid levels may reflect the genetic heterogeneity observed in macular telangiectasia type 2. Variations in environmental factors, such as diet, could also explain the variable penetrance of macular telangiectasia type 2 in families.
Although elevated levels of deoxysphingolipids are known to be cytotoxic,10,13,28,37 the mechanism of their toxicity is not fully understood. Deoxysphingolipids have been reported to disrupt mitochondrial function.16 In this study, we have shown that these lipids are toxic to human photoreceptors and identified deoxydihydroceramide as the main neurotoxic species. It accounted for nearly 90% of the hydrolyzed deoxysphinganine measurements in serum from patients. Further work is needed to understand the mechanism of retinal toxicity and the potential involvement of other sphingolipid metabolites. Although deoxysphinganine accumulation is observed systemically, it is not clear why the macular region of the retina and the extremities of the somatosensory nervous system are preferentially affected, but their unique metabolic demands could make them particularly vulnerable.
Abnormal serine metabolism, leading to increased levels of deoxysphingolipids, may represent a broader “serineopathy” class of diseases that includes both retinal and peripheral neuropathies, and so it might be expected that patients with macular telangiectasia type 2 would also be at risk for peripheral neuropathies. Elevated deoxysphingolipid levels have been reported in patients with diabetes and may contribute to diabetic retinopathy and peripheral neuropathy,28 which are common complications of diabetes. Although deoxysphingolipid levels can be reduced by systemically administering serine or fenofibrate,18,36 we caution against prescribing serine or fenofibrate to patients with macular telangiectasia type 2, given the complex genetic etiology of the condition and the genetic diversity in the patient population.
We have used a detailed patient registry, genetic and metabolomic analyses, and mouse and organoid models to uncover a link among macular telangiectasia type 2, HSAN1, and deoxysphingolipids. In so doing, we established a connection between central and peripheral neuropathies, as well as between plasma amino acid levels and these disorders.
Supplementary Material
Acknowledgments
Supported by the Lowy Medical Research Institute, with supplementary funding from the National Health and Medical Research Council of Australia (to Dr. Bahlo), a core grant from the National Eye Institute (EY14800, to Dr. Bernstein), the University of Melbourne Research Scholarship Program (to Mr. Bonelli), a grant from the National Science Foundation (1454425, to Dr. Metallo), and a grant from the National Institutes of Health (NCI R01CA188652, to Dr. Metallo). Heidelberg Engineering provided a Heidelberg Spectralis confocal imaging system to Dr. Bernstein free of charge for use in this study.
Disclosure forms provided by the authors are available at NEJM.org.
We thank the Lowy family for their support of the MacTel Project, the Lowy Medical Research Institute, and specifically, this study; Ms. Ellen Burns and the members of the Deater Foundation for their assistance in identifying patients with hereditary sensory neuropathy who were willing to participate in this study; Dr. Amanda Roberts and Ilham Polis of the Scripps Research Animal Models Core and Dr. Bernard Kok for their assistance with the studies in animals; Dr. Jessica Flowers for her assistance designing the amino acid animal diets; Drs. Alan Bird and Ferenc Sallo at the Moorfields Eye Hospital Reading Centre for their careful evaluation of clinical images; Dr. Aaron Lee of the University of Washington for his expertise in the use of artificial intelligence to evaluate the structure and function studies; Dr. Faith Barnett for her clinical insight regarding the connection with peripheral neuropathies; Drs. Traci Clemons and Gordon K. Smyth for their assistance with the statistical analyses; Dr. Robert Graham of the Victor Chang Cardiac Research Institute, Sydney, Dr. Joseph Gleeson of the University of California, San Diego, and Dr. Edward Holmes of the Sanford Consortium for Regenerative Medicine, La Jolla, for providing critical reviews of an earlier version of the manuscript; and Marti Lynn Moon, Jessica Orozco, Kelliann Farnsworth, Kelly Nelson, and Kimberley Wegner for administrative assistance.
REFERENCES
- 1.Chew EY, Clemons TE, Jaffe GJ, et al. Effect of ciliary neurotrophic factor on retinal neurodegeneration in patients with macular telangiectasia type 2: a randomized clinical trial. Ophthalmology 2019; 126: 540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gass JD, Blodi BA. Idiopathic juxtafoveolar retinal telangiectasis: update of classification and follow-up study. Ophthalmology 1993; 100: 1536–46. [PubMed] [Google Scholar]
- 3.Aung KZ, Wickremasinghe SS, Makeyeva G, Robman L, Guymer RH. The prevalence estimates of macular telangiectasia type 2: the Melbourne Collaborative Cohort Study. Retina 2010; 30: 473–8. [DOI] [PubMed] [Google Scholar]
- 4.Klein R, Blodi BA, Meuer SM, Myers CE, Chew EY, Klein BE. The prevalence of macular telangiectasia type 2 in the Beaver Dam eye study. Am J Ophthalmol 2010; 150(1): 55–62.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ronquillo CC, Wegner K, Calvo CM, Bernstein PS. Genetic penetrance of macular telangiectasia type 2. JAMA Ophthalmol 2018; 136: 1158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parmalee NL, Schubert C, Figueroa M, et al. Identification of a potential susceptibility locus for macular telangiectasia type 2. PLoS One 2012; 7(8): e24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parmalee NL, Schubert C, Merriam JE, et al. Analysis of candidate genes for macular telangiectasia type 2. Mol Vis 2010; 16: 2718–26. [PMC free article] [PubMed] [Google Scholar]
- 8.Scerri TS, Quaglieri A, Cai C, et al. Genome-wide analyses identify common variants associated with macular telangiectasia type 2. Nat Genet 2017; 49: 559–67. [DOI] [PubMed] [Google Scholar]
- 9.Duan J, Merrill AH Jr. 1-Deoxysphingolipids encountered exogenously and made de novo: dangerous mysteries inside an enigma. J Biol Chem 2015; 290: 15380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penno A, Reilly MM, Houlden H, et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 2010; 285: 11178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rotthier A, Auer-Grumbach M, Jans-sens K, et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet 2010; 87: 513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichler FS, Hornemann T, McCamp-bell A, et al. Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. J Neurosci 2009; 29: 14646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuellig RA, Hornemann T, Othman A, et al. Deoxysphingolipids, novel biomarkers for type 2 diabetes, are cytotoxic for insulin-producing cells. Diabetes 2014;6 3: 1326–39. [DOI] [PubMed] [Google Scholar]
- 14.Güntert T, Hänggi P, Othman A, et al. 1-Deoxysphingolipid-induced neurotoxicity involves N-methyl-d-aspartate receptor signaling. Neuropharmacology 2016; 110: Pt A: 211–22. [DOI] [PubMed] [Google Scholar]
- 15.Zitomer NC, Mitchell T, Voss KA, et al. Ceramide synthase inhibition by fumonisin B1 causes accumulation of 1-deoxysphinganine: a novel category of bioactive 1-deoxysphingoid bases and 1-deoxydihy-droceramides biosynthesized by mammalian cell lines and animals. J Biol Chem 2009; 284:4786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alecu I, Tedeschi A, Behler N, et al. Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction. J Lipid Res 2017; 58: 42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson ER, Kugathasan U, Abramov AY, et al. Hereditary sensory neuropathy type 1-associated deoxysphingolipids cause neurotoxicity, acute calcium handling abnormalities and mitochondrial dysfunction in vitro. Neurobiol Dis 2018; 117: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garofalo K, Penno A, Schmidt BP, et al. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest 2011; 121: 4735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauer L, Gensure RH, Hammer M, Bernstein PS. Fluorescence lifetime imaging ophthalmoscopy: a novel way to assess macular telangiectasia type 2. Ophthalmol Retina 2018; 2:587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 2001; 27: 309–12. [DOI] [PubMed] [Google Scholar]
- 21.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ernst D, Murphy SM, Sathiyanadan K, et al. Novel HSAN1 mutation in serine palmitoyltransferase resides at a putative phosphorylation site that is involved in regulating substrate specificity. Neuromolecular Med 2015; 17:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esaki K, Sayano T, Sonoda C, et al. L-serine deficiency elicits intracellular accumulation of cytotoxic deoxysphingolipids and lipid body formation. J Biol Chem 2015; 290:14595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heeren TFC, Kitka D, Florea D, et al. Longitudinal correlation of ellipsoid zone loss and functional loss in macular telangiectasia type 2. Retina 2018;38: Suppl 1: S20–S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peto T, Heeren TFC, Clemons TE, et al. Correlation of clinical and structural progression with visual acuity loss in macular telangiectasia type 2: MacTel Project Report No. 6 — the MacTel Research Group. Retina 2018; 38: Suppl 1: S8–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sallo FB, Peto T, Egan C, et al. “En face” OCT imaging of the IS/OS junction line in type 2 idiopathic macular telangiectasia. Invest Ophthalmol Vis Sci 2012; 53:6145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee D, Lad EM, Vann RR, et al. Correlation between macular integrity assessment and optical coherence tomography imaging of ellipsoid zone in macular telangiectasia type 2. Invest Ophthalmol Vis Sci 2017; 58: BIO291-BIO299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertea M, Rütti MF, Othman A, et al. Deoxysphingoid bases as plasma markers in diabetes mellitus. Lipids Health Dis 2010; 9: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clemons TE, Gillies MC, Chew EY, et al. Medical characteristics of patients with macular telangiectasia type 2 (MacTel Type 2) MacTel project report no. 3. Ophthalmic Epidemiol 2013;20: 109–13. [DOI] [PubMed] [Google Scholar]
- 30.Hammad SM, Pierce JS, Soodavar F, et al. Blood sphingolipidomics in healthy humans: impact of sample collection methodology. J Lipid Res 2010; 51: 3074–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maddocks OD, Berkers CR, Mason SM, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013; 493: 542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powner MB, Gillies MC, Zhu M, Vevis K, Hunyor AP, Fruttiger M. Loss of Müller’s cells and photoreceptors in macular telangiectasia type 2. Ophthalmology 2013; 120: 2344–52. [DOI] [PubMed] [Google Scholar]
- 33.Zhong X, Gutierrez C, Xue T, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun 2014; 5: 4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lone MA, Santos T, Alecu I, Silva LC, Hornemann T. 1-Deoxysphingolipids. Biochim Biophys Acta Mol Cell Biol Lipids 2019;1864: 512–21. [DOI] [PubMed] [Google Scholar]
- 35.Alecu I, Othman A, Penno A, et al. Cytotoxic 1-deoxysphingolipids are metabolized by a cytochrome P450-dependent pathway. J Lipid Res 2017; 58: 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Othman A, Benghozi R, Alecu I, et al. Fenofibrate lowers atypical sphingolipids in plasma of dyslipidemic patients: a novel approach for treating diabetic neuropathy? J Clin Lipidol 2015; 9: 568–75. [DOI] [PubMed] [Google Scholar]
- 37.Gorden DL, Myers DS, Ivanova PT, et al. Biomarkers of NAFLD progression: a lipidomics approach to an epidemic. J Lipid Res 2015; 56:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.