

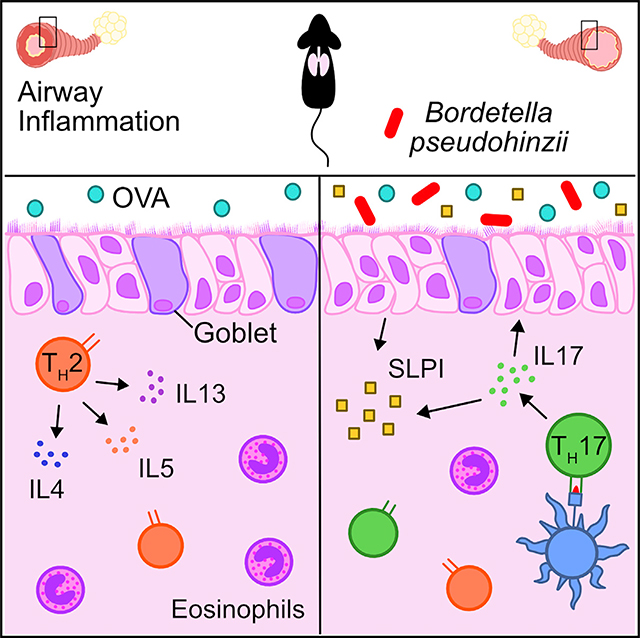
SUMMARY
Homeostatic mucosal immune responses are fine-tuned by naturally evolved interactions with native microbes, and integrating these relationships into experimental models can provide new insights into human diseases. Here, we leverage a murine-adapted airway microbe, Bordetella pseudohinzii (Bph), to investigate how chronic colonization impacts mucosal immunity and the development of allergic airway inflammation (AAI). Colonization with Bph induces the differentiation of interleukin-17A (IL-17A)-secreting T-helper cells that aid in controlling bacterial abundance. Bph colonization protects from AAI and is associated with increased production of secretory leukocyte protease inhibitor (SLPI), an antimicrobial peptide with anti-inflammatory properties. These findings are additionally supported by clinical data showing that higher levels of upper respiratory SLPI correlate both with greater asthma control and the presence of Haemophilus, a bacterial genus associated with AAI. We propose that SLPI could be used as a biomarker of beneficial host-commensal relationships in the airway.
In Brief
Asthma is known to be modified by airway microbes. Jaeger et al. use a murine-adapted bacterium to show that airway colonization evokes a Th17 response associated with increased SLPI, an antimicrobial peptide, and protection from lung inflammation. In people, SLPI was correlated with airway microbiota composition.
Graphical Abstract
INTRODUCTION
The airway microbiota is increasingly recognized for its diverse roles in respiratory inflammatory diseases, especially asthma. Microbiologic surveys of upper and lower airway samples have demonstrated that individuals with asthma harbor distinct microbial signatures associated with various facets of the disease. In early childhood, the presence of select bacteria within the upper airway, including Haemophilus, Streptococcus, and Moraxella (HSM) species, has been associated with later development of asthma (e.g., Bisgaard et al., 2007, 2010), although healthy individuals can also harbor these organisms, suggesting compensatory host responses that may mitigate asthma risk. In addition to predisposing human to asthma, the airway microbiota has been proposed as a factor that influences an individual’s clinical phenotype, including the immunologic markers associated with disease and response to therapy, referred to as an asthma endotype. Although the most established asthma endotype is associated with allergic T-helper 2 (Th2) responses, characterized by the production of interleukin-4 (IL-4), IL-5, and IL-13 cytokines, T-helper 17 (Th17) responses, defined by the production of IL-17A, is also of particular interest. In clinical studies, higher degrees of IL-17A expression have been associated with severe asthma and correlate with increased numbers of airway neutrophils and resistance to corticosteroid treatments (Chesné et al., 2014; Őstling et al., 2019). Animal models have additionally shown that the adoptive transfer of allergen-specific Th17 cells contributes to allergic airway inflammation (AAI) and drives airway neutrophilia (McKinley et al., 2008; Wakashin et al., 2008). In contrast, other studies suggest that IL-17A may directly antagonize Th2 responses and reduce AAI (Barlow et al., 2011; Choy et al., 2015; Newcomb et al., 2013; Schnyder-Candrian et al., 2006).
How airway microbes shape asthma endotypes and influence the particular clinical features of the disease is an area of active investigation. The importance of human airway microbes, such as Haemophilus influenzae (McCann et al., 2016), Streptococcus pneumoniae (Preston et al., 2007; Preston et al., 2011), and Moraxella catarrhalis (Alnahas et al., 2017), has been translated to animal models of asthma by demonstrating that respiratory exposure to these microbes directly affects AAI. Although these studies have confirmed the potential of select airway bacteria to modulate AAI, one important limitation of this approach is that mouse models using bacteria from the human airway may not establish a host-microbe interaction that adequately recapitulates important interactions observed in humans. This is especially relevant for asthma because many of the microbes implicated in worsening AAI from clinical studies only transiently colonize mice (Unhanand et al., 1992). These constraints on colonization limit our ability to use mice to model the effects of microbes that would be expected to persist in human airways. One possible solution to the challenge of modeling the effects of microbial exposure in murine models is to use natural members of the mouse microbiota. Experiments incorporating “wild” mouse microbes in a laboratory setting have been used to better align mouse models of endotoxemia (Rosshart et al., 2019), viral infections (Beura et al., 2016), and tumorigenesis (Rosshart et al., 2017) to human disease.
Here, we describe the host response to Bordetella pseudohinzii (Bph), a recently described murine respiratory microbe (Clark et al., 2016; Ivanov et al., 2016) that is also found in wild rodents (Loong et al., 2018), and its effects on a model of AAI. Although Bph persists in the respiratory tract of mice for months after inoculation (Dewan et al., 2019), the preponderance of infections do not lead to overt signs of illness (Hayashimoto et al., 2012). Others have reported that despite its apparent benign effects on mice, Bph can induce changes in pulmonary immune parameters (e.g., neutrophilia), leading to concerns about Bph possibly confounding murine pulmonary disease models (Clark et al., 2016, 2017; Perniss et al., 2018). On the other hand, we speculated that studying a bacterium within the context of AAI that evolved to reside in the murine respiratory tract and was not transplanted from humans may provide unique insights into how prolonged exposure to a respiratory microbe affects pulmonary inflammation. We found that Bph induces a marked Th17 response in mice that is associated with protection from AAI and stimulates the production of the antimicrobial protein secretory leukocyte protease inhibitor (SLPI). These findings are further supported by clinical data showing that increased nasal SLPI is correlated with greater asthma control and differences in bacterial composition within the upper airway microbiota.
RESULTS
Bph Colonizes the Mouse Respiratory Tract
We first wanted to contrast Bph to the “classical” bordetellae strains (Bordetella pertussis, Bordetella parapertussis, and Bordetella bronchiseptica) that are well-known causes of respiratory infections to better understand its virulence potential. To accomplish this, we compared known Bordetella reference strains and our own Bph isolate assemblies (designated 2–1 and 5–5) to a database of known bacterial virulence factors (Chen et al., 2016; Figure 1A). This analysis confirmed that Bph lacked the virulence factors associated with classical pathogenic Bordetella strains, including the genes for Pertussis toxin (Ptx), Filamentous Hemagglutinin (FHA), and the type III secretion system (T3SS).
Figure 1. Bordetella pseudohinzii (Bph) Induces a Th17 Response in the Lungs of Colonized Mice.

(A) Comparison of the abundance of virulence factor classes encoded in the genomes of members in the genus Bordetella. Virulence factors were identified by sequence alignment to the Virulence Factor Database (VFDB) and binned into functional groups defined by VFDB (Chen et al., 2016). Assemblies of Bph isolates described in this study (2–1 and 5–5) are also shown.
(B) Recovery of Bph from respiratory tract samples over a 184-day period. CFUs per section of tissue or mL of lavage fluid are shown. Box indicates 25th and 75th percentiles and whiskers are 1.5 × interquartile range. n = 2–6 mice per time point.
(C) Representative hematoxylin and eosin staining of mice that received either HK (top panel) or live (bottom panel) Bph taken 60 days after inoculation.
(D–F) Flow cytometry of lung tissue digests from mice 30 days after that received HK (blue) or live Bph (red) inoculation. n = 9–10 mice/group, combined from two independent experiments.
(D) Neutrophils as a percentage of live cells from lung. Neutrophils were defined as CD11b+Ly6G+.
(E) Percentage of Teff (CD4+TCRβ+FoxP3−CD44hiCD62Llo) cells from the lungs as a percentage of total T-helper cells.
(F) Percentages of IL-17A+IFNγ−-secreting T-helper 17 (Th17) cells from the lungs.
Statistical significance: Mann-Whitney U test. Horizontal lines indicate median values. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To directly assess the in vivo impact of Bph colonization on mice, we intranasally inoculated C57BL/6 6- to 8-week-old mice with ~104 colony-forming units (CFUs) of bacteria and collected tissues from groups of mice over a 6-month period. Bph was consistently recovered from the nasal lavage, trachea, and lung for 3 months after initial colonization (Figure 1B). Despite this persistent colonization, we did not observe significant weight loss in Bph-colonized mice compared to control mice over a 3-month period (Figure S1A), nor observe sneezing (Clark et al., 2016; Hayashimoto et al., 2012), tachypnea, poor grooming, or decreased activity. Consistent with earlier descriptions of Bph mouse colonization and the lack of virulence factors found in classical bordetellae, these results indicated that Bph colonization was persistent and associated with a mild phenotype (Clark et al., 2016; Dewan et al., 2019; Ivanov et al., 2016; Perniss et al., 2018).
Colonization with Bph Induces Th17 Cells in the Lung
To better understand the effects of Bph on the host, we performed microscopic examination of the lungs of wild-type (WT) mice, which demonstrated the formation of lymphoid aggregates, consistent with inducible bronchus associated lymphoid tissue (iBALT; Marin et al., 2019), that was first detectable 10 days post-colonization, present in all colonized mice by 28 days, and persistent out to 6 months after initial colonization (Figures 1C, S1B, and S1C). These results imply that Bph elicits an adaptive immune response that we further investigated by performing immunophenotyping on lung immune cell populations. After 28 days of colonization (Figure S1D), we observed a trend toward an increase in neutrophils in lung tissue, as previously observed (Clark et al., 2016), accompanied by a decrease in eosinophils in mice colonized with Bph compared to mice undergoing mock colonization with 104 CFU heat-killed (HK) bacteria (Figures 1D and S1E). This number of HK bacteria equals the number of bacteria used in the live inoculum and contains considerably (~10,000 fold) fewer HK bacteria than other models intended to elicit a pulmonary immune response (Amezcua Vesely et al., 2019; Chen et al., 2011). Among T cell subsets, effector T cells (Teff) and T regulatory (Treg) cells were increased in colonized mice compared to controls (Figures 1E and S1F). Additional characterization of CD4+ T cells by intracellular staining for cytokines showed that there was a marked increase in the percentages of IL-17A-expressing (TCRβ+CD4+IL-17A+IFNγ−, referred to here as Th17) but not interferon gamma (IFNγ)-expressing (Th1) T cells (Figures 1F, S1G, and S1L) in colonized mice compared to mock-colonized controls. Examination of the non-CD4+ T cell immune compartment showed no differences in IL-17A+ cells between mice that received live or HK Bph (Figure S1H), nor did we see an increase in transcripts for the constant regions of T cell receptor gamma (TCRg) or TCRδ (Table S1). Although we cannot absolutely exclude another in vivo source of IL-17A by ex vivo restimulation and intracellular cytokine staining, our data are most consistent with an expansion of Th17, rather than γδ-T cells, in response to Bph. These changes in immune cell subtypes within the lung were also accompanied by increases in Teff, Th17, and Treg cells in the spleens of Bph-colonized mice, demonstrating that airway colonization was also associated with immune changes detectable at distant tissues (Figures S1I, S1J, and S1K).
Bph Airway Colonization Induces a Th17 Immune Response
We next performed RNA sequencing (RNA-seq) on whole lungs from colonized and control mice 7 weeks after inoculation to determine the effects of Bph colonization on transcriptional regulation. Analysis of transcriptomic data using gene set enrichment analysis (GSEA) confirmed Bph markedly upregulated functions involved in the immune response in persistently colonized animals (Figure 2A). The most markedly upregulated pathway in colonized mice was associated with the intestinal immune response for immunoglobulin A (IgA) production. Examination of leading-edge genes (i.e., genes driving the enrichment score) from this pathway showed that many of the most strongly enhanced transcripts are also associated with iBALT formation, including genes for mucosal immune activation (Icos, Icosl, CD40, and CD40lg) and antibody generation (Aicda and Pigr; Figure 2B, top panel; Table S1). Genes involved in T cell differentiation pathways, including Th17 cell signaling and differentiation, were also enriched in colonized mice compared to controls, including Th17-associated cytokines Il17a and Il17f, (Figures 2A and 2B, middle panel), transcription factor (Rorc), and chemokines (Ccl20 and Ccr6; Figure S2), reinforcing the prominent Th17 signature we observed by immunophenotyping, (Table S1). Expression of Il22, another major Th17-associated cytokine, was not detected in the majority (80%) of samples and was not different between groups (Figure S2). In contrast, colonized mice demonstrated lower levels of gap-junction-associated gene transcripts, which may represent increased epithelial permeability in colonized mice resulting from immune activation (Figures 2A and 2B, bottom panel; Table S1). We additionally examined genes associated with T helper cell effector functions. We observed increases in transcriptional signatures of Th1 (Ifng and Tbx21) and Treg (Foxp3) cells but did not observe significant transcriptional changes in Th2-related genes (Il4, Il5, and Il13; Figure S2; Table S1).
Figure 2. Host Transcriptomic Changes Due to Bph Colonization.

(A) GSEA of KEGG pathways using whole-lung RNA-seq data from mice that received either live or HK Bph 52 days after inoculation. Only pathways with an adjusted p value of <0.05 are shown with their normalized enrichment score. Analysis was performed in R using the fgsea (Sergushichev, 2016) package. n = 5/group.
(B) Heatmaps and enrichment plots of selected KEGG pathways. On the left, enrichment plots for representative KEGG pathways are shown. To the right of each panel is a heatmap demonstrating normalized read counts and fold change of select leading-edge genes from each KEGG pathway. Average normalized read counts for HK (left column) and live (middle column) groups are shown in gray, and log2-fold change of each gene is shown in the rightmost column in blue and red. Genes that are significantly enriched are boxed. Analysis was performed in R using DEseq2.
Statistical significance: GSEA statistic as implemented in fgsea (Sergushichev, 2016) (A) or the Wald test with Benjamini-Hochberg (BH) correction as implemented in DESeq2 in (B).
In order to establish whether the expansion of Th17 cells to Bph was mediated by recognition of bacterial antigens displayed by antigen-presenting cells, we restimulated splenocytes from mice nasally inoculated with either HK or live Bph, stained them with carboxyfluorescein succinimidyl ester (CFSE), and then evaluated for cell division 72 h later. We observed an increase in the proliferation of activated T cells (CFSEloCD25+) when splenocytes isolated from mice colonized with live bacteria were co-incubated with Bph lysate compared to when splenocytes from mice that received a HK inoculum were co-incubated with Bph lysate (Figure 3A, bottom versus top rows). Co-incubation of cultures with an anti-major histocompatibility complex class II (MHC II) antibody blocked CD4+ T cell proliferation, confirming that Bph antigen requires MHC II to induce T cell proliferation (Figure 3A). Furthermore, ELISA quantification of IL-17A from the supernatants of restimulated cells from the lung demonstrated that CD4+ T cells from Bph-colonized mice produced IL-17A in an antigen- and MHC-class-II-dependent manner (Figure 3B). IL-17A was not detectable in the supernatant of T cell cultures from control mice.
Figure 3. The Th17 Response to Bph Is Antigen Specific and Aids Controlling Colonization.

(A) Left panel: Concatenated flow cytometry plots of cultures of splenocytes of five mice taken 52 days after inoculation with either HK or live Bph. Splenocytes were loaded with either no protein or Bph proteins from a HK culture. Cells were gated on CD4+TCRβ+ cells using CD25 as an activation marker and CFSE as a proliferation marker. Representative of 2 independent experiments. Right panel: Quantification of CD25+CFSElo T cells from splenocyte cultures as shown in (A). n = 9–10 mice/group, combined from 2 independent experiments.
(B) IL-17A ELISA of culture supernatants from lung CD4+ T cells co-cultured with antigen-loaded CD11c+ dendritic cells (DCs). CD4+ T cells were isolated from the lungs of five mice taken 43 days after inoculation with either HK or live Bph.
(C) CFU of Bph recovered from BALs of WT or RAG1−/− mice 14 days after colonization. n = 10 mice/group, combined from 2 independent experiments.
(D) Top: Schematic of the experimental approach to test the role of IL-17A during Bph colonization. Bottom: CFU of Bph recovered from lung homogenates (CFU/g), BAL, or nasal lavage (CFU/ml).
(E) ELISA showing SLPI protein expression in the lungs of mice inoculated with Bph and treated with anti-IL-17A monoclonal antibody compared to the isotype control.
Statistical significance: Kruskal-Wallis followed by post hoc one-tailed paired Wilcoxon rank-sum test with adjustment for multiple hypotheses using BH correction for (A) and (B) or two-tailed Wilcoxon rank-sum test for (C)–(E). Horizontal lines indicate median values. **p < 0.01; ***p < 0.001.
To further investigate the importance of an adaptive immune response in controlling bacterial abundance in the airways, we colonized WT and RAG1−/− C57BL/6J mice with Bph for 14 days and then sacrificed mice to assess the numbers of bacteria within bronchoalveolar lavage (BAL) fluid. We found that there was significantly greater Bph in the BALs of RAG1−/− than that of WT mice (Dewan et al., 2019), implicating an adaptive immune response in controlling the degree of bacterial colonization (Figure 3C). Interestingly, RAG1−/− mice colonized for 1 month with Bph did not demonstrate overt pneumonia (e.g., alveolar infiltrates) by histology (Figure S3A), nor significant weight loss compared to control mice that received a HK inoculum (Figure S3B), indicating that although an adaptive immune response helped control bacterial numbers, innate immune mechanisms were sufficient to prevent lethality. To further investigate the role of IL-17A in controlling Bph colonization, we treated mice with an anti-IL-17A blocking antibody before and during colonization (Figure 3D, top panel). Consistent with an important role for Th17 cells during Bph colonization, we found that mice treated with an anti-IL-17A blocking antibody had significantly higher titers of bacteria within the BAL and lung tissue than mice treated with an isotype control antibody (Figure 3D, bottom panel). Similar to RAG1−/− mice, although IL-17A aided in controlling Bph numbers, its blockade did not result in systemic effects, such as weight loss (Figure S3C).
Colonization with Bph Reduces Signatures of AAI
We next sought to test how long-term colonization with Bph and the Th17 response that it evokes would affect a concurrent allergic inflammatory response. To accomplish this, we colonized mice with Bph for 1 month before sensitizing and intranasally challenging mice to ovalbumin (OVA) (Figure 4A). Characterization of whole-lung tissue by flow cytometry demonstrated multiple alterations in immune cell populations that were attributable to Bph colonization. As previously seen in Bph-colonized mice (Figure 1), we noted an increased percentage of Teff populations (Figure S4A) and Th17 cells (Figure 4B) in mice undergoing OVA sensitization and challenge (OSC). However, in contrast to mice undergoing colonization alone, the percentage of Treg cells was reduced in colonized mice experiencing AAI (Figure S4B), and we noted no difference in neutrophils (Figure S4C).
Figure 4. Bph Protects from Allergic Airway Inflammation in an Ovalbumin Model.

(A) Schematic of the model to test the role of Bph in modulating AAI. Mice were inoculated with either HK or live Bph 4 weeks before starting OSC.
(B) Quantification of flow cytometry for intracellular cytokine staining of IL-17A+IFNγ− CD4+ T cells. n = 9–11 mice/group, combined from 2 independent experiments.
(C) Measurement of airway resistance in mice undergoing methacholine challenge after inducing AAI as shown in (A). Experiments were performed using a Flexivent FX1 system. Points represent mean ± SEM, n = 8 mice/group.
(D) Left: Representative PAS staining of mouse airways in mice initially receiving either an HK or live inoculum and then undergoing OSC. Right: Quantification of PAS-positive cells normalized to total bronchial epithelial area. n = 8–9 mice/group, combined from 2 independent experiments.
(E) Eosinophils as a percentage of live cells from lung. Eosinophils were defined as Ly6G−CD11b+SiglecF+MHC II−CD11c−SSChi cells. n = 9–11 mice/group, combined from 2 independent experiments.
(F) qRT-PCR of whole-lung RNA from mice undergoing OSC. Bars represent mean ± SEM, n = 7–8 mice/group.
(G) Top: Schematic of the experiment showing that mice were either inoculated with HK or live Bph 20 days before receiving 50,000 naive OTII T cells and then undergoing OSC. Bottom: Percentage of CD4+ T cells (CD4+) expressing the OT-II receptor (CD45.1+Vα2+Vβ5+, left panel) and eosinophils as percentage of live cell (right panel) recruited to the lung. n = 7–8 mice/group, combined from 2 independent experiments.
(H) Top: Schematic of mice inoculated with HK or live Bph receiving anti-IL-17A antibody or an isotype control during colonization and then OSC. Bottom: Percentage of Th2 cells (defined as TCRβ+CD4+CD25−CD44+St2+) as a percentage of CD4+ T cells.
(I) Percentage of Th17 cells (defined as TCRβ+CD4+CD25−CD44+CCR6+) as a percentage of CD4+ T cells. n = 5–10 mice/group.
Statistical significance: Mann-Whitney U test for (B), (E), and (G). Wilcoxon rank-sum test for (C), (D), and (F). Kruskal-Wallis test followed by post hoc Wilcoxon rank-sum test with adjustment for multiple hypotheses using BH correction in (H) and (I). Horizontal lines indicate median values. + p < 0.1; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
We gauged the effect of Bph on AAI by assessing airway hyperresponsiveness (AHR), a characteristic feature of asthma, in colonized and control mice during methacholine challenge. Colonized mice demonstrated less airway resistance in response to methacholine challenge at 6.25 mg/ml and higher doses, indicating reduced AHR in mice with Bph compared to that in control mice (Figure 4C). These changes in resistance were also associated with reduced elastance and tissue damping, consistent with decreased AAI in control mice (Figure S4D). We also examined the effects of Bph colonization on allergic inflammatory markers. Periodic-acid-Schiff (PAS) staining, which identifies goblet cells, of OVA-challenged mice demonstrated that colonized animals had fewer goblet cells, indicating reduced goblet cell metaplasia (Figure 4D). Likewise, an analysis of whole-lung tissue showed that eosinophils, a marker for allergic inflammation and severity in mouse models, were reduced by ~38% in control compared to those of colonized mice (Figure 4E). Both eosinophil recruitment and goblet cell metaplasia are mediated by cytokines produced by Th2 cells, including IL-4, IL-5, and IL-13. Quantitative RT-PCR (qRT-PCR) of these cytokines confirmed that Il4, Il5, and Il13 transcripts were reduced in OVA-sensitized and -challenged animals colonized with live Bph compared to those of HK controls (Figure 4F).
We next wanted to assess if the increased Th17 cells we observed in Bph-colonized, OVA-challenged mice represented bacterial-specific Th17 cells or if colonization created an environment that enhanced the generation of Th17 cells specific to OVA. As we observed in mice not exposed to OVA, splenocytes from colonized mice proliferated when stimulated with Bph lysates compared to those from mice exposed to a HK inoculum (Figure S3D). Similarly, restimulation of CD4+ T cells isolated from the lung also proliferated in response to Bph lysates, showing that Bph-specific T cells persisted in the lungs of colonized mice undergoing OSC (Figure S3E). The Th17 response that we observed in colonized mice was also selective to Bph compared to other related bacteria within the same phylum, as we did not observe any proliferation of T cells (Figure S3E) and detected only a minimal production of IL-17A in response to restimulation with another Proteobacterium, Escherichia coli (Figure S3F).
Exposure to Bph lysates also resulted in the production of IL-17A from CD4+ T cells isolated from the lung, spleen, mediastinal lymph node, and mesenteric lymph node of colonized mice. In contrast, IL-17A production by CD4+ T cells from control mice was markedly decreased compared to that from colonized mice, demonstrating that airway colonization resulted in the dissemination of Bph-specific, IL-17A-secreting T cells systemically (Figure S3G).
Given the persistence of a Bph-specific immune response during AAI, we wanted to assess whether a pre-existing immune response to an airway bacterium could reduce the degree of allergic sensitization to OVA and, consequently, prevent AAI. We first looked at the capacity of colonized and control mice to generate OVA-specific serum IgE after sensitization and challenge, by ELISA (Klaßen et al., 2017). We found that colonization with Bph did not significantly alter the amount of OVA-IgE in the serum compared to control mice (Figure S4I). We next examined the T cell response to OVA and found that both colonized and control mice responded similarly to the restimulation of splenocytes to OVA in vitro, demonstrating similar degrees of proliferation in response to OVA (Figures S4E and S4F). Furthermore, splenocytes from both colonized and control mice produced IL-17A in response to OVA, and although colonized mice produced slightly more IL-17A in response to restimulation, the baseline IL-17A production was also higher, suggesting that the difference between live and HK groups was not OVA dependent (Figure S4G). We observed a similar pattern of IL-17A production from CD4+ T cells isolated from the lung and restimulated with OVA, supporting the idea that the increased Th17 cells we observed in colonized mice undergoing OSC were primarily directed against Bph (Figure S4H).
Concurrent colonization with Bph could also decrease OVA presentation within the lung during allergic airway challenge, potentially accounting for the observed reduction in AAI. To assess if pre-existing colonization with Bph reduced OVA presentation in the lung, we injected colonized and control mice with CD45.1+CD4+ T cells expressing an OVA-specific T cell receptor (OTII) before sensitizing and challenging mice with OVA (Figure 4G, top panel). Both colonized and control mice had similar percentages and numbers of OTII cells in the spleen, suggesting that colonization did not systemically alter the response to OVA (Figures S5A and S5B). In the lungs of colonized mice receiving OTII cells, we again observed reduced eosinophils (Figure 4G, right), but the quantity of OTII T cells recruited to the lung was no different between colonized and control mice either by absolute counts (Figure S5C) or as a percentage of CD4+ T cells (Figure 4G, left). Thus, the antigen presentation of OVA was likely to be similar regardless of colonization status.
To directly assess the contribution of IL-17A to AAI, we treated groups of either Bph-colonized or control mice with anti-IL-17A antibody or isotype control, starting at the time of colonization and extending throughout OSC (Figures 4H and 4I, top panel). At the conclusion of the experiment, mice receiving both live Bph and anti-IL-17A antibody had increased titers of Bph from their BALs, but not nasal lavages, compared to mice receiving an isotype control antibody (Figures S5D and S5E). Along with this increase in bacterial abundance, mice receiving both Bph and anti-IL-17A lost significantly more weight than mice receiving the isotype control antibody, regardless of colonization status (Figure S5F). The increases in bacterial abundance, as well as the weight loss observed in Bph-colonized mice receiving anti-IL-17A antibody, imply a protective role of IL-17A in Bph-colonized mice undergoing allergic airway sensitization and challenge.
To assess the effect of anti-IL-17A treatment on allergic inflammation in the lung, we performed flow cytometry on lung immune cells using St2 expressed on Teff (TCRβ+CD4+CD25−CD44+St2+, referred to as Th2 cells hereafter; CD25 was used to exclude Treg cells expressing St2) as a marker for allergic inflammation. St2, or IL-33R, is known to be preferentially expressed on Th2 polarized cells (Löhning et al., 1998) and directly contributes to AAI by helping upregulate Th2 cytokine production (Schmitz et al., 2005). Affirming our earlier findings (Figures 4C–4F), we found that mice colonized with Bph and treated with the isotype control antibody had signs of reduced AAI, with lower percentages of Th2 cells than those of HK controls also receiving the isotype antibody (Figures 4H and S5G). Anti-IL-17A treatment reduced the severity of AAI, as measured by the percentage of Th2 cells in mice receiving a HK inoculum and treated with anti-IL-17A compared to isotype-control-antibody-treated mice. Blockade of IL-17A, however, did not significantly change the percentages of Th2 cells present in colonized mice, which were significantly reduced compared to isotype-treated HK mice.
To further understand these findings, we tested for differences in Th17 cells by examining Teff expressing CCR6, a marker for Th17 cells (Hirota et al., 2007) (TCRβ+CD4+CD25−CD44+CCR6+; referred to as Th17 cells hereafter). As shown previously (Figures 1F and 4B), mice colonized with Bph had higher levels of Th17 cells than HK mice (Figures 4I and S5H). Anti-IL-17A treatment reduced percentages of Th17 cells in HK mice when compared to isotype-treated HK controls. Yet, in Bph-colonized animals, the anti-IL-17A treatment did not significantly alter the percentages of Th17 cells, suggesting that blockade of IL-17A is insufficient to reverse the protective effect of Bph colonization. Although the percentages of Th17 cells in Bph-colonized mice was not different between anti-IL-17A and isotype control, the increase in bacterial abundance in the airway may provoke a compensatory immune response that complicates our interpretation of the role of IL-17A in our model of AAI. These results support the idea that Th17 cells induced by Bph are associated with protection from AAI but that monoclonal antibodies targeting IL-17A are inadequate to reverse their effect on AAI.
SLPI Is Regulated by Airway Colonization
To better understand how Bph reduces susceptibility to AAI, we examined the lung tissue transcriptional profile of mice undergoing OSC. This analysis corroborated our observation that Bph colonization resulted in a reduction in multiple markers of allergic inflammation (see Table S2; Figures 4F and 5A). Notably, the effect on these Th2 markers was only apparent in mice undergoing AAI because we noted no differences in expression of Th2-related cytokines in mice not undergoing sensitization and challenge (Figure S2; Table S1). Similarly, GSEA only identified two Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were significantly enriched in colonized mice undergoing OSC (Figures S6A and S6B), neither of which were enriched in colonized mice not undergoing AAI (Figure 2A). Examination of leading-edge genes from the IgA protection network and focal adhesion pathways identified in Figure 2A showed that these genes were no longer significantly different in mice undergoing OVA sensitization, consistent with the effects of AAI reducing the transcriptional signatures associated with colonization (Figure S6C).
Figure 5. SLPI Is Regulated by Colonization and Mediates Protection from AAI.

(A) Heatmap of immune genes regulated by Bph colonization in mice undergoing AAI. Average normalized read counts for each group are shown in gray, and log2-fold change in mice that received a live, compared to an HK, inoculum for each gene is shown in blue, white, and red. n = 3–5 mice/group.
(B) Volcano plot of whole-lung transcriptomic data from mice that received either an HK or live Bph inoculum and then underwent OSC (as shown in Figure 4A). Genes depicted as triangles were significantly enriched in colonized mice not undergoing AAI. Genes involved in an immune system process (defined by the Gene Ontogeny [GO] pathway, GO: 0002376) are shown in light blue and are also depicted in the heatmap shown in (A). n = 3–5 mice/group.
(C) ELISA showing SLPI protein expression in the lungs of mice inoculated with live Bph followed by AAI compared to those inoculated with HK. n = 7–8 mice/group.
(D) Correlation between SLPI protein expression and fold change in Il17a measured by qRT-PCR in the lungs of mice inoculated with HK or live Bph, followed by AAI. n = 7–8 mice/group
(E) qRT-PCR of SLPI from the whole lungs from germ-free, RAG1−/−, and conventionally raised WT mice. n = 9–10 mice/group.
(F) Transcription of Slpi in human alveolar epithelial cell line A549 in response to cytokine stimulation. Cells were treated with 1 ng/ml IL-1β, 100 ng/ml IL-17A, and/or 10 ng/ml TNF-α as shown. Experiment performed in n = 5 biological replicates, each representing the average of 3 technical replicates.
Statistical significance: Wald test with BH correction as implemented in DESeq2 in (A) and (B); Wilcoxon rank-sum test for (C); Spearman’s rank test in (D); or Kruskal-Wallis test followed by post hoc two-tailed paired Wilcoxon rank-sum test with adjustment for multiple hypotheses using BH correction in (E) and (F). Boxes indicate 25th and 75th percentiles and whiskers are 1.5 × interquartile range. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To find genes mediating protection from AAI, we sought to identify transcripts that were upregulated in colonized compared to control animals, both in mice undergoing colonization only (Table S1) and in mice undergoing OSC (Table S2). This analysis revealed that 44 genes were upregulated in both datasets by a minimum of a 2-log2-fold change with a maximum p value of 0.05 after correction for multiple hypotheses (Figure 5B, triangles). A total of 16 of these 44 transcripts originated from genes involved in immune system processes (Figures 5A and 5B, blue triangles). In addition to genes involved in germinal center formation (Nuggc) and antibody biosynthesis (Jchain), we noted that Slpi was significantly upregulated by Bph. We confirmed that the transcriptional upregulation of Slpi was also accompanied by increased SLPI protein in the lungs of Bph-colonized mice undergoing OSC (Figure 5C).
Slpi was first characterized as a constitutively expressed inhibitor of neutrophil elastase in the lung and has been demonstrated to be an important antimicrobial peptide in the airway, with efficacy against both Gram-negative and Gram-positive bacteria (Hiemstra et al., 1996; Wiedow et al., 1998). Animal models have directly demonstrated that mice lacking Slpi experience worsened AAI (Marino et al., 2011) and that endogenously overexpressed (Raundhal et al., 2015) or exogenously administered SLPI (Forteza et al., 2001) can mitigate AAI, indicating that Slpi may play a protective role in asthma. Although Slpi is known to be upregulated by innate immune activation through the functions of tumor necrosis factor alpha (TNF-α) and IL-1β (Sallenave et al., 1994), we noted that SLPI protein levels correlated with Il17a transcription in the lungs of mice undergoing AAI and may help to mediate the reduction of AAI observed in Bph-colonized mice (Figure 5D).
To test the idea that colonization with Bph increases the expression of SLPI within the lung through IL-17A, we first used qPCR to establish that microbial colonization of mice modulates the expression of Slpi in the lung by comparing conventionally raised WT, RAG1−/−, and germ-free (GF) C57BL/6 mice either undergoing 2 weeks of colonization with Bph or remaining non-colonized (Figure 5E). We first confirmed that the transcription of Slpi in WT mice was significantly upregulated in colonized mice compared to controls. In contrast, RAG1−/− mice did not demonstrate increased Slpi expression in response to Bph colonization, suggesting that the adaptive immune system helps regulate the expression of SLPI (Figure 5E). These results also showed that increased Slpi expression in the lung reflects local (rather than gut) microbial exposure since GF and non-colonized conventional mice had similar amounts of Slpi expression.
We next asked if the positive regulation of SLPI resulting from Bph colonization was mediated through IL-17A by treating a human lung epithelial cell line (A549) with TNF-α, IL-1β, and IL-17A. We found that although IL-1β and TNF-α alone were sufficient to upregulate Slpi, IL-17A did not significantly increase Slpi. Incubation of A549 cells with both TNF-α and IL-17A, however, demonstrated increased Slpi transcription in cells incubated with both cytokines compared to either one singly (Figure 5F). To confirm that IL-17A played a role in potentiating SLPI production in vivo, we examined mice colonized with Bph and treated them with a neutralizing monoclonal antibody to IL-17A for 2 weeks (Figure 3D). Compared to mice receiving an isotype control, colonized mice treated with an anti-IL-17A antibody showed reduced levels of SLPI in the lungs, as measured by ELISA (Figure 3E), supporting a stimulatory effect of IL-17A on SLPI production in the lung.
SLPI Shapes the Microbial Ecology of the Upper Airway in Humans
Our results demonstrate that SLPI is regulated by airway microbial colonization in mice and that higher amounts of SLPI are associated with reduced intensity of AAI we observe in Bph-colonized mice. In humans, lower expression of SLPI from epithelial brushings has previously been reported in individuals with severe asthma (Raundhal et al., 2015). To test the idea that microbial airway colonization could modulate the production of SLPI within the airway, we first selectively cultured nasal lavage specimens from adults and children with moderate to severe asthma or healthy controls (from the Microbiota in Asthma Research Study [MARS]; see Table S3 for demographic data) for Haemophilus, Streptococcus, and Moraxella airway bacteria previously associated with asthma. Overall, we noted that children demonstrated higher recovery of Haemophilus and Streptococcus (p < 0.0001 for each comparison, Fisher’s exact test) from nasal lavage than adults regardless of health status (Table 1). Consistent with prior reports of other respiratory microbiologic surveys in asthma (Bisgaard et al., 2007; Durack et al., 2017), we recovered Haemophilus more frequently from nasal lavages from adults and children with asthma than healthy controls (Table 1).
Table 1.
Recovery of Haemophilus, Streptococcus, and Moraxella from the Nasal Lavage of MARS Participants
| Genus | Adult and Pediatric | Adult | Pediatric | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Asthma (34) | Healthy (55) | p value | adjusted (adj.) p value | Asthma (16) | Healthy (35) | p value | adj. p value | Asthma (18) | Healthy (20) | p value | adj. p value | |
| Haemophilus | 76% | 35% | 0.00017* | 0.0015* | 50% | 11% | 0.0047* | 0.021* | 100% | 75% | 0.048* | 0.14 |
| Streptococcus | 65% | 49% | 0.19 | 0.28 | 38% | 31% | 0.75 | 0.84 | 89% | 80% | 0.66 | 0.84 |
| Moraxella | 12% | 2% | 0.068 | 0.15 | 0% | 0% | 1 | 1 | 22% | 5% | 0.17 | 0.28 |
Statistical significance: Fisher’s exact test with adjustment for multiple hypotheses using BH correction.
Denotes significance.
We did not observe any significant differences in nasal lavage SLPI abundances between healthy and asthmatic populations (Thijs et al., 2015; Figure S7A). However, we next performed an exploratory analysis to ask if nasal lavage SLPI corresponded with asthma control by comparing Asthma Control Test (ACT) scores, a clinically validated survey of symptom control in asthmatics (Nathan et al., 2004). This analysis showed that in adult asthmatics, better asthma control (corresponding to higher ACT scores) corresponded to higher nasal SLPI levels, consistent with a protective role for SLPI in asthma (Figure 6A). We also observed a relationship between SLPI and Haemophilus upper airway colonization across the adult and pediatric populations by showing a positive correlation between SLPI nasal concentration and the titers of Haemophilus recovered from colonized individuals (Figure 6B). These differences did not appear to be due to the amount of corticosteroid usage because we observed no difference in nasal SLPI levels in asthmatics between those with the highest inhaled corticosteroid doses and those with lower doses (p = 0.8, Wilcoxon rank-sum test; Table S3).
Figure 6. SLPI Levels in Human Upper Airways Are Regulated by the Microbiota.

(A) Correlation of ACT score to nasal lavage SLPI from adult asthmatics.
(B) Correlation of SLPI to recovery of Haemophilus in nasal lavage fluid. All individuals with and without asthma from both adult and pediatric cohorts that had Haemophilus colonization were included in this analysis.
(C) Oral lavage microbial community composition predicts SLPI levels. V4–16S data from healthy human children and adults were used to construct a Random Forest model to predict SLPI levels based on the abundance of 11 amplicon sequence variants (ASVs). Ten-fold cross-validation of this model performed comparably to the complete model (inset).
(D) The taxonomic assignments and mean increase in mean square error (MSE) of ASVs included in the Random Forest model depicted in (C). The mean increase in MSE is an estimate of the importance of each taxon to the Random Forest model.
Statistical significance: Spearman’s rank-order correlation for (A) and (B); or Pearson’s correlation in (C). Boxes indicate 25th and 75th percentiles and whiskers are 1.5 × interquartile range.
Together, these results show that SLPI abundance in the nasal passages is correlated both with the presence of Haemophilus and a clinical correlate of asthma control. Although SLPI levels in the airway are known to be modulated by bacterial colonization and infection (Parameswaran et al., 2011), we hypothesized that airway microbial community composition, as a whole, would also influence SLPI abundance. To test a relationship between SLPI levels and microbial community composition, we quantified SLPI and performed V4–16S rRNA sequencing on oral lavage samples from MARS participants. Similar to the nasal lavage specimens, we did not observe a difference in SLPI from oral lavage specimens (Figure S7B). 16S rRNA analysis showed that the composition of these oral microbial communities was consistent with prior surveys of oral microbial communities (Aas et al., 2005), showing that these communities were made up largely of Streptococcus, Veillonella, and Prevotella species (Figure S7C). We did not discern any differences in alpha diversity (Figure S7D) nor community composition (Figure S7E) in the microbial ecology of oral lavage specimens between the different cohorts. To test the idea that SLPI abundances in oral lavage samples would reflect microbial community composition, we built a supervised learning model from healthy adults and children (n = 55) to predict oral SLPI levels based on 16S rRNA abundance data. We first identified 11 taxa (Figure 6D) that were important for predicting SLPI abundances using random forest (Liaw and Wiener, 2002). These taxa were used to build a model that explained 40% of the variance in the oral lavage SLPI levels measured by ELISA (Figure 6C). This correlation was maintained after a 10-fold cross-validation, indicating that overfitting plays a limited role in the success of the model (Figure 6C, inset). Of the 11 taxa that we included in our model (Figure 6D), some of these bacteria or their close relatives have previously been noted to elicit, or be susceptible to, SLPI in vitro (e.g., Neisseria; Cooper et al., 2012).
DISCUSSION
Asthma is known to be shaped by a myriad of genetic factors, environmental factors, and—increasingly—microbial exposures. Although components of the airway microbiota have been previously associated with enhanced susceptibility to asthma, our findings point to a complex and potentially beneficial role of some bacteria within the airway. In this study, we show that airway colonization of mice with Bph, a murine-adapted bacterium, elicits a Th17 immune response that aids in controlling bacterial abundance in the airway. Furthermore, we show that when animals undergo AAI challenge, mice previously colonized with Bph demonstrate protection from AAI. Colonization with Bph does not appear to impact the degree of allergic sensitization nor the presentation of allergen in the lung, but our results do suggest an effect of colonization on Th2 polarization and blunting of allergic effector cell recruitment. In contrast to other studies that have emphasized the potentially deleterious roles of allergen-specific T cells polarized to Th17 in AAI (McKinley et al., 2008; Wakashin et al., 2008), our results show that immune responses to airway bacteria co-exist with immune responses to allergens and could counteract allergic responses. Although we were unable to demonstrate a direct protective role of IL-17A induced by Bph, this could be explained in part by the dual effect of anti-IL-17A on both bacterial colonization and AAI. Because anti-IL-17A treatment results in increased Bph titers, this worsened infection may offset the effects of IL-17A blockade by further stimulating a bacterial Th17 immune response, which is consistent with our observation that Th17 cells were not reduced in colonized mice treated with anti-IL-17A (Figure 4I). Alternatively, recent advances have shown that blocking IL-17A may lead to a compensatory upregulation of other Th17-related cytokines in some types of Th17 cells, which could also mitigate the effects of anti-IL-17A antibodies (Chong et al., 2020). Together, our data support previously described protective roles for Th17 in blunting allergic effector responses (Schnyder-Candrian et al., 2006) but additionally implicate immune responses to airway commensals as a potential source for this Th17 response.
During Bph colonization, we observed that the Th17 response was associated with increased expression of antimicrobial peptides, including SLPI, in the lung. Although SLPI is known to be regulated by TNF-α and IL-1β (Sallenave et al., 1994), we show that its expression is enhanced by IL-17A in vitro and in vivo. The potential of SLPI to mitigate AAI has been appreciated for decades (Wright et al., 1999), and low SLPI airway levels have been previously noted in severe asthma (Raundhal et al., 2015). A direct protective role of SLPI in AAI has been shown in animal models demonstrating that mice lacking SLPI have increased Th2 polarization markers, increased eosinophil recruitment, and worsened measures of AHR (Marino et al., 2011). The molecular and cellular targets of SLPI still require additional investigation. SLPI is known to have the ability to directly penetrate cells to inhibit nuclear factor κB (NF-κB) signaling (Taggart et al., 2005), and exogenously administered SLPI (Wright et al., 1999), as well as SLPI expressed from a transgene (Marino et al., 2011; Raundhal et al., 2015), reduces eosinophilic inflammation in models of AAI. Additionally, SLPI has been demonstrated to be an endogenous regulator of eosinophil and basophil function by inhibiting Toll-like receptor 4 (TLR4) signaling in allergic inflammation (Matsuba et al., 2017). However, more studies investigating the tissue-specific effects of SLPI resulting from airway bacterial colonization will be needed to address which cell types and molecular targets are affected by SLPI and mediate protection from AAI. Along with our findings that SLPI shapes upper airway microbial ecology and is enhanced by IL-17A, this suggests an important role for Th17 responses in regulating airway microbiota composition that may, in turn, affect AAI severity.
Our results help improve our understanding of the nature of airway microbe-immune interactions in human asthma and could have clinical ramifications. IL-17A and its associated effector functions, such as neutrophilia, are currently being investigated as both biomarkers for particular asthma endotypes and therapeutic targets. However, preliminary studies targeting IL-17A in asthma have not proven to be uniformly efficacious, potentially indicating varied roles of Th17 responses in asthma. Our findings directly demonstrate that although a blockade of IL-17A reduced markers of AAI in non-colonized mice, this protective response was lost upon bacterial airway colonization with Bph. This microbiota-mediated variability in response to anti-IL-17A therapy could be clinically relevant as better understanding the interplay between airway microbes and AAI could help define individuals that are likely to benefit from IL-17A-targeted therapies.
In our clinical study, we observed an increased prevalence of colonization with Haemophilus in asthmatics in both our pediatric and adult cohorts. Interestingly, the quantity of SLPI was associated both with individual bacteria in nasal specimens and overall microbial community composition from oral lavages. Combined with observations that SLPI abundance in the airway is modulated during infection (Parameswaran et al., 2011; Persson et al., 2017), we propose that regulation of SLPI expression is a crucial mechanism underlying homeostasis between the respiratory microbiota and the airway mucosal immune system. In addition to its correlation to Haemophilus in the upper airway, SLPI tended to be lower in adult subjects with poor asthma control. Although our finding that SLPI levels were increased with Haemophilus abundance may seem at odds with the previously described role of Haemophilus in severe asthma (Goleva et al., 2013; Simpson et al., 2016), we note that the correlation between SLPI and Haemophilus included both healthy and asthmatic individuals. One interpretation of our data is that the relationship between SLPI and Haemophilus may differ between healthy and asthmatic individuals, as evidenced by the ability of Haemophilus to alter the behavior of the immune system in the context of airway inflammation (Shilts et al., 2020). Furthermore, patients from the MARS study were limited to those with moderate to severe asthma without an ongoing exacerbation and do not represent the entire spectrum of asthma phenotypes and Haemophilus colonization. Nevertheless, our data suggest that SLPI expression is part of a regulatory response associated with Haemophilus colonization and that appropriate expression in the context of airway colonization is part of a beneficial response that leads to better controlled asthma.
A potential limitation of our study is that Bph itself is not a known human airway colonizer. Although other members of the Bordetella genus can colonize the airways, their relationship to asthma is less well characterized than HSM organisms. Nevertheless, in contrast to human airway microbes, the persistent colonization of Bph in the airways without inducing an overt pneumonia, combined with its potent ability to alter immunity, offers a perspective of respiratory bacteria as potentially beneficial, rather than detrimental, in asthma. Bph’s unique adaptation to the murine airway could make it a powerful tool to advance our understanding of airway microbe-immune interactions important in respiratory inflammatory diseases. An additional limitation from our human study was that we only observed a correlation between SLPI and asthma control in our adult cohort. This could be the result of the small sample size or differences in asthma control between our adult and pediatric cohorts (adults tended to have poorer asthma control), or it could indicate differences in the immune responses associated with asthma between adult and pediatric populations. In addition to addressing the role of SLPI in pediatric populations, future human studies will be needed to confirm the relationship between upper airway microbial ecology and SLPI using culture-independent methods and to determine the specific immune signatures, such as IL-17A, associated with SLPI production in asthma.
Overall, our findings support a key role of airway microbes in shaping the severity and character of AAI responses. In the context of our increasing recognition of asthma as a clinically and immunologically heterogeneous disease, our results emphasize the importance of taking the resident airway microbiota into account when evaluating immune responses in the context of asthma. Differentiating Th17 responses directed against exogenous allergens versus those directed at colonizing airway bacteria may prove to have an important impact on how we interpret the role of Th17 responses in asthma. We propose that SLPI’s multifactorial role as a microbiota-responsive, immune-modulatory protein makes it a biomarker for dissecting the relationships between airway microbial communities and asthma.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Kau (akau@wustl.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The accession number for the sequencing data from RNA-seq, genome sequencing assemblies, and 16S rRNA sequencing reported in this paper is European Nucleotide Archive: PRJEB36780.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
The study cohorts came from the Microbiota in Asthma Research Study (MARS), which was designed to investigate the contribution of the human airway and gut microbiota to asthma. Both an adult (ages 18–40 years) and a pediatric population (ages 6–10 years) were recruited from the St. Louis, Missouri area. Inclusion criteria for the asthmatic cohort included: (1) A physician diagnosis of moderate-to-severe asthma. We also used a prescription of either a medium to high dose inhaled corticosteroid, or a combination of inhaled corticosteroid with either a leukotriene antagonist or long-acting beta-agonist as evidence of moderate to severe asthma (This corresponds to step 3 of the Expert Panel Report 3 guidelines (National Asthma Education and Prevention Program, 2007). (2) Evidence of allergic sensitization with at least one positive skin prick test to a panel of aeroallergens or by the presence of aeroallergen-specific serum IgE. (3) A recent prescription of a course of oral corticosteroids within two years of enrollment. The healthy cohort included individuals without a self-reported history of wheezing or shortness of breath within 1 year of recruitment or a history of asthma, allergic rhinitis, food allergy or eczema. Additionally, both asthmatic and healthy study participants were excluded if they received antibiotics within 30 days of their study visit, if they had major surgery on the sinuses, lung or gastrointestinal tract or if they had another serious medical condition other than asthma. All samples described in this study were obtained during the enrollment visit when asthmatic subjects were not experiencing an exacerbation, as defined by the need for oral corticosteroids. Written informed consent documents were obtained from all patients or their legal guardians, and the protocol was approved by the Washington University Institutional Review Board (IRB ID# 201412035).
Oral lavage samples were obtained by having each subject swish 20 mL of sterile saline in their mouth for 30 s before spitting into a sterile container for further processing. Nasal lavage specimens were collected as described (Allen et al., 2013). Briefly, 5 mL of sterile saline was instilled into each nostril while the participant has his/her head tilted backward. After 5 s, the patient tilted their head forward and the nasal lavage fluid was collected into a sterile cup. To minimize the saline running into the back of subjects’ throats, we had patients repeat the sound “k-k-k” after administering saline. Separate oral and nasal lavage aliquots were either stored without additional processing (used for ELISA) or with a final concentration of 15% glycerol (for bacterial culture) and then stored at −80 C. As summarized in Table S3, a total of 93 participants were included in this analysis. Steroid dose was categorized based on previously published studies (Reddel et al., 2015). All available samples were used for each analysis.
Experimental Animals and Ethics
Animal experiments were reviewed by the Washington University Institutional Animal Care and Use Committee (Protocol #20180286). Male wild-type (WT) and RAG1−/− C57BL/6J mice were obtained from Jackson Laboratory (Bar Harbor, ME); OTII Rag1+/− Ly5.1+/− Foxp3-GFP and germ-free mice were bred in house for these experiments. Mice were maintained under either specific pathogen free (SPF) conditions or in a BSL-2 facility when colonized with B. pseudohinzii. Germ-free mice were maintained in a gnotobiotic facility using flexible plastic isolators and monitored monthly to ensure sterility. All animals were housed 5 animals per cage maximum and their welfare assessed daily after colonization. All mice were between 6 and 8 weeks old.
Bacterial Strains and Growth Conditions
Bordetella pseudohinzii strain 2–1 and 5–5 were isolated on Brain heart infusion (BHI) agar supplemented with 5% sheep’s blood from the bronchoalveolar lavage (BAL) of two different male C57BL/6J mice. We initially established that this strain was closely related to Bordetella hinzii by using selective PCR primers (Hayashimoto et al., 2012) and then conclusively identified this strain as Bph by whole genome sequencing (see below). For colonizations, Bph was grown overnight in BHI broth with shaking at 37°C. Bph strain 2–1 was used in all experiments unless otherwise noted. E. coli strain DH5a was grown in Luria Broth at 3C overnight with shaking.
Nasal lavage culture
We used a selective culture approach to quantify the amounts of live Streptococcus, Moraxella and Haemophilus present in nasal lavage fluid of asthmatic and non-asthmatic individuals. 100 μl of nasal lavage fluid from each participant were plated onto Streptococcus selective agar (BBL), M. catarrhalis selective agar (Remel), and Haemophilus selective agar (Remel). M. catarrhalis selective plates were incubated for 24 hours at 37°C, while Haemophilus and Streptococcus selective plates were grown for 24 h at 37°C in the presence of 5% CO2. We enumerated Streptococcus colonies that demonstrated alpha-hemolysis on Streptococcus selective plates; Moraxella as colonies that showed gamma-hemolysis on selective plates; and Haemophilus as colonies that were gamma-hemolytic on Haemophilus selective plates. The numbers of colonies were semiquantitatively assessed with the absence of growth as “0,” presence of 1–50 c.f.u. as “1+,” presence of 51–100 c.f.u. as “2+,” presence of 101–300 c.f.u. as “3+” and > 300 c.f.u. as “4+.” Contamination by other organisms was detected by 16S rRNA sequencing (see below) of plate sweeps of agar plates. If the presence of Moraxellaceae, Streptococcaceae, and Pasteurellaceae was not detected by 16S rRNA sequencing on their respective plate sweeps, growth was noted as “0.”
METHOD DETAILS
Colonization with Bph
Bph cultures were resuspended in PBS then diluted to approximately 10^5 CFU/ml. To nasally colonize mice, we anesthetized mice with ketamine/xylazine then pipetted a total of 50 μl of Bph into the nares of sedated animals with the goal of delivering ~104 CFU to each mouse. For mice receiving HK Bph, we autoclaved (125°C for 20 minutes) an aliquot of PBS Bph suspension to deliver a dose of ~104 CFU of HK Bph and confirmed the absence of viable bacteria by culture.
Bph Genome Sequencing
DNA was isolated from overnight cultures of Bph using bead-beating with phenol-chloroform extraction. The extracted and purified DNA was then sheared to 150 bp using a Covaris E220 sonicator. Barcoded sequencing adapters were then ligated to A-tailed, end-repaired DNA fragments (Chen et al., 2013) which were then amplified and sequenced using a MiSeq (Illumina) with paired-end 250 bp reads. Genome sequences were then assembled using SPAdes (Bankevich et al., 2012). Virulence factors in Bordetella genomes were identified by BLAST (Altschul et al., 1990) against a database of genes from the Virulence Factor Database (Chen et al., 2016).
Enumeration of Bph
BAL (Patnode et al., 2014) and nasal lavage were obtained as described (Puchta et al., 2014). Lungs were perfused with sterile PBS before being removed and 2 mm of tracheas were excised before homogenizing each tissue in 1 mL of PBS with 0.025% Triton X-100. Samples were then serially diluted then spotted onto BHI Blood Agar plates and colonies counted after 18h. Antibiotic markers were not available, so colony morphology was used to identify Bph. Additionally, we confirmed the presence of Bph in samples by V4 16S rRNA sequencing (see below). Typically, we observed no CFU from the BALs, lungs or tracheas of non-colonized animals. Nasal lavages from Bph colonized animals had ~10^3 more total CFU than non-colonized animals.
Asthma model
Allergic airway inflammation (AAI) was induced in mice using chicken egg ovalbumin (OVA), as previously described (Kuperman et al., 2005). Mice were sensitized on days 0, 7 and 14 by i.p. injections of OVA (50 mg, Sigma grade V) complexed with aluminum potassium sulfate (Imject Alum, Thermo Scientific) in a total volume of 200 μL in sterile PBS 1X. Mice were then challenged intranasally with OVA (1 mg in 50 μL of sterile PBS, or 20 mg/ml) on days 20–22 under anesthesia. Control mice were sensitized and challenged with PBS 1X unless otherwise noted. On day 23, mice were sacrificed and tissues were collected for further analysis.
Processing of mouse tissue for RNA and protein
We isolated total RNA from mouse tissue as previously described (Ridaura et al., 2013). Briefly, whole lungs were removed from mice and homogenized in 2 mL of Trizol reagent. Crude RNA was then extracted from a 0.3 mL portion of the homogenized tissue and then purified using the RNAeasy kit, according to the manufacturer’s protocol (QIAgen). If protein quantification in addition to RNA profiling was planned, mouse lungs were flash-frozen in liquid nitrogen at the time of sacrifice, then pulverized while still frozen then aliquoted and stored at −80°C until used for RNA extraction (as above) or protein quantification. For protein ELISAs pulverized aliquots were homogenized in PBS supplemented with protease inhibitor cocktail followed by centrifugation at 16,000 rcf. for 20 minutes and the supernatant retained. Samples were normalized to total protein quantitated using Qubit protein assay kit (Fisher Q33211)
Transcriptional profiling of mouse lungs
RNA quality was then assessed using a BioAnalyzer (Agilent) to ensure that all samples had an RNA integrity number greater than 8.0. Stranded, poly-A enriched libraries were then generated using the NEBnext Ultra II library prep kit according to the kit’s instructions and sequenced on two lanes of a HiSeq3000 using 1×50 bp chemistry. An average of 36,338,433 reads (with a standard deviation of 13.3 million reads) were obtained per sample. After demultiplexing these data, reads were mapped to the mouse genome downloaded from Ensembl (GRCm38.p6, release 98; Hunt et al., 2018) using bowtie2 (Langmead and Salzberg, 2012) and reads were quantified at the gene level using htseq (Anders et al., 2015). All technical replicates retained high similarity (Spearman rho > 0.93) and were combined for further analysis. Differentially expressed transcripts were identified using DESeq2 (Love et al., 2014). Transcripts were mapped to corresponding entrez ID using biomaRt (Durinck et al., 2009) and were used to identify genes (Figure 5B) and pathways of interest. Functional pathways altered during colonization and/or AAI were identified by gene set enrichment analysis using fgsea (Sergushichev, 2016) using the Kyoto Encyclopedia of Genes and Genomes database (Kanehisa and Goto, 2000) accessed through the gage R package (Luo et al., 2009).
Histopathology
Lung right lobe was collected in 4% PFA and tissue sections were prepared from paraffin block and stained with H&E. Whole lung was collected and stored in 4% PFA for 24 hours, then rinsed with 70% ethanol. Tissue sections were prepared from paraffin blocks and stained with PAS. Slides were analyzed on a Nanozoomer 2.0-HT at x20 and x40 objectives. The six largest airways were then analyzed using Aperio ImageScope software (Ge et al., 2016) positive pixel count algorithm. A ratio was calculated as the number of strong positive pixels (NSP) to the number of total pixels (Ntotal) (Ehlers et al., 2018).
16 s rRNA sequencing
DNA extracts were generated from fecal or cecal samples by phenol-chloroform extraction with bead beating as previously described (Kau et al., 2015). 16S amplicons were generated using indexed primers (Caporaso et al., 2011) and sequenced using a MiSeq with paired-end 250 bp reads. To process these data, we used DADA2 (version 1.10.1 in R) to generate amplicon sequence variants (ASVs) from the demultiplexed data (Callahan et al., 2016). Forward and reverse reads were merged, chimeras were removed, and taxonomy was assigned using the inbuilt DADA2 function for the RDP Classifier (Wang et al., 2007) with a custom database described in Kau et al. (2015) and minimum bootstrap support of 80%.
Analysis of 16S data was carried out using phyloseq (version 1.28.0) in R (version 3.6.1) (McMurdie and Holmes, 2013). Random forest was carried out using the randomForest (Liaw and Wiener, 2002) for feature selection as described in Rudnicki and Kursa (2009). Briefly, random forest was carried out in regression mode with SLPI levels measured by ELISA in oral lavage samples as the response and relative abundance of ASVs from 16S sequencing of oral lavage as predictors. Additionally, randomly permuted versions of each predictor, termed shadow variables, were included. After each iteration of Random Forest, the importance scores of the predictors were compared to the importance scores of their shadow variables. Predictors and their shadow variables were removed if the predictors were not more important than their shadows a significant number of times. After 100 iterations of Boruta, predictors that were selected more often than by chance alone were used in the final model.
Lymphocyte isolation from tissues
Mouse lung tissue was dissociated as previously reported (Patnode et al., 2014). Lungs were minced and incubated in digestion buffer (0.2 U/ml Liberase DL (Roche Applied Sciences) and 0.2 mg/ml DNase (Sigma) in Hank’s Buffered Salt Solution (without Ca2+/Mg2+) for 25 min at 37°C before being passed through a 70μm cell strainer. Spleen and lymph nodes were dissociated manually and passed through a 70μm cell strainer. Red blood cells were removed from lung and spleen samples by treating with ACK lysis buffer.
Restimulation assays
CD4+ lymphocytes from the lung, spleen and lymph nodes were purified via antibody-conjugated magnetic bead separation with the anti-CD4 kit from STEMCELL technologies (Easysep). Antigen-presenting cells (APCs) were harvested from the spleen of mice that had been injected 7 days previously with 1×106 B16-FLT3L expressing melanoma cells (Mach et al., 2000). APCs were isolated using the anti-CD11c kit from STEMCELL Technologies following the manufacture protocol. Bacterial antigens were prepared by resuspending bacteria from an overnight culture in PBS, autoclaving for 15 minutes, then normalizing to total protein content. 5×104 CD11c+ cells were incubated with 5 or 20 μg of Bph and E. coli antigen or 500 μg of OVA for 30 min. 1×105 CD4+ cells stained with Cell Trace FITC (Invitrogen™) were then added to the CD11c+ cells and incubated for 72 hours at 37°C. For restimulation of splenocytes, 7.5×105/ml cells were stained with Cell Trace FITC, then incubated with Bph proteins or OVA for 72 hours at 37°C. For MHC class II blocking experiments, CD11c+ cells were treated with 20 μg/mL anti-MHC class II blocking antibody for 30 minutes before adding bacterial proteins. All cells were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin and 100mM 2-mercaptoethanol.
FACS assays
For intracellular staining of cytokines, cells were stimulated for 4 h at 37°C with PMA (10ng/mL), ionomycin (200ng/mL), monensin (1:1000), and brefeldin A (1:1000). LIVE/DEAD Fixable Aqua Dead Cell Stain Kit was used to assess cell viability in all panels. Data were acquired on a FACS Canto II (BD Biosciences) equipped for the detection of eight fluorescent parameters or for Figures 4H and 4I, a five-laser Aurora (Cytek Biosciences). Data analysis was performed using FlowJo version 10 or higher software (Treestar, Ashland, OR).
Protein quantification by ELISA
IL-17A (Biolegend) and mouse SLPI (R&D Systems DY1735–05), and human SLPI (R&D Systems DY1274–05) ELISAs were performed according to the manufacturer’s protocol. Mouse serum OVA-specific IgE was quantified by sandwich ELISA (Zuberi et al., 2000). Briefly, plates were coated with 10μg/mL of OVA overnight at 4°C and then blocked with PBS 1x + 0.1% BSA. Serum (1:25 dilution) was placed in the wells and incubated for 2 h at room temperature. OVA-specific monoclonal IgE (Bio-Rad Cat No. MCA2259. Clone 2C6) was used as the standard curve. The bound IgE was detected by Rat anti-mouse IgE-HRP (Southern Biotech Cat No. 1130–05. Clone 23G3).
Quantitative PCR
RNA quality was assessed by gel electrophoresis and quantitated using Quant-it Ribogreen RNA assay kit (Invitrogen R11490). cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit at 500 ng RNA input (Fisher 4368814). qPCR was then performed using primers described in Table S4 using PowerSYBR Green PCR Master Mix (Fisher 4367659). Results were analyzed using the ddCT method (Livak and Schmittgen, 2001).
Measurement of Airway Hyperresponsiveness
We assessed murine airway physiology using a Flexivent with FX1 attachment. Mice were anesthetized using 10 mg/ml ketamine, 1 mg/ml xylazine cocktail and the trachea surgically cannulated (McGovern et al., 2013). After i.p. Injection of neuromuscular blockade with pancuronium (0.8 mg/kg) (Whitehead et al., 2014), we administered escalating doses of nebulized methacholine (0, 1.56, 3.125, 6.25, 12.5, 25 mg/ml) and assessed measures of respiratory mechanics as described in the main text using area under the curve measurements for each methacholine dose.
IL-17A neutralization
C57BL/6J mice were i.p. injected with a loading dose (500μg/mouse) of neutralizing anti-IL-17A antibody (BioXcell 17F3 BE0173) or IgG isotype control (BioXcell MOPC21 BE0083). Subsequent does (100mg/mouse) were administered 3 times weekly as depicted in the experimental schematics.
Adoptive transfer of naive T CD4+ cells
Lymphocytes from the spleen and lymph nodes of an OTII Rag1+/− Ly5.1+/− Foxp3-GFP mice were collected as described above. Naive (CD4+CD44−CD62L+ FoxP3−) cells were sorted into DMEM 10% using a FACS AriaII (BD Biosciences) and 5×104 cells per mouse were injected i.v. on day 15 of AAI model.
A549 cell culture
Human alveolar epithelial adenocarcinoma (A549) were obtained from ATCC (CCL-185) and maintained in Dulbecco’s Modified Eagles Medium (DMEM; GIBCOTM Cat. No. 31600–034) supplemented with 10% fetal bovine serum (FBS; GIBCO Cat. No. 16000–044) and 0.1 mg/mL of penicillin–streptomycin antibiotic (GIBCO Cat. No.15140122). Cells were maintained at 37°C and 5% CO2 in a humidified incubator with medium being replaced every 48 h.
To assess Slpi expression in response to cytokines, A549 cells (5×104) were plated on 24-well culture plates until they reached 50% confluence. Cells were starved (0.5% FBS) for 24 h then treated with IL-1β (1 ng/ml; Biolegend 579402), TNFα (10 ng/ml; Biolegend 570102), IL-17A (100 ng/ml; Biolegend 570502), TNFα plus IL-17A (10 ng/ml; 100 ng/ml) for 14 h at 37°C and 5% CO2. The supernatants were collected and the cells were lysed and stored in Trizol at −80°C. RNA was isolated from A549 cells stored in Trizol using the Macherey-Nagel Nucleospin RNA XS Kit according to the manufacturer’s protocol and qRT-PCR was performed as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Graphpad Prism 5.02 (GraphPad Software, La Jolla, CA) or R (R Development Core Team, 2011). Data are presented as the mean with error bars denoting SEM, box and whisker plots or scatterplots. Unless otherwise noted, statistically significant differences between groups with continuous data were determined by paired or unpaired Mann-Whitney U/Wilcoxon test, as indicated in the figures. For comparisons with multiple groups, we first performed a Kruskal-Wallace test and, if significant, performed a post hoc Wilcoxon with adjustment of p values for multiple hypotheses using BH correction. For categorical data, significance was determined using Fisher’s exact test. Associations were determined using Spearman’s Rank. In all figures, the following symbols were used to designate significance: n.s. = not significant, p > 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-mouse CD4 (Clone GK1.5) | Biolegend | Cat# 100406; RRID: AB_312691 |
| FITC anti-mouse CD11c (Clone N418) | Biolegend | Cat# 117306; RRID: AB_313775 |
| PE anti-mouse CD44 (Clone IM7) | BD Pharmigen™ | Cat# 553134; RRID: AB_394649 |
| PE anti-mouse SiglecF (Clone E50-2240) | BD Pharmigen™ | Cat# 562068; RRID: AB_10896143 |
| PE anti-mouse IL-17A (Clone TC11-18H0.1) | Biolegend | Cat# 506904; RRID: AB_315464 |
| PerCP-Cy™5.5 anti-mouse TCR β chain (Clone H57-597) | BD Pharmigen™ | Cat# 560657; RRID: AB_1727575 |
| PerCP anti-mouse CD45 (Clone 30-F11) | Biolegend | Cat# 103129; RRID: AB_893343 |
| PE/Cyanine7 anti-mouse IFNγ (Clone XMG1.2) | Biolegend | Cat# 505825; RRID: AB_1595591 |
| PE-Cy™7 anti-mouse CD11b (Clone M1/70) | BD Pharmigen™ | Cat# 552850; RRID: AB_394491 |
| PE/Cyanine7 anti-mouse CD62L (Clone MEL-14) | Biolegend | Cat# 104418; RRID: AB_313103 |
| APC anti-mouse Ly6G (Clone 1A8) | Biolegend | Cat# 127613; RRID: AB_1877163 |
| APC anti-mouse CD4 (Clone RM4-5) | Biolegend | Cat# 100516; RRID: AB_312719 |
| APC anti-mouse TNFα (Clone MP6-XT22) | BD Pharmigen™ | Cat# 561062; RRID: AB_2034022 |
| APC anti-mouse TCR β chain (Clone H57-597) | Biolegend | Cat# 109212; RRID: AB_313435 |
| APC anti-mouse CD45 (Clone 30-F11) | Biolegend | Cat# 103112; RRID: AB_312977 |
| APC/Cyanine7 anti-mouse TCR β chain (Clone H57-597) | Biolegend | Cat# 109220; RRID: AB_893624 |
| APC/Cyanine7 anti-mouse CD25 (Clone PC6) | Biolegend | Cat# 102026; RRID: AB_830745 |
| APC/Cyanine7 anti-mouse I-A/I-E (Clone M5/114.15.2) | Biolegend | Cat# 107627; RRID: AB_1659252 |
| eFluor450 anti-mouse FoxP3 (Clone FJK-16 s) | Thermo Fisher Scientific (eBioscience™) | Cat# 48-5773-82; RRID: AB_1518812 |
| eFluor450 anti-mouse IL-13 (Clone 13A) | Thermo Fisher Scientific (eBioscience™) | Cat# 48-7133-80; RRID: AB_11219690 |
| Brilliant Violet 421™ anti-mouse F4/80 (Clone BM8) | Biolegend | Cat# 123131; RRID: AB_10901171 |
| APC/Cyanine7 anti-mouse/human CD45R/B220 (Clone RA3-6B2) | Biolegend | Cat# 103224; RRID: AB_313007 |
| Brilliant Violet 421™ anti-mouse CD45.1 (Clone A20) | Biolegend | Cat# 110731; RRID: AB_10896425 |
| PE anti-mouse TCR Vα2 (Clone B20.1) | Biolegend | Cat# 127807; RRID: AB_1134184 |
| PerCP/Cyanine5.5 anti-mouse TCR Vα2 (Clone B20.1) | Biolegend | Cat# 127813; RRID: AB_1186118 |
| APC anti-mouse TCR Vβ5.1, 5.2 (Clone MR9-4) | Biolegend | Cat# 139505; RRID: AB_10897800 |
| PE/Cyanine7 anti-mouse TCR Vβ5.1, 5.2 (Clone MR9-4) | Biolegend | Cat# 139507; RRID: AB_2566020 |
| PE anti-mouse IL-4 (Clone 11B11) | Thermo Fisher Scientific (eBioscience™) | Cat# 12-7041-82; RRID: AB_466156 |
| Biotin anti-mouse CD11c (Clone N418) | Biolegend | Cat# 117304; RRID: AB_313773 |
| Biotin anti-mouse CD4 (Clone RM4-4) | Biolegend | Cat# 116010; RRID: AB_2561504 |
| Rat anti-mouse CD16/CD32 (Clone 2.4G2) | BD Bioscience | Cat# 553141; RRID: AB_394656 |
| LEAF TM Purified anti-mouse I-A/I-E (Clone M5/114.15.2) | Bioegend | Cat# 107610; RRID: AB_2813968 |
| Mouse anti-Ovalbumin antibody (Clone 2C6) | Bio-Rad | Cat# MCA2259; RRID: AB_2285753 |
| Rat Anti-Mouse IgE-HRP (Clone 23G3) | SouthernBiotech | Cat# 1130-05; RRID: AB_2794618 |
| InVivomAb anti-mouse IL-17A(Clone 17F3) | BioXcell | Cat# BE0173; RRID: AB_10950102 |
| InVivomAb mouse IgG1 isotype control, unknown specificity(Clone MOPC21) | BioXcell | Cat# BE0083; RRID: AB_1107784 |
| BUV737 Anti-mouse CD19 (Clone 1D3) | BD Biosciences | Cat# 612782; RRID: AB_2870111 |
| BUV395 Anti-mouse CD45 (Clone 30-F11) | BD Biosciences | Cat# 565967; RRID: AB_2739420 |
| BV750 Anti-mouse CD69 (Clone H1.2F3) | BD Biosciences | Cat# 747481; RRID: AB_2872156 |
| Pacific Blue™ Anti-mouse/human CD44 (IM7) | Biolegend | Cat# 103019; RRID: AB_493682 |
| Super Bright 436 Anti-mouse CD80 (Clone 16-10A1) | Thermo Fisher Scientific (eBioscience™) | Cat# 62-0801-80; RRID: AB_2716995 |
| Brilliant Violet 421™ Anti-mouse CD196 (CCR6) (Clone 29-2L17) | Biolegend | Cat# 129817; RRID: AB_10898320 |
| BD Horizon™ BV480 Anti-mouse CD103 (Clone M290) | BD Biosciences | Cat# 566201; RRID: AB_2739592 |
| Brilliant Violet 510™ Anti-mouse CD183 (CXCR3) (Clone CXCR3-173) | Biolegend | Cat# 126527; RRID: AB_2562204 |
| Brilliant Violet 570™ Anti-mouse Ly6G (Clone 1A8) | Biolegend | Cat# 127629; RRID: AB_10899738 |
| Brilliant Violet 605™ Anti-mouse CD8a (Clone 53-6.7) | Biolegend | Cat# 100743; RRID: AB_2561352 |
| Brilliant Violet 650™ Anti-mouse CD11b (Clone M1/70) | Biolegend | Cat# 101239; RRID: AB_11125575 |
| Brilliant Violet 711™ Anti-mouse I-A/I-E (MHCII) (Clone M5/114 15.2) | Biolegend | Cat# 107643; RRID: AB_2565976 |
| Brilliant Violet 785™ Anti-mouse CD11c (Clone N418) | Biolegend | Cat# 117335; RRID: AB_11219204 |
| Alexa Fluor 532 Anti-mouse CD4 (Clone RM4-5) | Thermo Fisher Scientific (eBioscience™) | Cat# 58-0042-82; RRID: AB_11218891 |
| PerCP-eFluor710 Anti-mouse CD64 (Clone X54-5/7.1) | Thermo Fisher Scientific (eBioscience™) | Cat# 46-0641-80; RRID: AB_2735015 |
| FITC Anti-mouse IgE (Clone RME-1) | Biolegend | Cat# 406905; RRID: AB_493288 |
| FITC Anti-mouse FcεRIα (Clone MAR-1) | Biolegend | Cat# 134305; RRID: AB_1626102 |
| PerCP/Cyanine5.5 Anti-mouse CD193 (CCR3) (Clone J073E5) | Biolegend | Cat# 144515; RRID: AB_2565741 |
| PE/Dazzle™ 594 Anti-mouse CD25 (Clone PC61) | Biolegend | Cat# 102047; RRID: AB_2564123 |
| PE/Cyanine5 Anti-mouse CD117 (ckit) (Clone 2B8) | Biolegend | Cat# 105809; RRID: AB_313218 |
| APC/Fire™ 750 anti-mouse TCR β chain (Clone H57-597) | Biolegend | Cat# 109245; RRID: AB_2629696 |
| Alexa Fluor 700 Anti-mouse NK 1.1 (Clone PK136) | Biolegend | Cat# 108729; RRID: AB_2074426 |
| APC Anti-mouse CD194 (CCR4) (Clone 2G12) | Biolegend | Cat# 131211; RRID: AB_1279135 |
| Anti-mouse IL-33Rα (St2) Biotin (Clone DIH9) | Biolegend | Cat# 145307; RRID: AB_2565735 |
| Anti-mouse CD11c (Clone N418) | STEMCELL Technologies (Easysep) | Cat# 60002BT.1 |
| Bacterial and Virus Strains | ||
| Bordetella pseudohinzii strain 2-1 and 5-5 | Isolated from two different male C57BL/6J mice bred in house | NA |
| Streptococcus, Moraxella and Haemophilus | Isolated from nasal lavage fluid of asthmatic and non-asthmatic individuals. Nasal lavage fluid were plated onto Streptococcus selective agar (BBL), M. catarrhalis selective agar (Remel), and Haemophilus selective agar (Remel). | NA |
| Escherichia coli (DH5α) | ATCC | Cat# 98489 |
| Biological Samples | ||
| Nasal and oral lavages were obtained from both an adult (ages 18–40 years) and a pediatric population (ages 6–10 years). | The study cohorts came from the Microbiota in Asthma Research Study (MARS | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI-1640 Medium | Milipore Sigma (Sigma Aldrich) | Cat# R8758 |
| DMEM, low glucose, pyruvate | Thermo Fisher Scientific (GIBCO™) | Cat# 11885084 |
| Trypsin | Thermo Fisher Scientific (GIBCO™) | Cat# 25300-54 |
| Fetal Bovine Serum | Thermo Fisher Scientific (GIBCO™) | Cat# 26140-079 |
| 2-Mercaptoethanol | Milipore Sigma (Sigma Aldrich) | Cat# M3148 |
| ACK Lysing Buffer | Thermo Fisher Scientific (GIBCO™) | Cat# A10492-01 |
| HBSS, no calcium, no magnesium, no phenol red | Thermo Fisher Scientific (GIBCO™) | Cat# 14175079 |
| Albumin from chicken egg white (Ovalbumin) - Grade V | Milipore Sigma (Sigma Aldrich) | Cat# A5503 |
| Imject™ Alum Adjuvant | Thermo Scientific™ | Cat# 77161 |
| Acetyl-β-methylcholine chloride | Milipore Sigma (Sigma Aldrich) | Cat# A2251 |
| DNase I | Milipore Sigma (Roche) | Cat# 10104159001 |
| Liberase™ DL Research Grade | Milipore Sigma (Roche) | Cat# 5401160001 |
| Defib sheep blood | HemoStat Laboratories | Cat# DSB050 |
| BD Difco Brain Heart Infusion Agar | Fisher Scientific (BD) | Cat# DF0418-07-9 |
| BD BBL Prepared Plated Media: Group A Selective Strep Agar with 5% Sheep Blood (ssA) | Fisher Scientific (BD) | Cat# L21779 |
| Remel Catarrhalis Selective Medium | Thermo Fisher Scientific | Cat# R01575 |
| Remel Haemophilus Isolation Agar w/bacitracin and horse blood | Thermo Fisher Scientific | Cat# R01470 |
| Phosphate Buffered Saline (PBS) 10X Powder | Fisher Scientific | Cat# BP665-1 |
| Phenol/Chloroform/Isoamyl Alcohol | Fisher Chemical | Cat# BP1752I400 |
| Triton™ X-100 | Milipore Sigma (Sigma Aldrich) | Cat# T8787 |
| TRIzol™ Reagent | Thermo Fisher Scientific (Invitrogen™) | Cat# 15596018 |
| 32% Paraformaldehyde | Fisher Scientific (Electron Microscopy Sciences) | Cat# 50-980-494 |
| Penicillin-Streptomycin | Milipore Sigma (Roche) | Cat# 11074440001 |
| Phorbol 12-myristate 13-acetate (PMA) | Milipore Sigma (Sigma Aldrich) | Cat# P8139 |
| Ionomycin from Streptomyces conglobatus | Milipore Sigma (Sigma Aldrich) | Cat# I9657 |
| Brefeldin A | Thermo Fisher Scientific | Cat# 00-4506-51 |
| Monensin | Thermo Fisher Scientific | Cat# 00-4505-51 |
| PowerSYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4367659 |
| Recombinant Human IL-1β | Biolegend | Cat# 579402 |
| Recombinant Human TNFα | Biolegend | Cat# 570102 |
| Recombinant Human IL-17A | Biolegend | Cat# 570502 |
| Streptavidin BUV563 | BD Biosciences | Cat# 612935 |
| Critical Commercial Assays | ||
| ELISA MAX Standard Set Mouse IL-17A | Biolegend | Cat# 432501 |
| ELISA MAX Standard Set Mouse IL-4 | Biolegend | Cat# 431101 |
| RNAeasy Mini kit | QIAgen | Cat# 74104 |
| Qubit™ Protein Assay Kit | Thermo Fisher Scientific | Cat# Q33211 |
| NEBNext® Ultra II Directional RNA Library Prep Kit for Illumina® | New England Biolabs | Cat# E7760S |
| Mouse SLPI DuoSet ELISA | R&D Systems | Cat# DY1735-05 |
| EasySep Mouse CD4 Positive Selection Kit II | STEMCELL technologies (Easysep) | Cat# 18952 |
| EasySep Mouse CD11c Positive Selection Kit II with Spleen Dissociation Medium | STEMCELL Technologies (Easysep) | Cat# 18781 |
| Human SLPI DuoSet ELISA | R&D Systems | Cat# DY1274-05 |
| Quant-it Ribogreen RNA assay kit | Invitrogen™ | Cat# R11490 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4368814 |
| NucleoSpin RNA XS, Micro kit | Macherey-Nagel | Cat# 740902.50 |
| CellTrace CFSE Cell Proliferation Kit | Thermo Fisher Scientific (Invitrogen™) | Cat# C34554 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit, for 405 nm excitation | Thermo Fisher Scientific (Invitrogen™) | Cat# L34966 |
| NucleoSpin RNA XS | Machery-Nagel | Cat# 740902.50 |
| Deposited Data | ||
| Sequencing data | This paper | European Nucleotide Archive, PRJEB36780 |
| Experimental Models: Cell Lines | ||
| A549 (Human alveolar epithelial adenocarcinoma) | ATCC | Cat# CCL-185 |
| B16-FLT3L expressing melanoma cells | Mach et al., 2000 | NA |
| Experimental Models: Organisms/Strains | ||
| Mouse: WT C57BL/6J: C57BL/6J | The Jackson Laboratory (Bar Harbor, ME) | Cat# 000664 |
| Mouse: RAG1−/− C57BL/6J: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory (Bar Harbor, ME) | Cat# 002216 |
| Mouse: OTII: OTII Rag1+/− Ly5.1+/− Foxp3-GFP | bred in house | NA |
| Mouse: Germ-free C57BL/6J: WTC57BL/6J | bred in house | NA |
| Software and Algorithms | ||
| R version 3.5.3 or higher | R Development Core Team, 2011 | https://www.r-project.org/ |
| FlowJo v10 | BD | RRID: SCR_008520, https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism 8 | GraphPad software | RRID: SCR_002798, https://www.graphpad.com/scientific-software/prism/ |
| BD FACSDiva™ software | BD Bioscience | RRID: SCR_001456 |
| SPAdes version 3.11.0 | Bankevich et al., 2012 | https://cab.spbu.ru/software/spades/ |
| BLAST version 2.6.0 | Altschul et al., 1990. | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Ensembl, GRCm38.p6, release 98 | Hunt et al., 2018 | ftp://ftp.ensembl.org/pub/release-98/fasta/mus_musculus/dna/ |
| bowtie2 version 2.3.4.1 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| htseq version 0.9.1 | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| DESeq2 version 1.22.2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| fgsea version 1.8 | Sergushichev, 2016 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| gage version 2.32.1 | Luo et. al 2009 | https://www.bioconductor.org/packages/release/bioc/html/gage.html |
| Aperio ImageScope software | Leica Byosystems | RRID:SCR_014311 |
| DADA2 (version 1.10.1 in R) | Callahan et al., 2016 | https://benjjneb.github.io/dada2/ |
| phyloseq (version 1.28.0) in R (version 3.6.1) | McMurdie and Holmes, 2013. | https://joey711.github.io/phyloseq/ |
| randomForest v 4.6-14 | Liaw and Wiener, 2002 | NA |
| Boruta version 6.0.0 | Rudnicki and Kursa, 2009 | https://cran.r-project.org/web/packages/Boruta/index.html |
| biomaRt version 2.38.0, ensmebl archive Sept 2019 | Durinck et al., 2009 | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
Highlights.
Bordetella pseudohinzii (Bph) colonizes the mouse airway and induces a Th17 response
Bph-colonized mice show reduced allergic airway inflammation (AAI) in an asthma model
The antimicrobial peptide SLPI is linked to diminished AAI and is regulated by IL-17A
In humans, upper airway microbiota composition was correlated with SLPI abundance
ACKNOWLEDGMENTS
We thank Tarisa Mantia, Caitlin O’Shaughnessy, and Shannon Rook for their assistance with patient recruitment and members of the Kau lab for helpful discussions. We would also like to thank MARS study participants and their families. The research was supported by the AAAAI Foundation Faculty Development Award (to A.L.K.), the American Asthma Foundation (to C.-S.H. and A.L.K.), and NIH grants T32 GM007200 (to A.L.R.), R01 AI136515 (to C.-S.H.) and K08 AI113184 (to A.L.K.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108331.
REFERENCES
- Aas JA, Paster BJ, Stokes LN, Olsen I, and Dewhirst FE (2005). Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 43, 5721–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen EK, Pitkäranta A, Mäki M, Hendley JO, Laakso S, Sale MM, and Winther B (2013). Bacteria in the nose of young adults during wellness and rhinovirus colds: detection by culture and microarray methods in 100 nasal lavage specimens. Int. Forum Allergy Rhinol. 3, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnahas S, Hagner S, Raifer H, Kilic A, Gasteiger G, Mutters R, Hellhund A, Prinz I, Pinkenburg O, Visekruna A, et al. (2017). IL-17 and TNF-α Are Key Mediators of Moraxella catarrhalis Triggered Exacerbation of Allergic Airway Inflammation. Front. Immunol. 8, 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Amezcua Vesely MC, Pallis P, Bielecki P, Low JS, Zhao J, Harman CCD, Kroehling L, Jackson R, Bailis W, Licona-Limon P, et al. (2019). Effector TH17 Cells Give Rise to Long-Lived TRM Cells that Are Essential for an Immediate Response against Bacterial Infection. Cell 178, 1176–1188.e1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JL, Flynn RJ, Ballantyne SJ, and McKenzie AN (2011). Reciprocal expression of IL-25 and IL-17A is important for allergic airways hyperreactivity. Clin. Exp. Allergy 41, 1447–1455. [DOI] [PubMed] [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bønnelykke K, Brasholt M, Heltberg A, Vissing NH, Thorsen SV, et al. (2007). Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 357, 1487–1495. [DOI] [PubMed] [Google Scholar]
- Bisgaard H, Hermansen MN, Bønnelykke K, Stokholm J, Baty F, Skytt NL, Aniscenko J, Kebadze T, and Johnston SL (2010). Association of bacteria and viruses with wheezy episodes in young children: prospective birth cohort study. BMJ 341, c4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, and Knight R (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 108, 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, Alcorn JF, Weaver CT, and Kolls JK (2011). Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35, 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SL, Wu M, Henderson JP, Hooton TM, Hibbing ME, Hultgren SJ, and Gordon JI (2013). Genomic diversity and fitness of E. coli strains recovered from the intestinal and urinary tracts of women with recurrent urinary tract infection. Sci. Transl. Med. 5, 184ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zheng D, Liu B, Yang J, and Jin Q (2016). VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44, D694–D697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesné J, Braza F, Mahay G, Brouard S, Aronica M, and Magnan A (2014). IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med. 190, 1094–1101. [DOI] [PubMed] [Google Scholar]
- Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, Wang W, Chen Z, Silver PB, Jittayasothorn Y, et al. (2020). The Cytokine IL-17A Limits Th17 Pathogenicity via a Negative Feedback Loop Driven by Autocrine Induction of IL-24. Immunity 53, 384–397.e385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, Jia G, Ohri CM, Doran E, Vannella KM, et al. (2015). TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci. Transl. Med 7, 301ra129–301ra129. [DOI] [PubMed] [Google Scholar]
- Clark SE, Purcell JE, Sammani S, Steffen EK, Crim MJ, Livingston RS, Besch-Williford C, and Fortman JD (2016). Bordetella pseudohinzii as a Confounding Organism in Murine Models of Pulmonary Disease . Comp. Med. 66, 361–366. [PMC free article] [PubMed] [Google Scholar]
- Clark SE, Purcell JE, Bi X, and Fortman JD (2017). Cross-Foster Rederivation Compared with Antibiotic Administration in the Drinking Water to Eradicate Bordetella pseudohinzii. J. Am. Assoc. Lab. Anim. Sci. 56, 47–51. [PMC free article] [PubMed] [Google Scholar]
- Cooper MD, Roberts MH, Barauskas OL, and Jarvis GA (2012). Secretory leukocyte protease inhibitor binds to Neisseria gonorrhoeae outer membrane opacity protein and is bactericidal. Am. J. Reprod. Immunol. 68, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewan KK, Taylor-Mulneix DL, Campos LL, Skarlupka AL, Wagner SM, Ryman VE, Gestal MC, Ma L, Blas-Machado U, Faddis BT, and Harvill ET (2019). A model of chronic, transmissible Otitis Media in mice. PLoS Pathog. 15, e1007696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durack J, Lynch SV, Nariya S, Bhakta NR, Beigelman A, Castro M, Dyer AM, Israel E, Kraft M, Martin RJ, et al. (2017). Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J. Allergy Clin. Immunol. 140, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers A, Xie W, Agapov E, Brown S, Steinberg D, Tidwell R, Sajol G, Schutz R, Weaver R, Yu H, et al. (2018). BMAL1 links the circadian clock to viral airway pathology and asthma phenotypes. Mucosal Immunol. 11, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forteza RM, Ahmed A, Lee T, and Abraham WM (2001). Secretory leukocyte protease inhibitor, but not alpha-1 protease inhibitor, blocks tryptase-induced bronchoconstriction. Pulm. Pharmacol. Ther. 14, 107–110. [DOI] [PubMed] [Google Scholar]
- Ge XN, Ha SG, Greenberg YG, Rao A, Bastan I, Blidner AG, Rao SP, Rabinovich GA, and Sriramarao P (2016). Regulation of eosinophilia and allergic airway inflammation by the glycan-binding protein galectin-1. Proc. Natl. Acad. Sci. USA 113, E4837–E4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goleva E, Jackson LP, Harris JK, Robertson CE, Sutherland ER, Hall CF, Good JT Jr., Gelfand EW, Martin RJ, and Leung DY (2013). The effects of airway microbiome on corticosteroid responsiveness in asthma. Am. J. Respir. Crit. Care Med. 188, 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashimoto N, Morita H, Yasuda M, Ishida T, Kameda S, Takakura A, and Itoh T (2012). Prevalence of Bordetella hinzii in mice in experimental facilities in Japan. Res. Vet. Sci. 93, 624–626. [DOI] [PubMed] [Google Scholar]
- Hiemstra PS, Maassen RJ, Stolk J, Heinzel-Wieland R, Steffens GJ, and Dijkman JH (1996). Antibacterial activity of antileukoprotease. Infect. Immun. 64, 4520–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, et al. (2007). Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 204, 2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SE, McLaren W, Gil L, Thormann A, Schuilenburg H, Sheppard D, Parton A, Armean IM, Trevanion SJ, Flicek P, and Cunningham F (2018). Ensembl variation resources. Database (Oxford) 2018, bay119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov YV, Linz B, Register KB, Newman JD, Taylor DL, Boschert KR, Le Guyon S, Wilson EF, Brinkac LM, Sanka R, et al. (2016). Identification and taxonomic characterization of Bordetella pseudohinzii sp. nov. isolated from laboratory-raised mice. Int. J. Syst. Evol. Microbiol. 66, 5452–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I, Manary MJ, Liu T-C, Stappenbeck TS, Maleta KM, et al. (2015). Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy . Sci. Transl. Med 7, 276ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaßen C, Karabinskaya A, Dejager L, Vettorazzi S, Van Moorleghem J, Lühder F, Meijsing SH, Tuckermann JP, Bohnenberger H, Libert C, and Reichardt HM (2017). Airway Epithelial Cells Are Crucial Targets of Glucocorticoids in a Mouse Model of Allergic Asthma. J. Immunol. 199, 48–61. [DOI] [PubMed] [Google Scholar]
- Kuperman DA, Lewis CC, Woodruff PG, Rodriguez MW, Yang YH, Dolganov GM, Fahy JV, and Erle DJ (2005). Dissecting asthma using focused transgenic modeling and functional genomics. J. Allergy Clin. Immunol. 116, 305–311. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A, and Wiener M (2002). Classification and Regression by randomForest. R News 2, 18–22. [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Löhning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, Levinson D, Radbruch A, and Kamradt T (1998). T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc. Natl. Acad. Sci. USA 95, 6930–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loong SK, Che-Mat-Seri NA, Abdulrazak O, Douadi B, Ahmad-Nasrah SN, Johari J, Mohd-Zain SN, and Abubakar S (2018). Recovery of Bordetella bronchiseptica sequence type 82 and B. pseudohinzii from urban rats in Terengganu, Malaysia. J. Vet. Med. Sci. 80, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, and Dranoff G (2000). Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 60, 3239–3246. [PubMed] [Google Scholar]
- Marin ND, Dunlap MD, Kaushal D, and Khader SA (2019). Friend or Foe: The Protective and Pathological Roles of Inducible Bronchus-Associated Lymphoid Tissue in Pulmonary Diseases. J. Immunol. 202, 2519–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino R, Thuraisingam T, Camateros P, Kanagaratham C, Xu YZ, Henri J, Yang J, He G, Ding A, and Radzioch D (2011). Secretory leukocyte protease inhibitor plays an important role in the regulation of allergic asthma in mice. J. Immunol. 186, 4433–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuba S, Yabe-Wada T, Takeda K, Sato T, Suyama M, Takai T, Kikuchi T, Nukiwa T, and Nakamura A (2017). Identification of Secretory Leukoprotease Inhibitor As an Endogenous Negative Regulator in Allergic Effector Cells . Front. Immunol. 8, 1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann JR, Mason SN, Auten RL, St Geme JW 3rd, and Seed PC (2016). Early-Life Intranasal Colonization with Nontypeable Haemophilus influenzae Exacerbates Juvenile Airway Disease in Mice. Infect. Immun. 84, 2022–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern TK, Robichaud A, Fereydoonzad L, Schuessler TF, and Martin JG (2013). Evaluation of respiratory system mechanics in mice using the forced oscillation technique . J. Vis. Exp, e50172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, Henry A, Irvin CG, Piganelli JD, Ray A, and Kolls JK (2008). TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 181, 4089–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan RA, Sorkness CA, Kosinski M, Schatz M, Li JT, Marcus P, Murray JJ, and Pendergraft TB (2004). Development of the asthma control test: a survey for assessing asthma control. J. Allergy Clin. Immunol. 113, 59–65. [DOI] [PubMed] [Google Scholar]
- National Asthma Education and Prevention Program (2007). Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007. J. Allergy Clin. Immunol. 120, S94–S138. [DOI] [PubMed] [Google Scholar]
- Newcomb DC, Boswell MG, Reiss S, Zhou W, Goleniewska K, Toki S, Harintho MT, Lukacs NW, Kolls JK, and Peebles RS Jr. (2013). IL-17A inhibits airway reactivity induced by respiratory syncytial virus infection during allergic airway inflammation. Thorax 68, 717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Östling J, van Geest M, Schofield JPR, Jevnikar Z, Wilson S, Ward J, Lutter R, Shaw DE, Bakke PS, Caruso M, et al. (2019). IL-17-high asthma with features of a psoriasis immunophenotype. J. Allergy Clin. Immunol. 144, 1198–1213. [DOI] [PubMed] [Google Scholar]
- Parameswaran GI, Sethi S, and Murphy TF (2011). Effects of bacterial infection on airway antimicrobial peptides and proteins in COPD. Chest 140, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnode ML, Bando JK, Krummel MF, Locksley RM, and Rosen SD (2014). Leukotriene B4 amplifies eosinophil accumulation in response to nematodes. J. Exp. Med. 211, 1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perniss A, Schmidt N, Gurtner C, Dietert K, Schwengers O, Weigel M, Hempe J, Ewers C, Pfeil U, Gärtner U, et al. (2018). Bordetella pseudohinzii targets cilia and impairs tracheal cilia-driven transport in naturally acquired infection in mice. Sci. Rep. 8, 5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson LJ, Aanerud M, Hardie JA, Miodini Nilsen R, Bakke PS, Eagan TM, and Hiemstra PS (2017). Antimicrobial peptide levels are linked to airway inflammation, bacterial colonisation and exacerbations in chronic obstructive pulmonary disease. Eur. Respir. J. 49, 1601328. [DOI] [PubMed] [Google Scholar]
- Preston JA, Essilfie AT, Horvat JC, Wade MA, Beagley KW, Gibson PG, Foster PS, and Hansbro PM (2007). Inhibition of allergic airways disease by immunomodulatory therapy with whole killed Streptococcus pneumoniae. Vaccine 25, 8154–8162. [DOI] [PubMed] [Google Scholar]
- Preston JA, Thorburn AN, Starkey MR, Beckett EL, Horvat JC, Wade MA, O’Sullivan BJ, Thomas R, Beagley KW, Gibson PG, et al. (2011). Streptococcus pneumoniae infection suppresses allergic airways disease by inducing regulatory T-cells. Eur. Respir. J. 37, 53–64. [DOI] [PubMed] [Google Scholar]
- Puchta A, Verschoor CP, Thurn T, and Bowdish DM (2014). Characterization of inflammatory responses during intranasal colonization with Streptococcus pneumoniae . J. Vis. Exp, e50490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2011). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, Huff R, Pilewski J, Holguin F, Kolls J, et al. (2015). High IFN-γ and low SLPI mark severe asthma in mice and humans. J. Clin. Invest. 125, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddel HK, Bateman ED, Becker A, Boulet LP, Cruz AA, Drazen JM, Haahtela T, Hurd SS, Inoue H, de Jongste JC, et al. (2015). A summary of the new GINA strategy: a roadmap to asthma control. Eur. Respir. J. 46, 622–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, et al. (2019). Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, Hickman HD, McCulloch JA, Badger JH, Ajami NJ, et al. (2017). Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 171, 1015–1028.e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki WR, and Kursa MB (2009). Feature Selection with the Boruta Package . J. Stat. Softw. 36, 1–13. [Google Scholar]
- Sallenave JM, Shulmann J, Crossley J, Jordana M, and Gauldie J (1994). Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase-specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am. J. Respir. Cell Mol. Biol. 11, 733–741. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al. (2005). IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490. [DOI] [PubMed] [Google Scholar]
- Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, and Schnyder B (2006). Interleukin-17 is a negative regulator of established allergic asthma. J. Exp. Med. 203, 2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev AA (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 10.1101/060012. [DOI] [Google Scholar]
- Shilts MH, Rosas-Salazar C, Turi KN, Rajan D, Rajagopala SV, Patterson MF, Gebretsadik T, Anderson LJ, Peebles RS Jr., Hartert TV, and Das SR (2020). Nasopharyngeal Haemophilus and local immune response during infant respiratory syncytial virus infection. J. Allergy Clin. Immunol, S0091–6749(20)30952–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JL, Daly J, Baines KJ, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Hugenholtz P, Willner D, and Gibson PG (2016). Airway dysbiosis: Haemophilus influenzae and Tropheryma in poorly controlled asthma. Eur. Respir. J. 47, 792–800. [DOI] [PubMed] [Google Scholar]
- Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, Low TB, O’neill SJ, and McElvaney NG (2005). Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J. Exp. Med. 202, 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijs W, Janssen K, van Schadewijk AM, Papapoulos SE, le Cessie S, Middeldorp S, Melissant CF, Rabe KF, and Hiemstra PS (2015). Nasal Levels of Antimicrobial Peptides in Allergic Asthma Patients and Healthy Controls: Differences and Effect of a Short 1,25(OH)2 Vitamin D3 Treatment. PLoS One 10, e0140986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unhanand M, Maciver I, Ramilo O, Arencibia-Mireles O, Argyle JC, McCracken GH Jr., and Hansen EJ (1992). Pulmonary clearance of Moraxella catarrhalis in an animal model. J. Infect. Dis. 165, 644–650. [DOI] [PubMed] [Google Scholar]
- Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Iwakura Y, et al. (2008). IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am. J. Respir. Crit. Care Med. 178, 1023–1032. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead GS, Thomas SY, and Cook DN (2014). Modulation of distinct asthmatic phenotypes in mice by dose-dependent inhalation of microbial products. Environ. Health Perspect. 122, 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedow O, Harder J, Bartels J, Streit V, and Christophers E (1998). Antileukoprotease in human skin: an antibiotic peptide constitutively produced by keratinocytes. Biochem. Biophys. Res. Commun. 248, 904–909. [DOI] [PubMed] [Google Scholar]
- Wright CD, Havill AM, Middleton SC, Kashem MA, Lee PA, Dripps DJ, O’Riordan TG, Bevilacqua MP, and Abraham WM (1999). Secretory leukocyte protease inhibitor prevents allergen-induced pulmonary responses in animal models of asthma. J. Pharmacol. Exp. Ther. 289, 1007–1014. [PubMed] [Google Scholar]
- Zuberi RI, Apgar JR, Chen SS, and Liu FT (2000). Role for IgE in airway secretions: IgE immune complexes are more potent inducers than antigen alone of airway inflammation in a murine model. J. Immunol. 164, 2667–2673. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data from RNA-seq, genome sequencing assemblies, and 16S rRNA sequencing reported in this paper is European Nucleotide Archive: PRJEB36780.