

Abstract
We provide a protocol for generating forebrain structures in vivo from mouse embryonic stem cells (ESCs) via neural blastocyst complementation (NBC). We developed this protocol for studies of development and function of specific forebrain regions, including the cerebral cortex and hippocampus. We describe a complete workflow, from methods for modifying a given genomic locus in ESCs via CRISPR/Cas9-mediated editing to the generation of mouse chimeras with ESC-reconstituted forebrain regions that can be directly analyzed. The procedure begins with genetic editing of mouse ESCs via CRISPR/Cas9, which can be accomplished in about four to eight weeks. We provide protocols to achieve fluorescent labeling of ESCs in about two to three weeks, which allows tracing of the injected, ESC-derived donor cells in chimeras generated via NBC. Once modified ESCs are ready, NBC chimeras are generated in about three weeks via injection of ESCs into genetically programmed blastocysts that are subsequently transferred into pseudopregnant fosters. Our in vivo brain organogenesis platform is efficient, allowing functional and systematic analysis of genes and other genomic factors in as little as three months, in the context of a whole organism.
EDITORIAL SUMMARY
Embryonic stem cells undergo CRISPR/Cas9-mediated editing and are then used to reconstitute forebrain regions in mouse chimeras via neural blastocyst complementation.
INTRODUCTION
Although organoid-based approaches for neurobiological studies are gaining acceptance, genetically engineered mice remain critically important for studies of the cerebral cortex and hippocampus in vivo and for neurobehavioral studies that require a whole organism. Technologies for generating genetically modified mice have been informed by studies of the early mouse embryo (Figure 1A) and involve embryonic stem cells (ESCs) and embryo manipulation. Mouse ESCs were first isolated from the inner cell mass (ICM) of early embryos called blastocysts1,2. These ICM-derived ESCs are pluripotent and retain the capacity to differentiate into any germ layer (mesoderm, endoderm, and ectoderm), and thus can generate all cell types present in the adult organism3. Mouse ESCs have a high capacity for self-renewal, and can be grown in culture without undergoing senescence4. ESCs injected into 3.5 days post coitum (dpc) blastocysts can contribute to soma and germline of mouse chimeras3. These features have made mouse ESCs attractive tools for gene targeting and generation of mouse models5. However, the resulting chimeras need to be bred for germline transmission of the modified allele(s), which makes the generation of genetically modified mice time consuming and costly6.
Figure 1. Overview of mouse early embryonic development and the NBC organogenesis approach.

(A) Key steps in early mouse development from zygote to blastocyst are illustrated: Fertilization involves the fusion of two haploid gametes to form a zygote. The zygote (0.5 dpc) undergoes cleavages to give rise to an eight-cell embryo by 2.5 dpc. Blastomeres then become compacted and polarized, forming a 16-cell embryo (morula), and subsequently the blastocyst. Differentiation of the blastomeres sets up embryonic and extra-embryonic lineages via formation of the inner cell mass (ICM), which gives rise to the embryo, and the trophectoderm, which gives rise to the placenta24. NBC involves injection of ESCs into 3.5 dpc blastocysts. (B) Schematic overview of the NBC organogenesis approach. Blastocyst injection of mouse ESCs into genetically programmed blastocysts restores forebrain structures in resulting chimeras. Schematic modified from6. (C) CRISPR/Cas9-based gene targeting enables rapid engineering and testing of complex genotypes, which can then directly be analyzed in vivo. Mouse brain schematics were generated based on a reference image from the Allen Mouse Brain Atlas.
Beyond generation of genetically modified mice, methods involving injection of mouse ESCs into host blastocysts have also been used for organogenesis via “blastocyst complementation”. The term “blastocyst complementation” refers to the replacement of ablated cells deriving from the genetically-modified host embryo by progeny of the injected ESCs, thus generating ESC-derived cells, tissues, and organs6–9. The concept of blastocyst complementation was first conceived and implemented in the context of the adaptive immune system: injection of ESCs into Rag2-deficient blastocysts resulted in the generation of mice with ESC-derived mature B and T cells8,9. Based on this fundamental discovery, blastocyst complementation was subsequently used to generate other cells and organs, including the ocular lens, pancreas, thymus, kidney, and heart7,10–14.
We recently developed neural blastocyst complementation (NBC), a blastocyst complementation approach to facilitate neurobiological in vivo studies of the mouse forebrain6. Here we present a protocol describing how to perform NBC studies.
Overview of the protocol
NBC involves injection of mouse ESCs into genetically-engineered, blastocyst-stage embryos that are programmed to ablate dorsal telencephalic progenitors (DTPs) at around embryonic day (E) 9.5 (Figure 1B–C). This is accomplished by Emx1-Cre mediated, conditional expression of attenuated diphtheria toxin subunit A (DTA) from the Rosa26 locus. Without ESC injection, Emx1-Cre; R26-DTA blastocysts generate embryos with extensive ablation of hippocampal, neocortical, and olfactory bulb structures6. Moreover, these mice show incompletely developed jaws15. Upon ESC injection into Emx1-Cre; R26-DTA blastocysts, repopulation of the vacant dorsal telencephalic niche by progeny of the injected ESCs restores the ablated forebrain regions and jaw development6. Restored jaw development is easily visible in NBC-derived embryos and thus provides a convenient visual readout of ESC-mediated reconstitution. ESC-reconstituted NBC chimeras can be used for direct analysis or can be bred for germline transmission of the modified allele(s), if desired6.
Applications of the method
The NBC protocol described here facilitates in vivo studies of cerebral cortex and hippocampus, enabling questions related to neurodevelopment, neural function, and the cell type of origin for disease phenotypes to be answered. NBC allows analysis of mice at ages ranging from the early embryonic period up to at least two years of age6. NBC can be used to rapidly and reliably assess even subtle developmental brain phenotypes, as demonstrated by our studies of Doublecortin (Dcx) function via NBC: use of Dcx knockout ESCs for NBC fully recapitulates the reported phenotypes of germline Dcx knockout mice6. By allowing facile generation of multiple, genetic alterations in ESCs and subsequent analysis in vivo without the need of time-consuming intercrossing of segregating alleles, NBC can substantially accelerate functional analysis of genes (and other genomic elements) in cortical and hippocampal development and physiology. NBC can also be applied to reveal whether genetic mutations in the host- or donor-derived cells drive disease phenotypes. Specifically, donor cells in this protocol reproducibly generate cortical and hippocampal excitatory neurons and astrocytes, while host cells contribute mainly cell types such as cortical inhibitory neurons, microglia, and blood vessel endothelial cells. Thus, knock-in alleles and complex mutations that otherwise cannot easily be induced in a cell type-specific fashion can now be compared between cell types in both host and donor contexts, which could have applications in studies of many neuropsychiatric disorders and neurodegenerative diseases in which candidate genes are expressed across a range of cell types.
Thus far NBC has involved injection of mouse ESCs into genetically-engineered, blastocyst-stage embryos that are programmed to ablate dorsal telencephalic progenitors (DTPs) at around embryonic day (E) 9.5 through Emx1-Cre mediated, conditional expression of attenuated diphtheria toxin subunit A (DTA) from the Rosa26 locus. By utilising alternative Cre-driver lines, this protocol could be modified and utilized to modulate the extent of donor cell contribution to other brain regions and cell types.
Expertise needed to implement the protocol
NBC involves techniques that fall within the technical repertoire of many laboratories addressing neurobiological and developmental questions. However, combined expertise across a relatively wide range of disciplines is required to successfully implement the protocol. Fundamental expertise in working with mouse ESCs, gene targeting, and mouse genetics will be helpful. Experience with generation of mutant mouse models will further facilitate establishment of the approach in a given lab. Any core facility familiar with generation of transgenic mice via blastocyst injection should be able to easily adapt our approach. Increasingly, core facilities also offer assistance with gene targeting. Use of such facilities would be a good option if the requisite expertise is not available in a given lab.
Limitations of the NBC approach
NBC chimeras show consistently high donor cell contribution in forebrain regions, yet cells derived from non-dorsal telencephalic progenitors (such as interneurons that migrate tangentially into the cortex, resident macrophage-derived microglia, and blood vessel endothelial cells) can still be chimeric, i.e., derived from both donor ESCs and host blastocyst cells6. This can be easily assessed by fluorescence analysis as host blastocyst-derived cells express DsRed.T36 (Figure 2). We routinely detect a very small fraction of host-derived cells across forebrain regions6 and presence of such cells should be taken into consideration when interpreting phenotypes. Moreover, non-neural tissues in NBC chimeras are frequently chimeric, similar to what is observed in conventional chimeras. Such chimerism outside the brain may impact neurobiological phenotypes via non-cell autonomous systemic effects, as in conventional mouse models in which the mutation of interest is present in non-neural cell types (e.g., in whole body knockouts) and should be considered. Genetic alterations that impede cell survival or ability to differentiate during development would prevent analysis via NBC. Thus, cell proliferation and viability of mutant ESCs compared to wild-type ESCs should be assessed prior to NBC. Last, the sex of the ESCs used for NBC should be considered. Many established ESC lines, including TC1 and JM8.F6, are male. This can provide a distinct advantage for genetic factors located on the sex chromosomes because only one allele needs to be modified—as in the case of Dcx6. However, injection of male ESCs into mouse blastocysts can cause the embryo to develop into a male, thus causing a high proportion of males instead of an equal ratio of both sexes in the progeny16. Should this be a cause of concern, female ESCs could be used. Alternatively, following initial characterization via NBC, a subset of NBC chimeras could be bred for germline transmission of the mutant allele, which will enable generation of both female and male mice for analysis.
Figure 2. Schematic of the breeding crosses used to generate DsRed-labeled NBC host blastocysts.

(A) Stud males are homozygous Emx1-Cre; hemizygous DsRed.T3. (B) Stud males are heterozygous Emx1-Cre; hemizygous DsRed.T3. Tg, transgene. Expected Mendelian ratios are listed in percent.
Experimental Design
ESC Culture (general):
Optimal ESCs culture technique is crucial. This requires close monitoring of ESC morphology and density. Growth medium should be replaced daily, and passaging times should be carried out based on confluency—rather than fixed schedules—and may vary between ESC lines. We have successfully used various ESC lines for NBC, including 129Sv-derived TC16 (a gift from P. Leder, Harvard Medical School), 129Sv:C57BL/6 F1 hybrid EF1 (derived in-house)6, and C57BL/6 JM8.F6. Because increased ESC passage number can cause genomic instability and impair pluripotency, it is important to keep low-passage stocks and limit passage number as much as possible. It is critical that ESC lines are free of mycoplasma and other pathogens.
CRISPR/CAS9-MEDIATED GENOME EDITING IN MOUSE EMBRYONIC STEM CELLS (ESCs) (steps 1–17)
The procedure starts with the modification of ESCs. We describe a plasmid-based approach for introducing CRISPR/Cas9-mediated modifications into ESCs by using generation of a Doublecortin (Dcx) knockout allele as an example6. We also describe an alternative approach involving Cas9/gRNA ribonucleoprotein (RNP) complexes that is based on commercially available reagents. Although both approaches give good results when used as described below6, based on our preliminary data, the RNP complex-based approach is slightly faster as it does not require plasmid generation. It also eliminates the risk of random integration of CRISPR/Cas9 expression plasmid sequences into the ESC genome.
FLUORESCENT LABELING OF DONOR ESCs (steps 18–39)
In NBC, donor ESC progeny and host blastocyst cell contribution can be traced throughout the embryo via detection of fluorescent markers. We use lentiviral vectors for constitutive H2B-eGFP expression in donor ESCs and progeny (Figure 3), and a CAG-DsRed.T3 transgene to label host blastocyst-derived cells6. This approach results in dual-labeled chimeras in which donor ESC-derived cells (H2B-eGFP; green) and host blastocyst-derived cells (DsRed.T3; red) can be easily distinguished. Steps 26–39 describe generation of stably H2B-eGFP-expressing ESCs via lentiviral transduction. We have also successfully used a lentiviral hUbc-H2B-mCherry vector (available from Addgene #132333) for NBC but have not yet published our results using this. This vector can be used for labeling donor ESCs in red (but note that it will not work in combination with DsRed.T3 blastocysts because both host- and donor-derived cells will be red fluorescent).
Figure 3. Lentiviral transduction of ESCs.

(A) Diagram of the lentiviral vector FUGW-H2B-eGFP (adapted from25) used to fluorescently label ESCs. Only the relevant portions of the plasmid are shown. (B) Representative bright field and fluorescence microscopy images of wild-type TC1 ES cells and Dcx-/y ES cells before and after lentiviral integration of H2B–eGFP (original magnification, 100×). The ES cells shown stably express H2B–eGFP and were repeatedly imaged during the study with similar results. Also note the ESC morphology. The ESC clones shown are approximately 100–150 μm long. Images were acquired with a Nikon Diaphot 300 inverted fluorescence phase contrast microscope.
Note that we typically engineer the desired mutation first and then fluorescently label multiple, independently generated ESC clones. We follow this experimental sequence because this approach is fast and provides flexibility in terms of the marker used. Another advantage is that the gene targeted, unlabeled ESCs are available to generate genetically modified mice via conventional germline transmission of the mutation, if desired. However, although we have not yet experienced this issue, there is a chance that the lentiviral integration site may affect cellular function. We control for this by always labeling multiple clones and deriving multiple fluorescent ESC subclones. For each genotype, we choose at least two different, lentivirally transduced eGFP+ ESC clones to control for potential effects of insertional mutagenesis. Alternatively, gene targeting can be used to insert and express a fluorescent marker from a defined “safe harbor” locus in the genome. For example, Rosa26 is a well characterized “safe harbor” locus that can be easily targeted. Reagents to insert fluorescent markers into the Rosa26 locus are widely available (for example, Addgene #217149).
CULTURE AND PREPARATION OF ESCs FOR BLASTOCYST INJECTION (steps 40–47)
Culture and preparation of mutant and wild-type ESCs is required for blastocyst injection.
NEURAL BLASTOCYST COMPLEMENTATION (steps 48–67)
This section describes generation of blastocysts and pseudopregnant fosters, ESC injection into blastocysts, and transfer of injected blastocyst into pseudopregnant fosters. To this end, homozygous R26-DTA females are superovulated and set with homozygous Emx1-Cre males (or heterozygous Emx1-Cre; hemizygous DsRed.T3 males) to generate blastocysts. Pseudopregnant fosters are generated by setting CD-1 females with vasectomized CD-1 stud males.
MATERIALS
REAGENTS
DNA oligonucleotides (Integrated DNA Technologies, Inc. (IDT) or other suppliers)
FastDigest BbsI and 10× FastDigest buffer (Thermo Scientific, cat. no. FD1014)
Fast alkaline phosphatase (Thermo Scientific, cat. no. EF0654)
T4 polynucleotide kinase (PNK) and 10× T4 PNK buffer (NEB, cat. no. M0201S)
LigaFast Rapid DNA Ligation System (contains T4 DNA ligase and 2× Rapid ligation buffer, Promega, cat. no. M8221)
10 mM ATP (NEB, cat. no. P0756S)
Plasmid-Safe DNAse and 10× PlasmidSafe Buffer (Lucigen, cat. no. E3101K)
Taq DNA Polymerase (250 U) and CoralLoad PCR 10× Buffer (Qiagen, cat. no. 201203)
Universal 67-mer tracrRNA (IDT, cat. no. 1072533)
Nuclease-free duplex buffer (IDT, cat. no. 11-01-03-01)
Alt-R S. pyogenes Cas9 Nuclease V3 (IDT, cat. no. 1081058)
PureLink Quick Plasmid Miniprep Kit (Invitrogen, cat. no. K210011)
Colcemid (KaryoMAX solution, Invitrogen, cat. no. 15212-012)
Methanol (Fisher Chemical, cat. no. A412-4)
Glacial acetic acid (Fisher Chemical, cat. no. A38-500)
KCl (Fisher Chemical, cat. no. P217-500)
DAPI Fluoromount-G (Southern Biotech, cat. no. 0100-20)
Puromycin (Gibco, cat. no. A1113803)
TransIT-293 (Mirus, cat. no. MIR 2705)
Dulbecco’s phosphate-buffered saline (DPBS) (Gibco, cat. no. 14190-144)
DMEM with L-glutamine, 4.5 g/L glucose and sodium pyruvate (Corning, cat. no. 10-013-CV)
Heat-inactivated fetal bovine serum (Atlanta Biologicals, cat. no. S11050H)
CRITICAL: Fetal bovine serum should be tested for use with embryonic stem cell culture in-house or embryonic stem cell-grade fetal bovine serum should be used.
Leukemia inhibitory factor (LIF; Chemicon, cat. no. ESG1107)
L-Glutamine:Penicillin:Streptomycin solution (Gemini Bio Products, cat. no. 400-110)
L-Glutamine (Gibco, cat. no. 25030081)
Non-essential amino acids (Gibco, cat. no. 11140-050)
β-Mercaptoethanol (Sigma, cat. no. M-3148)
Gelatin (Sigma, cat. no. G-2500)
Trypsin-EDTA (0.5%, 10×) (Gibco, cat. no. 15400-054)
Polybrene (hexadimethrine bromide, Sigma, cat. no. H9268)
PEG-8000 (molecular biology grade, Promega, cat. no. V3011)
Cryovial Screw Cap microtube (Sarstedt, cat.no. 72.694.006)
Breeder diet for foster females (LabDiet, cat. no. Picolab mouse diet 20)
Standard mouse diet (LabDiet, cat. no. Prolab Isopro RMH3000)
Pregnant mare serum gonadotropin (PMSG) (Biovendor, cat. no. RP1782725000)
Human chorionic gonadotropin (hCG; Sigma, cat. no. CG10-1VL)
! CAUTION: Potential reproductive toxicity. Wear standard lab personal protective equipment (PPE).
Proteinase K (Bioline Meridian Bioscience, cat. no. BIO-37039)
1 M HEPES solution (Corning, cat. no. 25-060-CI)
Penicillin-Streptomycin solution (Gibco, cat. no. 15-070-063)
EmbryoMax KSOM mouse embryo medium (Millipore, cat. no. MR-121-D)
CRITICAL: Store the KSOM medium at −20 °C. After thawing, keep it at 4 °C and use it within two weeks.
Light paraffin mineral oil (Fisher Scientific, cat. no. 0121-4, CAS 8042-47-5)
Ketamine-HCl (KetaVed, VedCo Inc., NDC 50989-161-06)
! CAUTION: Controlled substance. Follow institutional guidelines and practices.
Xylazine (AnaSed, Akorn Animal Health, NDC 59399-110-20)
Eye lubricant (Puralube vet ointment, NDC 17033-211-38)
Buprenorphine (0.3 mg/mL in 5% (wt/vol) dextrose; NDC 12496-0757-5)
! CAUTION: Controlled substance. Follow institutional guidelines and practices.
Plasmids
pX330-U6-Chimeric_BB-CBh-hSpCas9 vector (Addgene #42230)
psPAX2 (Addgene #12260)
pMD2.G (Addgene #12259)
FUGW-H2B-eGFP (lentiviral hUbc-H2B-eGFP vector; gift from Guo-Liang Xu, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences)
FUGW-H2B-mCherry (lentiviral hUbc-H2B-mCherry vector; Addgene #132333)
Cells
γ-irradiated murine embryonic fibroblasts (prepared in-house as per standard protocols17 or purchased from various suppliers, such as Thermo Scientific or Lonza)
Mouse embryonic stem cells (such as TC1 (RRID:CVCL_M350); gift from Philip Leder, Harvard Medical School)
293T cells (ATCC, cat. no. CRL-3216; RRID:CVCL_0063)
Mice
! CAUTION: All experiments involving mice must be conducted in compliance with all relevant Institutional Animal Care and Use Committee (IACUC) and governmental regulations.
CD-1 vasectomized stud males (purchased at nine weeks of age; Charles River Laboratories, cat. no. 24101152)
CD-1 females (purchased at five weeks of age; Charles River Laboratories, cat. no. 24101136)
Homozygous R26-DTA females (The Jackson Laboratory, JAX stock no. 010527; 129Sv:C57BL/6)
Homozygous Emx1-Cre males (The Jackson Laboratory, JAX stock no. 005628; C57BL/6)
Heterozygous Emx1-Cre; hemizygous DsRed.T3 males (generated from JAX stock nos. 005628 and 006051; C57BL/6)
EQUIPMENT
6-well plates (Corning Costar, cat. no. 3516)
24-well plates (Corning Costar, cat. no. 3526)
35-mm dishes (Corning Falcon, cat. no. 353001)
Petri dishes (35 × 10 mm; Falcon, cat no. 351008)
60-mm dishes (Corning Falcon, cat. no. 353002)
100-mm dishes (Corning Falcon, cat. no. 353003)
150-mm dishes (Corning Falcon, cat. no 351058)
50-mL conical tubes (Sarstedt, cat. no. 62.547.100)15-mL conical tubes (Sarstedt, cat. no 62.554.100)1.5-mL microcentrifuge tubes (Sarstedt, cat. no 72.690.301)
Steriflip-GP sterile centrifuge tube top filter unit (Millipore, cat. no. SCGP00525)
Stericup-GP sterile vacuum filtration system (Millipore, cat. nos. SCGPU02RE and SCGPU05RE)
Steritop threaded bottle top filter (Millipore, cat. no. SCGPS02RE)
QIAquick gel extraction kit (Qiagen, cat. no. 28706)
Amaxa 4D-nucleofector core unit (Lonza, cat. no. AAF-1002B) with X unit (Lonza, cat. no. AAF-1002X)
P3 primary cell 4D-Nucleofector X kit S (Lonza, cat. no. V4XP-3032)
Vortex Genie 2 (VWR, cat no. 14216-184)
Superfrost Plus microscope slides (25 × 75 mm; Fisher Scientific, cat. no. 12-550-15)
Coverslips (24 × 40 mm; Fisher Scientific; cat. no. 12-518-108B)
Standard fluorescence microscope capable of visualizing DAPI-stained chromosomes
Screw cap micro tube (2 mL, Sarstedt, cat. no. 72.694.006)
Parafilm M (Sigma, cat. no. P7793)
Beckman ultracentrifuge (e.g., Beckman Optima XPN, cat. no. A94469)
SW-41 Ti swinging-bucket rotor (Beckman Coulter, cat. no. 331336)
Ultra-Clear centrifuge tubes (14 × 89 mm; Beckman Coulter, cat. no. BK344059)
Countess Automated Cell Counter (Invitrogen) or hemocytometer
Kimwipes (Kimberly-Clark Professional, cat. no. 34256)
3-mL Luer-Lok syringe (Becton Dickinson, cat. no. BD309657)
1-mL syringe with 26-G needle (Becton Dickinson, cat. no. BD 309597)
30-G needle (Becton Dickinson, BD Precision Glide Needle, cat. no. BD305106)
! CAUTION: Be careful when handling sharps. Refer to institutional guidelines.
0.45-μm syringe filters (GE healthcare, cat. no. 6780-2504)
Sterile alcohol prep pads (Fisherbrand, cat. no. 22-363-750)
Povidone-iodine swabsticks (Medline, cat. no. MDS 093901)
Suture material (Surgical Specialties Corporation; Look Suture, cat. no. 495B)
Michel suture clips (7.5 × 1.75 mm, stainless steel; Fine Science Tools, cat. no. 12040-01)
9-in. borosilicate glass Pasteur pipette (VWR; cat. no. 14672-412 CTPL)
Straight-tube Bunsen burner
!CAUTION: Fire hazard. Use caution near open flame.
Bunsen burner striker
Borosilicate glass (O.D. 1.0 mm, I.D. 0.75 mm; Sutter Instrument Company, cat. no. B100-75-10)
Suction mouthpiece for micropipette (HPI Hospital Products, Medtech, cat. no. 1501 (B4036-2)
Tygon tubing for micropipette setup (1/8 in. I.D., 1/4 in. O.D., 1/16 in. wall; Tygon B44-4X)
Filter unit for micropipette setup (Millex-GP filter unit 0.22 μm, Millipore cat. no.
SLGP033RB)
Oster cordless trimmers (Walmart, cat. no. 550155276)
Sterilization indicator strips (Propper Manufacturing Co., Inc., cat. no. 264103)
! CAUTION: Use sterilization indicator strips that are appropriate for the type of sterilization used (steam or chemical) for sterilization of surgical tools.
Mayo-Hegar needle holders (Roboz Surgical Instrument Co., cat. no. RS-7912)
Microdissection forceps (Biomedical Research Instruments, Inc., cat. no. 10-2850)
Graefe forceps (Roboz Surgical Instrument Co., cat. no. RS-5139)
Stevens microdissection scissors (Roboz Surgical Instrument Co., cat. no. RS-5940)
Johns Hopkins clamp (Roboz Surgical Instrument Co., cat. no. RS-7440)
Dumont #55 forceps (Roboz Surgical Instrument Co., cat. no. RS-5063)
Gross anatomy probe (for plug checking) (Fine Science Tools, cat.no. 10088-15)
Nikon SMZ U 1:10 stereo microscope with ED Plan 1× objective and SMZ-U UW10xA/24 eyepiece containing microruler
Nikon SMZ 1500 C-DSS115 stereo microscope equipped with a Nikon P-FMD 1007009 focusing mount
Nikon Diaphot 300 inverted fluorescence phase contrast microscope
Nikon Eclipse TE2000-s microscope (4×, 10×, and 20× objectives) equipped with Leitz manipulator arms
Nikon T12-U microscope (4×, 10×, 20×, and 40× objectives) with Narashige manipulator arms
Nikon inverted routine microscope Eclipse TS2 (for ES cell clone picking) with plain stage (4×, 10×, 20×, and 40× objectives)
Olympus BX61 fluorescence microscope
Benchtop protector sheets (Kimberly-Clark, Professional Kimtech Science, Fisher Scientific, cat.no. 15-235-101)
Tissue culture incubator (Forma Scientific, model no. 3158)
Flaming/Brown Micropipette Puller P-87 (Sutter Instrument Company)
Micro-forge MF-1 (TPI Technical Products International)
Heat Therapy Pump HTP-1500 (Adroit Medical Systems, Inc.)
Glass bead sterilizer (Inotech Steri-350, Inotech Biosciences LLC, cat. no. IS-350)
! CAUTION: Use care when handling hot surfaces.
Anti-vibration table (Technical Manufacturing Corporation Ametek Inc., cat. no. MICRO-g 63-541)
Medical NF Grade Nitrogen (for anti-vibration table; Airgas, cat. no. NI NF200)
REAGENT SETUP
PMSG. Dissolve PMSG powder in sterile DPBS to generate a 50 IU/mL solution. Store aliquots of PMSG solution frozen at −20 °C for up to one month (or at −80 °C for up to four months).
hCG. Dissolve hCG powder in sterile DPBS to generate a 50 U/mL solution. Store hCG aliquots frozen at −20 °C for up to one month (or at −80 °C for up to four months).
LIF. Prepare a 100,000 U/mL stock in DMEM supplemented with 10% (v/v) FBS. Store in aliquots at −20°C. Avoid freeze-thaw cycles. Thawed solution can be kept at 4 °C for up to one week.
β-mercaptoethanol. Generate a 100 mM β-mercaptoethanol solution by adding 70 μL β-mercaptoethanol (14.3 M stock) to 10 mL of sterile DPBS. Store at 4 °C for up to 2 weeks.
CRITICAL: β-mercaptoethanol is hazardous. Handle it with gloves and dispense waste according to local institutional guidelines.
Gelatin solution. Generate a 0.2% (w/v) gelatin solution by dissolving 1 g gelatin powder in 500 mL of DPBS. Swirl to mix and autoclave on 30-min cycle to fully dissolve and sterilize. Store gelatin solution at room temperature (22–25 °C) for up to three months.
Polybrene. Dissolve polybrene (hexadimethrine bromide) powder in sterile, tissue-culture grade water to generate an 8 mg/mL solution. Filter sterilize (0.2-μm) and store frozen in aliquots at −20 °C for up to six months. Avoid freeze-thaw cycles.
4× PEG concentration solution (40% (w/v) PEG-8000, 1.2 M NaCl). To 80 mL sterile water, add 80 g PEG-8000 powder, 14 g NaCl, and 20 mL 10× DPBS. Vortex to mix, heat gently if necessary, until solids are dissolved. Adjust pH to 7.0–7.2 and bring total volume to 200 mL with sterile water. Filter sterilize (0.2-μm) and store solution at 4 °C for up to six months.
ESC lysis buffer 100 mM Tris-HCl, pH 8.0, 5 mM EDTA, 0.2% (w/v) SDS, 200 mM NaCl, 0.2 mg/mL proteinase K. Store without proteinase K at room temperature for up to 6 months. Add proteinase K immediately before use.
TE for genomic DNA 10 mM Tris-Cl, pH 8, 0.1 mM EDTA. Store at room temperature for up to one year.
Carnoy’s fixative 75% (v/v) methanol, 25% (v/v) glacial acetic acid. Prepare right before use.
293T growth medium DMEM with 10% (v/v) fetal bovine serum and 2 mM L-glutamine; add penicillin-streptomycin if desired. Store at 4 °C for up to one month.
Blastocyst flushing medium 435 mL DMEM with L-glutamine, 4.5 g/L glucose and sodium pyruvate, 50 mL heat-inactivated FBS, 10 mL 200 mM HEPES, and 5 mL 5,000 U/mL penicillin-streptomycin. Blastocyst flushing medium should be 0.22-μm filtered and can be stored for one month at 4 °C.
Tail lysis buffer (50 mM NaCl, 10 mM Tris-HCl (pH 8.0), 5 mM EDTA, 0.1% (w/v) SDS) containing 150 μg of proteinase K. Store without proteinase K at room temperature for up to 6 months. Add proteinase K immediately before use.
EQUIPMENT SETUP
Preparation of injection micropipette needles and holding micropipette needles.
Prepare injection micropipette needles from borosilicate glass (O.D. 1.0 mm, I.D. 0.75 mm). To do this, pull injection micropipette needles on a Flaming/Brown Micropipette Puller using the following settings: H450, P200, V100, T50, pressure 20. This should result in an internal diameter of 10–20 μm. Hand-cut pulled injection micropipette needles under a stereo microscope to create a curved, shovel scoop tip. Next, bend injection micropipette needles 5° to 20° on a Micro-forge MF-1 or equivalent (See Step 54 for details).
Prepare holding micropipette needles by hand-pulling borosilicate glass (O.D. 1.0 mm, I.D. 0.75 mm) over a Bunsen burner flame to blunt break to a diameter that is half the diameter of a blastocyst (20–50 μm). Then fire polish the tip of the holding micropipette needle using the micro-forge and bend the needle 5° to 20°. Alternatively, holding micropipettes can be commercially purchased (Origio, cat. no. MPH-MED-20)
PROCEDURE
Genetic editing of mouse ESCs.
1. Option A describes generation of Cas9/sgRNA expression plasmids. Option B describes generation of Cas9/gRNA ribonucleoprotein complexes.
CRITICAL: We design at least two pairs of sgRNAs for each locus to be modified. We then generate and characterize independent ESC clones from each of these pairs. This is to reduce the likelihood of common off-target mutations that might be present in clones derived from only one pair of sgRNAs. We do not recommend using more than one pair of sgRNAs simultaneously in one nucleofection reaction as that may increase the probability of off-target genomic alterations.
Option A. Generate Cas9/sgRNA expression plasmids - TIMING ~ 5 d
CRITICAL Steps ii–vii for generating Cas9/sgRNA-expressing plasmids are based on protocols from Feng Zhang’s lab (MIT)18.
i. Design sgRNAs by entering the genomic region of interest into a tool for CRISPR guide design (for example, CHOPCHOP, http://chopchop.cbu.uib.no/; CRISPOR, http://crispor.tefor.net/; or GPP sgRNA Designer https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). To generate a Dcx knockout allele, we designed a pair of sgRNA sequences to target genomic regions upstream of exon 2 and downstream of exon 3.
CRITICAL: We design at least two pairs of sgRNAs for each locus to be modified. We then generate and characterize independent ESC clones from each of these pairs. This is to reduce the likelihood of common off-target mutations that might be present in clones derived from only one pair of sgRNAs. We do not recommend using more than one pair of sgRNAs simultaneously in one nucleofection reaction as that may increase the probability of off-target genomic alterations.
ii. Synthesize each sgRNA-encoding sequence from a pair of oligonucleotides containing overhangs that allow direct insertion into the pX330 vector (Addgene #42230). Ns indicate the 20 nucleotides corresponding to the genomic target region in the table below.
| Oligonucleotides | Sequence |
|---|---|
| Oligonucleotide 1 | 5’ CACCGNNNNNNNNNNNNNNNNNNNN 3’ |
| Oligonucleotide 2 | 3’ CNNNNNNNNNNNNNNNNNNNNCAAA 5’ |
iii. Digest and gel extract the pX330 vector. Incubate the reaction below at 37 °C for 30 min, then gel purify the linearized plasmid using the QIAquick Gel Extraction Kit or a similar product.
| Component | Amount (per reaction, μL) | Final |
|---|---|---|
| pX330 (1 μg/μL) | 1 | 1 μg |
| 10× FastDigest buffer | 2 | 1 × |
| FastDigest BbsI | 1 | 10 U |
| Fast Alkaline Phosphatase | 1 | 1 U |
| ddH2O | 15 | - |
| Total volume | 20 | |
iv. Generate oligonucleotide duplex by incubating the reaction detailed below in a thermocycler using the following settings: 37 °C for 30 min, 95 °C for 5 min, temperature gradient from 95 °C to 20 °C (5 °C/min). Once completed, dilute annealed oligonucleotide duplexes 1:250 with ultrapure water suitable for molecular biology.
| Component | Amount (per reaction, μL) | Final |
|---|---|---|
| Oligonucleotide 1 (100 μM stock) | 1 | 10 μM |
| Oligonucleotide 2 (100 μM stock) | 1 | 10 μM |
| 10 mM ATP | 1 | 1 mM |
| 10× T4 PNK buffer | 1 | 1× |
| T4 polynucleotide kinase (PNK) | 0.5 | 10 U |
| ddH2O | 5.5 | - |
| Total volume | 10 | |
v. Ligate the annealed oligonucleotide duplex into the pX330 plasmid using the reaction detailed below. Mix by pipetting, centrifuge briefly (10,000g, 3 s), and ligate at room temperature for 10 min.
| Component | Amount (per reaction, μL) | Final |
|---|---|---|
| BbsI-digested pX330 (20 ng/μL) | 2.5 | 50 ng |
| Annealed oligonucleotide duplex (1:250) | 1 | 4 nM |
| 2× Rapid ligation buffer | 5 | 1× |
| T4 DNA ligase | 1 | 3 U |
| ddH2O | 1.5 | - |
| Total volume | 11 | |
vi. Treat the ligated plasmid with Plasmid-Safe ATP-dependent DNase as detailed below and incubate at 37 °C for 30 min.
| Component | Amount (per reaction, μL) | Final |
|---|---|---|
| Ligation reaction (from Step v above) | 11 | - |
| 10 mM ATP | 1.5 | 1 mM |
| 10× Plasmid-Safe Buffer | 1.5 | 1× |
| Plasmid-Safe DNase | 1 | 10 U |
| Total volume | 15 | |
vii. Transform 2 μL of ligation reaction into competent bacteria and plate onto LB agar plates containing ampicillin or carbenicillin as per standard protocols. Incubate plates at 37 °C overnight and inoculate two to four colonies for plasmid DNA miniprep (PureLink Quick Plasmid Miniprep Kit) the next day. Verify that the sgRNA-encoding sequences are free of mutations by sequencing the plasmids from a U6 primer (5′-ACTATCATATGCTTACCGTAAC-3′).
Option B. Design and generate Cas9/gRNA ribonucleoprotein complexes - TIMING ~ 1 d
i. Design and order crRNAs for use with the Alt-R CRISPR-Cas9 System as per guidelines provided by IDT (www.idtdna.com/pages/products/crispr-genome-editing/alt-r-crispr-cas9-system). Generate gRNA complexes and RNP complexes as described in Step 5, Option B i-iii, immediately prior to nucleofection.
Nucleofection of ESCs with CRISPR-Cas9 expression vectors or CRISPR-Cas9 ribonucleoprotein complexes - TIMING 6–8 d
CRITICAL: We use the Amaxa 4D-nucleofector core unit with X unit and the P3 primary cell kit for ESC nucleofection. The following steps describe how to use these components.
2. Thaw ESCs quickly and plate in ESC medium (DMEM medium supplemented with 15% (v/v) fetal bovine serum, 20 mM HEPES (pH 7.4), 2 mM L-glutamine, 50 U/mL penicillin/streptomycin, 0.1 mM MEM non-essential amino acids, 50 μM β-mercaptoethanol, 500 U/mL LIF) onto γ-IR inactivated mouse embryonic fibroblast (MEF) feeders. We generally start cells in one well of a 6-well plate in 4 mL of ESC medium. It is possible to scale down to smaller well plates. Check that the ESCs form colonies with distinct boundaries and display healthy morphology without signs of differentiation or cell death. ESCs will need to be fed at least daily; never let the ESC medium turn yellow. We grow ESCs at 37 °C, 5% CO2 in a standard tissue culture incubator.
CRITICAL STEP: We refer readers unfamiliar with stem cell culture to the many excellent protocols (for example: 17,19) on general aspects of mouse ESC culture, cryopreservation, and preparation of MEF feeder cells.
3. Passage the ESCs at least once before nucleofection: feed ESCs (and one recipient well with feeder cells) by replacing medium with 4 mL of pre-warmed ESC medium about one to two hours before passaging. Rinse ESCs with 5 mL of DPBS, then add 0.5 mL of 0.05% (v/v) trypsin-EDTA/DPBS. Incubate for 5 min at 37 °C. Triturate ESC suspension gently with a P1000 pipette tip. Add 2 mL of ESC medium to inactivate trypsin. Centrifuge the cell suspension at 180 × g for 5 min at 4 °C. Remove supernatant from cell pellet and resuspend thoroughly (but gently) in 1 mL of fresh ESC medium. Triturate again with a P1000 pipette tip to generate a single-cell suspension. Passage ESC suspension 1:6 into a feeder well containing 4 mL ESC medium.
4. On the day of nucleofection, trypsinize ESCs and generate a single-cell suspension (as described in previous step). Add cell suspension to a gelatinized 60-mm dish (pre-treated for at least 15 min with 0.2% (w/v) gelatin/DPBS). Leave cells in this plate for at least 40 min (but not more than 1 h) to deplete the MEF feeder cells.
CRITICAL: The MEF feeder cells can be depleted from the ESCs by the differential adhesion method. 40–60 min of incubation are sufficient to allow most of the MEFs to loosely adhere to the gelatinized plate, whereas the ESCs can still easily be resuspended by gently tapping or swirling plates. Longer incubation times (>1 h) may cause loss of ESCs due to adhesion.
5. Prepare the nucleofection mixture as shown in option A for plasmid-based Cas9/gRNA expression. Alternatively, prepare Cas9/gRNA ribonucleoprotein complexes and transfect as described in option B.
A) Plasmid-based CRISPR-Cas9 delivery
i) Combine the following components:
| Component | Amount (μL) |
|---|---|
| pX330-sgRNA 1 (1 μg/μL) | 1 |
| pX330-sgRNA 2 (1 μg/μL) | 1 |
| Nucleofector solution P3 | 98 |
CRITICAL: Include 0.2 μg of a plasmid containing a puromycin resistance marker in the nucleofection reaction to transiently allow for enrichment of nucleofected cells.
CRITICAL: Do not linearize plasmids before nucleofection as that may result in random integration of plasmid sequences into the ESC genome20.
B) Ribonucleoprotein complex-mediated CRISPR-Cas9 delivery
i) Generate gRNA complexes by preparing 200 μM stocks of crRNA and tracrRNA (IDT, cat. no. 1072533) in nuclease-free duplex buffer (IDT, cat. no. 11-01-03-01). For each gRNA complex, mix 5 μL 200 μM crRNA and 5 μL 200 μM tracrRNA. Heat mixture at 95 °C for 5 min in a PCR machine. Remove from heat and let cool to RT on bench top.
ii) Prepare RNP complexes (per 100 μL nucleofection reaction): by mixing 2.4 μL annealed gRNA duplex (from previous step), 3.3 μL of 61 μM Alt-R S.p. Cas9 Nuclease V3 (IDT, cat. no. 1081058), and 4.3 μL DPBS (total volume is 10 μL). Incubate for 20 min at RT, then place on ice.
iii) Combine the following:
| Component | Amount (μL) |
|---|---|
| RNP complex 1 (genomic site A) | 10 |
| RNP complex 2 (genomic site B) | 10 |
| Nucleofector solution P3 | 80 |
CRITICAL: This example is for generating a deletion via creating CRISPR-Cas9-mediated generation of DNA double-strand breaks at two genomic sites. If editing only one genomic site, use only one RNP complex and increase the volume of nucleofector solution P3 to 90 μL in order to keep the total volume to 100 μL.
6. Collect the ESCs from the supernatant (Step 4 above) and determine cell number by using a hemocytometer. Per nucleofection, place 2 × 106 ESCs into a fresh 15-mL conical tube and centrifuge cells at 180 × g for 5 min at 4 °C.
7. Discard the supernatant and immediately resuspend the ESC pellet in the appropriate nucleofection mixture listed in Step 5 A i or B iii and immediately transfer to a nucleofection cuvette. Gently tap to make sure sample covers the cuvette bottom. Avoid generation of air bubbles. Place the cuvette into the 4D-nucleofector X unit. Use program CG-104 for nucleofection.
8. Upon completion of nucleofection, remove the cells immediately from the cuvette by using the supplied plastic Pasteur pipette and place into 10 mL of prewarmed ESC medium in a 15-mL conical tube. Resuspend cells and aliquot 0.2 mL, 2 mL, and 7.8 mL of cell suspension into three 60-mm dishes seeded with feeders. Return the dishes quickly to the incubator.
9. 12 h after nucleofection, remove the ESC medium completely, and add 3–4 mL of ESC medium. If plasmid-based CRISPR-Cas9 has been used (Step 5 option A), use medium containing 1 μg/mL puromycin for selection.
10. Perform a full medium change every day (including puromycin if appropriate) and monitor ESC dishes daily for appearance of ESC clones. Depending on the growth rate of the ESC line used, and the efficiency of the nucleofection, the dish seeded at the highest density (7.8 mL) should have many colonies three to four days after nucleofection. On the dishes seeded at lower density (0.2 mL and 2 mL), individual colonies should become visible under a microscope (4× or 10× objective) five or six days after nucleofection.
Isolation and validation of mutant ESC clones - TIMING 12–14 d
11. To obtain an estimate of gene modification efficiency, collect genomic DNA from the plate seeded at the highest density on day three or four. Perform a genotyping PCR to determine if the desired genomic modification is present (see Step 16 and Step 17 section A for details). If it is, proceed to pick clones from the dishes seeded at lower density (as described in the next step), usually six to eight days after nucleofection.
12. ESC colonies are ready for picking when they have the approximate size of the opening of a standard P20 pipette tip. Place the ESC dish under a light microscope equipped with a 10× objective. Use a P20 pipette tip to aspirate individual ESC colonies and transfer them one-by-one to individual 96-well plate wells containing 10 μL of DPBS (See Figure 4A for an example of a clone picking setup inside a biosafety cabinet). When identifying colonies to pick, select those with a 3D morphology (i.e., non-flattened) and clear, distinct edges that do not show signs of differentiation (See Figures 3B and 4B).
Figure 4. ESC clone picking.
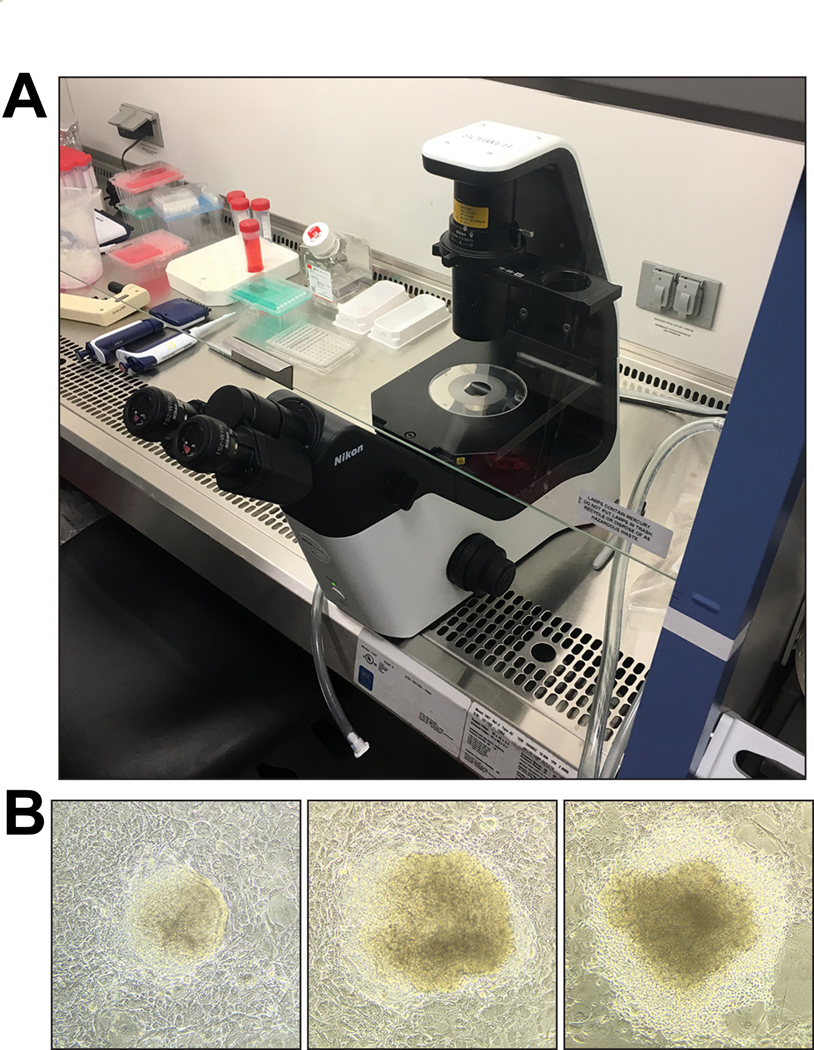
(A) Example of setup for picking ESC clones under a microscope inside a biosafety cabinet. An inverted routine microscope (for example, Nikon TS2 Eclipse with plain stage) can easily be moved into a biosafety cabinet for clone picking. ESC clones are picked with sterile, P20 filter pipette tips under a 10× objective. (B) When picking, domed clones with clear, distinct borders should be preferred over clones with flattened appearance and signs of differentiation. Three clones—ordered from better (left) to worse (right) morphology—are shown as examples. Approximate sizes of ESC clones shown are 200 μm (left), 360 μm (middle), and 400 μm (right). Images were acquired with an Apple iPhone 6S attached to a Nikon Eclipse TS2 inverted routine microscope.
13. After 8 to 12 clones have been picked and transferred into the 96-well plate, add 50 μL 0.1% (v/v) trypsin-EDTA/DPBS to each well. Incubate for 5 min at 37 °C, then triturate with a P200 pipette tip to dissociate the colony. A multichannel pipette can be used to simultaneously process 8–12 clones.
14. Use the multichannel pipette to transfer the single-cell suspensions to a fresh 96-well plate that has been seeded with feeders and contains 200 μL ESC medium per well. Pipette gently to mix. Return plate to tissue culture incubator.
15. Repeat Steps 12–14 until the desired number of ESC clones has been collected. We typically collect at least 96 ESC clones (and sometimes several hundred).
16. After three to four days of culture, split the 96-well plate into two 96-well plates, one gelatinized but without feeders and one containing feeders.
CRITICAL STEP The plate gelatinized but without feeders is to be used for genomic DNA preparation and a genotyping PCR to identify positive clones that contain the desired genomic modification (detailed in Step 17 section A; the other plate (containing feeders) is to be used to maintain the cells and for karyotyping (Step 17 section B).
Characterisation of ESC
17. Follow Step 17A(i-xii) to carry out a genotyping PCR to identify positive clones that contain the desired genomic modification. Follow Step 17B(i-ix) for karyotypic analysis.
A) Genotyping PCR and Southern blotting
i) Remove 96-well plate with ES cells from tissue culture incubator (process one plate at a time), remove lid and invert plate to discard medium. Blot the plate onto a stack of paper towels to completely remove the medium (see Figure 5A). No medium should be left on the cells (see Figure 5A).
Figure 5. DNA isolation from ESC clones and karyotyping.

(A) Left, medium is discarded, and plates are inverted and gently blotted against a stack of paper towels. Right, 96-well plate with ESCs after removal of medium before addition of lysis buffer. (B) Examples of normal (top) and abnormal (bottom) karyotypes. Sizes of chromosomes shown range from approximately 3 μm to 7 μm. Images were acquired with an upright Olympus BX61 fluorescence microscope.
ii) Use a multichannel pipet to add 50 μL of ESC lysis buffer to each well.
iii) Place the 96-well plate into a tissue culture incubator overnight at 37 °C.
PAUSEPOINT Cells should be lysed and digested for at least 18 h but can be left for 48 h.
iv) Remove plate from incubator and let cool to room temperature. Use a multichannel pipet to add 100 μL of ice-cold 100% ethanol per well. Let the 96-well plate sit on the bench for 45–60 min at RT. DNA fibers should become visible. It is not necessary to shake the plate during this incubation time.
v) Carefully tilt the plate to pour off the ethanol. Genomic DNA fibers should be visible on bottom and remain attached to the wells. Once most of the ethanol has been poured off, invert the plate and touch the plate gently against a stack of paper towels to remove the ethanol completely.
vi) Use a multichannel pipet to add 200 μL of ice-cold 75% (v/v) ethanol per well and pour off. Repeat for a total of two washes.
vii) After pouring off most of the ethanol, gently invert the plate onto a stack of paper towels to remove any residual ethanol. Gently blot away excess ethanol.
viii) Let plates air dry for about 40 min at room temperature inside a tissue culture biosafety cabinet. The wells should now look entirely dry.
ix) Add 100 μL TE for genomic DNA pre-warmed to 55 °C. Let plate sit at room temperature for about 30 min. Wrap each plate with Saran wrap and incubate at 4 °C over night.
x) Use a multichannel pipet to resuspend the DNA in each well (with P200 tip).
xi) Identify positive ESC clones by using an appropriate PCR-based genotyping strategy (for details and examples see6,16). Expand positive clones from the 96-well plate to a 6-well plate to collect genomic DNA for Southern blotting for further validation, and to freeze down ESC clones for future use, as per standard protocols.
? TROUBLESHOOTING
xii) Validate positive ESC clones by Southern blotting with specific probes (see standard molecular biology protocols for details on Southern blotting and hybridization21). If plasmid-based CRISPR-Cas9 delivery is used, we also routinely perform Southern blotting with probes against the pX330 plasmid backbone and the puromycin resistance plasmid to confirm that these sequences have not integrated into the genome of positive clones.
CRITICAL STEP: Although we consider Southern blot analysis the gold standard, alternate approaches can be used. For example, depending on the nature of the mutation and the gene targeting approach, a combination of long-range PCR and qPCR assays can be used to validate ESC clones if Southern blotting analysis is not possible. Ryder et al.22 provide a description of quality control criteria and assays for ESC validation.
B) Metaphase karyotyping of ESC clones - TIMING 2–3 d
i) Passage validated ESC clones to be karyotyped into one gelatinized well of a 6-well plate (or a gelatinized 35-mm dish). When cells reach about 70–80% confluency, treat with 0.05 μg/mL colcemid for 6 h in the tissue culture incubator.
ii) Trypsinize cells (as in Step 3) and transfer cell suspension to a 15-mL conical tube. Collect cells by centrifugation at 200 × g for 5 min at 4 °C. Discard supernatant and wash the cell pellet once with 5 mL DPBS.
iii) Pre-warm 75 mM KCl solution to 37 °C. Add 5 mL of 75 mM KCl solution dropwise (very slowly) to resuspend the ESC pellet while shaking the tube gently on a vortex mixer set to the lowest speed. Make sure to prevent aggregation. Flick tube after 1 mL of 75 mM KCl solution has been added, then add the remaining 4 mL of 75 mM KCl solution. Bring the volume up to 12 mL with more 75 mM KCl solution. Incubate at 37 °C for 20 min.
iv) Collect cells by centrifugation at 90 × g for 8 min at 4 °C. Discard supernatant. Loosen the cell pellet by gently flicking the tube, then slowly (about 1 drop per second) add 5 mL of freshly prepared, ice-cold Carnoy’s fixative while shaking the tube gently on a vortex mixer set to the lowest speed. Bring the volume to 12 mL by adding more Carnoy’s fixative. Collect cells by centrifugation at 90 × g for 8 min at 4 °C. Discard supernatant.
v) Repeat the fixation (Step iv) two more times, for a total of three fixation steps. On the final fixation step, do not centrifuge the cells but store them in 12 mL of Carnoy’s fixative at −20 °C overnight.
vi) The next day, collect cells by centrifugation at 90 × g for 8 min at 4 °C. Discard the supernatant, leaving a small volume (~0.5 mL) of Carnoy’s fixative in the tube. Flick the tube to gently resuspend the pellet.
vii) Use a P10 pipette tip to drop 7–10 μL of the cell suspension onto a Superfrost Plus microscope slide. Position the pipette about an inch above the glass slide for optimal metaphase dropping. Allow slides to dry overnight at room temperature.
viii) Place a drop of DAPI Fluoromount-G mounting medium onto the slide and add a 24 × 40 mm coverslip. PAUSEPOINT Store slides protected from light at 2–8 °C and analyze within one week.
ix) Visualize the metaphase chromosomes on a microscope under UV light. Use a 63× (or 100×) oil immersion objective to acquire images of chromosomes. Analyze chromosome spreads in widely separated fields and acquire at least 50 metaphase spreads and count the chromosomes. Euploid mouse ESCs contain 40 chromosomes. See examples of normal and abnormal karyotypes in Figure 5B.
Lentivirus production - TIMING 4 d
! CAUTION: All steps involving lentiviruses must be performed in accordance with your local regulations and guidelines regarding work with lentiviral vectors. We do not comment on operational/safe work practices as they may differ between institutions. Please contact your local biosafety officer for biological agent risk assessment and biosafety operational practices prior to initiating work with lentiviral vectors.
18. Plate approximately 5.5 × 106 293T cells per 100-mm dish in 10 mL of 293T growth. At time of transfection (approximately 18 to 24 h later) cells should be at about 70% confluency.
19. Per 100-mm dish to be transfected, mix 30 μL TransIT-293 with 570 μL DMEM (without serum or any additives) in a sterile 1.5-mL microcentrifuge tube (see Mirus TransIT-293 manual for details). Vortex briefly or pipet to mix thoroughly. Incubate for 10 min at room temperature (RT) while preparing the following three-plasmid mix in sterile 1.5-mL microcentrifuge tubes:
| Component | Amount (μL) | Final (μg) |
|---|---|---|
| psPAX2 (1 μg/μL) | 4.5 | 4.5 |
| pMD2.G (1 μg/μL) | 0.5 | 0.5 |
| hUbc-H2B-eGFP (1 μg/μL) | 5 | 5 |
20. Add 600 μL of TransIT-293/DMEM mix to tubes containing 10 μL of the three-plasmid mix. Mix gently by pipetting with a P1000 pipette tip and incubate for 30 min at room temperature.
21. Add transfection mix dropwise to 293T cells. Rock dish gently back and forth while adding the transfection complexes. Place 100-mm dish inside of a 150-mm dish with a lid to prevent spills and return to tissue culture incubator.
22. 12 h after addition of transfection complexes, carefully remove the growth medium (discard into a container with 10% (v/v) bleach) and add 10 mL of fresh 293T medium.
23. 24 h after medium change, collect virus-containing supernatant (harvest 1) and transfer to a sterile 50-mL conical tube. Collect supernatant carefully because 293T cells detach easily at this point. Gently add 10 mL of fresh 293T growth medium to the 100-mm dish and return to incubator (inside of a 150-mm dish with lid). Store harvest 1 in a 50-mL conical tube at 4 °C until harvest 2 is collected the next day.
24. 24 h after collection of harvest 1, collect supernatant (harvest 2) and add to 50-mL conical tube used in Step 23.
Lentivirus concentration - TIMING 1–2 d
25. Concentrate lentiviruses for transduction of ESCs for use in NBC either by ultracentrifugation (Option A) or by PEG-8000 precipitation (Option B). The latter method is based on a published protocol23. Commercial lentivirus concentrator solutions are also available (for example, Lenti-X Concentrator, Takara Bio Inc.).
Lentivirus concentration by ultracentrifugation
! CAUTION: Ultra-Clear SW41 tubes cannot be autoclaved. Sterilize by filling with 70% (v/v) ethanol and incubating in biosafety cabinet for 10 min. Discard ethanol and let tubes air dry. You must use SW41 Ti rotor buckets with intact seals.
CRITICAL: Autoclave rotor buckets before use. Also autoclave Kimwipes on dry cycle for later steps.
i. Centrifuge virus-containing supernatant (pooled harvest 1 and 2, Step 24) at 300 × g for 5 min, 4 °C. Filter through a 0.45-μm syringe filter into a fresh 50-mL conical tube.
ii. Add clarified viral supernatant to Ultra-Clear SW41 tubes (maximum volume per tube is about 12 mL; add DPBS to fill tube to about 5 mm from top to prevent tubes from collapsing during ultracentrifugation). Weigh tubes to ensure they are balanced.
iii. Centrifuge viral supernatant for 90 min at 107,000 × g(max) (25,000 RPM) at 4 °C.
iv. Transfer centrifuge buckets to biosafety cabinet before opening. Remove tubes from rotor and discard supernatant. Invert tube onto sterile (autoclaved) Kimwipe tissue for 1 min. Wipe tube walls with sterile Kimwipe.
v. Resuspend pellet with 100 μL DPBS by pipetting up and down about 20 times with a P200 filter pipette tip. Seal top of SW41 tube with Parafilm M, place it into a sterile 50-mL conical tube, and incubate overnight at 4 °C to let viral particles resuspend.
vi. After overnight incubation, prepare 20-μL aliquots of concentrated virus suspension in sterile screw cap micro tubes.
PAUSEPOINT Virus can be stored at −80 °C (for up to one year).
B. Lentivirus concentration by precipitation
i. Centrifuge viral supernatant (see Step 24) at 800 × g, for 10 min at room temperature to remove debris. Transfer supernatant to a fresh conical tube.
ii. Add 1 volume of 4× PEG concentration solution to 3 volumes of viral supernatant (e.g., 10 mL 4× PEG solution to 30 mL viral supernatant). Mix by pipetting. Gently invert tubes to mix for 1 min. Incubate for >4 hours at 4 °C on rotator/rocker (overnight incubation at 4 °C may increase yield).
iii. Centrifuge tube for 1,600 × g, for 30–60 min at 4 °C. Remove supernatant without disturbing the whitish pellet. Centrifuge tube briefly again and remove all residual supernatant from pellet.
iv. Resuspend viral pellet in DPBS (use about 1/10th to 1/20th of the original volume). Pipette gently to resuspend. Let sit for 10 min at RT (or o/n at 4 °C) to resuspend.
v. Remove protein debris by pipetting 20 times (avoid bubbles which may inactivate the virus) and transfer to cryovials. Centrifuge at 20,000 × g for 3 min at 4 °C to pellet protein debris. Collect the supernatant containing the virus. Aliquot virus preparation into 1.5-mL cryovials.
PAUSEPOINT store virus at −80 °C for up to a year.
Lentiviral transduction of ESCs - TIMING 12–14 d
26. Grow ESCs in ESC medium (see Step 2) on feeder cells in a 35-mm dish to about 60–70% confluency.
27. Four hours prior to lentiviral transduction, aspirate the ESC medium and add 4 mL of pre-warmed, fresh ESC medium. Return cells to tissue culture incubator.
28. Gelatinize one 60-mm dish and three 35-mm dishes by covering them with 0.2% (w/v) gelatin/DPBS solution. Incubate for at least 10 min in tissue culture incubator (or for at least 20 min at room temperature in the biosafety cabinet).
29. Four hours after feeding (Step 27), aspirate medium from ESCs, rinse cells twice with 2 mL DPBS and incubate with 0.5 mL 0.05% (v/v) trypsin-EDTA/DPBS solution for 5 min in tissue culture incubator. Inactivate trypsin by adding 3 mL of ESC medium. Centrifuge the cell suspension at 180 × g for 5 min at 4 °C.
30. Resuspend cell pellet in 5 mL of fresh ESC medium by pipetting up and down several times to obtain a single cell suspension. Plate the dissociated ESCs in a gelatinized 60-mm dish and incubate for 30 min in the tissue culture incubator to deplete the feeder cells (as in Step 4).
31. Carefully (without dislodging the settled feeder cells) recover ESCs from the supernatant. Pipet up and down several times to obtain a single-cell suspension. Determine cell number with an automated cell counter or by using a hemocytometer.
32. Plate single ESCs onto three gelatinized 35-mm dishes: Plate 10,000 ESCs per dish in 2 mL of fresh ESC medium containing 8 μg/mL polybrene.
33. Remove frozen aliquots of concentrated virus preparation (from step 25) from the −80 °C freezer. Quickly thaw aliquots in 37 °C water bath.
34. Briefly centrifuge the tube (10,000g, 3 s) containing the lentiviral particles. Add 10 μL, 20 μL, or 40 μL of lentiviral preparation to each of the 35-mm dishes containing ESCs and polybrene. Gently rock the dishes to distribute cells and virus evenly. Incubate cells in the incubator for 12–16 h.
35. Remove virus-containing medium from ESCs and gently rinse cells with 2 mL of DPBS. Remove DPBS and add 2 mL pre-warmed ESC medium per dish. Return cells to incubator.
36. Prepare five 60-mm dishes with feeder cells. Continue to culture the ESCs for another 24 h on the gelatinized 35-mm dishes.
37. Check eGFP expression under a fluorescence microscope. Choose the dish with the highest number of eGFP-positive ESC for further passaging.
38. Collect transduced ESCs from the selected dish by trypsinization (as in Step 29). Resuspend ESCs in 1 mL of fresh ESC medium and dilute the resulting single-cell suspension at ratios of 1:25, 1:50, 1:100, 1:200, and 1:400, respectively, into the five 60-mm dishes containing feeder cells and 4 mL ESC medium (prepared in Step 36).
39. Regularly observe cells under a microscope. Isolated colonies should become visible three to four days later. Observe dishes under a fluorescence microscope and mark position of eGFP-positive clones on the dish with a marker pen. Pick eGFP-positive colonies into a 24-well plate with feeder cells (see Steps 12–15 for details on clone picking). Passage the clones twice and select clones with good ESC morphology and stable eGFP expression for further experiments. All ESC clones should be confirmed by karyotyping (Step 17 option B).
Preparation for blastocyst injection
40. Both ESCs (Step 40A(i-xi)) and mice (Step 40B(i-v)) need to be prepared in parallel by carrying out the following steps on the indicated days (before blastocyst injection).
A) Culture of ESCs for injection - TIMING 9 d
i) On Day −9 (before blastocyst injection), prepare one 6-well plate containing feeder cells. This will provide enough feeder cells for culturing the ESCs until the day of blastocyst injection.
CRITICAL STEP Time frame can be adjusted and ESCs can be grown in smaller wells or dishes if desired. ESCs should be passaged once or twice before injection into blastocysts.
ii) On Day −8, thaw a frozen vial of ESCs and plate ESCs into one well of a 6-well plate with feeder cells.
iii) On Day −7 (about 18 h after initial ESC plating), replace growth medium with 4 mL of fresh ESC medium.
iv) Subculture ESCs on Day −6 (Passage 1): feed cells by replacing medium with 4 mL of pre-warmed ESC medium about one to two hours before passaging. Also replace medium on one well with feeder cells with 4 mL of fresh ESC medium.
v) Rinse ESCs with 5 mL of DPBS, then add 0.5 mL of 0.05% (v/v) trypsin-EDTA/DPBS. Incubate for 5 min at 37 °C. Triturate ESC suspension gently with a P1000 pipette tip. Add 2 mL of ESC medium to inactivate trypsin. Centrifuge the cell suspension at 180 × g for 5 min.
CRITICAL: It is essential to thoroughly dissociate ESCs after trypsinization.
vi) Remove supernatant from cell pellet and resuspend thoroughly (but gently) in 1 mL of fresh ESC medium. Triturate again with P1000 pipette tip to generate a single-cell suspension. Passage ESC suspension 1:6 into the feeder well containing 4 mL ESC medium.
CRITICAL: The success of NBC injection strongly depends on the culture conditions and overall quality of the ESCs. We recommend to closely observe ESC morphology and plating density every time before and after replacing growth medium. The ESC colonies should form oval, domed structures with clear boundaries (see Figures 3B and 4B for examples). Never use flat-looking ESCs for NBC injection. The density of ESCs is one of the most important factors. To achieve appropriate density, ESCs should be split at ratios between 1:5 and 1:6 every two days. In our experience, these splitting ratios apply to the vast majority of the ESCs lines we have used, including TC1 and JM8.F6 ESCs.
vii) On Day −5, replace growth medium with 4 mL of fresh ESC medium.
viii) Passage cells on Day −4 into another feeder well of a 6-well plate (same procedure as in Steps iv–vi).
ix) On Day −3, replace growth medium with 4 mL of fresh ESC medium.
x) On Day −2 (Passage 3), feed ESCs with 4 mL of pre-warmed ESC medium about one to two hours before passaging. Passage cells (as in Steps iv–vi above) but split cells at 1:3 and 1:6 ratios into two new wells with feeders.
xi) On Day −1, replace growth medium with 4 mL of fresh ESC medium.
B) Generation of blastocysts for ESC injection and pseudopregnant fosters - TIMING 6 d
! CAUTION: All work involving mice must be carried out in compliance with protocols and guidelines established by your local IACUC and Institutional Biosafety Committee (IBC), as well as governing agencies. All experiments described were authorized by the IACUC and IBC committees at the University of California, San Francisco, and Boston Children’s Hospital.
CRITICAL: Maintain all mice on a 14-h light, 10-h dark schedule in a temperature-controlled environment, with food and water provided ad libitum.
i) At 12 p.m. (six days before blastocyst injection), superovulate 4- to 5-week-old homozygous R26-DTA donor females by intraperitoneal (i.p.) injection of 5 U (0.1 mL) of 50 IU/mL pregnant mare serum gonadotropin (PMSG). Use a 1-mL syringe with a 26-G needle.
ii) 48 h later (12 p.m., four days before blastocyst injection), administer 5 IU (0.1 mL) of 50 U/mL human chorionic gonadotropin (hCG) via i.p. injection. Immediately set females 1:1 with homozygous Emx1-Cre males or heterozygous Emx1-Cre; hemizygous DsRed.T3 males (Figure 2). We usually set up 10–20 mating pairs for blastocyst generation. We recommend using males that are at least 8 weeks old for this step and replace males when they are more than 8 months old.
iii) Before 9 a.m. the next morning (three days before blastocyst injection), check superovulated females for presence of coital mating plugs and separate the plugged females from the males. Females displaying a coital mating plug are considered 0.5 days post coitum (dpc) and are group-housed at up to 10 mice per large mouse cage until blastocyst isolation three days later.
CRITICAL: PMSG and hCG stocks should not be stored for more than one month at −20 °C (or up to four months at −80 °C) to avoid loss of activity. Do not refreeze PMSG and hCG stocks and use immediately after thawing. Superovulated females not displaying a coital mating plug can be used for natural mating about 10 to 14 days later. Superovulation can be carried out a second time, but is not considered as reliable16.
iv) To obtain pseudopregnant fosters, set up CD-1 females with vasectomized CD-1 stud males at a ratio of 1:1 three days before blastocyst injection. We routinely set up 50 males with 50 females.
CRITICAL: CD-1 vasectomized stud males (purchased at nine weeks of age from Charles River Laboratories or prepared in-house) should be single-housed before use. Users may wish to test new batches of vasectomized males to verify their sterility. This is accomplished by mating them with females; none of the plugged females should subsequently become pregnant, confirming sterility of the vasectomized males. We recommend replacing vasectomized CD-1 stud males every six months. CD-1 females are replaced weekly (or less frequently if doing fewer NBC experiments). Unplugged CD-1 females can be kept and reused after 10 days, if desired. CD-1 pseudopregnant females are used as foster recipients at 2.5 dpc (if not enough females with plugs are obtained, 0.5 dpc CD-1 females can be used as fosters). Foster mothers are fed a breeder diet (Picolab mouse diet 20) during pregnancy and while nursing.
v) Before 9 a.m. the next morning, check CD-1 foster females for presence of a coital mating plug and separate them from the males. Females displaying a coital mating plug are group-housed at up to 10 mice per large mouse cage until the day of transfer surgery.
? TROUBLESHOOTING
Harvest of ESCs for blastocyst injection - TIMING 2–3 h
41. At about 8 a.m. on the blastocyst injection day (Day 0), warm 15-mL aliquots of ESC medium and DPBS to 37 °C. Bring 0.05% (v/v) trypsin-EDTA/DPBS to room temperature.
42. Select the well of ESCs to be used for injection based on cell morphology. Feed cells to be used for injection by replacing medium with 4 mL of pre-warmed ESC medium.
43. 1 h after feeding, remove ESC medium and rinse ESCs once with 5 mL DPBS (warmed to 37 °C) per well. Add 0.5 mL 0.05% (v/v) trypsin-EDTA/DPBS and incubate for 5 min in tissue culture incubator.
44. Pipette up and down with P1000 pipette tip to triturate ESCs (set pipette to 450 μL to avoid air bubbles). Stop the trypsin reaction by adding 4 mL of pre-warmed ESC medium. Produce a single-cell suspension, but do not pipette more than necessary or too harshly. Centrifuge the cell suspension at 180 × g for 5 min at 4 °C. Remove supernatant from cell pellet and resuspend thoroughly (but gently) in 1 mL of fresh ESC medium. Triturate again with a P1000 pipette tip to generate a single-cell suspension. Add another 3 mL of pre-warmed ESC medium to the ESC suspension.
45. To deplete feeder cells, add the cell suspension to a gelatinized 60-mm dish and return to the tissue culture incubator for 30 min. Most feeder cells will become loosely attached to the dish bottom whereas the ESCs will mostly remain in suspension (as in Step 4).
46. Very carefully tilt the dish and collect medium containing ESCs. Centrifuge the cell suspension at 130 × g for 3 min at 4 °C.
47. Remove supernatant and resuspend the cell pellet thoroughly (but gently) in 800 μL of ice-cold ESC medium. Transfer the suspension to a 15-mL conical tube containing 5 mL of fresh, ice-cold ESC medium and place tube on ice. ESCs are now ready for blastocyst injection. The cells should now be kept on ice. Cells should be injected (Step 56) within two hours of collection.
Blastocyst collection– TIMING 2 h
48. For blastocyst collection, prepare two to four 35-mm Petri dishes containing up to six 20-μL drops of EmbryoMax KSOM mouse embryo medium each and cover with light paraffin mineral oil (Figure 6A). Use enough oil to completely cover the height of the 20-μL drops (we use about 3 mL oil per 35-mm dish). Pre-equilibrate dishes in a 5% CO2, 37 °C tissue culture incubator for at least 15 min. Dishes can be placed into a tissue culture incubator for up to several hours before they are used (in injection, Step 56).
Figure 6. Collection of blastocysts.
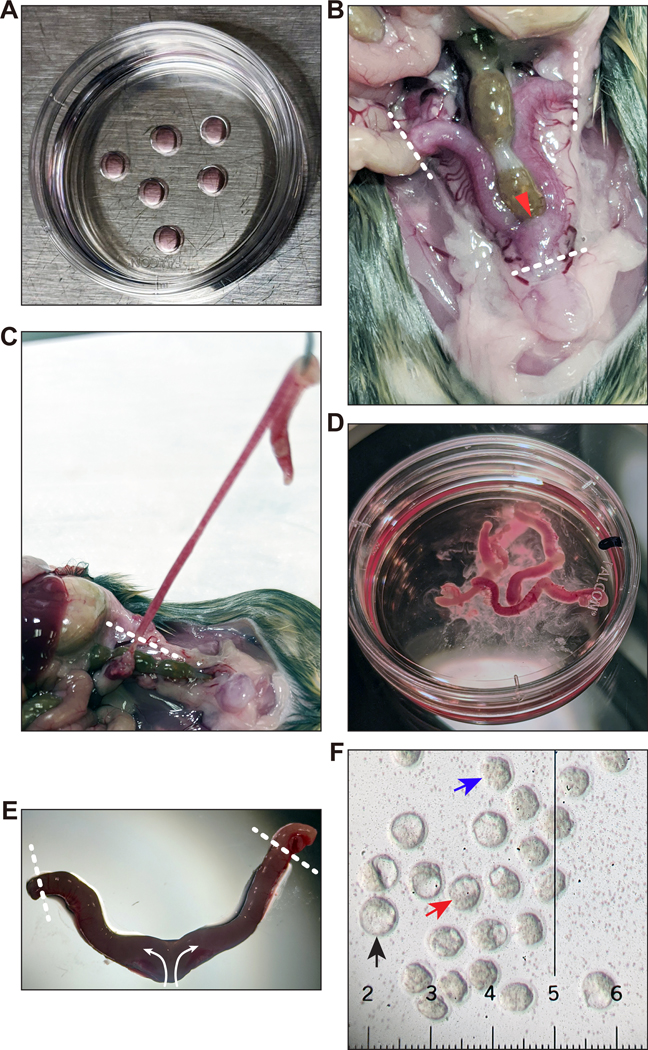
(A) Preparation of 35-mm Petri dishes with 20-μL drops of KSOM under a thin layer of mineral oil for placement of collected blastocysts. These dishes must be kept in a tissue culture incubator after preparation. (B) Frontal overview of the uterus and uterine horns in situ. Dissection tools are used to lift and cut at the urogenital junction, then remove the uterine horns by cutting at each oviduct-uterine junction. Regions where cuts should be made are indicated by white dotted lines. The red arrowhead indicates where to grasp the junction. (C) Removal of uterine horns. Use dissection tools to cut at the oviduct-uterine junction depicted by the white dotted line. Cutting the horn too short may cause embryos to leak out. Supplementary Video 1 shows how to remove the uterine horns. (D) The dissected uterine horns are kept in a 35-mm Petri dish containing blastocyst flushing media to keep them hydrated prior to individual flushing. If the horns were cut too short, a few embryos may leak out and end up in this dish. After flushing is complete, you may place the uterine horns in another dish and rinse with flushing media to see if any embryos were stuck to the exterior of the uterus. (E) Dissected uterine horns at higher magnification. Fine forceps are used to hold the uterine horn, while a syringe of flushing media with a needle is used to cut the ends of the horns (to allow embryos to exit). Then, the needle is placed inside the junction (either through the large holes from dissection or by making a new hole with the needle). With the flowing of the media, each horn is flushed of embryos. The white dotted lines show where to cut with the needle; the arrows indicate the flow direction of the flushing media. For further details on how to flush the uterine horns, see Supplementary Video 2. (F) Recovered blastocysts and morulae. The black arrow points to a fully expanded blastocyst that is ready to be injected before hatching from the Zona pellucida occurs. The red arrow points to a blastocyst that should be cultured longer to develop to the fully expanded stage. The blue arrow indicates a compacted morula that would need to be cultured longer to become a fully expanded blastocyst. The smallest ticks on the ruler are 10 μm apart; distance between each large tick is 0.1 mm. Images in (E) and (F) were acquired with a Nikon SMZ U 1:10 stereo microscope with SMZ-U UW10xA/24 eyepiece containing a microruler. Appropriate institutional regulatory board permission was obtained for all experiments involving mice.
49. Between 8 a.m. and 10 a.m. at 3.5 dpc (three days after Step 40 B iii), euthanize superovulated blastocyst donor females. We use CO2 asphyxiation.
CAUTION You must follow local IACUC guidelines and approved protocols.
50. Place donor females on their backs (supine). Clean the abdominal area with sterile alcohol prep pads. Expose the abdominal cavity, remove uterine horns and transfer them to a 35-mm Petri dish (Falcon 351008) containing 2 mL of blastocyst flushing medium. See Figure 6B–D and Supplementary Video 1 for details.
51. While viewing under a light microscope, ensure the oviduct ends of both uterine horns are open by transversely slicing with a 26-G or 30-G needle (Figure 6E). Next, place the tip of a 26-G or 30-G (0.5-in.) needle attached to a 3-mL syringe containing blastocyst flushing medium inside the uterine horn (Figure 6E). Flush embryos from each uterine horn from cervical end towards oviductal ends of uterine horns with about 0.5 mL blastocyst flushing medium into a fresh 35-mm Petri dish (Supplementary Video 2).
52. While viewing under a light microscope (Figure 6F), collect blastocysts and morulae by using a 9-in. borosilicate glass Pasteur transfer pipette (hand-pulled using a Bunsen burner to about 50–100 μm diameter; see Supplementary Video 3 and Figure 7) equipped with a suction mouthpiece, Tygon tubing (1/8 in. I.D., 1/4 in. O.D., wall 1/16 in.), and a 0.22-μm filter (Millex-GP filter unit) under a Nikon SMZ U 1:10 stereo microscope, using an ED Plan 1× objective at highest power.
Figure 7. Transfer pipette setup for blastocyst collection and uterine transfer surgery of injected blastocysts and microinjection holding needle.
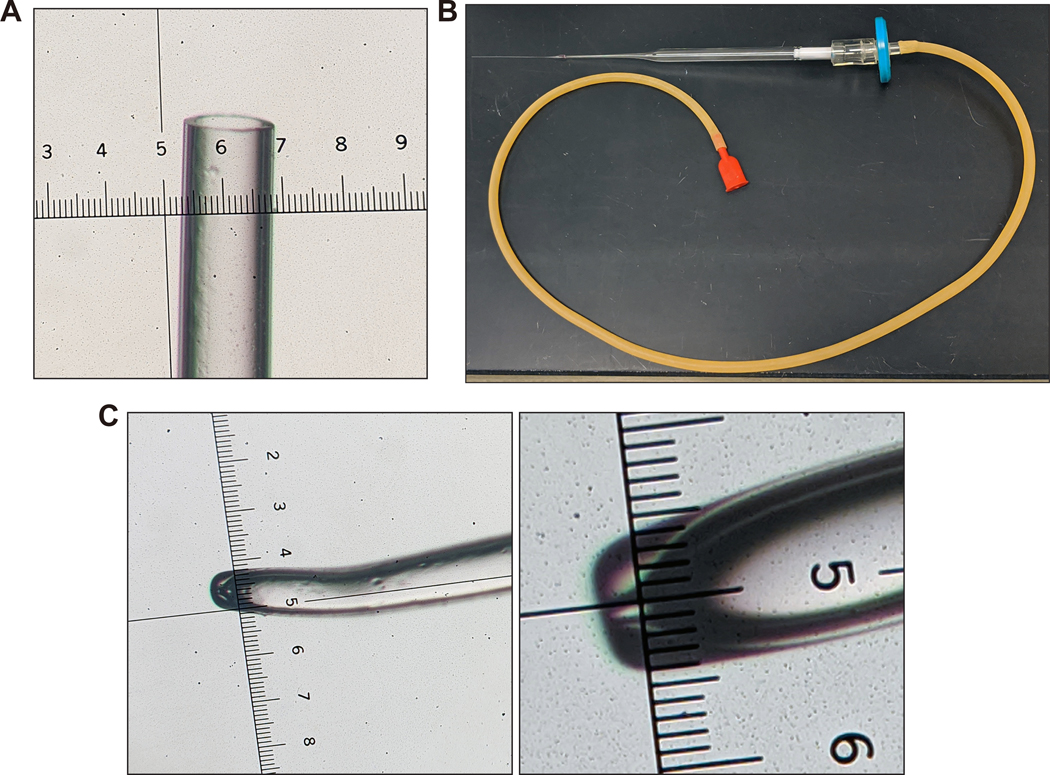
(A) The transfer pipettes are made by hand-pulling a cotton-plugged, 9-inch glass Pasteur pipette on a Bunsen burner (See Supplementary Video 3). The pipette is held in the flame of a Bunsen burner for about 5–7 s. The pipette is then removed from the heat and immediately pulled to about shoulder width apart. The end is then cut blunt so that the diameter is about 0.1–0.2 mm, equal to that of a blastocyst, as shown (smallest ticks on ruler in image are 10 μm apart; distance between each large tick is 0.1 mm). It is important that the end is blunt as jagged edges could damage the blastocyst. (B) Overview of the assembled transfer pipette setup. (C) The holding needle is prepared by pulling the borosilicate glass on the Bunsen burner and hand-cutting it to the desired size, then polishing and bending on the Microforge. The distance between the smallest ticks on ruler in left and right images is 10 μm. Note that microinjection holding needles can also be purchased commercially (for example, cat. no. MPH-MED-20, Origio Inc.). Images in (A) and (C) were taken with a Nikon SMZ U 1:10 stereo microscope with SMZ-U UW10xA/24 eyepiece containing a microruler.
53. Place up to 20 collected blastocysts or morulae (sorted into different drops, i.e., one drop with blastocysts and another drop with morulae) per each 20-μL drop in 35-mm Petri dishes (prepared in Step 48). Record the total number of embryos.
? TROUBLESHOOTING
CRITICAL: It is essential to use rigorous sterile techniques when collecting blastocysts. All working areas should be entirely free of dust and covered with fresh benchtop protector sheets.
Institutional animal facility required PPE should be worn. If available, a biosafety cabinet can be used.
ESC injection into blastocysts– TIMING 3 h
54. Prepare injection micropipette needles and holding micropipette needles as described in Equipment Setup (See Figure 8 and Supplementary Video 4 for details) immediately prior to use.
Figure 8. Microinjection needle, ES cell injection, and overview of microinjection setup.
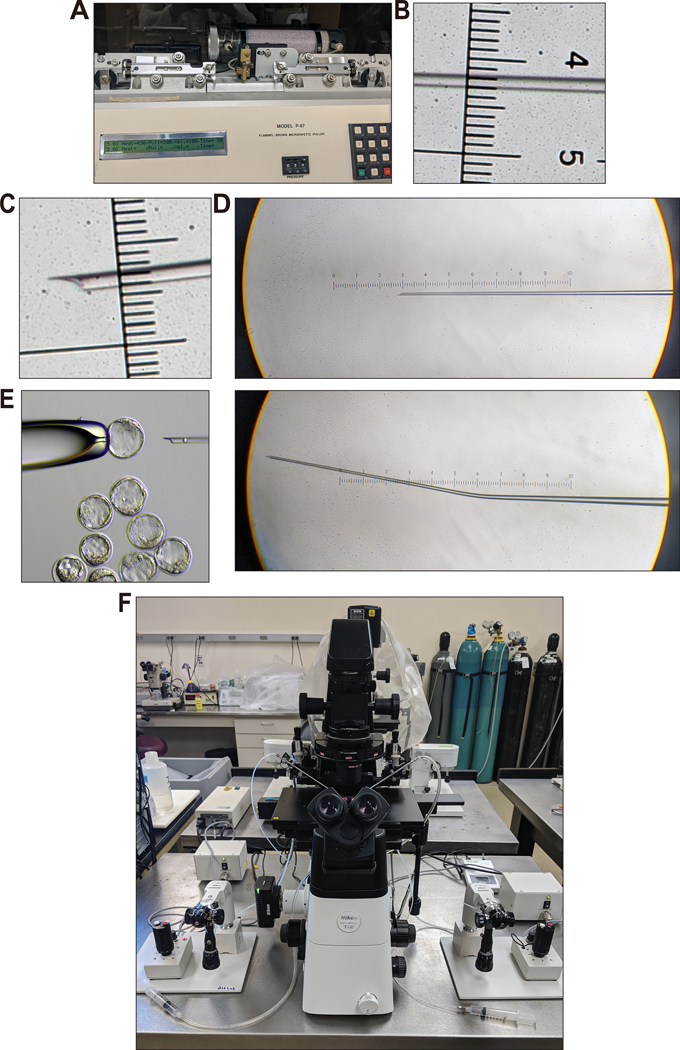
(A) Borosilicate glass (O.D. 1.0 mm, I.D. 0.75 mm; Sutter Instrument Co. B100-75-10) is pulled on a micropipette puller and cut by hand using a razorblade under the light microscope (see Supplementary Video 4). (B) The pulled microinjection needle viewed under a light microscope. The distance between the smallest ticks on ruler in image is 10 μm. (C) The cut microinjection injection needle as viewed under a light microscope. Note the shape of the tip. No rough edges that could damage ES cells or blastocysts during injection should be present. Notice that the diameter is about the diameter of a single ES cell. This helps align the ES cells in a single file when loading the needle for injection. (D) Images of the injection needle before (top) and after (bottom) bending on the microforge. Bending the microinjection injection needle 5–20° allows the needle to lay flat along the bottom of the microinjection dish. (E) ES cell injection into a blastocyst attached to the holding needle. Image was acquired with a Nikon Eclipse TE2000-s microscope. (F) See Experimental Setup and Steps 54–57 for details. Images in (B–D) were taken with a Nikon SMZ U 1:10 stereo microscope with SMZ-U UW10xA/24 eyepiece containing a microruler.
55. Perform blastocyst injection on either a Nikon Eclipse TE2000-s microscope equipped with Leitz manipulator arms or a Nikon T12-U microscope with Narashige manipulator arms (or use an equivalent setup) situated on an anti-vibration lab table (Figure 8F). Place the lid of a 35-mm Petri dish containing 2 mL of blastocyst flushing medium onto the stage. Lower injection needle and holding needle into the medium, then overlay the medium with 2 mL of light paraffin mineral oil. Use the Pasteur transfer pipette to add 10 blastocysts at a time. Add an aliquot of about 10 μL ESC suspension (stored on ice, within 2 h of collection from Step 47) by using the same transfer pipette.
56. Inject 10–15 blastocysts with 12 ESCs each (we have found a range of 10–12 ESCs per blastocyst to be optimal for NBC). Use a 20× objective (200×) magnification for blastocyst injection. Blastocysts should not be left in the injection dish for more than 30 min (see Supplementary Video 5).
57. Use a transfer pipette equipped with a suction mouthpiece (see Step 52) to place injected blastocysts back into EmbryoMax KSOM mouse embryo medium under light paraffin mineral oil. Incubate in a 5% CO2, 37 °C tissue culture incubator until transfer surgery about two to three hours later.
CRITICAL STEP: With optimal technique, about 25 blastocysts can be injected in one hour. Presence of feeder cells, low quality of ESCs, issues with the injection equipment, and blastocyst quality can significantly reduce this number. It should be kept in mind, however, that injection quality is more important than the number of blastocysts injected16.
CAUTION Be careful whilst mouth pipetting. Mouth pipetting is the standard in the field. It gives optimal results and so alternatives cannot be recommended.
? TROUBLESHOOTING
Blastocyst transfer surgery – TIMING 2 h
58. Anesthetize 2.5-dpc pseudopregnant recipient females (or, if necessary, 0.5-dpc pseudopregnant recipient females) by i.p. injection of a mix of 100–120 mg/kg Ketamine and 5–10 mg/kg Xylazine and check for desired plane of anesthesia
CAUTION For these steps you must follow local IACUC guidelines and approved protocols.
59. Place eye lubricant in each eye. Shave fur completely from an approximately 1 × 1.5 in. rectangle on the back by using an Oster Pocket Cordless Trimmer or similar device. Disinfect skin by alternating application of sterile alcohol prep pads and Povidone-iodine three times, ending with Povidone-iodine.
60. Transfer mouse in ventral position (i.e., in sternal recumbency) to a sterile surgical area. Use microdissection scissors to make a 1- to 2-cm midline incision on the back and a 0.5-cm incision in the peritoneal wall over the ovaries. Use Graefe forceps to remove the ovarian fat pad and hold with a Johns Hopkins clamp to expose the ovary, oviduct, and uterine horns.
61. Place the mouse on the sterile area on the base of a Nikon SMZ 1500 C-DSS115 stereo microscope equipped with a Nikon P-FMD 1007009 focusing mount. While holding the uterine horn securely with Dumont #55 forceps, pierce a small hole into the uterus with a 30-G needle. Use the hand-pulled transfer mouth pipette (see Step 52) to transfer five to eight ESC-injected blastocysts into each uterine horn of the 2.5 dpc pseudopregnant recipients. Alternatively, if using 0.5 dpc pseudopregnant fosters, pierce a small hole into the ampullae of the oviduct and transfer five to eight blastocysts.
62. Reintroduce uterus, oviduct, and fat pad into the intraperitoneal cavity. Close the body wall with dissolvable suture material (Look Suture, Cat. No. 495B, PolySyn suture, 18 in., size 5–0, 3/8 circle, precision reverse cutting) with Mayo-Hegar needle holders and Graefe forceps. Repeat this process on the other oviduct or uterine horn. Close the skin with 7.5 × 1.75 mm stainless steel Michel suture clips. Administer 0.1 mg/kg Buprenorphine (0.3 mg/mL in 5% (wt/vol) dextrose) subcutaneously (or other IACUC-approved post-op analgesic) and place the mouse into a small cage. Place the cage on a circulating water cage warmer (Heat Therapy Pump HTP-1500). Monitor each mouse until fully conscious before returning it to the holding room. Sterilize all surgical tools between female recipients by using a glass bead sterilizer.
Chimeric pups birth – TIMING 17–19 days
63. Pups will be born 17 days after blastocyst transfer to uterus (or 19 days after transfer to oviduct). Follow the same postnatal procedures you do for conventional mouse pups. Analysis of embryos at various prenatal developmental stages is also an option.
CRITICAL: Pregnant females should be housed in an optimal environment after transfer surgery. For example, noise, vibration, or odors can negatively impact the pregnant females, inducing abortion or increasing the likelihood of cannibalizing pups. Cages with pregnant females should not be manipulated within two days of delivery, i.e., bedding changes should be carried out in advance, and nesting materials should be provided before the anticipated delivery date, to decrease cannibalism16. On average, about 50% of ESC-injected blastocysts transferred into fosters should develop into newborn mice.
? TROUBLESHOOTING
Genotyping – TIMING 2 d
CRITICAL: When using heterozygous Emx1-Cre; hemizygous DsRed.T3 males and homozygous R26-DTA donor females to generate the host blastocysts (Figure 2B), it is essential to perform genotyping with gene-specific primers to distinguish conventional chimeras from reconstituted NBC chimeras (Figures 2A and 9) before analyzing mouse pups at various prenatal developmental stages.
Figure 9. PCR genotyping of NBC chimeras and representative photograph of newborn mice.

(A) PCR genotyping of NBC chimeras. Representative, ethidium bromide-stained agarose gel images show genotyping PCR results. NBC chimeras are positive for both R26-DTA and Emx1-Cre. Littermate controls (no ablation) lack Emx1-Cre. (B) From left to right: wild-type, non-chimeric pups from a normal mouse breeding cross; conventional chimeras generated by injecting donor ES cells into a wild-type host blastocyst; NBC chimeras without reconstitution of the forebrain using donor ES cells (‘ablated’); and NBC chimeras with reconstitution of the forebrain using donor ES cells (‘reconstituted’); adapted from6. The lack of a milk spot in non-reconstituted NBC chimeras is consistent with the inability to suckle as a result of incomplete jaw development. Appropriate institutional regulatory board permission was obtained for all experiments involving mice.
64. Take ~2 mm piece of tail biopsies from postnatal mouse pups. Incubate the tail tips overnight at 56 °C in 400 μL of tail lysis buffer.
65. Precipitate DNA with an equal volume of isopropanol, briefly wash with 70% (v/v) ethanol, dry, and dissolve in 200 μL TE buffer at 56 °C for at least 2 h. To determine the genotype, PCR-amplify the extracted DNA using the gene-specific primers listed below under the following conditions indicated in the following tables:
| Primers | Sequence (5’−3’) | Expected results | |
|---|---|---|---|
| oIMR8052_ Mutant | GCGAAGAGTTTGTCCTCAACC | Mutant = 340 bp | |
| oIMR8545_Common | AAAGTCGCTCTGAGTTGTTAT | Heterozygote = 340 bp and 650 bp | |
| oIMR8546_WT | GGAGCGGGAGAAATGGATATG | Wild type = 650 bp | |
| oIMR1084_Mutant | GCGGTCTGGCAGTAAAAACTATC | Mutant = 102 bp | |
| oIMR1085_Mutant | GTGAAACAGCATTGCTGTCACTT | ||
| oIMR4170_WT | AAGGTGTGGTTCCAGAATCG | Heterozygote = 102 bp and 378 bp | |
| oIMR4171_WT | CTCTCCACCAGAAGGCTGAG | Wild type = 378 bp | |
| DsRed.T3-F | GGTGATGTCCAGCTTGGAGT | ||
| DsRed.T3-R | CCCCGTAATGCAGAAGAAGA | DsRed transgene = 208 bp | |
| Component | Amount (μL) | Final |
|---|---|---|
| Tail DNA (~80 ng/μl) | 2 | ~160 ng |
| CoralLoad 10× buffer | 2.5 | 1× |
| dNTPs (2.5 mM each) | 2.5 | 100 μM |
| Forward primer (2.5 μM) | 0.5 | 0.1 μM |
| Reverse primer (2.5 μM) | 0.5 | 0.1 μM |
| Taq DNA polymerase (5U/ μL) | 0.2 | 1 U |
| ddH2O | 16.8 | - |
| Total | 25 | |
CRITICAL: We do not always measure DNA concentration before setting up the genotyping reactions as in our experience the method is robust over a wide range of DNA concentrations (50–500 ng).
| Cycle number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 95 °C, 5 min | ||
| 2–36 | 95 °C, 30 s | 60 °C, 30 s | 72 °C, 40 s |
| 37 | 72 °C, 5 min | ||
66. Separate the amplified DNA products by gel electrophoresis on a 1.5% (w/v) agarose gel in 1× TAE buffer. See Figure 9A for representative gel images of genotyping PCR results.
Phenotypic analysis – TIMING depends on assays and research questions
67. Examine and phenotype mice generated by NBC using standard procedures established for conventional mouse studies. Choice of studies will depend on the research question. For example, we have successfully used NBC for neurodevelopmental and behavioral studies in mice from the early embryonic period up to two years of age6.
TIMING
Steps 1–17, Genome editing in ESCs and validation: ~4 to 8 weeks
Steps 18–39, Fluorescent labeling of donor ESCs: ~2 to 3 weeks
Steps 40–47, Expansion of mutant and parental wild-type ESCs for NBC (simultaneous generation of blastocysts and fosters): 9 days
Steps 48–62, Blastocyst collection, ESC injection, and transfer into fosters: 1 day
Step 63, Chimeric pups born: 17 to 19 days
Steps 64–66, Genotyping: 2 days
Step 67, Phenotypic analysis: variable
TROUBLESHOOTING
Step 17. A xi. ESC clones do not contain the desired mutation.
If ESC clones with the desired mutation are not present, consider re-designing the CRISPR-Cas9 strategy. Also verify that the PCR screening strategy is working correctly.
Step 40. B v. Low number of plugged females.
If only a few females with vaginal plugs are present, several aspects should be considered: age of stud males should be two- to six months and ideally stud males should be replaced after six months. Consider also how often stud males have been mated and ensure that stud males are not mated more than twice weekly. For troubleshooting also check the quality of the hormones used: verify that they were prepared and stored correctly and that they are not expired (see instructions for preparing and storing hormones in this protocol). Verify that the light-dark cycle of the mouse holding room is correctly adjusted. Mice should not be disturbed during the dark cycle to ensure optimal mating. Factors such as construction noise and vibrations, or fluctuating room temperature, can negatively affect mating.
Step 53. Low blastocyst yield.
Verify that hormones have been prepared and stored appropriately and that they are not expired. Make sure that the age of females used for blastocyst production is within the recommended age range and that vaginal plug check is carried out correctly. If less than about 10 blastocysts are recovered per mouse, make sure that recovery technique is optimal. Make sure there are no problems with the light cycle, hormones, and general housing environment.
Step 57. Difficult ESC microinjection.
Care should be taken to optimally prepare ESCs for injection. Incorrect preparation can make cells difficult to handle and cause significant problems for the injectionist. Feeder cells should be depleted as described, as they may clog the injection needle. Further, prolonged exposure of ESCs to trypsin can make cells sticky, which can also make injection very difficult. To avoid this, ESCs should be handled very gently before injection and be kept on ice.
Step 63. Low rate of pregnancy after blastocyst transfer.
Potential causes for this include incorrect techniques related to blastocyst handling, ESC injection, and transfer surgery. Moreover, verify that pseudopregnant fosters were plug checked and timed correctly as described in the relevant sections of this protocol. As indicated in Troubleshooting for Step 40B(v), verify that the mouse holding room and environment is optimal. A low rate of pregnancy after blastocyst transfer can also be caused by low quality ESCs. Make sure that recommendations on ESC culture are followed and that the optimal number of ESCs is injected per blastocyst (not more than 15 ESCs and ideally 10–12 ESCs). ESCs should also be karyotyped to ensure they have not become aneuploid.
ANTICIPATED RESULTS
The NBC approach described in this protocol should greatly facilitate many neurobiological studies of the cerebral cortex and hippocampus. By using this protocol, we have generated a large number of chimeras and have found that the approach is very reliable over a large number of experiments. As reported6, compared to wild-type and conventional chimeras (Figure 9B), non-chimeric and low chimerism NBC pups also survive birth. Non-chimeric NBC pups show extensive ablation of hippocampal, neocortical and olfactory bulb structures, whereas the thalamus, midbrain, hindbrain and cerebellum appear normal, consistent with the specificity of Emx1-Cre recombination6. Non-reconstituted NBC pups show incomplete jaw development6, which allows visual assessment of overall reconstitution (Figure 9B). Reconstituted NBC chimeras show complete restoration of hippocampal and neocortical structures, normal jaws, develop normally after birth, and show no increased mortality up to at least 24 months of age6. Both male and female NBC chimeras are fertile, and female chimeras display normal nurturing behaviour towards their pups6. We find consistent rates of pup survival, ESC-mediated reconstitution, and litter sizes. Detailed summaries of recovery rates of blastocyst injections, survival rates, and litter sizes for multiple ESC lines have been reported6.
The dual labeling approach used for NBC enables reliable detection of both donor- and host-derived cells. By using this dual labeling approach, we find that the vast majority of cells in the cortex and hippocampus of NBC chimeras are derived from donor ESCs6. The very small fraction of host-derived cells in NBC forebrain regions likely corresponds to non-dorsal telencephalon-derived cells of host origin (such as interneurons that migrate tangentially into the cortex, microglia that are derived from resident macrophages, and endothelial cells from blood vessels) as would be expected, given Emx1-mediated DTP ablation in the host embryo. Our studies of Dcx biology via NBC show that the approach can substantially accelerate in vivo studies of gene function6.
Supplementary Material
Dissection of uterine horns. Super-ovulated donor females are euthanized according to protocol, then the ventral skin is cut and pulled back. After opening the body wall, organs are moved to the side and the uterine horns are dissected out as shown. Appropriate institutional regulatory board permission was obtained for all experiments involving mice.
Flushing of uterine horns for embryo collection. The uterine horns are held with forceps, a syringe of flushing media with a needle is used to cut the ends of the horns, and the embryos are gently flushed out. Appropriate institutional regulatory board permission was obtained for all experiments involving mice.
Pulling and blunting the transfer pipette. See Step 52.
Cutting the microinjection needle after pulling. The microinjection injection needle is cut by hand with a conventional razor blade under a light microscope. The needle is rested on a silicone mat to prevent damage to the microscope stage.
ACKNOWLEDGMENTS
We thank members of the Alt and Schwer labs and Diana Laird for discussions, Pei-Yi Huang for help with blastocyst injections, Hwei-Ling Cheng for advice and help with ESC culture. This work was supported by the Howard Hughes Medical Institute, the BCH Department of Medicine (DOM) Support Fund, the BCH DOM Anderson Porter Fund, a major grant from the Charles H. Hood Foundation, Inc., Boston, MA (F.W.A), and a generous gift from the Dabbiere family (B.S.). B.S. is a Kimmel Scholar of The Sidney Kimmel Foundation, supported by the UCSF Brain Tumor SPORE Career Development Program, the American Cancer Society, the UCSF Program for Breakthrough Biomedical Research (which is partially funded by the Sandler Foundation), the Andrew McDonough B+ Foundation, the Shurl and Kay Curci Foundation, and a Martin D. Abeloff V Scholar award of The V Foundation for Cancer Research. B.S. holds the Suzanne Marie Haderle and Robert Vincent Haderle Endowed Chair, UCSF. H-Q.D. is a fellow of the Cancer Research Institute of New York. F.W.A. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
DATA AVAILABILITY
All data referred to or analyzed are included in Chang et al. Nature, 2018, doi: 10.1038/s41586-018-0586-0.
REFERENCES
- 1.Martin GR Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 78, 7634–7638 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans MJ & Kaufman MH Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 (1981). [DOI] [PubMed] [Google Scholar]
- 3.Bradley A, Evans M, Kaufman MH & Robertson E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature 309, 255–256 (1984). [DOI] [PubMed] [Google Scholar]
- 4.Miura T, Mattson MP & Rao MS Cellular lifespan and senescence signaling in embryonic stem cells. Aging Cell 3, 333–343 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Bradley A, Hasty P, Davis A & Ramirez-Solis R. Modifying the mouse: design and desire. Biotechnology (NY) 10, 534–539 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Chang AN et al. Neural blastocyst complementation enables mouse forebrain organogenesis. Nature 563, 126–130 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi T et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 142, 787–799 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Lansford R, Stewart V, Young F & Alt FW RAG-2-deficient blastocyst complementation: an assay of gene function in lymphocyte development. Proc. Natl. Acad. Sci. USA 90, 4528–4532 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J et al. Generation of normal lymphocyte populations by Rb-deficient embryonic stem cells. Curr. Biol 3, 405–413 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Usui J et al. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am. J. Pathol 180, 2417–2426 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Liégeois NJ, Horner JW & DePinho RA Lens complementation system for the genetic analysis of growth, differentiation, and apoptosis in vivo. Proc. Natl. Acad. Sci. USA 93, 1303–1307 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanger BZ, Tanaka AJ & Melton DA Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 445, 886–891 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Wu J et al. Stem cells and interspecies chimaeras. Nature 540, 51–59 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Wu J et al. Interspecies Chimerism with Mammalian Pluripotent Stem Cells. Cell 168, 473–486.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorski JA et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci 22, 6309–6314 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behringer R, Gertsenstein M, Nagy KV & Nagy A Manipulating the mouse embryo: a laboratory manual. (Cold Spring Harbor Laboratory Press, 2014). [Google Scholar]
- 17.Conner DA Mouse embryo fibroblast (MEF) feeder cell preparation. Curr. Protoc. Mol. Biol 23.23.2 (2001) [DOI] [PubMed] [Google Scholar]
- 18.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conner DA Mouse embryonic stem (ES) cell culture. Curr. Protoc. Mol. Biol 23.23.3 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Yang H, Wang H & Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc 9, 1956–1968 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Brown T. Southern blotting. Curr. Protoc. Immunol. 10.10.6A (2001). [DOI] [PubMed] [Google Scholar]
- 22.Ryder E et al. Molecular characterization of mutant mouse strains generated from the EUCOMM/KOMP-CSD ES cell resource. Mamm. Genome 24, 286–294 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutner RH, Zhang X-Y & Reiser J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc 4, 495–505 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Nissen SB et al. Four simple rules that are sufficient to generate the mammalian blastocyst. PLoS Biol. 15, e2000737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lois C, Hong EJ, Pease S, Brown EJ & Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dissection of uterine horns. Super-ovulated donor females are euthanized according to protocol, then the ventral skin is cut and pulled back. After opening the body wall, organs are moved to the side and the uterine horns are dissected out as shown. Appropriate institutional regulatory board permission was obtained for all experiments involving mice.
Flushing of uterine horns for embryo collection. The uterine horns are held with forceps, a syringe of flushing media with a needle is used to cut the ends of the horns, and the embryos are gently flushed out. Appropriate institutional regulatory board permission was obtained for all experiments involving mice.
Pulling and blunting the transfer pipette. See Step 52.
Cutting the microinjection needle after pulling. The microinjection injection needle is cut by hand with a conventional razor blade under a light microscope. The needle is rested on a silicone mat to prevent damage to the microscope stage.