

Abstract
As the field of mass spectrometry imaging continues to grow, so too do its needs for optimal methods of data analysis. One general need in image analysis is the ability to classify the underlying regions within an image, as healthy or diseased, for example. Classification, as a general problem, is often best accomplished by supervised machine learning strategies; unfortunately, conducting supervised machine learning on MS imaging files is not typically done by mass spectrometrists because a high degree of specialized knowledge is needed. To address this problem, we developed a fully open-source approach that facilitates supervised machine learning on MS imaging files, and we demonstrated its implementation on sets of cancer spheroids that either had or had not undergone chemotherapy treatment. These supervised machine learning studies demonstrated that metabolic changes induced by the monoclonal antibody, Cetuximab, are detectable but modest at 24 hours, and by 72 hours, the drug induces a larger and more diverse metabolic response.
Graphical Abstract
Introduction
Imaging mass spectrometry is becoming a mature field. An early report published in 1991 by Oak Ridge National Laboratory scientists showed images reconstructed from MS data from an “organic ion microprobe” technique that relied on secondary ion mass spectrometry to image surfaces up to 2 cm in diameter.(1) Caprioli’s group first successfully demonstrated MALDI imaging mass spectrometry by collecting images of a rat splenic pancreas in 1997.(2) Since these early days, the imaging MS field extended its reach to take on many diverse and exciting challenges. For example, the Eberlin group has demonstrated that lipid distributions can be used in diagnoses to determine tumor margins.(3) On a larger scale, the Dorrestein group has cleverly adapted imaging protocols to map the distribution of compounds across the surface of entire human beings.(4) Our lab has applied imaging workflows to organoids derived from patient biopsies to determine patient-specific drug metabolism.(5) As MS imaging studies become more prevalent, the demand for tools to differentiate the regions within and between images is also growing.
Currently, unsupervised machine learning is the most common approach for classifying different regions in imaging mass spectrometry data. A notable example includes a highly-regarded study by Fonville and coauthors, who used Principal Components Analysis (PCA) to assess many different pre-processing methods to improve image classification.(6) In another example, Bemis et al. highlighted the utility of spatial shrunken centroids, another unsupervised method, and facilitated its implementation in an innovative R package, Cardinal.(7) The unsupervised machine learning approach, CLARA, was also used to perform several classifications on spheroids (8); this application overlaps most closely with the work presented herein. Unsupervised classification of MS imaging data is a burgeoning field, and significant advances have been expertly summarized in a recent review.(9)
While unsupervised machine learning of MS image data is flourishing, supervised machine learning has the potential to do a better job of discriminating groups, since the class labels are used to train the model on the important differences between the groups. In a recent elegant example, mathematicians modified a common unsupervised method, Non-negative Matrix Factorization, by introducing class labels of the training data into the algorithm, therefore making it a supervised machine learning method; when they applied the approach to MS images of lung cancer, the supervised model outperformed the unsupervised one.(10) Supervised machine learning in image analysis is typically only carried out by those with a high level of specialized knowledge. For example, Random Forest approaches used for imaging in general (11) and MS imaging specifically (8, 12) have been implemented previously as well; yet, all three of these research projects relied on the use of MatLab, and they required workflows or scripts that needed to be developed in-house.
Other types of supervised machine learning also have potential to be useful in MS image analysis. Indeed, both Support Vector Machines (SVM), and k-Nearest Neighbor classifiers (k-NN) are more commonly implemented than Random Forest classifiers in certain other types of image analysis problems.(13) And new tools continue to emerge, including the Aristotle Classifier, a tool we recently developed.(14, 15) Unfortunately, researchers who want to implement any of these tools, or even the Random Forest classifiers used earlier, need to develop their own workflows, potentially combining several licensed and open-source products.
To address this technology gap, we have built a fully open-source pipeline to conduct supervised machine learning on MS imaging data sets. The approach is fully described herein, and all functionality is accomplished in RStudio, an open-source software package that is commonly implemented in data science. This workflow is meant to complement the already developed tool, Cardinal, which greatly facilitates a limited number of machine learning options.(7) Our approach relies on some of Cardinal’s functionality but also provides users with flexibility in using any machine learning algorithms they like for classification.
To demonstrate how the pipeline works, we have conducted several classification experiments on cancer spheroids. Spheroids are three-dimensional cell cultures, first developed by Sutherland.(16) The spheroids described in this manuscript are composed of human epithelial colon carcinoma cells. Colon spheroids grow in a radially symmetric fashion, with cells sequentially dividing and adding to the outer layers. As the spheroid grows, chemical gradients develop as a result of reduced diffusion in and out of the spheroid. For example, while the pH on the outside of the spheroid will match the cell culture media and remain around 7, the pH in the center of the spheroid will decrease. Lactic acid and the build-up of other cellular waste products reduces the pH to 5 in the center of the spheroid. Other species, most critically, oxygen, will also develop gradients. These chemical gradients affect the health of the cells and resulting pathophysiological gradients will also develop, with healthy proliferating cells residing on the outside and dying cells living in the inhospitable, acidic central core.
Spheroids have been used as a valuable tumor model systems and are especially useful for the evaluation of new drugs.(17) Given the recent advances that have been made in the field of immunotherapy, we evaluated mass spectrometry imaging for examining the impact of the drug Cetuximab in colon spheroids. Cetuximab, also known by its clinical name Ertibux, targets the epidermal growth factor receptor (EGFR) to halt division of cancer cells. It is one of the most frequently prescribed immunotherapies for colorectal cancer and thus, a good test case for mass spectrometry imaging analysis.
To compare spheroids that were or were not treated with Cetuximab, we applied the machine learning workflow first described herein. In this workflow, we used a new supervised machine learning tool, the Aristotle Classifier, which has recently been described and applied to classifying glycomics samples(14) and bacteria from closely related species.(15) We were able to detect early metabolic changes in the spheroids treated with the drug and study the differences in the spheroids that had been treated for 24 vs 72 hours.
Experimental
The mass spectrometry data used in this manuscript had been collected as part of an earlier study. See reference 18. Details about the samples are provided here, and readers can refer to the earlier manuscript if further details are needed. The spheroids were prepared with HT-29 colon carcinoma cells, obtained from American Type Culture Collection (ATCC, Manasses, VA). HT-29 cells were grown with McCoy’s 5A media (Life Technologies, Carlsbad, CA). The media were supplemented with 10% fetal bovine serum (FBS; Thermo Scientific, Waltham MA). Spheroids were prepared by seeding 7000 cells from either cell line in 200 μL cell culture media in agarose-coated 96-well plates. The spheroids were allowed to grow for four days and then one-half volume media changes were completed every two days thereafter. On day thirteen, the spheroids were incubated with cetuximab in PBS (1 mg/mL), while the control spheroids were dosed with the same volume of PBS. The spheroids were harvested either 24 or 72 hours later.
All data analysis steps were performed in R, version 3.5.1. In addition, the R package, Cardinal, version 2.0.4, was installed,(7) as was the R package “pROC”.(19) MALDI-MS files were saved as imzML files and converted into a Cardinal-readable format as described in the online Cardinal documentation.(20) The MS data matrices were extracted and normalized as described in more detail in the workflow section below. Supervised machine learning was conducted using the Aristotle Classifier, using the version provided in Supplemental Materials of reference 14. During supervised classification, all the pixels in a first spheroid were selected as a training group 1, and all the pixels in a second spheroid were selected as training group 2. All the pixels in the remaining spheroids were used as the test set. The aggregating replicates (k), an adjustable parameter in the Aristotle Classifier, was set to 100, and the classification scores for all the pixels in all the spheroids, from both the training sets and test sets, were used for generation of the data herein.
After the classification experiments were compete, a separate set of experiments was conducted to identify the most important features at each timepoint. In these experiments, all the pixels from all the spheroids at a given timepoint were used. A vector containing the class label for each pixel was generated, and the area under the receiver-operating curve (AUC) was determined for each m/z (each feature) in the data set, using the R package, pROC, and the function, auc. The fifty features with the highest AUC at each timepoint (72 hours and 24 hours) were reported.
Results and Discussion
The overall goal of this research project is to develop a fully open-source approach for applying supervised machine learning algorithms to mass spectrometry imaging data. The challenges to be overcome include: efficiently extracting the relevant data into the appropriate format that is machine-learning ready, preprocessing the data for maximal classification accuracy, choosing an optimal machine learning strategy, and visualizing the results. We pictorially delineate the necessary steps to accomplish all these goals in Figure 1 and offer extensive details below.
Figure 1.
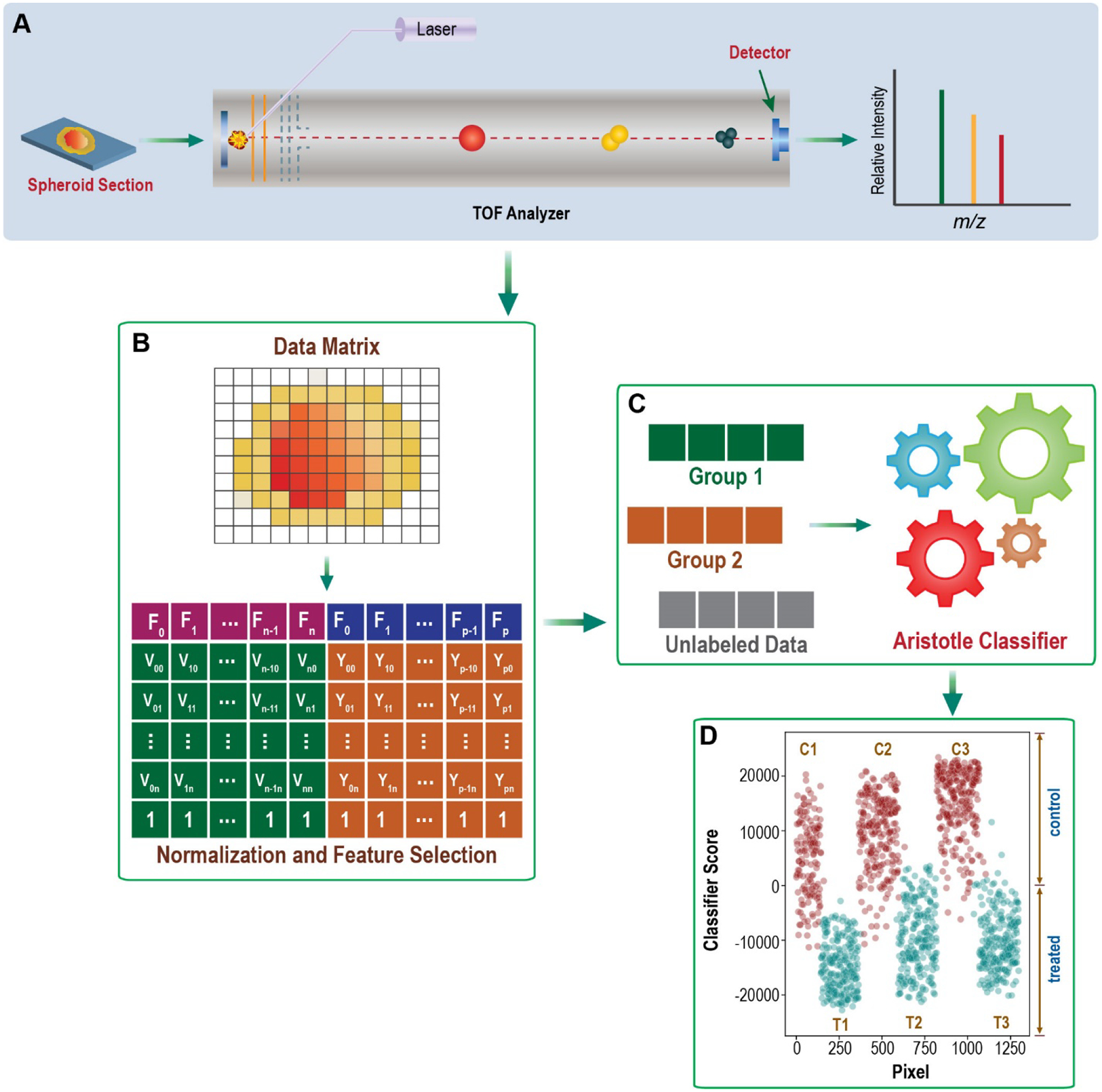
General workflow pictorially describing the steps needed for machine learning on imaging mass spectrometry data.
From image files to matrices for supervised machine learning.
This work pre-assumes that researchers have collected mass spectrometry imaging data, as this is an obvious prerequisite. After doing so, the first requirement for performing machine learning on a data set is that the data be available in a matrix containing “samples” and “features”. For MS imaging files, the samples are individual pixels where imaging data was collected. The features are the intensities of each of the m/z’s acquired. A widely used programming language for machine learning is R, and it is also fully open source, so we built our workflow to be fully executable in the R environment. To quickly get the data in the appropriate matrix format, and to later take advantage of some of the utilities in the open-source R package, Cardinal, we obtained the required matrices by first reading the imzML files into the R package, Cardinal. After the files are read, extracting the necessary matrix for machine learning is easy. For example, the required data matrix is immediately accessible by executing the single command:
as.matrix(spectra(NameOfCardinalDataFile))
When the goal is to perform classification on different regions within a single imaged file, the above command will be useful for constructing a matrix that contains all the pixels in that image. When multiple imaging sets --from different spheroids, for example -- are to be compared with each other, all the files need to be extracted first; then they can be assembled into a single matrix. We extracted the requisite matrix from each desired spheroid and combined them into a single, larger matrix using simple R commands.
Data processing.
This step includes both pre-processing of the spectral data, which is almost always required in MALDI-MS imaging experiments, and feature selection, which is sometimes done to improve classification accuracy but is not always necessary. Since ionization efficiency is not very consistent from one MALDI mass spectrum to the next, especially on imaged samples, data normalization is almost always beneficial. We normalized the data by dividing each ion abundance by the sum of the ion abundances for the given pixel; this is a very common normalization strategy.(6) Other preprocessing steps could also be done directly in R, or by using features of the Cardinal package, but none were needed for the applications herein. Reference 6 provides a useful discussion of preprocessing options. Feature selection can also be done at this point. At the end of the manuscript, we give one example showing how to extract the most discriminating features, but many other strategies are possible. To demonstrate that feature selection is not always needed, we performed the supervised classification experiments below on all 3500 features between m/z 210.3 and m/z 994.5.
Supervised Classification.
Supervised classification differentiates itself from unsupervised classification in that labeled data are used to train the model on the important trends that distinguish the groups. Therefore, at least some of the data need to be labeled for use in model training. In the examples we show below, we use all the pixels in one treated spheroid and one control spheroid for model training; all the pixels in the remaining four spheroids from the same timepoint are treated as unlabeled data. They are classified based on the trained model. Alternatively, a subset of pixels within one image can be selected and used as labeled data. The easiest way to do this is to take advantage of the functionalities in the Cardinal package, as described in that tool’s documentation
In the examples provided below, supervised machine learning was accomplished with the Aristotle Classifier.(14, 15) This is a new classifier that we developed for glycomics data (14) and later applied to bacterial typing(15). A text file containing the R codes to execute the Aristotle Classifier are provided in the Supplemental Materials of reference 15. Required inputs include the labeled training samples, the number of aggregating replicates, k, which we set to 100, and the matrix of samples (pixels) and features (normalized intensities of the different m/z’s.) More information about the use of the Aristotle Classifier can be found in references 14 and 15. The data matrix extracted from Cardinal, as described above, is already in the appropriate dimension and orientation to be input directly into the Aristotle Classifier, so it is ready for input after data processing. Other supervised machine learning tools could also be used after a processed matrix is obtained, but they typically require that the data be transformed so that the individual pixels are in rows and the intensities for each m/z (each feature) are in columns.
Visualizing results.
In many instances, the results of a machine learning classifier can be used without further manipulation to understand underlying trends in the data. We provide examples in a subsequent section showing this to be the case. However, sometimes, to quickly convey a point to audiences interested in the results but less familiar with the classification methods being used, the classification results can instead be displayed directly as an image with the same dimensions as a larger version of original imaged object. When supervised machine learning algorithms output numeric results, these can easily be overlayed onto the original object dimensions and visualized. For example, the Aristotle Classifier’s output is a numerical score, so it is particularly amenable to displaying the output as an image.
The steps to displaying classifier output as an image are as follows: 1) Determine the dimensions of the pixels in the original data set. 2) Build a matrix with the same x and y dimensions as the imaged object. This matrix will be a rectangle with x rows and y columns. 3) Determine the location of each of the imaged pixels in the original rectangle. 4) Fill the matrix that was built in Step 1 with the classifier results, in the locations identified in Step 2. 5) Image the matrix. These steps can be done with five very short lines of R code, as shown below:
>dim(c1@imageData) #The second and third dimension returned are the dimensions of the object, c1.
>NewMatrix<-matrix(0,18,14) #This builds a matrix of the appropriate size; in this case, the original object was 18 by 14 pixels.
>data<-(which(c1@imageData[1, , ]>(−100))) # This command identifies where each of the pixels are located in the original image file, named “c1” and lists them in a vector called “data”.
>NewMatrix[data]<-Results[1:158] #This code populates the matrix to be visualized as an image with the classifier’s results for the original image. In this case, there were 158 pixels to be populated.
>image(NewMatrix) #This command displays the results as an image.
The general approach we describe here is not limited to machine learning using the Aristotle Classifier. If one were using a Support Vector Machine classifier, for example, the probabilities for each pixel could be visualized as an image instead. As a second example, class labels generated from a random forest algorithm could be visualized as an image.
Machine Learning Examples.
To demonstrate the workflow shown in Figure 1, we used MS data sets of cancer spheroids that were either dosed or not dosed with an anticancer drug, Cetuximab. Earlier MS imaging experiments on these spheroids confirmed that the drug had penetrated the spheroids, and it was detectable inside the spheroids at 72 hours after dosing.(18) Changes in ATP levels were also detectable inside the treated spheroids,(18) but global metabolomic changes were not investigated. Herein, we use supervised machine learning techniques to first determine whether the treated spheroids’ global metabolic profiles uniquely identify the spheroids as either being treated or untreated at two different time points (24 and 72 hours post treatment.) Additionally, we used feature selection techniques to explore the underlying metabolomic changes.
The first classification example contains six spheroids that had been subjected to MALDI imaging: three control spheroids and three that had been treated with the antibody, Cetuximab, for 72 hours. After the imaging data were acquired for all the spheroids, the IMZml files were read into Cardinal in R, so the matrices containing the abundance of each m/z (the features) for each pixel could be used as input data for classification. The data were normalized, as described above, and they were then classified using the Aristotle Classifier.
Supervised machine learning requires that the model be trained on labeled data, so in this case, the classifier was trained on the MS data from Control Spheroid #2 and Treated Spheroid #2. A total of 3500 different m/z’s, ranging from m/z 210.3 to 994.5, were used as features, and the “samples” to be compared were all the MS data from each pixel from “Control 2” and “Treated 2.” All the pixels in the remaining four spheroids were treated as “unknown” test samples, and their response to treatment was classified based solely on the training data from Control 2 and Treated 2. Note that a certain degree of within-spheroid heterogeneity exists; the inner core, for example, is known to be more acidic and hypoxic. We did not treat the inner and outer components of the spheroids separately in the classifications shown herein. We made this decision because preliminary attempts at classifying the different regions of the spheroid separately (comparing only inner cores to inner cores and outer regions to outer regions, for example) did not improve classification accuracy.
The Aristotle Classifier assigned nearly all the pixels in the “Control 2” sample a value of >0, indicating that each pixel is more similar to the control sample profile than the treated sample profile; see Figure 2. Pixels with values of <0 indicate that the mass spectrometry data for that pixel is more similar to the “treated” group overall. The “Treated 2” sample has negative values for almost all of its pixels, indicating that each of the individual locations within the treated spheroid look similar to the global “treated” profile.
Figure 2.
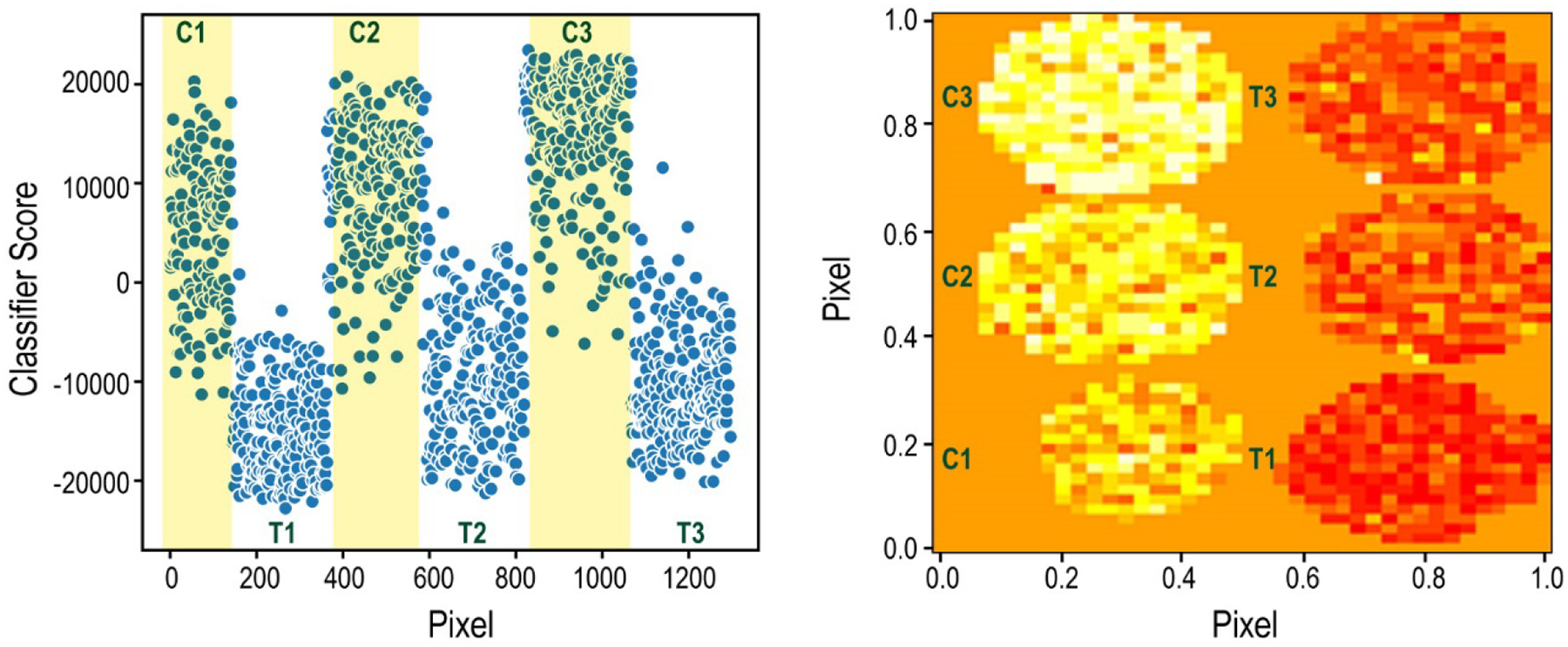
Supervised classification results comparing the spheroids either treated for 72 hours with Cetuximab or their corresponding controls. Left: Raw data from the Aristotle Classifier. Each point represents the classifier score for a pixel from one of the spheroids. The classifier’s output for the pixels from the spheroid controls, C1, C2, and C3, are in the yellow shaded regions. The classifier’s output for the treated samples (T1, T2, T3) are in unshaded regions. Note: This plot (left) does not maintain the spatial arrangement of the pixels; rather, the Y dimension is Classifier Score. The X dimension, Pixel, contains the pixels from each spheroid as they were represented in the original data matrices. Right: Numeric data from the classifier imaged in the same dimensions as the original spheroids. In this plot, each pixel is represented in the orientation in which it existed on the MALDI target. Both X and Y dimensions, are “Pixel”. The color intensity correlates to the classifier score: More red is more negative, indicating the pixels classify as “treated”. More yellow is more positive, indicating the pixels classify as “control”.
While Spheroid 2 Control obviously produces different results than Spheroid 2 Treated, it is important to determine whether the differences between these two spheroids are due to the treatment itself or are simply due to natural metabolic variability from one spheroid to another. To demonstrate that the differences in the metabolomic profile that the classifier identified in these two spheroids are specific to the treatment-induced changes, we classified each of the pixels in four additional spheroids (Treated 1, Control 1, Treated 3, Control 3) using only spheroid set 2 as the training data. In this case, all four test spheroids classified according to their treatment group, as shown in Figure 2, illustrating that the changes detected in the classified spheroids are, in fact, due to the treatment, and that these changes are universally apparent in other spheroids obtained under the same conditions. Figure 2 shows both the raw results from the Aristotle Classifier for both the labeled and unlabeled samples on the left, along with the results overlaid as images with dimensions of the original spheroids on the right. For the Aristotle Classifier results, pixels with a value of greater than zero are classified as control samples. The color variation in the image on the right indicates the similarity to the control group (yellow color) or the treated group (red color.)
After successfully determining that the spheroids at 72 hours-post treatment had substantially different metabolic profiles, and that the metabolic differences were replicable in all three sets of spheroids tested, we decided to undertake a more aggressive challenge: Is it also possible to use the spheroids’ metabolic profiles to classify them after just 24 hours of treatment? Earlier work had shown that the drug can penetrate the cells within 24 hours, but little if any cellular changes have occurred at that time point: KI-67, a cell proliferation marker, did not indicate a difference in the 24 hour treated samples vs. controls, while a large difference in this marker was present at 72 hours.(18) Furthermore, ATP, which was substantially upregulated at 72 hours (p<0.01), was only beginning to be up-regulated at 24 hours.(18) Thus, prior to the studies herein, it was unknown whether or not global metabolomic changes within the 24-hour treatment group were reliably measurable.
The data in Figure 3 show the classifier results after attempting to classify the spheroids based on their metabolic profiles after exposure to drug treatment for 24 hours. The first spheroid set (control 1 and treated 1) was used for training, and the parameters that differentiated these two spheroids were used to test the remaining four spheroids, to see if they were more like the control or the treated sample. In this case, the initial two spheroids could be somewhat distinguished from each other – most of the pixels in the “Control 1” spheroid had classifier results >0, indicating that these pixels were more like the control group. A complementary trend was seen for the treated spheroid, where the majority of the pixels received classifier scores of <0, indicating its difference from the control sample. Additionally, the majority of pixels in three of the four test spheroids were assigned to their correct groups. Only the Treated 3 spheroid was mis-classified; the majority of its pixels had positive scores, indicating it is overall more similar to the control spheroids.
Figure 3.
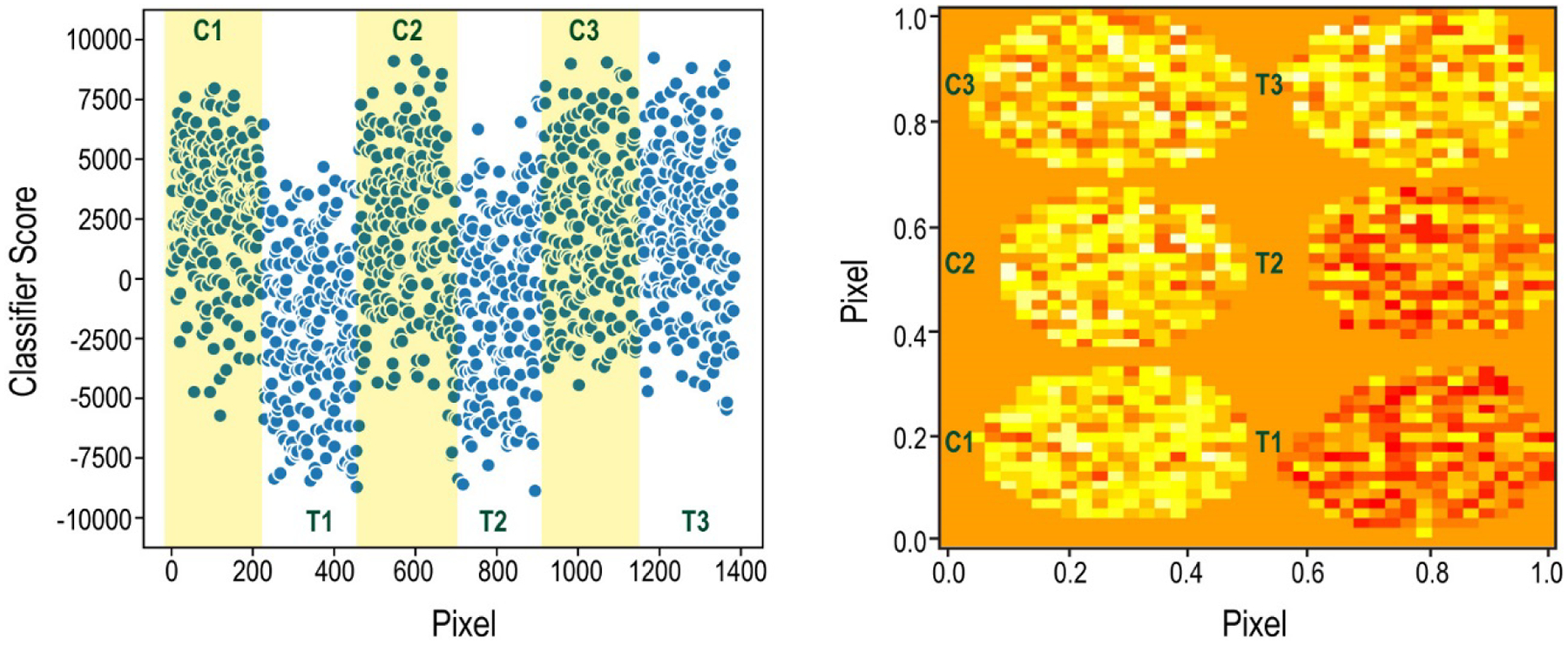
Supervised classification results comparing the spheroids either treated for 24 hours with Cetuximab or their corresponding controls. Left: Raw data from the Aristotle Classifier. Each point represents the classifier score for a pixel from one of the spheroids. The classifier’s output for the pixels from the spheroid controls, C1, C2, and C3, are in the yellow shaded regions. The classifier’s output for the treated samples (T1, T2, T3) are in unshaded regions. Note: This plot (left) does not maintain the spatial arrangement of the pixels; rather, the Y dimension is Classifier Score. The X dimension, Pixel, contains the pixels from each spheroid as they were represented in the original data matrices. Right: Numeric data from the classifier imaged in the same dimensions as the original spheroids. In this plot, each pixel is represented in the orientation in which it existed on the MALDI target. Both X and Y dimensions, are “Pixel”. The color intensity correlates to the classifier score: More red is more negative, indicating the pixels classify as “treated”. More yellow is more positive, indicating the pixels classify as “control”.
Why didn’t the third treated sample classify as a treated sample? One possible explanation is that some spheroids take slightly longer than others to begin undergoing metabolic changes: Perhaps the 24-hour time point is near the beginning of metabolic change, and the third treated spheroid had not yet begun substantial changes. Several pieces of evidence suggest that the 24-hour time point is a very early timepoint for considering metabolic change. First, earlier experiments demonstrated that KI-67 changes were not visible at 24 hours;(18) this indicates that the 24-hour time point is perhaps too early to detect many changes that the spheroids would eventually undergo. Second, by comparing the 24-hour classification data side-by-side with the replicate experiment at 72 hours, as shown in Figure 4, it is clear that the extent of change at 72 hours is substantial compared to that at 24 hours. For the spheroids within the data set representing the 72-hour experiment, all six spheroids are clearly assignable to their respective group, and most of the pixels within each spheroid are correctly classified. For the spheroids within the 24-hour data set, the global differences are more subtle. The overall signal magnitude in the y direction, which indicates the number of features that are changing and the extent to which they change, is much smaller in the 24- hour samples than the 72-hour samples. Additionally, many pixels within the samples for the first and second treated spheroids in the 24-hour data set are classifying as untreated. This likely indicates that the effects due to treatment are just beginning and not uniformly detectable throughout the spheroids.
Figure 4:
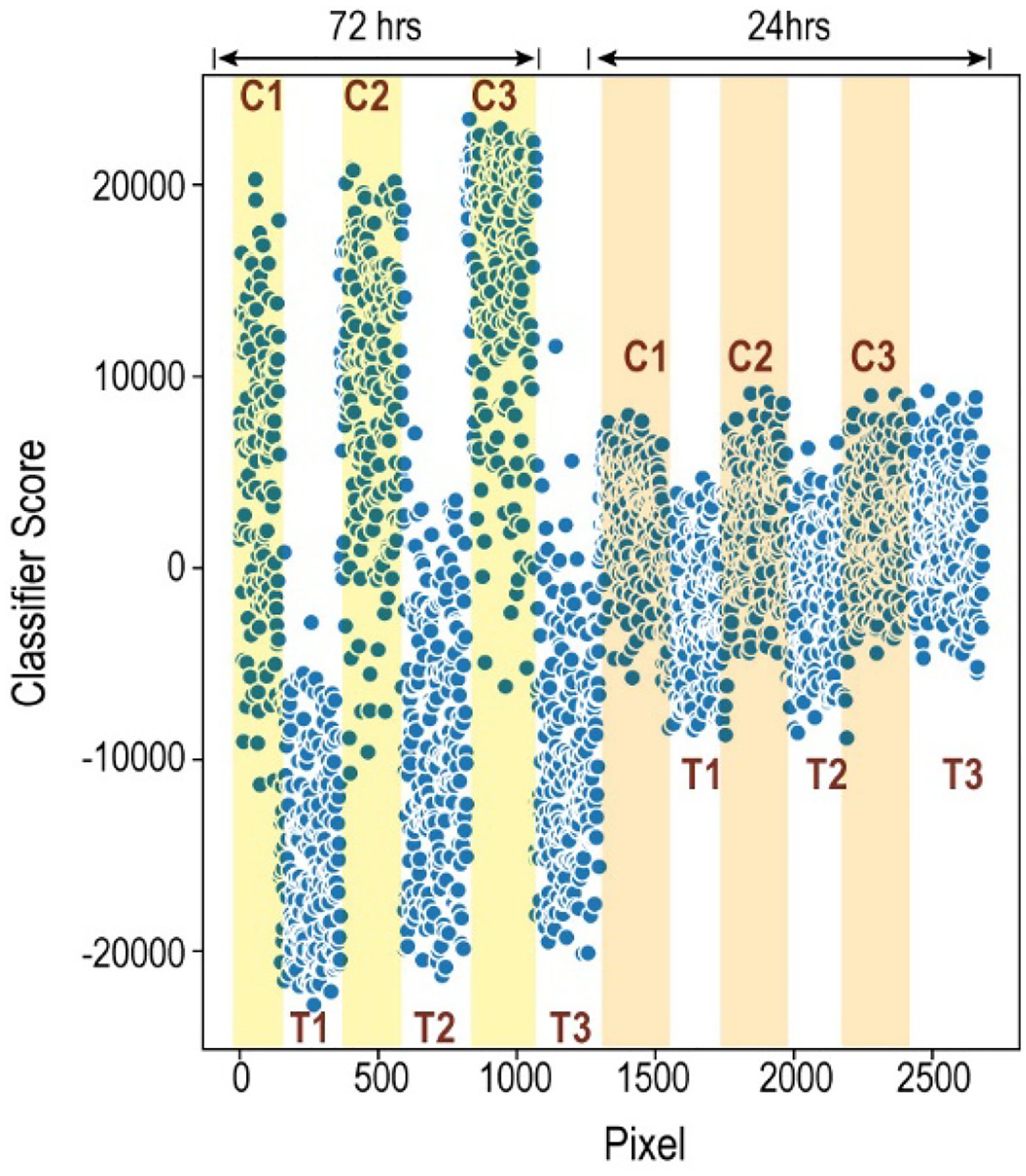
Comparison of the results for the supervised classification at 24 and 72 hours. Each point represents the classifier score for a pixel from one of the spheroids. The plot does not maintain the spatial arrangement of the pixels; rather, the Y dimension is Classifier Score. The X dimension, Pixel, contains the pixels from each spheroid as they were represented in the original data matrices. Pixels from controls are shaded (72 hrs: yellow; 24 hrs: orange.) Pixels from treated samples are unshaded. The increased separation of the treated vs control groups, and the larger signal magnitude for the 72-hour data set, indicates that the global metabolic profile between the treated and untreated spheroids is more pronounced at 72 hours than at 24 hours.
Part II: Studying the metabolic changes.
In part I, we built a fully open-source workflow that allows researchers to use any desired machine learning technique for image analysis. We demonstrated that this workflow can be used to classify spheroids, and the results obtained were consistent with earlier reports: that by 72 hours, substantial change had taken place, but at 24 hours, the spheroids had undergone very modest changes. After accomplishing these goals, we used the tools in the machine learning workflow to further study the 24-hour spheroid set with the goal of identifying the treatment-related effects that begin at this earliest time point. Additionally, we investigated whether the treatment-related differences that begin at the 24-hour timepoint are the same as those occurring at the 72-hour timepoint.
Using the entire group of six spheroids in the 24-hour set, we determined which m/z’s (which features) best differentiated the two groups, treated vs. untreated. In this case, we considered each feature independently and selected the ones that changed the most between the control and treated groups. To determine which ones changed the most, we measured each feature’s area under a receiver-operating curve (ROC). The higher the area, the better that feature does at discriminating the two groups. The area under the curve (AUC) for any feature in a data set is easily obtained in R using the open-source package, pROC. Figures 5A to 5D show four plots for the top four features (m/z’s) with the highest AUC’s for the 24-hour data set. Note: the plots in Figure 5 contain the intensities of the m/z for each of the pixels in each of the spheroids at both 24 and 72 hours. While the spheroids at 72 hours were not used to extract the features with highest AUC at 24 hours, including them in Figure 5 provides useful information about whether these same features are still differentially abundant in the spheroids at 72 hours. In the case of the top four features that discriminate the 24-hour spheroids, differences in these features are also quite important at 72 hours. In some cases, the differences at 72 hours are even more pronounced than the differences at 24 hours. This comparison reaffirms that at the 24-hour time-point, the metabolomic changes are just beginning to emerge.
Figure 5:
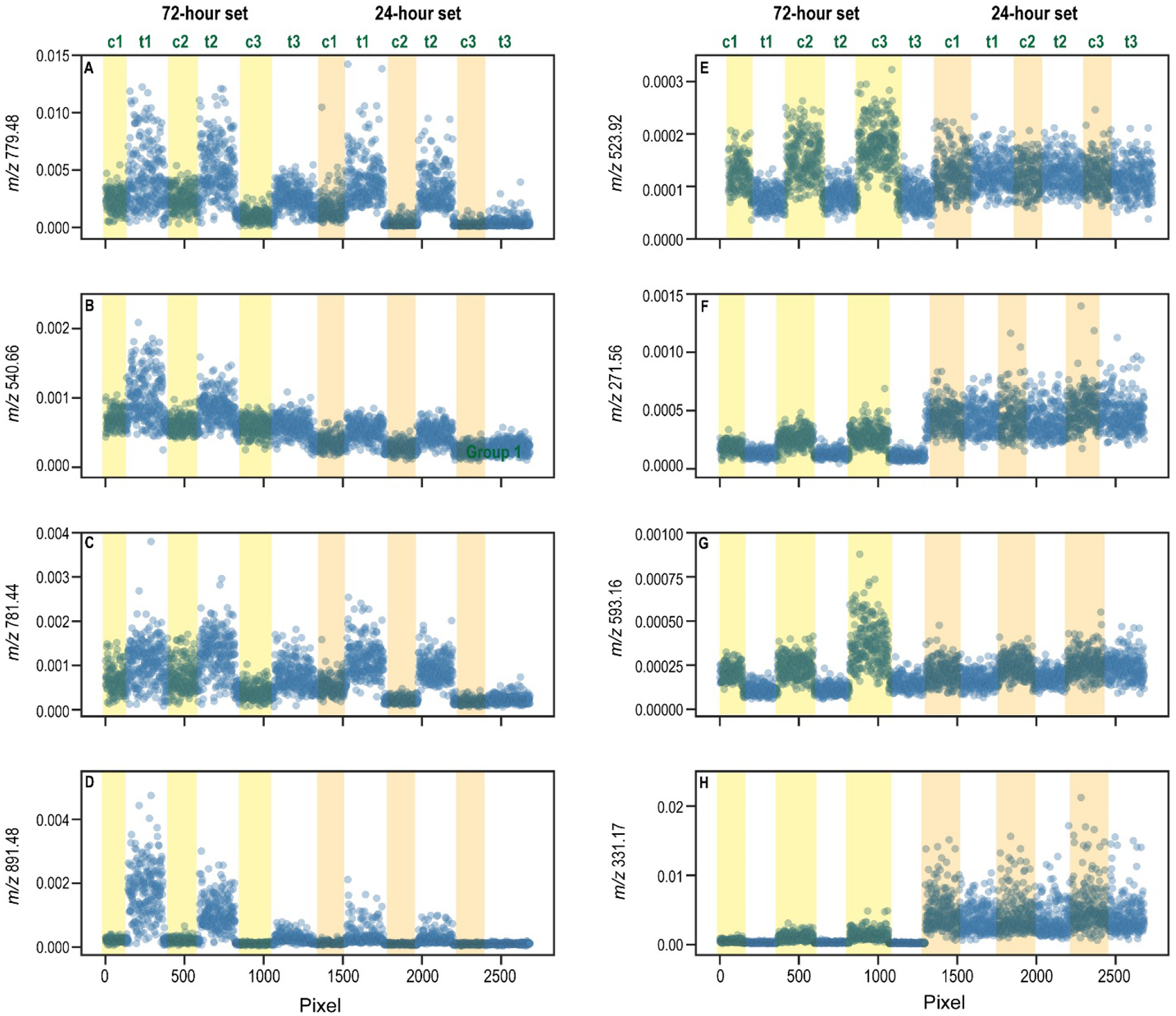
Plots of the mass spectrometry data for the top four features discriminating each data set. Each pixel in each spheroid is included on the x axis; the y axis shows the intensity of the m/z of interest for the corresponding pixel. Figures A to D represent data for the top four features discriminating the 24-hour data set. Figures E to H correspond to the top four features discriminating the 72-hour data set.
We similarly investigated the metabolomic changes at the 72-hour time point, using the same strategy; we calculated the AUC for each of the 3500 features (3500 m/z’s) used to classify the spheroids, by considering the ion abundances for every pixel in every spheroid in the 72-hour data set. The data for the four top features is plotted in Figure 5E to 5H. Again, these data show the intensity of the feature for each pixel in the 72-hour data set and the 24-hour data set. Please note: while the pixels from the 24-hour data set are in the plot, these values were not used in extracting the AUC’s for Figures 5E to 5H. Rather, they are plotted to demonstrate whether the most important changes at 72 hours are also apparent at 24 hours. In these four plots, the most important changes at 72 hours are either completely indiscernible (plots 5E and 5F) or just beginning (5G and 5H) at 24 hours. In sum, at 72 hours the drug has more of an impact on the metabolic pathways and it impacts more pathways overall than at 24 hours. Twelve additional plots, including the next six most discriminating features for each data set, are provided in Supplemental Data, Figures S1 and S2. These data similarly indicate that the important changes at 72 hours are generally not observable at 24 hours.
Further exploring the identity of the treatment-related features will be useful in mapping out the early metabolomic changes and identifying potential unintended effects of the drug. As a first step, the relevant features’ m/z’s can be determined. This is most readily accomplished by leveraging the built-in commands in Cardinal, where the m/z’s for any feature in a matrix can be determined by a command like this: mz(DatasetName)[1308]. In this case, the 1308th feature in the data set “DatasetName” would be reported. This approach was used to determine the m/z’s for the features in Figure 5, and the m/z’s are labeled on the y axes. In the future, these peaks could be further explored by searching within metabolomics databases and conducting MS/MS experiments to determine the peaks’ identities. The m/z’s for the 50 most-discriminating features for each group --the 24-hour group and the 72-hour group-- are provided in the Supplemental Data section. By comparing these two lists of features, which do not have a significant degree of overlap, one can again observe that the spheroids at 72 hours are not simply undergoing a greater response to the drug; rather, their response is also changed, since the most significant features do not overlap very heavily between the two lists.
Conclusion
We provided the MS-imaging community with a simple-to-use, completely open-source workflow to facilitate supervised machine learning analyses on MS imaging data. We demonstrated the utility of the workflow by studying the metabolic changes induced by the anti-cancer drug, Cetuximab. At 72 hours post treatment, the drug’s effects were substantial, and the metabolic profiles for the spheroids that had been treated universally replicated a deviation from the untreated controls. These findings support earlier conclusions, that this drug induces substantial functional changes at 72 hours. Machine learning was also used to glean new information about the early metabolic impact of this drug on cancer cells. We showed that at 24 hours, the impact of the drug is substantially lower than at 72 hours, and that many important metabolic changes that are detectable at 72 hours are not yet different between the treated and untreated groups at 24 hours. Further exploring these key metabolites could provide new insight into the drug’s mechanism of action and/or its off-target effects.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R35GM130354 to HD and R01-GM110406-06 to ABH.
Footnotes
Supporting Information
Figures S1 and S2 contain plots of pixel-vs-intensity for the 5th to the 10th most discriminating features at 24 hours and 72 hours respectively.
Tables S1 and S2 contain lists of the 50 most discriminating m/z’s from the 24-hour and 72-hour data sets respectively.
References
- 1.Grimm CC; Short RT; Todd PJ “A Wide-angle Secondary Ion Probe for Organic Ion Imaging.” J Am Soc Mass Spectrom. 1991, 2, 362–371. [DOI] [PubMed] [Google Scholar]
- 2.Caprioli RM; Farmer TB; Gile J “Molecular Imaging of Biological Samples: Localization of Peptides and Proteins Using MALDI-TOF MS.” Anal. Chem 1997, 69(23), 4751–4760. [DOI] [PubMed] [Google Scholar]
- 3.Sans M; Gharpure K; Tibshirani R; Zhang J; Liang L; Liu J; Young JH; Dood RL; Sood AK; Eberlin LS “Metabolic Markers and Statistical Prediction of Serous Ovarian Cancer Aggressiveness by Ambient Ionization Mass Spectrometry Imaging.” Cancer Res. 2017, 77(11), 2903–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouslimani A; Porto C; Rath CM; Wang M; Guo Y; Gonzalez A; Berg-Lyon D; Ackermann G; Moeller Christensen GJ; Nakatsuji T; Zhang L; Borkowski AW; Meehan MJ; Dorrestein K; Gallo RL; Bandeira N; Knight R; Alexandrov T; Dorrestein PC; “Molecular Cartography of the Human Skin Surface in 3D.” Proc Natl Acad Sci USA. 2015, 112(17), E2120–E2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X; Flinders C; Mumenthaler SM; Hummon AB “MALDI Mass Spectrometry Imaging for Evaluation of Therapeutics in Colorectal Tumor Organoids.” J Am Soc Mass Spectrom. 2018, 29(3), 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fonville JM; Carter C; Cloarec O; Nicholson JK; Lindon JC; Bunch J; Holmes E “Robust Data Processing and Normalization Strategy for MALDI Mass Spectrometric Imaging.” Anal. Chem 2012, 84, 1310–1319. [DOI] [PubMed] [Google Scholar]
- 7.Bemis KD; Harry A; Eberlin LS; Ferreira C; van de Ven SM; Mallick P; Stolowitz M; Vitek O “Cardinal: an R Package for Statistical Analysis of Mass Spectrometry-Based Imaging Experiments.” Bioinformatics, 2015, 31(14), 2418–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian X; Zhang G; Zou Z; Yang Z “Anticancer Drug Affects Metabolomic Profiles in Multicellular Spheroids: Studies Using Mass Spectrometry Imaging Combined with Machine Learning.” Anal. Chem 2019, 91, 5802–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verbeeck N; Caprioli RM; Van de Plas R “Unsupervised Machine Learning for Exploratory Data Analysis in Imaging Mass Spectrometry.” Mass Spec. Rev 2019, 00, 1–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leuschner J; Schmidt M; Fernsel P; Lachmund D; Boskamp T; Maass P “Supervised Non-Negative Matrix Factorization Methods for MALDI Imaging Applications.” Bioinformatics, 2019, 35(11), 1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heylman C; Datta R; Sobrino A; George S; Gratton E “Supervised Machine Learning for Classification of the Electrophysiological Effects of Chronotropic Drugs on Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes.” PLoS One, 2015, 10(12), e0144572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanselmann M; Kothe U; Kirchner M; Renard BY; Amstalden ER; Glunde K; Heeren RMA; Hamprecht FA “Toward Digital Staining using Imaging Mass Spectrometry and Random Forests.” Journal of Proteome Research, 2009, 8, 3558–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma L; Li M; Ma X; Cheng L; Du P; Liu Y “A Review of Supervised Object-Based Land-Cover Image Classification.” ISPRS Journal of Photogrammetry and Remote Sensing, 2017, 130, 277–293. [Google Scholar]
- 14.Hua D; Patabandige MW; Go EP; Desaire H “The Aristotle Classifier: Using the Whole Glycomic Profile to Indicate a Disease State.” Anal Chem. 2019, 91(17), 11070–11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desaire H; Hua D “Adaption of the Aristotle Classifier for Accurately Identifying Highly Similar Bacteria Analyzed by MALDI-TOF MS.” Anal. Chem 2020, 92(1), 1050–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutherland RM; Sordat B; Bamat J; Gabbert H; Bourrat B; Mueller-Klieser W “Oxygenation and Differentiation in Multicellular Spheroids of Human Colon Carcinoma.” Cancer Res. 1986, 46(10), 5320–5329. [PubMed] [Google Scholar]
- 17.Friedrich J; Seidel C; Ebner R; Kunz-Schughart LA “Spheroid-Based Drug Screen: Considerations and Practical Approach.” Nat Protoc. 2009, 4(3), 309–324. [DOI] [PubMed] [Google Scholar]
- 18.Liu X; Lukowski JK; Flinders C; Kim S; Georgiadis RA; Mumenthaler SM; Hummon AB “MALDI-MSI of Immunotherapy: Mapping the EGFR-Targeting Antibody Cetuximab in 3D Colon-Cancer Cell Cultures.” Anal. Chem 2018, 90, 14156–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robin X; Turck N; Hainard A; Tiberti N; Lisacek F; Sanchez JC; Muller M “pROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves.” BMC Bioinformatics, 2011, 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.http://bioconductor.org/packages/release/bioc/html/Cardinal.html (accessed May 27, 2020)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.