

Johne’s disease (JD) is an economically important infectious disease in livestock farming caused by Mycobacterium avium subsp. paratuberculosis. As an alternative to serological tests, which are used mainly for the screening of whole herds, we developed a novel ResoLight-based real-time PCR (RL-PCR) assay with pooled fecal samples for the detection of fecal shedders in cattle herds. The RL-PCR assay included an internal amplification control (IC) which was amplified using the same primer pair as the target molecule M. avium subsp.
KEYWORDS: Johne’s disease, screening test, pooled feces, real-time PCR, internal control
ABSTRACT
Johne’s disease (JD) is an economically important infectious disease in livestock farming caused by Mycobacterium avium subsp. paratuberculosis. As an alternative to serological tests, which are used mainly for the screening of whole herds, we developed a novel ResoLight-based real-time PCR (RL-PCR) assay with pooled fecal samples for the detection of fecal shedders in cattle herds. The RL-PCR assay included an internal amplification control (IC) which was amplified using the same primer pair as the target molecule M. avium subsp. paratuberculosis IS900 and differentiated based on melting temperatures. Individual fecal suspensions were pooled and concentrated by centrifugation to avoid a loss of sensitivity by the dilution effect. Combined with a DNA extraction kit (Johne-PureSpin; FASMAC), no inhibition of PCR amplification was observed with up to 15 fecal samples in a pool. The detection limit of RL-PCR at a pool size of 10 was 10 M. avium subsp. paratuberculosis organisms per gram of feces, which was comparable to that of individual testing. A total of 2,654 animals in 12 infected herds were screened by individual antibody-enzyme-linked immunosorbent assay (ELISA) and the RL-PCR assay using pooled feces. Fifty animals were diagnosed with JD through the screening by RL-PCR, compared with only 5 by ELISA (which were also positive in RL-PCR). In 7 JD-free herds, the results of 4 out of 327 pools (1.2%) were invalid due to the lack of IC amplification, and then animals were confirmed negative individually. Our results suggest that implementation of herd screening by pooled RL-PCR would advance the monitoring and control of JD in cattle herds.
INTRODUCTION
Johne’s disease (JD) is a chronic granulomatous enteritis in ruminants caused by Mycobacterium avium subsp. paratuberculosis. It is an economically important infectious disease in livestock farming, and control programs at the regional and or national level have been implemented in many countries (1–3). Animals infected with M. avium subsp. paratuberculosis become infectious primarily by starting the fecal excretion of the pathogen, and thereby they are a risk for transmission of infection within the herd mainly via the fecal-oral route. It has been suggested that the most effective strategy to control JD is preventing transmission by breaking infection routes based on hygienic measures supported by test-based culling of infected animals (4). However, in a herd with JD, it is assumed that the number of infected animals at the subclinical stage is several times more than that in the clinical stage (5), and the diagnostic test performance is known to be different depending on the stage of infection (6).
Isolation of M. avium subsp. paratuberculosis from shedding animals by fecal culture has been considered definitive for the diagnosis of JD. The most considerable disadvantage of this method is that it requires several months to obtain results due to the extremely slow growth of M. avium subsp. paratuberculosis. During the long incubation period of fecal culture, a steady contamination by the fecal shedders may increase the risk of transmission. Detection of antibodies against M. avium subsp. paratuberculosis by enzyme-linked immunosorbent assay (ELISA) is another frequently used diagnostic test that is generally less expensive, faster, and easier to perform than fecal culture (7). However, the development of humoral responses in M. avium subsp. paratuberculosis-infected animals is correlated with the stage of the disease and usually does not occur until the later stage (6, 8); therefore, the overall sensitivity of ELISA has been estimated to be under 30% compared with fecal culture (9). If ELISA is the only test used, animals which are ELISA negative but shedding M. avium subsp. paratuberculosis in their feces create a high risk of undetected transmission.
It has been demonstrated that a direct fecal quantitative real-time PCR (qPCR) assay enables rapid and sensitive detection of animals shedding M. avium subsp. paratuberculosis (10, 11). A longitudinal study conducted previously in sheep suggested that a qPCR-based test could detect signs of M. avium subsp. paratuberculosis infection earlier in the course of infection than fecal culture and serum ELISA (12). However, for the screening of whole herds, PCR-based tests of individual animals are labor-intensive and more costly than serology, which is commonly used for this purpose despite the lack of sensitivity. Although testing pooled fecal samples could overcome the limitation of qPCR-based diagnostic tests, general fecal pooling protocols are designed by dilution of feces or fecal suspensions, which may have lower sensitivity as M. avium subsp. paratuberculosis in an infected sample becomes diluted by uninfected samples in the same pool. In a previous study, an effective fecal pooling and DNA extraction method combined with a qPCR assay was reported. To avoid a loss of sensitivity by the dilution effect, individually prepared fecal suspensions were pooled and then concentrated by centrifugation (13). In this fecal pooling method, samples can be pooled without a loss of M. avium subsp. paratuberculosis in infected feces.
Due to the effect of residual PCR inhibitors, nucleic acid amplification and detection of microbial pathogens in fecal samples may lead to false-negative results (14, 15), and the chance of PCR inhibition increases in a pool containing more than 10 fecal samples (16). In the fecal pooling protocol described by Mita et al., PCR inhibitors may also increase as a result of the concentrated fecal materials processed for DNA extraction (13). Inhibition of qPCR for the detection of M. avium subsp. paratuberculosis in feces resulted in higher quantification cycle (Cq) values, lower DNA quantity, or no amplification (17). To confirm if the reaction was completed without inhibition, the inclusion of an internal amplification control (IC) in the assay is recommended (18, 19). In a real-time PCR assay with a DNA-binding dye, it is possible to include an IC which melts at a different temperature from the target amplicons (19). ResoLight dye is a saturating DNA-binding dye designed for high-resolution melting analysis of PCR amplicons (20). Due to the higher fluorescence signal generated by ResoLight dye, the difference between the melting temperature (Tm) peaks of the target and the IC could be detected more clearly than other commonly used dyes such as SYBR green I (20).
The objective of this study was to develop a novel real-time PCR-based screening test for JD with pooled fecal samples, while maintaining test sensitivity and managing PCR inhibition. A field evaluation of the test for the detection of fecal shedders in a cattle herd was also conducted in JD-infected and uninfected herds.
MATERIALS AND METHODS
Mycobacterial strains and purification of genomic DNA.
A total of 59 mycobacterial strains, including cattle (C) and sheep (S) strains of M. avium subsp. paratuberculosis, were used to evaluate the analytical specificity of the ResoLight-based real-time PCR (RL-PCR) assay (Table 1). Bacterial DNA was prepared using a commercial DNA extraction kit, (Johne-Spin; FASMAC, Kanagawa, Japan), which employed mechanical disruption of cells with bead beating followed by DNA purification using a spin column. The purity and the concentration of each DNA sample was determined by spectrophotometry at 260 nm, and 10 pg of DNA from each sample was applied in the RL-PCR assay.
TABLE 1.
Mycobacterial strains included in this study
| Taxon by groupa | No. of strains tested | RL-PCR result |
|---|---|---|
| MAC | ||
| M. avium subsp. paratuberculosis C-type | 2 | + |
| M. avium subsp. paratuberculosis S-type | 1 | + |
| M. avium subsp. avium | 10 | − |
| “M. avium subsp. hominissuis” | 9 | − |
| M. avium subsp. silvaticum | 1 | − |
| M. intracellulare | 18 | − |
| MTC | ||
| M. bovis | 3 | − |
| M. tuberculosis | 4 | − |
| AFB | ||
| M. scrofulaceum | 5 | − |
| M. smegmatis | 1 | − |
| M. kansasii | 1 | − |
| M. fortuitum | 1 | − |
| M. phlei | 1 | − |
| M. genavense | 1 | − |
| Mycobacterium sp. 2333 | 1 | − |
| Total | 59 |
MAC, Mycobacterium avium complex; MTC, Mycobacterium tuberculosis complex; AFB, acid-fast bacillus.
Fecal pooling and DNA extraction.
Fecal suspensions were prepared individually as described previously (21). According to the fecal pooling protocol reported previously (13), 1 ml of each suspension was collected and pooled in a fresh 15-ml tube; a 50-ml tube was used instead if the pool size was more than 10. After centrifugation at 900 × g for 30 min, all but approximately 1 ml of the supernatant was removed. The fecal pellet was resuspended in the remaining supernatant and transferred to a tube containing zirconia beads provided with a Johne-PureSpin kit (FASMAC). For individual testing, 1 ml of fecal suspension prepared individually was transferred directly to the bead tube.
DNA extraction using a Johne-PureSpin kit was conducted according to the manufacturer’s instructions. Briefly, the bead tube containing individual or pooled fecal suspension was centrifuged at 20,000 × g for 5 min. After the supernatant was carefully removed, 400 μl of lysis buffer 1-A was added and agitated at 4,600 rpm for 3 min using a homogenizer (Micro Smash MS-100; Tomy Seiko Co., Ltd., Tokyo, Japan). Following centrifugation at 20,000 × g for 5 min, the supernatant was transferred to a 1.5-ml tube containing 200 μl of lysis buffer 1-B and 75 μl of extraction buffer 2. After centrifugation at 20,000 × g for 10 min, 500 μl of the supernatant was mixed with 400 μl of binding buffer 3. The mixture was transferred to a spin column and centrifuged at 13,000 × g for 1 min. The column was washed once with 600 μl of washing buffer 4 by centrifugation at 13,000 × g for 1 min and then placed onto a new 1.5-ml tube. DNA samples were eluted with 50 μl of elution buffer 5 by centrifugation at 13,000 × g for 1 min and then stored at –20°C until PCR analyses were performed.
Determination of the acceptable number of fecal samples in a pool.
Known M. avium subsp. paratuberculosis-free bovine feces were collected from 2 Japanese black adult animals, which were sourced from a farm with no JD history and confirmed uninfected by fecal culture, fecal qPCR, and ELISA. Fecal suspensions required for the following experiments were prepared as described above, and the mixture of fecal suspensions was used as a pool of negative fecal suspensions. One millilter, 5 ml, 10 ml, 12 ml, 15 ml, and 20 ml of the negative fecal suspension were processed for DNA extraction in triplicates, which simulated pool sizes of individual, 5, 10, 12, 15, and 20, respectively. Using the same sets of fecal suspensions, DNA samples were also prepared by the Johne-Spin (FASMAC) method as described previously (13, 21).
Preparation of M. avium subsp. paratuberculosis-spiked feces.
M. avium subsp. paratuberculosis strain K-10 (ATCC BAA-968) grown on a Middlebrook 7H10 agar-based slant (21) was harvested into saline plus 0.1% (vol/vol) Tween 80 and 0.1% (vol/vol) bovine serum albumin (BSA-Tween saline) followed by filtration through a 5-μm filter. The total number of filtered cells was counted by light microscopy, and a stock bacterial suspension was prepared with BSA-Tween saline. The stock suspension was diluted 10-fold in the negative fecal suspension to yield a final concentration ranging from 105 to 100 M. avium subsp. paratuberculosis cells/g of feces, which is equivalent to 5 × 103 to 5 × 10−2 cells/ml of suspension. One milliliter of each dilution was mixed with 9 ml of negative fecal suspension to provide a pool size of 10. For individual testing, 1 ml of each spiked suspension was directly processed for DNA extraction. These sets of spiked individual and pooled fecal suspensions were prepared to enable testing in triplicate.
RL-PCR assay.
Primers targeting M. avium subsp. paratuberculosis IS900 were designed by referring to the sequence of Mycobacterium sp. strain 2333 which harbors one copy of an IS900-like sequence in the genome (22) (Table 2).
TABLE 2.
Primers for the detection of M. avium subsp. paratuberculosis IS900 used in this study
| Primer name | Sequence (5′–3′) | Assay | Reference |
|---|---|---|---|
| MP10-1 | ATGCGCCACGACTTGCAGCCT | SYBR green-based qPCR | 10 |
| MP11-1 | GGCACGGCTCTTGTTGTAGTCG | ||
| IS900-3 | GCCGGGCAGCGGCTGCTTTATA | RL-PCR | This study |
| IS900-32 | GCGCGCAGAGGCTGCAAGTCGT |
The DNA used as an internal amplification control (IC) in the RL-PCR assay was synthesized at Nippon Gene Co., Ltd. (Tokyo, Japan). The synthetic DNA contained IS900-3 and IS900-32 priming sites with foreign DNA designed based on the sequence of the lambda phage (GenBank accession no. U39286.1; nucleotide range, 29364 to 29541) (23); it is amplified using the same primer pair as that of the target molecule M. avium subsp. paratuberculosis IS900. The synthetic IC was cloned into pUC19, and the plasmid DNA was diluted with Tris-EDTA (TE) buffer containing 5 ng/μl ColE1 DNA (Nippon Gene) as carrier DNA.
The reaction mixture containing 5 μl of template DNA, 25 μl of 2× GeneAce RL qPCR mix (Nippon Gene), 1 μl of 25 pmol of forward (IS900-3) and reverse (IS900-32) primers, 1 μl of 0.4 fg/μl IC, and 0.5 μl of 1 U/μl uracil-DNA glycosylase (UDG; Nippon Gene) was made up to 50 μl with nuclease-free water. Based on the result of a titration study (data not shown), the lowest reproducible amount of IC was added in the PCR master mix. PCR runs were performed on a LightCycler480 system II (Nippon Genetics, Tokyo, Japan), and the reaction program was designed as follows: initial incubation for UDG at 50°C for 2 min, followed by an activation step of 10 min at 95°C, and then 45 cycles of PCR amplification at 95°C for 30 sec and 68°C for 60 sec. After PCR amplification, the dissociation curve data were collected for analyzing melting temperature (Tm) peaks. The Tm peaks for the target (91.5 ± 1.0°C) indicated a presence of M. avium subsp. paratuberculosis IS900, while the Tm peak for the IC (85.5 ± 1.0°C) indicated a reaction without inhibition (Fig. 1). If neither Tm peak was detected, the result was interpreted as invalid due to PCR inhibition. Positive/negative criteria for each sample in the RL-PCR assay are shown in Fig. 1.
FIG 1.

Result interpretation for the RL-PCR assay. +ve, positive; −ve, negative.
Conventional SYBR green-based qPCR assay.
A SYBR green-based qPCR assay to detect and quantify M. avium subsp. paratuberculosis IS900 in feces was performed as described previously (10) with some modifications (21). All DNA templates were tested in duplicate, and the sample was defined as positive if either well was positive. For quantification, a standard curve was created by using genomic DNA extracted from M. avium subsp. paratuberculosis strain 42-13-1 (field isolate IS900 RFLP C1 type). The diagnostic cutoff point was set at 0.001 pg/well of M. avium subsp. paratuberculosis DNA with reference to the results of fecal culture (11). If 0.001 pg or more DNA was detected in at least one replicate, the animal was diagnosed as JD positive.
Serum antibody ELISA.
Serum samples collected during the field study were tested by a commercial antibody detection ELISA kit (Johne screening-Pourquier; Kyoto Biken Laboratories, Inc., Kyoto, Japan) according to the manufacturer’s instructions. Any sample with an S/P value equal to or greater than 60% was defined as positive.
Fecal samples and cattle herds.
Archived fecal samples used in this study were collected from naturally and experimentally infected cattle, including both dairy (Holstein) and beef (Japanese black) cattle. A total of 64 feces samples that were positive by the conventional SYBR green-based qPCR assay were selected to evaluate the effect of pooling on the sensitivity of the RL-PCR assay. Fecal suspensions were prepared individually, and 1 ml of each suspension was mixed with 9 ml of known negative fecal suspension prepared above to provide a pool size of 10.
Twelve JD-infected dairy and beef herds (herd size, 90 to 500) were selected (Table 3). Whole herd screening for JD was conducted using the RL-PCR assay and individual ELISA as summarized in Fig. 2. For the RL-PCR assay, up to 10 individual fecal samples were pooled at random and tested. If the result of pooled RL-PCR was positive or invalid, fecal samples in the pool were tested individually by RL-PCR to identify the positive animal(s). Serum samples were also collected and tested individually by ELISA. Animals that had positive results in either screening test were confirmed by the conventional SYBR green-based qPCR assay individually. Animals were diagnosed with JD if M. avium subsp. paratuberculosis DNA quantity more than the cutoff value of 0.001 pg/well was detected in the fecal sample.
TABLE 3.
JD-infected herds screened by RL-PCR and ELISA in this study
| Herd ID | Herd type | No. of animals screened | Screened by RL-PCR |
Screened by ELISA |
|||||
|---|---|---|---|---|---|---|---|---|---|
| % of positive pools | No. of positive animals | No. of SYBRa-positive animals (no. diagnosed) | Prevalence (%)b | No. of positive animals | No. of SYBRa-positive animals (no. diagnosed) | Prevalence (%)b | |||
| P-1 | Dairy | 90 | 11.1 | 1 | 1 (1) | 1.11 | 6 | 0 | 0 |
| P-2 | Dairy | 246 | 4.0 | 1 | 1 (1) | 0.41 | 0 | 0 | 0 |
| P-3 | Dairy | 386 | 100 | 265 | 243 (18) | 4.66 | 4 | 3 (1) | 0.26 |
| P-4 | Dairy | 133 | 35.7 | 10 | 7 (0) | 0 | 2 | 0 | 0 |
| P-5 | Dairy | 88 | 22.2 | 2 | 1 (0) | 0 | 0 | 0 | 0 |
| P-6 | Dairy | 112 | 75.0 | 21 | 17 (5) | 4.46 | 3 | 2 (1) | 0.89 |
| P-7 | Dairy | 498 | 10.0 | 6 | 6 (4) | 0.80 | 0 | 0 | 0 |
| P-8 | Dairy | 335 | 60.0 | 35 | 22 (9) | 2.69 | 1 | 1 (1) | 0.30 |
| P-9 | Dairy | 255 | 53.8 | 23 | 17 (10) | 3.92 | 1 | 1 (1) | 0.39 |
| P-10 | Dairy | 150 | 26.7 | 20 | 17 (0) | 0 | 0 | 0 | 0 |
| P-11 | Beef | 149 | 26.7 | 6 | 6 (2) | 1.34 | 2 | 1 (1) | 0.67 |
| P-12 | Beef | 212 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Total | 2,654 | 390 | 338 (50) | 20 | 8 (5) | ||||
Conventional SYBR green-based qPCR assay.
Herd prevalence of JD based on the number of diagnosed animals detected through the screening test.
FIG 2.

Summary of whole-herd screening by RL-PCR and ELISA in JD-infected herds.
Seven JD-free herds (herd size, 120 to 800) were also selected and screened by pooled RL-PCR (Table 4). Up to 10 individual fecal samples were pooled, and if the result of pooled RL-PCR was invalid, fecal samples in the pool were tested individually.
TABLE 4.
JD-uninfected herds included in this study
| Herd ID | Herd type | No. of animals | No. of pools | No. of positive pools | No. of invalid pools (%) | No. of positive animals |
|---|---|---|---|---|---|---|
| N-1 | Beef | 206 | 26 | 0 | 0 | 0 |
| N-2 | Beef | 811 | 84 | 0 | 0 | 0 |
| N-3 | Beef | 408 | 44 | 0 | 3 (6.8) | 0 |
| N-4 | Beef | 391 | 41 | 0 | 0 | 0 |
| N-5 | Beef | 124 | 13 | 0 | 0 | 0 |
| N-6 | Beef | 444 | 49 | 0 | 0 | 0 |
| N-7 | Dairy | 691 | 70 | 0 | 1 (1.4) | 0 |
| Total | 3,075 | 327 | 0 | 4 (1.2) | 0 |
Statistical analysis.
The relationship between the pool size and the mean Cq value was assessed by correlation analysis in two commercial DNA extraction kits, namely, Johne-PureSpin and Johne-Spin. The Cq values for the unamplified samples were defined to be >45 cycles. The Cq values in each pool size were compared with those individual testing using one-way analysis of variance (ANOVA) followed by pairwise comparison using the Bonferroni method. As a predictor of within-herd prevalence in JD-infected herds, whole herd screening by pooled RL-PCR and ELISA were compared using linear regression. The association between the percentage of positive pools in each herd and the reduction in the number of testing per herd was also analyzed by linear regression in 12 JD-infected herds. All statistical analyses were performed by R version 3.6.3 (24).
RESULTS
Analytical specificity of the RL-PCR assay.
The specificity of the RL-PCR assay was examined for 59 mycobacterial strains, including 3 M. avium subsp. paratuberculosis strains. The Tm peak for the target IS900 was detected in both C and S strains of M. avium subsp. paratuberculosis, while only the Tm peak for the IC was detected in 56 non-M. avium subsp. paratuberculosis mycobacterial strains, including the IS900-like sequence-containing strain 2333 (Table 1).
Comparison of DNA extraction kits for PCR inhibition related to the pool size.
Two DNA extraction kits, namely, Johne-PureSpin and Johne-Spin, were compared for PCR inhibition related to the pool size. The acceptable number of fecal samples in a pool was determined using fecal suspension prepared from negative bovine feces. The mean Cq values for DNA samples extracted by Johne-Spin increased in relation to the pool size (r = 0.8142), and the IC was not amplified in any of the replicates in a pool size of 20 (Fig. 3). In contrast, by using Johne-PureSpin, significant inhibition of PCR amplification was not observed with up to 15 fecal samples in a pool (r = 0.6496) (Fig. 3). With a pool size of 20, slight inhibition was indicated by higher Cq values and a reduction in the height of the Tm peaks for the IC.
FIG 3.
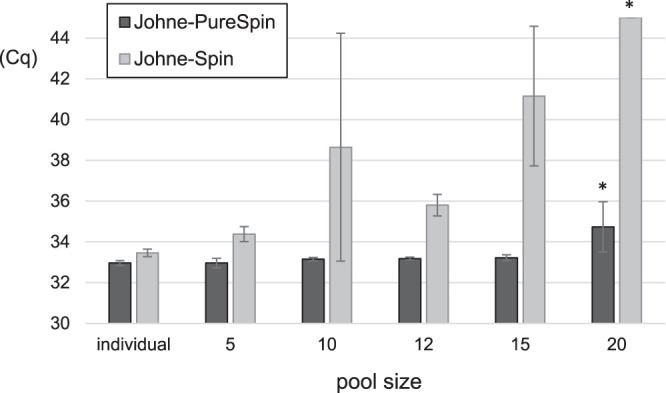
Comparison of DNA extraction kits for the PCR inhibition related to the pool size. The mean Cq values in each pool size were compared to individual testing. The Cq values for the unamplified samples were defined to be >45 cycles. *, difference in comparison to individual testing (P < 0.05) (Bonferroni method).
Analytical sensitivity of pooled and individual RL-PCR.
The analytical sensitivity of the RL-PCR assay was evaluated using negative feces spiked with dilutions of M. avium subsp. paratuberculosis cells. The detection limit of the RL-PCR at pool size of 10 was 10 M. avium subsp. paratuberculosis organisms in 1 gram of feces, which was comparable to the limit of individual testing (Table 5).
TABLE 5.
Analytical sensitivity of pooled and individual RL-PCR in M. avium subsp. paratuberculosis-spiked feces
| No. of cells/g of feces | No. of cells per extraction | No. positive/no. tested |
|
|---|---|---|---|
| Individual | Pool of 10 | ||
| 104 | 500 | 3/3 | 3/3 |
| 103 | 50 | 3/3 | 3/3 |
| 102 | 5 | 3/3 | 3/3 |
| 101 | 0.5 | 1/3 | 1/3 |
| 100 | 0.05 | 0/3 | 0/3 |
| 0 | 0 | 0/3 | 0/3 |
Effect of pooling on the sensitivity of RL-PCR using feces that tested positive in the conventional SYBR green-based qPCR assay.
A total of 64 qPCR-positive feces was tested by pooled RL-PCR. At a pool size of 10, simulating 1 infected plus 9 uninfected, the RL-PCR assay detected 95.1% of the positive feces containing over the diagnostic cutoff point of 0.001 pg/well of M. avium subsp. paratuberculosis DNA (Table 6). The positive rate was 65.2% in the feces containing less than 0.001 pg/well (Table 6). The overall sensitivity was 84.4% (Table 6).
TABLE 6.
Effect of pooling on the sensitivity of RL-PCR in qPCR-positive feces
| Pooled RL-PCRa | Values by DNA quantity (pg/well)b
of: |
Total | |
|---|---|---|---|
| <0.001 | ≥0.001 | ||
| No. tested | 23 | 41 | 64 |
| No. positive (%) | 15 (65.2) | 39 (95.1) | 54 (84.4) |
Tested at a pool size of 10 (1 positive plus 9 negative fecal samples).
DNA quantity detected by the conventional SYBR green-based qPCR assay.
Herd screening by RL-PCR.
A total of 2,654 animals in 12 JD-infected herds were screened by the RL-PCR assay and individual antibody-ELISA (Fig. 2). Through the screening test using pooled RL-PCR followed by identification of positive animals by individual RL-PCR, 390 animals from 11 herds were detected, while 20 from 8 herds were positive by ELISA (Table 3). Of the 390 animals detected by RL-PCR, 338 (86.7%) were positive by the conventional SYBR green-based qPCR assay and 50 of them were diagnosed with JD due to the detection of M. avium subsp. paratuberculosis DNA more than the cutoff value of 0.001 pg/well. On the other hand, 8 of 20 (40%) animals detected by ELISA were positive in the qPCR assay. Only 5 animals were diagnosed with JD, which were also detected through the screening by RL-PCR (Table 3).
The prevalence of JD in each herd was calculated based on the number of animals diagnosed with JD through the respective screening tests, RL-PCR and ELISA (Table 3). The within-herd prevalence estimated through screening by ELISA was lower than that by pooled RL-PCR (Table 3). In screening by the pooled RL-PCR assay, the percentage of positive pools in each herd was correlated with the prevalence of diagnosed animals (P < 0.01) (Fig. 4A), while the antibody prevalence did not reflect the prevalence of fecal shedders (P = 0.703) (Fig. 4B).
FIG 4.

The relationship between the within-herd prevalence of diagnosed animals and the screening test used in the herd. The herd prevalence calculated based on the number of diagnosed animals detected through each screening test is shown on the y axes. (A) Screened by pooled RL-PCR at a pool size of 10; (B) screened by ELISA.
The positive rate in pooled RL-PCR in each herd was 0% to 100% (Table 3). There was a significant relationship between the percentage of positive pools in the herd and the reduction in the number of tests per herd compared with those of individual testing (Fig. 5).
FIG 5.

The association between the percentage of positive pools and the total number of tests in each of the 12 JD-infected herds.
In the 7 JD-free herds, fecal samples from 3,075 animals were tested by pooled RL-PCR (327 pools), and no positive results were obtained (Table 4). The results of 4 out of 327 pools (1.2%) from 2 herds were invalid due to the lack of IC amplification. All the animals in these 4 pools were confirmed negative individually.
DISCUSSION
Although a direct fecal qPCR assay enables rapid and sensitive detection of animals shedding M. avium subsp. paratuberculosis (10, 11), PCR-based tests of individual animals are labor-intensive for the screening of whole herds. This study presents a novel real-time PCR-based screening test for JD with pooled fecal samples and the evaluation of the test for the detection of fecal shedders in a cattle herd.
The RL-PCR assay developed in this study included an IC to confirm if the PCR was completed without inhibition. As the IC used in this assay was amplified with the same primer set as the for the target M. avium subsp. paratuberculosis IS900, some competition between the target and the IC was expected (25, 26). When a competitive IC is used, it is recommended that the length of the IC should be longer than the target sequence to reduce the amplification efficiency compared with the smaller target fragment (19). The size of the IC was 222 bp, while that of the target was 146 bp in the RL-PCR assay. The concentration of the IC is also critical for the detection limit of the PCR assay (19, 26). The smallest reproducible amount of IC DNA was added in the PCR master mix. The analytical sensitivity of individual RL-PCR was 10 M. avium subsp. paratuberculosis organisms in 1 g of feces (equivalent to 0.5 M. avium subsp. paratuberculosis per extraction; note that IS900 is a multicopy element) (Table 5), which was comparable to the limit of the conventional SYBR green-based qPCR assay described previously (10).
During the RL-PCR, amplification of the target and the IC was monitored on a real-time basis using a fluorescent DNA binding dye, ResoLight. The amplicons were further analyzed by collecting the dissociation curve data and then identified as either the target M. avium subsp. paratuberculosis IS900 or the IC based on the melting temperature. ResoLight dye is a DNA intercalating dye, which can be used at saturating concentrations leading to enhanced sharpness of fluorescent signals compared with other commonly used DNA-binding dyes, e.g., SYBR green I. In the RL-PCR assay, the approximate Tm values for the target and IC were 91.5°C and 85.5°C, respectively. By using ResoLight dye, the melting profiles of amplicons were analyzed at high resolution, and 2 clearly separated peaks corresponding to the target and the IC were observed (Fig. 1).
The RL-PCR assay indicated high analytical specificity for the detection of M. avium subsp. paratuberculosis (Table 1). The Tm peak for the target IS900 was detected in M. avium subsp. paratuberculosis strains but not in the other mycobacterial strains, including Mycobacterium sp. 2333, in which there is one copy of an IS900-like sequence with 94% homology to M. avium subsp. paratuberculosis IS900 (22). During the field study in JD-free herds, the RL-PCR assay also demonstrated high diagnostic specificity: none of the pools were positive and all the animals were ultimately confirmed as negative (Table 4). As well as designing specific primers for M. avium subsp. paratuberculosis IS900, the inclusion of the competitive IC possibly assisted to avoid the formation of primer dimers or other nonspecific amplifications and consequently improved the specificity.
It is well known that a variety of components in feces act as so-called PCR inhibitors, adversely affecting the amplification and detection of nucleic acid in PCR assays (14, 15). Through the fecal pooling process applied in this study, it was expected that PCR inhibitors were also concentrated (13). To remove PCR inhibitors from pooled fecal samples, the use of Johne-Spin as a DNA extraction kit has been suggested (13). However, among the DNA samples prepared with Johne-Spin and tested in the RL-PCR assay, amplification of the IC was inhibited by increases in the pool size (Fig. 3). In contrast, by using Johne-PureSpin, no significant inhibition of PCR amplification was observed with up to 15 fecal samples in a pool (Fig. 3). Owing to the high concentration of PCR inhibitors in pooled fecal samples, the selection of a DNA extraction kit is critical to achieve sufficient diagnostic sensitivity. PCR inhibitors were efficiently removed from DNA templates obtained using Johne-PureSpin.
In view of PCR inhibitors in feces, PCR-based diagnostic tests with pooled samples are generally designed for diluted feces or fecal suspensions. It was also suggested that the dilution of extracted DNA could be effective for improving test sensitivity (17). In a previous study, a higher sensitivity at a pool size of 10 was obtained by 5-fold dilution of the DNA extracts (16). However, the dilution of feces/fecal suspensions or DNA templates may lead to false-negative results if the sample contains only a small amount of M. avium subsp. paratuberculosis/M. avium subsp. paratuberculosis DNA. The fecal pooling protocol followed by the DNA extraction and PCR assay used in this study has theoretically equal sensitivity compared with that of individual testing (13). The analytical sensitivity of the RL-PCR assay examined in M. avium subsp. paratuberculosis-spiked feces suggested that the detection limit at a pool size of 10 was comparable to that of individual testing (Table 5). It was demonstrated that the RL-PCR assay combined with DNA extraction by Johne-PureSpin sufficiently detected M. avium subsp. paratuberculosis DNA in pooled fecal samples without a loss of diagnostic sensitivity at least up to a pool size of 10.
The effect of pooling on the sensitivity of the test was further evaluated in bovine feces containing different levels of M. avium subsp. paratuberculosis DNA. Although the overall sensitivity was 84.4% at a pool size of 10, the test sensitivity in pooled feces depended on the quantity of M. avium subsp. paratuberculosis DNA in the positive feces (Table 6). The diagnostic cutoff point in the conventional SYBR green-based qPCR assay was set at 0.001 pg/well based on the results of fecal culture. In this study, 95.1% of the pools, including diagnostically positive feces were positive at a pool size of 10 (Table 6), suggesting that most of the animals diagnosed with JD by the individual qPCR test can be detected through screening by the pooled RL-PCR assay.
During the field validation conducted in JD-infected herds, the RL-PCR assay with up to 10 fecal samples in a pool demonstrated higher diagnostic sensitivity than that of ELISA: 50 animals were diagnosed with JD through the screening by RL-PCR, while only 5 were detected by ELISA (all 5 were also positive in the RL-PCR) (Table 3). The sensitivity of ELISAs is dependent on the stage of the disease and the age of the animals (6, 8). Fecal shedding is generally observed earlier in the course of infection than the antibody response. Moreover, although young animals have not been considered a major source of within-herd transmission of M. avium subsp. paratuberculosis, tentative shedding in calves at an early stage of infection and therefore calf-to-calf/calf-to-environment transmission have been reported (27–30). Our results suggest that whole-herd screening based on ELISAs is likely to fail to detect antibody-negative infectious animals which have a risk of transmission without being detected.
Although PCR inhibition was not observed in the pools from JD-infected herds, the results of 4 pools (1.2%) from 2 JD-free herds were invalid due to the lack of IC amplification (Table 4). All the animals in the undetermined pools were confirmed negative by detection of the IC in the DNA template extracted individually. Particularly for diagnostic PCR using clinical samples, monitoring PCR inhibition by a suitable IC has been recommended (18). Inclusion of IC in the RL-PCR assay ensured that the negative results were truly negative. For identification of shedding animals in positive pools, individual RL-PCR was conducted prior to the diagnostic confirmation using the conventional SYBR green-based qPCR assay. Although the SYBR green-based qPCR assay has been accepted as a rapid and sensitive diagnostic test for JD and circumvents time-consuming fecal culture (10, 11), it possibly overlooks false-negative results. The follow-up individual test combined with pooled RL-PCR could be improved in further study, namely, a qPCR assay for detection and quantification of M. avium subsp. paratuberculosis DNA and monitoring of PCR inhibition simultaneously.
Testing pooled samples was expected to save time, labor, and cost compared with those of individual testing (13). However, the number of tests per herd depended on the number of positive pools which needed to be followed up by individual testing for identification of the positive animal(s). In our field study in JD-infected herds, the reduction in the number of tests per herd with pooled RL-PCR was significantly correlated with the rate of positive pools (Fig. 5). As a whole-herd screening test for detection of shedding animals, the RL-PCR assay with pooled fecal samples performed more effectively in herds with low prevalence than in highly contaminated herds. The follow-up individual test for animals in positive pools should be considered carefully particularly in highly contaminated herds because not only infected animals but also animals passively shedding M. avium subsp. paratuberculosis or M. avium subsp. paratuberculosis DNA in the feces are detected. Passive fecal shedding by ingestion of M. avium subsp. paratuberculosis in the contaminated environment is a known phenomenon in JD, and PCR-based diagnostic tests cannot distinguish passive shedders from infected active shedders (31).
Besides the detection of fecal shedders, it was shown that the results of pooled RL-PCR reflected the prevalence of shedding animals in the herd. The percentage of positive pools in the herd was correlated with the prevalence of diagnosed animals (Fig. 4A), suggesting that whole-herd screening by pooled RL-PCR could be useful to estimate the within-herd prevalence. In contrast, the within-herd prevalence calculated based on the seroprevalence was likely to be imprecise and to underestimate true prevalence. Although fecal culture has been used in order to obtain more accurate estimation of within-herd prevalence, bacterial culture of pooled feces or environmental samples have been reported for this purpose as being more cost-effective testing strategies (32–34). Compared with culture methods which usually require several months to obtain results, whole-herd screening by pooled RL-PCR enables rapid estimation of the within-herd prevalence. Longitudinal herd monitoring using pooled RL-PCR may provide useful information to assess the progress of disease control in the herd.
In conclusion, the RL-PCR assay combined with effective DNA extraction from pooled fecal samples demonstrated comparable sensitivity to individual testing up to a pool size of 10. PCR inhibition was clearly monitored based on the IC amplification detected using ResoLight dye. For the detection of shedding animals, whole-herd screening by pooled RL-PCR was significantly more sensitive than ELISA screening. The RL-PCR assay with pooled fecal samples performed more effectively in herds with lower prevalence when the follow-up individual test was conducted for all the animals in positive pools. The rate of positive pools in the herd reflected the within-herd prevalence of fecal shedders, suggesting that implementation of herd screening by pooled RL-PCR would be an advance for monitoring and control of JD in cattle herds.
ACKNOWLEDGMENTS
We thank Bo Xiao, Shiho Nishino, and Toshiko Oki for laboratory assistance and technical support. Sota Kobayashi (Division of Bacterial and Parasitic Disease, National Institute of Animal Health, NARO, Japan) is also appreciated for his statistical advice. Richard Whittington (Faculty of Veterinary Science, The University of Sydney, Australia) is gratefully acknowledged for helpful comments on a draft of the manuscript.
This research was supported by grants from the Project of the NARO Bio-oriented Technology Research Advancement Institution (the special scheme project on vitalizing management entities of agriculture, forestry, and fisheries).
REFERENCES
- 1.Geraghty T, Graham DA, Mullowney P, More SJ. 2014. A review of bovine Johne’s disease control activities in 6 endemically infected countries. Prev Vet Med 116:1–11. doi: 10.1016/j.prevetmed.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi S, Tsutsui T, Yamamoto T, Nishiguchi A. 2007. Epidemiologic indicators associated with within-farm spread of Johne's disease in dairy farms in Japan. J Vet Med Sci 69:1255–1258. doi: 10.1292/jvms.69.1255. [DOI] [PubMed] [Google Scholar]
- 3.Whittington R, Donat K, Weber MF, Kelton D, Nielsen SS, Eisenberg S, Arrigoni N, Juste R, Sáez JL, Dhand N, Santi A, Michel A, Barkema H, Kralik P, Kostoulas P, Citer L, Griffin F, Barwell R, Moreira MAS, Slana I, Koehler H, Singh SV, Yoo HS, Chávez-Gris G, Goodridge A, Ocepek M, Garrido J, Stevenson K, Collins M, Alonso B, Cirone K, Paolicchi F, Gavey L, Rahman MT, de Marchin E, Van Praet W, Bauman C, Fecteau G, McKenna S, Salgado M, Fernández-Silva J, Dziedzinska R, Echeverría G, Seppänen J, Thibault V, Fridriksdottir V, Derakhshandeh A, Haghkhah M, Ruocco L, Kawaji S, et al. . 2019. Control of paratuberculosis: who, why and how. A review of 48 countries. BMC Vet Res 15:198. doi: 10.1186/s12917-019-1943-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kudahl AB, Østergaard S, Sørensen JT, Nielsen SS. 2007. A stochastic model simulating paratuberculosis in a dairy herd. Prev Vet Med 78:97–117. doi: 10.1016/j.prevetmed.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 5.Magombedze G, Magombedze G, Ngonghala CN, Lanzas C. 2013. Evaluation of the “iceberg phenomenon” in Johne's disease through mathematical modelling. PLoS One 8:e76636. doi: 10.1371/journal.pone.0076636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen SS, Toft N. 2008. Ante mortem diagnosis of paratuberculosis: a review of accuracies of ELISA, interferon-g assay and faecal culture techniques. Vet Microbiol 129:217–235. doi: 10.1016/j.vetmic.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Kalis CHJ, Barkema HW, Hesselink JW, van Maanen C, Collins MT. 2002. Evaluation of two absorbed enzyme-linked immunosorbent assays and a complement fixation test as replacements for fecal culture in the detection of cows shedding Mycobacterium avium subspecies paratuberculosis. J Vet Diagn Invest 14:219–224. doi: 10.1177/104063870201400305. [DOI] [PubMed] [Google Scholar]
- 8.Harris NB, Barletta RG. 2001. Mycobacterium avium subsp paratuberculosis in veterinary medicine. Clin Microbiol Rev 14:489–512. doi: 10.1128/CMR.14.3.489-512.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins MT, Wells SJ, Petrini KR, Collins JE, Schultz RD, Whitlock RH. 2005. Evaluation of five antibody detection tests for diagnosis of bovine paratuberculosis. Clin Diagn Lab Immunol 12:685–692. doi: 10.1128/CDLI.12.6.685-692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawaji S, Taylor DL, Mori Y, Whittington RJ. 2007. Detection of Mycobacterium avium subsp. paratuberculosis in ovine faeces by direct quantitative PCR has similar or greater sensitivity compared to radiometric culture. Vet Microbiol 125:36–48. doi: 10.1016/j.vetmic.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Plain KM, Marsh IB, Waldron AM, Galea F, Whittington AM, Saunders VF, Begg DJ, de Silva K, Purdie AC, Whittington RJ. 2014. High-throughput direct fecal PCR assay for detection of Mycobacterium avium subsp. paratuberculosis in sheep and cattle. J Clin Microbiol 52:745–757. doi: 10.1128/JCM.03233-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawaji S, Begg DJ, Plain KM, Whittington RJ. 2011. A longitudinal study to evaluate the diagnostic potential of a direct faecal quantitative PCR test for Johne’s disease in sheep. Vet Microbiol 148:35–44. doi: 10.1016/j.vetmic.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 13.Mita A, Mori Y, Nakagawa T, Tasaki T, Utiyama K, Mori H. 2016. Comparison of fecal pooling methods and DNA extraction kits for the detection of Mycobacterium avium subspecies paratuberculosis. Microbiologyopen 5:134–142. doi: 10.1002/mbo3.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monteiro L, Bonnemaison D, Vekris A, Petry KG, Bonnet J, Vidal R, Cabrita J, Megraud F. 1997. Complex polysaccharides as PCR inhibitors in feces: Helicobacter pylori model. J Clin Microbiol 35:995–998. doi: 10.1128/JCM.35.4.995-998.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thornton CG, Passen S. 2004. Inhibition of PCR amplification by phytic acid, and treatment of bovine fecal specimens with phytase to reduce inhibition. J Microbiol Methods 59:43–52. doi: 10.1016/j.mimet.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Ly A, Dhand NK, Sergeant ESG, Marsh I, Plain KM. 2019. Determining an optimal pool size for testing beef herds for Johne’s disease in Australia. PLoS One 14:e0225524. doi: 10.1371/journal.pone.0225524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acharya KR, Dhand NK, Whittington RJ, Plain KM. 2017. PCR inhibition of a quantitative PCR for detection of Mycobacterium avium subspecies Paratuberculosis DNA in feces: diagnostic implications and potential solutions. Front Microbiol 8:115. doi: 10.3389/fmicb.2017.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoorfar J, Cook N, Malorny B, Wagner M, De Medici D, Abdulmawjood A, Fach P. 2003. Making internal amplification control mandatory for diagnostic PCR. J Clin Microbiol 41:5835. doi: 10.1128/jcm.41.12.5835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoorfar J, Malorny B, Abdulmawjood A, Cook N, Wagner M, Fach P. 2004. Practical considerations in design of internal amplification controls for diagnostic PCR assays. J Clin Microbiol 42:1863–1868. doi: 10.1128/jcm.42.5.1863-1868.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ. 2003. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem 49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 21.Kawaji S, Nagata R, Mori Y. 2013. Detection and confirmation of Mycobacterium avium subsp. paratuberculosis in direct quantitative PCR positive fecal samples by the manual fluorescent MGIT culture system. J Vet Med Sci 76:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Englund S, Bolske G, Johansson KE. 2002. An IS900-like sequence found in a Mycobacterium sp. other than Mycobacterium avium subsp. paratuberculosis. FEMS Microbiol Lett 209:267–271. doi: 10.1111/j.1574-6968.2002.tb11142.x. [DOI] [PubMed] [Google Scholar]
- 23.Pham DG, Madico GE, Quinn TC, Enzler MJ, Smith TF, Gaydos CA. 1998. Use of lambda phage DNA as a hybrid internal control in a PCR-enzyme immunoassay to detect Chlamydia pneumoniae. J Clin Microbiol 36:1919–1922. doi: 10.1128/JCM.36.7.1919-1922.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 25.Siebert PD, Larrick JW. 1992. Competitive PCR. Nature 359:557–558. doi: 10.1038/359557a0. [DOI] [PubMed] [Google Scholar]
- 26.Jiang W, He S, Wang J, Chen C, Sha Y, Sun H, Yuan X, Liu X, Wang C. 2019. The impact of sharing primer, the quantity of the internal control gene and the primer dimer on reaction system in duplex PCR. Clin Lab 65:2341–2348. doi: 10.7754/Clin.Lab.2019.190441. [DOI] [PubMed] [Google Scholar]
- 27.van Roermund HJ, Bakker D, Willemsen PT, de Jong MC. 2007. Horizontal transmission of Mycobacterium avium subsp. paratuberculosis in cattle in an experimental setting: calves can transmit the infection to other calves. Vet Microbiol 122:270–279. doi: 10.1016/j.vetmic.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Mortier RA, Barkema HW, Orsel K, Wolf R, De Buck J. 2014. Shedding patterns of dairy calves experimentally infected with Mycobacterium avium subspecies paratuberculosis. Vet Res 45:71. doi: 10.1186/s13567-014-0071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf R, Orsel K, De Buck J, Barkema HW. 2015. Calves shedding Mycobacterium avium subspecies paratuberculosis are common on infected dairy farms. Vet Res 46:71. doi: 10.1186/s13567-015-0192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto T, Murai K, Hayama Y, Kobayashi S, Nagata R, Kawaji S, Osaki M, Sakakibara SI, Tsutsui T. 2018. Evaluation of fecal shedding and antibody response in dairy cattle infected with paratuberculosis using national surveillance data in Japan. Prev Vet Med 149:38–46. doi: 10.1016/j.prevetmed.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Kralik P, Pribylova-Dziedzinska R, Kralova A, Kovarcik K, Slana I. 2014. Evidence of passive faecal shedding of Mycobacterium avium subsp. paratuberculosis in a Limousin cattle herd. Vet J 201:91–94. doi: 10.1016/j.tvjl.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Raizman EA, Wells SJ, Muñoz-Zanzi CA, Tavornpanich S. 2011. Estimated within-herd prevalence (WHP) of Mycobacterium avium subsp. paratuberculosis in a sample of Minnesota dairy herds using bacterial culture of pooled fecal samples. Can J Vet Res 75:112–116. [PMC free article] [PubMed] [Google Scholar]
- 33.Lavers CJ, McKenna SL, Dohoo IR, Barkema HW, Keefe GP. 2013. Evaluation of environmental fecal culture for Mycobacterium avium subspecies paratuberculosis detection in dairy herds and association with apparent within-herd prevalence. Can Vet J 54:1053–1060. [PMC free article] [PubMed] [Google Scholar]
- 34.Corbett CS, Naqvi SA, De Buck J, Kanevets U, Kastelic JP, Barkema HW. 2018. Environmental sample characteristics and herd size associated with decreased herd-level prevalence of Mycobacterium avium ssp. paratuberculosis. J Dairy Sci 101:8092–8099. doi: 10.3168/jds.2018-14661. [DOI] [PubMed] [Google Scholar]