

Neisseria meningitidis is a leading cause of bacterial meningitis and sepsis worldwide and an occasional cause of meningococcal urethritis. When isolates are unavailable for surveillance or outbreak investigations, molecular characterization of pathogens needs to be performed directly from clinical specimens, such as cerebrospinal fluid (CSF), blood, or urine. However, genome sequencing of specimens is challenging because of low bacterial and high human DNA abundances. We developed selective whole-genome amplification (SWGA), an isothermal multiple-displacement amplification-based method, to efficiently enrich, sequence, and de novo assemble N. meningitidis DNA from clinical specimens with low bacterial loads.
KEYWORDS: N. meningitidis, selective whole-genome amplification, outbreak, meningitis, genomic epidemiology, culture-free sequencing, meningitis outbreaks, meningitis surveillance, Neisseria meningitidis, targeted enrichment, targeted sequencing
ABSTRACT
Neisseria meningitidis is a leading cause of bacterial meningitis and sepsis worldwide and an occasional cause of meningococcal urethritis. When isolates are unavailable for surveillance or outbreak investigations, molecular characterization of pathogens needs to be performed directly from clinical specimens, such as cerebrospinal fluid (CSF), blood, or urine. However, genome sequencing of specimens is challenging because of low bacterial and high human DNA abundances. We developed selective whole-genome amplification (SWGA), an isothermal multiple-displacement amplification-based method, to efficiently enrich, sequence, and de novo assemble N. meningitidis DNA from clinical specimens with low bacterial loads. SWGA was validated with 12 CSF specimens from invasive meningococcal disease cases and 12 urine specimens from meningococcal urethritis cases. SWGA increased the mean proportion of N. meningitidis reads by 2 to 3 orders of magnitude, enabling identification of at least 90% of the 1,605 N. meningitidis core genome loci for 50% of the specimens. The validated method was used to investigate two meningitis outbreaks recently reported in Togo and Burkina Faso. Twenty-seven specimens with low bacterial loads were processed by SWGA before sequencing, and 12 of 27 were successfully assembled to obtain the full molecular typing and vaccine antigen profile of the N. meningitidis pathogen, thus enabling thorough characterization of outbreaks. This method is particularly important for enhancing molecular surveillance in regions with low culture rates. SWGA produces enough reads for phylogenetic and allelic analysis at a low cost. More importantly, the procedure can be extended to enrich other important human bacterial pathogens.
INTRODUCTION
Neisseria meningitidis is an obligate commensal that normally colonizes the upper respiratory tract mucosa of 10 to 30% of the healthy human population, a phenomenon known as carriage (1). This pathogen can also cause life-threatening, contagious invasive meningococcal disease, with incidence rates varying from 1 to 1,000 cases per 100,000 (2). The highest incidence is reported in the African meningitis belt, with periodic meningococcal epidemics (3). N. meningitidis also causes urethritis, and a recent increase in the number of cases reported from sexually transmitted disease clinics in the United States has been attributed to a specific clade of nongroupable N. meningitidis, the U.S. NmNG urethritis clade (4).
Rapid and accurate identification and characterization of N. meningitidis and other bacterial pathogens is critical for timely case management, disease surveillance, and control strategies. N. meningitidis strains and the genetic similarity between them are commonly characterized by multilocus sequencing typing (MLST), which determines the alleles of seven housekeeping genes (abcZ, adk, aroE, fumC, gdh, pgm, and pghC) (5). In addition, two variable regions (VR1 and VR2) of porin A (PorA) with an additional VR defined for ferric enterobactin transport (FetA) have been used for fine typing (6) to increase the typing resolution in conjunction with MLST. Three genes (fHbp, nadA, and nhbA) are of interest because they encode vaccine antigens that are included in the two serogroup B meningococcal vaccines (7). Typing of these vaccine genes has been used to estimate strain coverage by these vaccines (8). Isolation of the pathogen from clinical specimens, such as cerebrospinal fluid (CSF) from meningitis cases or urine from urethritis cases, allows extensive strain characterization but is not always possible due to antibiotic administration prior to specimen collection, suboptimal handling of clinical specimens before culture is attempted, and lack of culture capacity (9). Therefore, culture-independent methods are needed for testing of clinical specimens. Although real-time PCR (RT-PCR) can efficiently detect N. meningitidis in clinical specimens (10), it cannot comprehensively characterize its genetic features, as it provides information only about presence or absence of specific markers. Therefore, whole-genome data are required to understand N. meningitidis transmission, investigate the impact of genomic variation on the clinical outcome of disease (11), and monitor genomic epidemiology (12). Specifically, whole-genome sequencing (WGS) is increasingly being used for molecular typing of N. meningitidis, allowing allelic or single nucleotide polymorphism (SNP)-based whole-genome analyses to improve typing resolution (13, 14).
When an N. meningitidis genome from a clinical specimen is sequenced, the number of bacterial reads is often low due to the high abundance of human DNA, preventing identification of typing loci. Thus, several commercial kits have been developed to enrich bacterial DNA in clinical specimens by removing human DNA based on the differences between bacterial and human cell envelopes (15) or between bacterial and human DNA (16). An alternative enrichment strategy uses sequence-specific oligonucleotides to selectively pull down bacterial DNA, thus increasing the bacterial/human DNA ratio, as has been demonstrated with N. meningitidis RNA bait libraries (17). DNA enrichment can also be achieved using an isothermal multiple displacement amplification reaction with phi29 polymerase and primers designed to selectively amplify a microbial genome. This targeted enrichment strategy, called selective whole-genome amplification (SWGA), was successfully applied to Wolbachia pipientis from infected Drosophila melanogaster (18, 19), Plasmodium reichenowi and Plasmodium gaboni from ape whole-blood specimens (20), and Plasmodium falciparum (21) and Plasmodium vivax (22) from human dried blood spots.
Here, we demonstrate how targeted enrichment using phi29 polymerase and selective primers provides an inexpensive, simple, and efficient method for sequencing and de novo assembly of N. meningitidis genomes from clinical specimens, enabling full molecular typing during meningococcal outbreak investigations.
MATERIALS AND METHODS
Primer design.
To design primers that would selectively amplify the N. meningitidis genome, we identified a set of heptamer sequences that have high density in the N. meningitidis FAM18 reference genome but not in the human h38 reference genome (Table S1). Density was calculated by counting k-mers with DSK v2.1.0 (23) and dividing by the total genome size (2.13 Mb for FAM18; 2.95 Gb for h38). The k-mers with the highest binding bias score (ratio of density in the N. meningitidis genome to density in the human genome) were selected to create a set of heptamer primers with overall binding densities greater than 17/kb, while limiting it to heptamers occurring at least 1,000 times in the N. meningitidis genome. A total of 25 heptamers met these criteria (Table S1). The heptamer primer sequences in the N. meningitidis genome are separated by at most 3,457 nucleotides, and 99.8% of gaps between primers are less than 1 kb long (Fig. S1). Density distributions of the heptamers calculated in silico on 6 N. meningitidis targets (FAM18, MC58, NM53442, Z2491, alfa14, and NEM8013) are similar (Fig. S2). Primers were synthesized by Integrated DNA Technologies, Inc., with two consecutive 3′ phosphodiester bonds replaced by phosphorothioate bonds to protect the primers against phi29 exonuclease activity. Each primer was dissolved at 1 mM in water, and then all primers were mixed in equimolar proportions.
Specimen selection and use.
Three sets of specimens were used in the study. The mock specimens were prepared by mixing N. meningitidis and human DNA (described below) and then used for the optimization of the SWGA procedure. The validation specimens included 12 CSF specimens collected through surveillance of invasive meningococcal disease cases in 2015 in Burkina Faso, Mali, Uganda, Chad, and Ghana, as well as 12 urine specimens collected from meningococcal urethritis cases at the Columbus, OH, Sexual Health Clinic in 2017 to 2019. Isolates were available for comparison from 11 of these urethritis cases. The outbreak investigation specimens were from two meningitis outbreaks in 2019 in Burkina Faso and Togo; these were evaluated after validation of the SWGA method.
DNA extraction from specimens.
To prepare mock clinical specimen DNA mixtures, DNA was extracted from N. meningitidis strain M28467 using the Gentra Puregene Yeast/Bact kit (Qiagen). N. meningitidis DNA was mixed with human genomic DNA (Promega) in a 1:100 ratio, with 0.31 pg/μl of N. meningitidis DNA and 31 pg/μl of human DNA. Assuming an N. meningitidis genome size of 2.2 Mb, the number of genomes presented in 1 μl of such a mixture is about 130 copies. sodC is used as a marker to measure the relative abundance of N. meningitidis DNA by real-time PCR (RT-PCR) (24). The cycle threshold (CT) value of sodC for the mixture is 28. This relationship and an amplification efficiency of 2 allow translation of any CT value of DNA specimen into N. meningitidis genome copy number according to the following formula: 130 copies/μl × 228−CT, where CT is the CT value of sodC for the specimen.
DNA was extracted from 200-μl CSF specimens using a QIAamp DNA minikit (Qiagen).
To extract DNA from urine, the corresponding specimens (2 to 10 ml) were spun down for 4 min at 13,200 rpm. The pellets were resuspended in 25 μl of 10 mM Tris buffer (pH 7.5) and processed using the Gentra Puregene blood kit (Qiagen) with the following minor modifications: addition of 20 μl of Qiagen proteinase K to the specimen instead of 1.5 μl of Puregene proteinase K and overnight incubation at −20°C in 50% isopropanol to improve precipitation instead of 5 min at room temperature.
Amplification of N. meningitidis DNA.
The REPLI-g single-cell kit (Qiagen) is used for whole-genome amplification (WGA) with nonselective primers. In this study, two protocols modified from this kit were used for selective whole-genome amplification (SWGA) to enrich N. meningitidis DNA.
Protocol 1 was slightly modified from the REPLI-g single-cell kit procedure. A 2.5-μl portion of extracted DNA was used according to the manufacturer’s instructions, and 5 μl of N. meningitidis heptamers (Table S1) replaced 5 μl of water in the final reaction mixture of 50 μl, which was then incubated at the tested temperature for 16 h.
Protocol 2 used a reconstituted reaction mixture with alkali denaturation. A 7.75-μl portion of DNA was mixed with 7.75 μl of DLB buffer diluted according to the manufacturer’s instructions (reference no. 1068797; Qiagen). The mixture was incubated for 3 min and neutralized with 15.5 μl of stop solution (reference no. 1032393; Qiagen). The resulting 31-μl solution was mixed with 5 μl of phi29 buffer (Thermo Fisher), 5 μl of a deoxynucleoside triphosphate (dNTP) mixture (10 mM concentration of each type [Roche]), 5 μl of N. meningitidis heptamers (Table S1), 3 μl of phi29 (Thermo Fisher), and 1 μl of inorganic pyrophosphatase (Thermo Fisher) and incubated at the tested temperature for 16 h.
Both protocols were optimized using a mock specimen at three incubation temperatures (30°C, 35°C, and 40°C).
Protocol 1 was first used for DNA from clinical specimens, since it requires low DNA input (2.5 μl). If selective amplification by protocol 1 did not sufficiently enrich N. meningitidis DNA as assessed by RT-PCR (see below), protocol 2, which used high DNA input (7.75 μl), was used to improve the amplification. Upon completion of SWGA or nonselective WGA, DNA was purified from the reaction mixture using Agencourt AMPure XP beads (Beckman Coulter) according to the manufacturer’s instructions, eluted into 50 μl of 10 mM Tris buffer (pH 7.5), quantified by Qubit (Invitrogen), and saved for library preparation, sequencing, and RT-PCR. The final DNA concentration was in the range 500 to 1,000 ng/μl.
Assessing N. meningitidis DNA enrichment by SWGA.
Enrichment of N. meningitidis DNA was assessed by RT-PCR using PerfeCTa SYBR green SuperMix (Quanta Biosciences) and 7 gene markers (Table S2). Each PCR mixture includes 2 μl of tested DNA and specific primers and probe targeting one of the 6 N. meningitidis gene markers, including sodC. The gene coding for RNase P (rnp) was used as a marker to assess the enrichment for human DNA. The amplification for each marker was quantified as the difference in CT values before and after the SWGA reaction, as illustrated by the following equation: ΔCT(marker) = CT(unenriched marker) − CT(enriched marker). A higher ΔCT value indicates a higher degree of DNA amplification. The degree of selective N. meningitidis amplification was quantified by the difference between amplification of the human DNA marker and amplification of the N. meningitidis marker, as follows: ΔΔCT = ΔCT(N. meningitidis marker) − ΔCT(human marker). A higher ΔΔCT indicates a higher degree of selective amplification and enrichment for N. meningitidis DNA.
DNA sequencing and genome sequence assembly.
DNA extracted from urine and CSF validation specimens (see “Specimen selection and use”) was sequenced after they had been amplified with SWGA (“enriched”) or sequenced directly without any amplification (“unenriched”). DNA was quantified by Qubit; 1 μg enriched DNA or 1 to 100 ng unenriched DNA was used for library preparation. The DNA solution was brought to 55 μl with 10 mM Tris buffer (pH 7.5) and fragmented with a Covaris ultrasonicator (Covaris, USA). The libraries were prepared with a NEBNext Ultra II DNA Library Prep kit (New England Biolabs) using selection for 400- to 500-bp fragments and PCR amplification for 7 to 11 cycles. Multiplex sequencing of DNA libraries (4 to 12 libraries per run) was performed on an Illumina MiSeq system using the 500-cycle V2 kit (Illumina, USA) in accordance with the manufacturer’s protocol.
For the outbreak investigation, the DNA from CSF specimens (see “Specimen selection and use”) collected from Togo and Burkina Faso was sequenced without enrichment if the CT value of sodC was ≤19. SWGA was performed if CT values were >19 or if sequencing of unenriched DNA was not successful. Sequencing was performed on a HiSeq system using a rapid SBS 500-cycle V2 kit (Illumina, USA) as part of a batch run with 73 libraries per run.
Following sequencing, human DNA sequences were removed by mapping reads to the hg38 reference genome with Bowtie2 (v2.2.9; local alignment mode) (25). PCR duplicates were removed with bbtools-clumpify (v37.41) (26), and then TruSeq adapter sequences and low-quality base calls were trimmed from reads with cutadapt (v1.8.3; options n = 5, trim-n = True, m = 50) (27). Reads from the N. meningitidis genome were identified using kSLAM (v1.0) with the default bacterial genome database (28).
Genome coverage was assessed by mapping N. meningitidis reads to a reference N. meningitidis genome for each specimen (29). For urine specimens from which an isolate was obtained, the isolate’s genome was used as the reference. For CSF specimens from the African countries, a reference genome was selected by comparing the N. meningitidis reads to the collection described previously (30) and selecting the most similar genome according to Mash 2.1 (using 10,000 32-mers with a minimum of 2 observations in the N. meningitidis reads) (31).
The N. meningitidis reads were assembled using SPAdes v3.12 with default settings (32). Prior to assembly, the read depth was normalized using bbtools-bbnorm (v37.41; bits = 16, prefilter = True) (26), with the targeted depth being the greater of 50× or the expected mean depth, which was calculated as the number of nucleotides in the reads divided by 2.2 Mb. Specimens generating less than 5 Mb of N. meningitidis sequence data (≤2.3× coverage per base) were not assembled. The workflow for bench work and data analysis is shown in Fig. S3.
Molecular typing and phylogenetic analysis.
Molecular typing loci were evaluated in the assembled genomes using BLAST to identify 1,605 loci from N. meningitidis cgMLST (core genome MLST) scheme (v1.0) and 14 other loci defined in the PubMLST database (33) (https://pubmlst.org/neisseria). The 14 molecular typing loci included 7 MLST loci and 7 peptide sequences: PorA VR1, PorA VR2, FetA, PorB, and three vaccine antigens, NadA, NhbA, and FHbp. The PorA variable regions were treated as separate loci for this purpose because they are identified using separate BLAST searches. A locus was considered to be present in the genome assembly if BLAST returned an alignment that included at least 90% of any known allele for that locus. The accuracy of molecular typing results from specimens was assessed by comparing these results with those obtained from isolates from the same disease cases. Isolates for 11 urethritis cases were available and sequenced (34). The alleles identified from specimens were considered correct if they matched the allele identified from the matching isolate. The molecular typing accuracy was defined as the number of loci with correct alleles divided by the total number of loci assessed in the specimen genome assembly. Accuracy was calculated separately for the cgMLST loci and the 14 molecular typing loci. The effect of sequence data abundance on molecular typing results was assessed by randomly subsampling raw reads using seqtk (v1.0-r31) (35) to create a series of 46 data sets for each specimen, with sizes ranging from 20,000 to 1,280,000 N. meningitidis read pairs, each data set being 10% larger than the previous one.
Phylogenetic analysis of N. meningitidis DNA sequences from urine and CSF specimens was performed using the evolutionary placement algorithm (EPA) implemented in RAxML v8.2.9 (36). The EPA uses partial genome sequences obtained from specimen sequencing to place the bacteria from each specimen on an existing phylogenetic tree that is based on a whole-genome alignment; we created the phylogenetic trees from a recombination-filtered core SNP alignment of isolate genome sequences using RAxML (34). The accuracy of EPA results was assessed using data from 11 urethritis cases where both urine specimens and isolates were available. The phylogenetic tree included the 11 isolate genomes along with an additional 213 genomes of meningococcal urethritis isolates (34), and EPA accuracy was measured as the distance on the tree between the specimen and its matched isolate.
To compare strains from Togo and Burkina Faso outbreaks with those from other African countries, the phylogenetic tree was constructed, including 125 CC10217 isolates collected in the African meningitis belt (30) and the largest N. meningitidis genome assembly obtained from unenriched specimens in each outbreak. CC10217 genome sequences were aligned to the complete genome of the BL16188 carriage isolate (37).
Ethical declaration.
Evaluation of urine specimens was approved by the Ohio State University Biomedical Sciences Institutional Review Board (no. 2017H0121). In accordance with CDC human research protection procedures, CDC involvement was determined to not constitute engagement in human subject research (project determination identifier 2017_DBD_Wang_227). As to CSF specimens from African countries, the project determination (identifier 2017_DBD_Wang_411) states that personal identification information was not shared with CDC.
Data availability.
Unmapped reads are available at SRA BioProject under accession no. PRJNA590135.
RESULTS
SWGA optimization.
To optimize the conditions for SWGA, DNA from a mock specimen was amplified using heptamer primer sets at three temperatures (30°C, 35°C, or 40°C) according to SWGA protocol 1 (low DNA input) or protocol 2 (high DNA input). The extent of selective amplification was assessed using one human and six N. meningitidis markers. The greatest selective amplification for all 6 N. meningitidis markers was obtained at 35°C (Fig. 1). No selective amplification for N. meningitidis markers was observed using nonselective WGA. Increasing the temperature to 40°C greatly reduced amplification of all markers. Therefore, 35°C was selected as the optimal condition for testing specimens.
FIG 1.

The extent of amplification (ΔCT) for human (rnp) and N. meningitidis (sdhA, NEIS2381, NEIS0417, NEIS2065, NEIS1699, and sodC) markers (A) and the relative extent of selective amplification (ΔΔCT) for N. meningitidis markers (B). Amplification conditions (temperature and protocol) are given at the bottom of the figure, where, e.g., 30_1 indicates SWGA at 30°C using protocol 1. WGA, nonselective whole-genome amplification.
SWGA and sequencing of clinical specimens.
DNA extracted from 24 N. meningitidis-positive clinical specimens (12 CSF specimens from patients with invasive meningococcal disease and 12 urine specimens from urethritis patients) were enriched by SWGA at 35°C. The DNA samples were first enriched using SWGA protocol 1, which requires less DNA input (2.5 μl of DNA sample). The extent of selective amplification was assessed by RT-PCR targeting N. meningitidis marker sodC and human marker rnp. The majority of specimens (8/12 urine samples and 8/12 CSF samples) showed a profound decrease in the CT value for sodC but not for the human marker rnp, with at least 3.3 cycles of relative extent of selective amplification for sodC (Table 1).
TABLE 1.
Analysis of unenriched and SWGA-enriched specimens by RT-PCR and sequencing
| Specimen | RT-PCR |
% N. meningitidis readsa |
Fold enrichmentb | % of N. meningitidis genome with ≥10-fold coverage depth |
||||
|---|---|---|---|---|---|---|---|---|
| ΔCT, sodC | ΔCT, rnp | ΔΔCT | Unenriched | Enriched | Unenriched | Enriched | ||
| Ur-1 | 11.5 | −1.1 | 12.6 | 0.4 | 66.1 | 165 | 1.8 | 35.5 |
| Ur-2 | 17.9 | 0.9 | 17.0 | 0.1 | 90.5 | 905 | 0.03 | 97.6 |
| Ur-3 | 15.5 | −0.8 | 16.3 | 0.05 | 67.3 | 1,346 | 0.01 | 51.8 |
| Ur-4 | 11.1 | −5.2 | 16.3 | 0.03 | 57.2 | 1,907 | <0.01 | 48.9 |
| Ur-5 | 15.6 | 0 | 15.6 | 0.4 | 77.7 | 194 | 0.6 | 97.6 |
| Ur-6 | 10.2 | 1.8 | 8.4 | 0.7 | 1.1 | 2 | 7.4 | 14.1 |
| Ur-7 | 14.5 | −1.4 | 15.9 | 0.4 | 61.9 | 155 | 0.6 | 93.3 |
| Ur-8 | 16.1 | −1.8 | 17.9 | 1.6 | 65.9 | 41 | 36.9 | 40.0 |
| Ur-9 | 3.3 | −3.3 | 6.6 | 0.02 | 3.7 | 185 | <0.01 | 89.6 |
| Ur-10 | 5.4 | −3.4 | 8.8 | 0.04 | 6.2 | 155 | <0.01 | 76.3 |
| Ur-11 | 17.2 | 10.4 | 6.8 | 0.09 | 21.1 | 234 | <0.01 | 90.0 |
| Ur-12 | 7.6 | −0.2 | 7.8 | 0.04 | 2.2 | 55 | 0.01 | 3.0 |
| CSF-1 | 10.5 | 3.2 | 7.3 | 1 | 33.3 | 33 | 7.4 | 93.5 |
| CSF-2 | 1.9 | 1.5 | 0.4 | 0.2 | 0.7 | 4 | 0.4 | 9.1 |
| CSF-3 | 1.7 | 1.2 | 0.5 | 0.9 | 1 | 1 | 20.7 | 16.9 |
| CSF-4 | 12.5 | 7 | 5.5 | 0.2 | 4.5 | 23 | 0.04 | 74.9 |
| CSF-5 | 11.2 | 5.1 | 6.1 | 0.4 | 19.7 | 49 | 0.7 | 91.8 |
| CSF-6 | 4.4 | −0.4 | 4.8 | 0.3 | 6.8 | 23 | 0.1 | 90.8 |
| CSF-7 | 9.0 | 3.8 | 5.2 | 2.7 | 61 | 23 | 77.7 | 97.9 |
| CSF-8 | 1.8 | 2.4 | −0.6 | 0.9 | 27.4 | 30 | 3.6 | 56.0 |
| CSF-9 | 9.4 | −0.2 | 9.6 | 0.5 | 65.7 | 131 | 1.1 | 96.0 |
| CSF-10 | 5.9 | 2.8 | 3.1 | 2.4 | 22.6 | 9 | 50.0 | 85.8 |
| CSF-11 | 7.7 | 1.9 | 5.8 | 0.05 | 15.6 | 312 | <0.01 | 83.7 |
| CSF-12 | 10.2 | −5.6 | 15.8 | 0.3 | 86.5 | 288 | 0.3 | 97.8 |
N. meningitidis read pairs as a percentage of total read pairs obtained from the specimen.
Calculated as percent enriched N. meningitidis reads divided by percent unenriched N. meningitidis reads. Expanded data from RT-PCR and sequencing analysis are available in Table S3.
The specimens Ur-2, -4, -6, and -11 and CSF-2, -3, -8, and -10 were poorly amplified. All except CSF-3 (no DNA remained) were retested using SWGA protocol 2, which allows high DNA input. N. meningitidis DNA from specimens Ur-2, -4, -6, and -11 and CSF-10 were successfully amplified and enriched (Table 1).
We sequenced both the unenriched DNA and SWGA-enriched DNA from each of the 24 specimens. After SWGA, the mean proportion of N. meningitidis reads increased from 0.48% to 21.1% for CSF specimens and from 0.086% to 57.2% for urine specimens. The median enrichment was greater than 100-fold in both specimen types (Table 1). The unenriched specimens had 0.02 to 2.7% N. meningitidis reads, while the enriched specimens had 0.7 to 90.5% N. meningitidis reads.
SWGA enrichment increased the number of N. meningitidis reads obtained from all 24 specimens (Table 1). A high depth of coverage (defined as at least 10-fold for this analysis) is required for high-confidence base calling (29, 38). Prior to SWGA enrichment, only 2 specimens had >40% of the N. meningitidis genome with 10-fold coverage. However, after SWGA enrichment, 19 of 24 specimens had >40% of the N. meningitidis genome with 10-fold coverage (Table 1).
Molecular typing of N. meningitidis.
After SWGA, 23 of 24 enriched specimens had enough sequence data to generate genome assemblies (versus 9 unenriched). More than 1,200 core genome MLST (cgMLST) loci were identified in 15 enriched specimens (versus 4 unenriched). As a comparison, >1,500 cgMLST loci can be obtained from genome assemblies of an N. meningitidis isolate. All 7 MLST loci were identified in 16 enriched specimens (versus 3 unenriched), and 4 additional typing loci (PorA VR1, PorA VR2, FetA, and PorB) were identified in 12 specimens (versus 5 unenriched). FHbp, NhbA, and NadA were identified in 16, 18, and 13 specimens, respectively (versus 5, 2, and 3 in unenriched specimens, respectively) (Table 2; Table S4).
TABLE 2.
Molecular typing results for assembled specimen sequence dataa
| Specimen | Genome assembly size (kb) |
No. (%) of cgMLST loci present |
cgMLST accuracy (%)b
|
Typing accuracy |
No. of MT locic |
No. of MLST loci |
MLST resultd |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unenr | Enr | Unenr | Enr | Unenr | Enr | Unenr | Enr | Unenr | Enr | Unenr | Enr | Unenr | Enr | |
| Ur-1 | 1,100 | 866 | 209 (13) | 584 (36.4) | NA | NA | NA | NA | 3 | 4 | 1 | 2 | — | — |
| Ur-2 | — | 2,092 | — | 1,561 (97.3) | — | 99.2 | — | 14/14 | 0 | 14 | 0 | 7 | — | ST-11; CC11 |
| Ur-3 | — | 1,249 | — | 842 (52.5) | — | 90.3 | — | 5/5 | 0 | 5 | 0 | 4 | — | — |
| Ur-4 | — | 1,115 | — | 776 (48.3) | — | 92.1 | — | 5/5 | 0 | 5 | 0 | 3 | — | — |
| Ur-5 | — | 2,095 | — | 1,562 (97.3) | — | 99.6 | — | 14/14 | 0 | 14 | 0 | 7 | — | ST-11; CC11 |
| Ur-6 | 1,495 | 769 | 410 (25.5) | 365 (22.7) | 79.7 | 86.5 | 6/7 | 4/5 | 8 | 5 | 4 | 2 | — | — |
| Ur-7 | — | 2,043 | — | 1,519 (94.6) | — | 98.5 | — | 13/13 | 0 | 13 | 0 | 7 | — | ST-11; CC11 |
| Ur-8 | 1,926 | 994 | 1,262 (78.6) | 634 (39.5) | 93.8 | 90.8 | 12/13 | 5/6 | 13 | 6 | 7 | 3 | New ST; CC11 | — |
| Ur-9 | — | 2,044 | — | 1,535 (95.6) | — | 97.7 | — | 13/13 | 0 | 13 | 0 | 7 | — | ST-11; CC11 |
| Ur-10 | — | 2,015 | — | 1,482 (92.3) | — | 95.7 | — | 14/14 | 0 | 14 | 0 | 7 | — | ST-11; CC11 |
| Ur-11 | — | 1,980 | — | 1,474 (91.8) | — | 98.2 | — | 14/14 | 0 | 14 | 0 | 7 | — | ST-11; CC11 |
| CSF-1 | 1,848 | 2,058 | 874 (54.5) | 1,554 (96.8) | NA | NA | NA | NA | 9 | 14 | 3 | 7 | — | ST-11; CC11 |
| CSF-2 | — | 1,641 | — | 828 (51.6) | NA | NA | NA | NA | 0 | 8 | 0 | 5 | — | — |
| CSF-3 | 1,988 | 1,664 | 1,235 (76.9) | 894 (55.7) | NA | NA | NA | NA | 9 | 13 | 3 | 7 | — | ST-11; CC11 |
| CSF-4 | — | 1,882 | — | 1,377 (85.8) | NA | NA | NA | NA | 0 | 11 | 0 | 7 | — | ST-181; CC181 |
| CSF-5 | 1,257 | 2,010 | 343 (21.4) | 1,517 (94.5) | NA | NA | NA | NA | 2 | 12 | 0 | 7 | — | ST-12446; CC10217 |
| CSF-6 | — | 2,040 | — | 1,518 (94.6) | NA | NA | NA | NA | 0 | 13 | 0 | 7 | — | ST-10217; CC10217 |
| CSF-7 | 2,080 | 2,036 | 1,527 (95.1) | 1,553 (96.8) | NA | NA | NA | NA | 12 | 13 | 7 | 7 | ST-12446; CC10217 | ST-12446; CC10217 |
| CSF-8 | 1,476 | 1,696 | 452 (28.2) | 164 (10.2) | NA | NA | NA | NA | 9 | 5 | 6 | 3 | — | — |
| CSF-9 | — | 2,064 | — | 1,512 (94.2) | NA | NA | NA | NA | 0 | 13 | 0 | 7 | — | ST-181; CC181 |
| CSF-10 | 2,024 | 2,032 | 1,413 (88) | 1,369 (85.3) | NA | NA | NA | NA | 13 | 12 | 7 | 7 | ST-181; CC181 | New ST; CC181 |
| CSF-11 | — | 1,901 | — | 1,391 (86.7) | NA | NA | NA | NA | 0 | 14 | 0 | 7 | — | ST-11; CC11 |
| CSF-12 | — | 2,092 | — | 1,570 (97.8) | NA | NA | NA | NA | 0 | 14 | 0 | 7 | — | ST-11; CC11 |
Enr, enriched; Unenr, unenriched; MT, molecular typing; NA, no matching isolate was available for the comparison. A dash indicates failure of genome assembly due to poor sequencing data and as consequence nonavailability of molecular typing results. No assembly was produced for 15 unenriched and 1 enriched specimen. Full data are available in Table S4.
Percentage of present cgMLST loci that were identical to those from the matching isolate.
Out of 14 molecular typing loci identified.
“New ST” indicates a previously unidentified MLST profile.
To assess the accuracy of SWGA, 11 urine specimens that had a matching isolate were examined. Ten of the 11 urine specimens produced genome assemblies after SWGA, which were compared with the genome sequences of the matching isolates. The genome assemblies from enriched specimens had an overall allele calling accuracy of 94.4% for the 1,605 cgMLST loci, as determined by comparing the allele calls from the enriched specimens and the matching isolate (Table 2). The allele calling accuracy for the 14 molecular typing loci was 98.1% (Table 2).
High cgMLST allele calling accuracy (>95%) was obtained for genome assemblies from 6 urine specimens (Table 2), with >1,400 cgMLST loci being detected. The assemblies for 6 CSF specimens allowed detection of >1,400 cgMLST loci as well, though their accuracy could not be measured, as no matching isolate was available. The genome assemblies created from the other 11 clinical specimens (urine or CSF) had fewer than 1,000 cgMLST loci present (Table 2).
Quality metrics for SWGA-enriched specimens.
To test if the number of cgMLST loci present in a genome assembly is a reliable indicator of allele calling accuracy, we produced a series of genome assemblies based on random subsets of sequencing reads from each of the 6 enriched specimens that produced assemblies with allele calling accuracy greater than 95%. The number of cgMLST loci present in the genome assemblies increased with the number of reads used for the assembly (Fig. 2A) and was strongly correlated with the accuracy of the cgMLST allele calls (Fig. 2B) (r = 96.8). Genome assemblies with more than 1,400 cgMLST loci generally had over 95% accuracy at those loci. The correlation between the number of loci present and allele calling accuracy was strong despite variation in the number reads necessary to produce a genome assembly with at least 1,400 cgMLST loci, ranging from 88,000 read pairs for Ur-9 to 305,000 read pairs for Ur-11 (Fig. 2).
FIG 2.

Dependence of quality of N. meningitidis genome assemblies on the number of provided N. meningitidis read pairs. (A) The number of cgMLST loci present in genome assemblies as a function of the number of N. meningitidis read pairs used for the assembly. To produce a range of data set sizes, reads were randomly subsampled from the reads produced from the 6 enriched specimens for which genome assemblies had over 95% allele calling accuracy. Empty circles, Ur-2; empty diamonds, Ur-5; filled squares, Ur-7; filled triangles, Ur-9; empty triangles, Ur-10; filled inverse triangles, Ur-11. (B) Correlation between accuracy of allele calling for cgMLST loci and the number of loci present. The arrows shows correspondence between minimum tolerated allele calling accuracy (95%) and the minimum number of N. meningitidis read pairs or cgMLST loci required to provide that level of accuracy.
Reproducibility of SWGA.
The reproducibility of SWGA was tested by performing 12 independent reactions on the same specimen, CSF-12 (Table 2), and sequencing the resulting DNA products on a single MiSeq run. N. meningitidis DNA was enriched to 80.7 to 83.5% (the unenriched specimen was 0.3%) in all replicates, and the 12 assemblies included 1,490 to 1,521 cgMLST loci (out of 1,605). Identical results were obtained across all replicates for the 14 molecular typing loci. The rate of allele call discrepancies across cgMLST loci ranged from 0.12 to 0.73%.
Phylogenetic placement of strains detected in clinical specimens.
SWGA generally increased the fraction of the genome with at least 10-fold coverage (Table 1), which is a common threshold for high-confidence base calls using reference mapping (29, 38). We used the evolutionary placement algorithm (EPA) to test if SWGA generated enough sequence data to place N. meningitidis from 11 urine specimens at the same location as the matching isolates from the same cases on a phylogenetic tree containing isolates of the U.S. NmNG urethritis clade (Fig. 3). SWGA improved evolutionary placement results for 9 of the 11 specimens, as determined by the distance (substitutions per site) between the specimens and isolates in the evolutionary tree (Table S5). Four specimens (Ur-3, -5, -8, and -10) were at the same location as the matching isolates after SWGA. Sufficient sequencing data were also obtained from unenriched specimen Ur-8. One enriched specimen (Ur-6) was placed farther from the isolate than the corresponding unenriched specimen. CSF specimens were not assessed because a matching isolate was not available.
FIG 3.
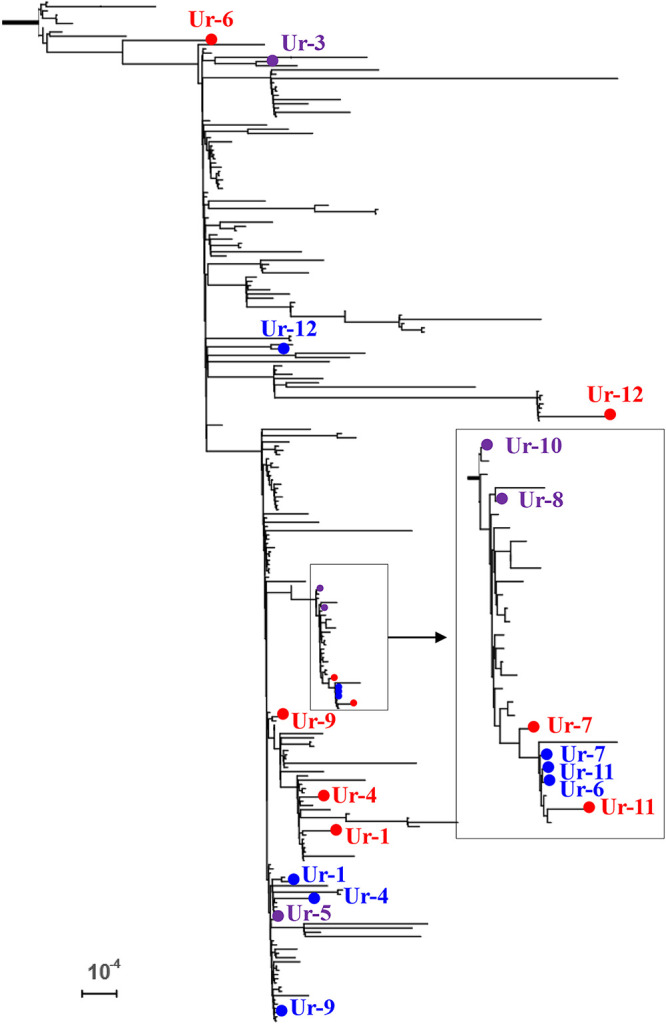
Evolutionary placement of N. meningitidis DNA sequences from enriched urine specimens (red dots) on a phylogenetic tree of the U.S. NmNG urethritis clade that includes isolates (blue dots) from the same cases as the specimens. Purple dots indicate that the specimen was placed with the isolate on the tree. The inset shows detail for the boxed branch. Bar, 10−4 substitutions per site. Distances are reported in Table S5.
SWGA for investigation of two meningitis outbreaks.
Outbreaks caused by serogroup C meningococcal disease were reported in both Burkina Faso and Togo in 2019. To further characterize the outbreak strains and assess their similarity to other epidemic-prone serogroup C strains in Africa, we tested 38 CSF specimens from confirmed N. meningitidis serogroup C cases: 17 from Burkina Faso (BF_1 through BF_17) and 21 from Togo (Togo_1 through Togo_21). Eleven specimens with CT values of <19 were directly sequenced by shotgun sequencing without enrichment, and the remaining 27 specimens, which had lower bacterial loads (CT > 19), were enriched by SWGA prior to sequencing. Ten of 11 unenriched specimens produced genome assemblies, with >1,400 cgMLST loci and all 14 molecular typing loci (Table 3). One unenriched specimen missed one vaccine antigen gene and was retested with SWGA, bringing the total of SWGA specimens to 28. Genome assemblies were obtained from 26 of 28 SWGA-enriched specimens, with two Togo specimens (Togo_20 and Togo_21) failing to assemble. In total, 12 SWGA specimens (6 from Burkina-Faso and 6 from Togo) had >1,400 cgMLST loci and all 14 molecular typing loci identified (Table 3). The N. meningitidis strains from Togo and Burkina Faso specimens belonged to ST-9367 (CC10217) and ST-10217 (CC10217), respectively (Table 3).
TABLE 3.
Molecular typing of N. meningitidis in specimens from the 2019 Togo and Burkina Faso meningitis outbreaka
| Specimen | Sequencing method | CT, sodC | Genome assembly size (kb) | No. of cgMLST loci identified | No. of molecular typing loci | No. of MLST loci | Sequence type; clonal complexb |
|---|---|---|---|---|---|---|---|
| BF_1 | Unenr | 17 | 2,136,395 | 1,559 | 14 | 7 | ST-10217; CC10217 |
| BF_2 | Unenr | 19 | 2,132,672 | 1,556 | 14 | 7 | ST-10217; CC10217 |
| BF_3 | Unenr | 17 | 2,132,682 | 1,557 | 14 | 7 | ST-10217; CC10217 |
| BF_4 | Unenr | 16 | 2,064,493 | 1,560 | 14 | 7 | ST-10217; CC10217 |
| BF_5 | Unenr | 16 | 2,063,009 | 1,564 | 14 | 7 | ST-10217; CC10217 |
| BF_6 | Unenr | 19 | 2,052,439 | 1,559 | 14 | 7 | ST-10217; CC10217 |
| BF_7 | Unenr | 18 | 2,064,961 | 1,556 | 14 | 7 | ST-10217; CC10217 |
| BF_8 | Unenr | 19 | 2,069,327 | 1,563 | 14 | 7 | ST-10217; CC10217 |
| BF_9 | Unenr | 16 | 2,069,218 | 1,560 | 14 | 7 | ST-10217; CC10217 |
| BF_10 | Unenr/SWGA_1c | 17 | 2,093,455 | 1,553 | 14 | 7 | ST-10217; CC10217 |
| BF_11 | SWGA_1 | 21 | 2,104,298 | 1,530 | 14 | 7 | ST-10217; CC10217 |
| BF_12 | SWGA_1 | 30 | 2,059,934 | 1,509 | 14 | 7 | ST-10217; CC10217 |
| BF_13 | SWGA_1 | 25 | 1,351,960 | 534 | 9 | 5 | — |
| BF_14 | SWGA_1 | 30 | 2,133,478 | 1,535 | 14 | 7 | ST-10217; CC10217 |
| BF_15 | SWGA_1 | 22 | 2,064,379 | 1,520 | 13 | 6 | ST-ND; CC10217 |
| BF_16 | SWGA_1 | 33 | 2,095,986 | 1,551 | 14 | 7 | ST-10217; CC10217 |
| BF_17 | SWGA_1 | 26 | 2,085,832 | 1,549 | 14 | 7 | ST-10217; CC10217 |
| Togo_1 | Unenr | 17 | 2,121,115 | 1,552 | 14 | 7 | ST-9367; CC10217 |
| Togo_2 | SWGA_1 | 20 | 2,508,337 | 1,222 | 13 | 6 | — |
| Togo_3 | SWGA_1 | 33 | 1,081,654 | 442 | 4 | 2 | — |
| Togo_4 | SWGA_1 | 25 | 2,084,690 | 1,517 | 14 | 7 | ST-9367; CC10217 |
| Togo_5 | SWGA_1 | 32 | 1,077,209 | 410 | 4 | 2 | — |
| Togo_6 | SWGA_1 | 30 | 1,923,570 | 1,375 | 12 | 7 | ST-9367; CC10217 |
| Togo_7 | SWGA_1 | 22 | 2,114,107 | 1,539 | 14 | 7 | ST-9367; CC10217 |
| Togo_8 | SWGA_1 | 26 | 2,153,980 | 1,236 | 14 | 7 | ST-9367; CC10217 |
| Togo_9 | SWGA_1 | 25 | 2,114,641 | 1,533 | 14 | 7 | ST-9367; CC10217 |
| Togo_10 | SWGA_2 | 26 | 2,210,580 | 1,263 | 11 | 7 | New |
| Togo_11 | SWGA_1 | 22 | 1,679,000 | 952 | 10 | 4 | — |
| Togo_12 | SWGA_1 | 23 | 2,116,610 | 1,540 | 14 | 7 | ST-9367; CC10217 |
| Togo_13 | SWGA_1 | 28 | 2,003,579 | 1,413 | 12 | 6 | ST-ND; CC10217 |
| Togo_14 | SWGA_1 | 27 | 2,014,369 | 1,424 | 11 | 5 | ST-ND; CC10217 |
| Togo_15 | SWGA_1 | 21 | 2,107,985 | 1,544 | 14 | 7 | ST-9367; CC10217 |
| Togo_16 | SWGA_2 | 26 | 842,874 | 159 | 4 | 0 | — |
| Togo_17 | SWGA_1 | 23 | 2,365,275 | 1,377 | 14 | 7 | New |
| Togo_18 | SWGA_1 | 22 | 1,828,557 | 1,224 | 12 | 7 | New |
| Togo_19 | SWGA_1 | 20 | 2,061,090 | 1,557 | 14 | 7 | ST-9367; CC10217 |
Specimens which did not meet an accuracy cutoff of 1,400 in cgMLST are highlighted with gray shading. Unenr, unenriched; SWGA_1, SWGA protocol 1; SWGA_2, SWGA protocol 2.
ND, not determined (not enough information to identify the ST for the strain). “New” indicates a new sequence type (ST) or clonal complex (CC). A dash indicates nonavailability of ST and CC due to poor sequencing data. Full data are available in Table S6.
All but one locus was identified by sequencing of not enriched specimen. The missing locus (NhbA) was determined from the corresponding SWGA-enriched backup of the specimen.
EPA was used to test if the N. meningitidis sequences from each outbreak belonged to a single phylogenetic group. The reference tree for EPA (Fig. 4) consisted of 125 previously published genomes of CC10217 isolates from the African meningitis belt (30) as well as the best genome assembly from each outbreak (specimens Togo-1 and BF-1 in Table 3, with 1,559 and 1,552 cgMLST loci, respectively). The N. meningitidis sequences from all 16 Burkina Faso specimens were placed on the branch with the BF-1 genome assembly with high confidence (likelihood weight = 100%). Likewise, the N. meningitidis sequences from all 20 Togo specimens (including two that did not produce assemblies) were placed on the branch with the Togo-1 genome assembly with high confidence (likelihood weight = 100%).
FIG 4.

Evolutionary placement of N. meningitidis from Togo and Burkina Faso outbreaks. Previously published CC10217 genomes from African countries are also included in the tree. The recombination-corrected phylogenetic tree was produced from an alignment of 125 CC10217 genomes and one genome assembly to represent each outbreak (BF-1 and Togo-1). N. meningitidis genomes from Togo and Burkina Faso outbreaks in 2019 are in large type. Triangles indicate clades that were collapsed for display; the number of isolates in the clade is noted in parentheses. Bootstrap support values greater than 50% are shown. Bar, 10−5 substitutions per site.
DISCUSSION
Increased WGS use has greatly improved disease surveillance and outbreak response by providing more accurate and in-depth information on the causative pathogens. High-quality WGS data for genetic characterization of bacterial pathogens is most reliably produced from isolates, but these are not always available. Characterization of bacterial pathogens through direct sequencing from clinical specimens is highly appealing for clinical and public health laboratories but often requires enrichment prior to sequencing (15–17). SWGA has been successfully developed and used for pathogen genome sequencing in a number of applications for various types of specimens (18–22). In this study, we demonstrated that SWGA allows generation of sufficient data for molecular typing of a very important human pathogen, N. meningitidis, directly from clinical specimens such as CSF and urine based on de novo genome assemblies.
The quality of molecular typing results was strongly associated with the completeness of the draft genome assembly (Fig. 2), demonstrating that the number of cgMLST loci in an assembly can serve as a quality control criterion. Similarly, the evolutionary placement algorithm produced more accurate placements for sequence data that covered a larger percentage of a reference genome (Table 2; Table S5); SWGA-enriched specimens that had 10× depth of coverage on >50% of the reference genome (Table 2) were all placed within 1.1 × 10−5 substitutions/site of their corresponding isolate sequence in the reference phylogeny (Table S5), while specimens with sufficient coverage on smaller portions of the reference genome were typically more distant from their corresponding isolate. This level of accuracy is sufficient to identify groups of bacteria, such as the U.S. NmNG urethritis clade or the ST-10217 clade, but not to confidently identify the closest relatives within those groups (Fig. 3).
SWGA uses customized primer design to favorably amplify targeted organisms and therefore can be applied to many other pathogens and improve disease surveillance, outbreak investigations, and other epidemiological investigations. Molecular surveillance data on bacterial meningitis are very limited in regions with low culture rates, especially in countries of the African meningitis belt, which hinders epidemiological investigations and responses. SWGA may tremendously enhance the representativeness of molecular data through sequencing of N. meningitidis directly from clinical specimens, thus improving surveillance and outbreak responses, as demonstrated by our investigation of the recent Togo and Burkina Faso outbreaks. SWGA-based sequencing revealed that the strains that had caused the outbreak in Togo and Burkina Faso belonged to the same clonal complex, CC10217, but were two different sequence types (STs). The Burkina Faso outbreak strain (ST-10217) is genetically closer to the strains that caused the large outbreaks and epidemics in Niger and Nigeria during 2013 to 2016 (30). The Togo outbreak strain (ST-9367) is very close to the carriage isolate (ST-9367) from Burkina Faso, which was believed to be the origin of the emerging ST-10217 in Africa (37).
Cost-effectiveness.
SWGA-based sequencing provides an effective and affordable method for sequencing targeted pathogens directly from clinical specimens. Selectively enriching N. meningitidis DNA and reducing human DNA proportion improve N. meningitidis-specific sequencing efficiency. Based on our experience, any specimens with a CT value for the sodC marker below 30 can undergo SWGA to improve N. meningitidis sequencing data and analysis. The enrichment process is a straightforward procedure which includes only one reaction step with overnight incubation. The estimated cost for SWGA-based sequencing is about $150 per specimen when 12 specimens are multiplexed using the MiSeq platform, while the RNA bait method, which was recently used to generate good-quality culture-free N. meningitidis genomes (17), is up to $500 per specimen (39). In order to make cost-effective decisions about whether to proceed with the costly sequencing step, a rapid and inexpensive RT-PCR assay can be used to assess whether N. meningitidis DNA was sufficiently enriched by SWGA to allow successful genome assembly. We found that ΔΔCT values greater than 3 were an effective cutoff, representing a roughly 10-fold extent of selective amplification.
SWGA-based sequencing is also less expensive than shotgun sequencing of unenriched specimens, which can test only 4 to 6 specimens per MiSeq cartridge. These features may allow its easy implementation in public health and research laboratories in comparison to other, slower, and more costly DNA enrichment procedures.
Limitations.
SWGA for N. meningitidis has some limitations. Primer design for SWGA is constrained by the optimal temperature range, 30 to 35°C, of phi29 polymerase, which is used in the isothermal multiple-displacement amplification reaction. Increasing the length of primers may improve binding specificity for N. meningitidis, but the primer melting temperatures will be higher than the optimal temperature for phi29, which will inhibit the amplification.
The heptamers were designed to distinguish N. meningitidis from human DNA in clinical specimens from patient sterile sites that contain only one or, rarely, very few bacterial pathogens; they may not apply to specimens collected from nonsterile sites (such as nasopharyngeal swabs) that contain a wide spectrum of bacterial species. Further validation is required if SWGA is to be used for the detection of N. meningitidis from these types of specimens.
Some regions of the N. meningitidis genome are not well represented in SWGA-enriched DNA, most notably the capsule locus, which is an important virulence factor of N. meningitidis (40). This is likely due to the low GC content of the capsule locus (40), while the heptamers used in the study have high GC content (Table S1). The entire capsule locus was revealed in some SWGA-enriched specimens from the studied outbreaks, but in the majority of specimens, one or more capsule locus genes were incomplete or missing, allowing only the serogroup C backbone to be identified (Table S7).
Template-independent amplification (TIA) is a well-known phenomenon during WGA that diminishes the efficiency of template amplification (41). While an increase of unidentified “junk” DNA (neither human nor N. meningitidis) was observed following SWGA enrichment, it was not found to degrade the quality of genome assemblies or molecular typing results, likely due to the bioinformatic identification of N. meningitidis reads prior to assembly (data not shown).
In conclusion, SWGA provides a relatively inexpensive and robust method to enrich N. meningitidis DNA from different types of clinical specimens (e.g., CSF and urine) for effective sequencing. Continued development of similar methods for other important pathogens will enable genomic characterization of diverse pathogens, thereby strengthening public health surveillance for various diseases worldwide.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nadav Topaz for his critical discussion on the molecular epidemiology of the meningitis belt and Fang Hu, Caelin Potts, and Daya Marasini for their assistance with DNA extraction and WGS library preparations.
WDS, Inc., and IHRC, Inc., are contracted to the Centers for Disease Control and Prevention.
No conflicts of interest are declared.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Christensen H, May M, Bowen L, Hickman M, Trotter CL. 2010. Meningococcal carriage by age: a systematic review and meta-analysis. Lancet Infect Dis 10:853–861. doi: 10.1016/S1473-3099(10)70251-6. [DOI] [PubMed] [Google Scholar]
- 2.MacNeil JR, Blain AE, Wang X, Cohn AC. 2018. Current epidemiology and trends in meningococcal disease–United States, 1996–2015. Clin Infect Dis 66:1276–1281. doi: 10.1093/cid/cix993. [DOI] [PubMed] [Google Scholar]
- 3.Agier L, Martiny N, Thiongane O, Mueller JE, Paireau J, Watkins ER, Irving TJ, Koutangni T, Broutin H. 2017. Towards understanding the epidemiology of Neisseria meningitidis in the African meningitis belt: a multi-disciplinary overview. Int J Infect Dis 54:103–112. doi: 10.1016/j.ijid.2016.10.032. [DOI] [PubMed] [Google Scholar]
- 4.Bazan JA, Turner AN, Kirkcaldy RD, Retchless AC, Kretz CB, Briere E, Tzeng YL, Stephens DS, Maierhofer C, Del Rio C, Abrams AJ, Trees DL, Ervin M, Licon DB, Fields KS, Roberts MW, Dennison A, Wang X. 2017. Large cluster of Neisseria meningitidis urethritis in Columbus, Ohio, 2015. Clin Infect Dis 65:92–99. doi: 10.1093/cid/cix215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jolley KA, Brehony C, Maiden MC. 2007. Molecular typing of meningococci: recommendations for target choice and nomenclature. FEMS Microbiol Rev 31:89–96. doi: 10.1111/j.1574-6976.2006.00057.x. [DOI] [PubMed] [Google Scholar]
- 7.Lucidarme J, Comanducci M, Findlow J, Gray SJ, Kaczmarski EB, Guiver M, Vallely PJ, Oster P, Pizza M, Bambini S, Muzzi A, Borrow R. 2010. Characterization of fHbp, nhba (gna2132), nadA, porA, and sequence type in group B meningococcal case isolates collected in England and Wales during January 2008 and potential coverage of an investigational group B meningococcal vaccine. Clin Vaccine Immunol 17:919–929. doi: 10.1128/CVI.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muzzi A, Brozzi A, Serino L, Bodini M, Abad R, Caugant D, Comanducci M, Lemos AP, Gorla MC, Křížová P, Mikula C, Mulhall R, Nissen M, Nohynek H, Simões MJ, Skoczyńska A, Stefanelli P, Taha M-K, Toropainen M, Tzanakaki G, Vadivelu-Pechai K, Watson P, Vazquez JA, Rajam G, Rappuoli R, Borrow R, Medini D. 2019. Genetic Meningococcal Antigen Typing System (gMATS): a genotyping tool that predicts 4CMenB strain coverage worldwide. Vaccine 37:991–1000. doi: 10.1016/j.vaccine.2018.12.061. [DOI] [PubMed] [Google Scholar]
- 9.Feagins AR, Vuong J, Fernandez K, Njanpop-Lafourcade BM, Mwenda JM, Sanogo YO, Paye MF, Payamps SK, Mayer L, Wang X. 2019. The strengthening of laboratory systems in the meningitis belt to improve meningitis surveillance, 2008–2018: a partners' perspective. J Infect Dis 220:S175–S181. doi: 10.1093/infdis/jiz337. [DOI] [PubMed] [Google Scholar]
- 10.Vuong J, Collard JM, Whaley MJ, Bassira I, Seidou I, Diarra S, Ouedraogo RT, Kambire D, Taylor TH Jr, Sacchi C, Mayer LW, Wang X. 2016. Development of real-time PCR methods for the detection of bacterial meningitis pathogens without DNA extraction. PLoS One 11:e0147765. doi: 10.1371/journal.pone.0147765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chokshi DA, Parker M, Kwiatkowski DP. 2006. Data sharing and intellectual property in a genomic epidemiology network: policies for large-scale research collaboration. Bull World Health Organ 84:382–387. doi: 10.2471/blt.06.029843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Retchless AC, Fox LM, Maiden MCJ, Smith V, Harrison LH, Glennie L, Harrison OB, Wang X. 2019. Toward a global genomic epidemiology of meningococcal disease. J Infect Dis 220:S266–S273. doi: 10.1093/infdis/jiz279. [DOI] [PubMed] [Google Scholar]
- 13.Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. 2013. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whaley MJ, Joseph SJ, Retchless AC, Kretz CB, Blain A, Hu F, Chang HY, Mbaeyi SA, MacNeil JR, Read TD, Wang X. 2018. Whole genome sequencing for investigations of meningococcal outbreaks in the United States: a retrospective analysis. Sci Rep 8:15803. doi: 10.1038/s41598-018-33622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Street TL, Barker L, Sanderson ND, Kavanagh J, Hoosdally S, Cole K, Newnham R, Selvaratnam M, Andersson M, Llewelyn MJ, O’Grady J, Crook DW, Eyre DW. 2019. Optimizing DNA extraction methods for nanopore sequencing of Neisseria gonorrhoeae directly from urine samples. J Clin Microbiol 58:e01822-19. doi: 10.1128/JCM.01822-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feehery GR, Yigit E, Oyola SO, Langhorst BW, Schmidt VT, Stewart FJ, Dimalanta ET, Amaral-Zettler LA, Davis T, Quail MA, Pradhan S. 2013. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One 8:e76096. doi: 10.1371/journal.pone.0076096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark SA, Doyle R, Lucidarme J, Borrow R, Breuer J. 2018. Targeted DNA enrichment and whole genome sequencing of Neisseria meningitidis directly from clinical specimens. Int J Med Microbiol 308:256–262. doi: 10.1016/j.ijmm.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Clarke EL, Sundararaman SA, Seifert SN, Bushman FD, Hahn BH, Brisson D. 2017. swga: a primer design toolkit for selective whole genome amplification. Bioinformatics 33:2071–2077. doi: 10.1093/bioinformatics/btx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leichty AR, Brisson D. 2014. Selective whole genome amplification for resequencing target microbial species from complex natural samples. Genetics 198:473–481. doi: 10.1534/genetics.114.165498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sundararaman SA, Plenderleith LJ, Liu W, Loy DE, Learn GH, Li Y, Shaw KS, Ayouba A, Peeters M, Speede S, Shaw GM, Bushman FD, Brisson D, Rayner JC, Sharp PM, Hahn BH. 2016. Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nat Commun 7:11078. doi: 10.1038/ncomms11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oyola SO, Ariani CV, Hamilton WL, Kekre M, Amenga-Etego LN, Ghansah A, Rutledge GG, Redmond S, Manske M, Jyothi D, Jacob CG, Otto TD, Rockett K, Newbold CI, Berriman M, Kwiatkowski DP. 2016. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J 15:597. doi: 10.1186/s12936-016-1641-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cowell AN, Loy DE, Sundararaman SA, Valdivia H, Fisch K, Lescano AG, Baldeviano GC, Durand S, Gerbasi V, Sutherland CJ, Nolder D, Vinetz JM, Hahn BH, Winzeler EA. 2017. Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of Plasmodium vivax from unprocessed clinical samples. mBio 8:e02257-16. doi: 10.1128/mBio.02257-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizk G, Lavenier D, Chikhi R. 2013. DSK: k-mer counting with very low memory usage. Bioinformatics 29:652–653. doi: 10.1093/bioinformatics/btt020. [DOI] [PubMed] [Google Scholar]
- 24.Dolan Thomas J, Hatcher CP, Satterfield DA, Theodore MJ, Bach MC, Linscott KB, Zhao X, Wang X, Mair R, Schmink S, Arnold KE, Stephens DS, Harrison LH, Hollick RA, Andrade AL, Lamaro-Cardoso J, de Lemos AP, Gritzfeld J, Gordon S, Soysal A, Bakir M, Sharma D, Jain S, Satola SW, Messonnier NE, Mayer LW. 2011. sodC-based real-time PCR for detection of Neisseria meningitidis. PLoS One 6:e19361. doi: 10.1371/journal.pone.0019361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bushnell B. BBTools software package. https://jgi.doe.gov/data-and-tools/bbtools/.
- 27.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 28.Ainsworth D, Sternberg MJE, Raczy C, Butcher SA. 2017. k-SLAM: accurate and ultra-fast taxonomic classification and gene identification for large metagenomic data sets. Nucleic Acids Res 45:1649–1656. doi: 10.1093/nar/gkw1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seemann T. Snippy: rapid haploid variant calling and core SNP phylogeny. https://github.com/tseemann/snippy.
- 30.Topaz N, Caugant DA, Taha MK, Brynildsrud OB, Debech N, Hong E, Deghmane AE, Ouedraogo R, Ousmane S, Gamougame K, Njanpop-Lafourcade BM, Diarra S, Fox LM, Wang X. 2019. Phylogenetic relationships and regional spread of meningococcal strains in the meningitis belt, 2011–2016. EBioMedicine 41:488–496. doi: 10.1016/j.ebiom.2019.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. 2016. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17:132. doi: 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jolley KA, Bray JE, Maiden MCJ. 2018. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124–124. doi: 10.12688/wellcomeopenres.14826.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Retchless AC, Kretz CB, Chang HY, Bazan JA, Abrams AJ, Norris Turner A, Jenkins LT, Trees DL, Tzeng YL, Stephens DS, MacNeil JR, Wang X. 2018. Expansion of a urethritis-associated Neisseria meningitidis clade in the United States with concurrent acquisition of N. gonorrhoeae alleles. BMC Genomics 19:176. doi: 10.1186/s12864-018-4560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H. seqtk: toolkit for processing sequences in FASTA/Q formats. https://github.com/lh3/Seqtk.
- 36.Berger SA, Krompass D, Stamatakis A. 2011. Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst Biol 60:291–302. doi: 10.1093/sysbio/syr010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brynildsrud OB, Eldholm V, Bohlin J, Uadiale K, Obaro S, Caugant DA. 2018. Acquisition of virulence genes by a carrier strain gave rise to the ongoing epidemics of meningococcal disease in West Africa. Proc Natl Acad Sci U S A 115:5510–5515. doi: 10.1073/pnas.1802298115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katz LS, Griswold T, Williams-Newkirk AJ, Wagner D, Petkau A, Sieffert C, Van Domselaar G, Deng X, Carleton HA. 2017. A comparative analysis of the Lyve-SET phylogenomics pipeline for genomic epidemiology of foodborne pathogens. Front Microbiol 8:375. doi: 10.3389/fmicb.2017.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai W, Nunziata S, Rascoe J, Stulberg MJ. 2019. SureSelect targeted enrichment, a new cost effective method for the whole genome sequencing of Candidatus Liberibacter asiaticus. Sci Rep 9:18962. doi: 10.1038/s41598-019-55144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrison OB, Claus H, Jiang Y, Bennett JS, Bratcher HB, Jolley KA, Corton C, Care R, Poolman JT, Zollinger WD, Frasch CE, Stephens DS, Feavers I, Frosch M, Parkhill J, Vogel U, Quail MA, Bentley SD, Maiden MC. 2013. Description and nomenclature of Neisseria meningitidis capsule locus. Emerg Infect Dis 19:566–573. doi: 10.3201/eid1904.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson JR. 2014. Random-primed, Phi29 DNA polymerase-based whole genome amplification. Curr Protoc Mol Biol 105:Unit 15 13. doi: 10.1002/0471142727.mb1513s105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unmapped reads are available at SRA BioProject under accession no. PRJNA590135.