

Abstract
Abstract
Although vaccine development is being undertaken at a breakneck speed, there is currently no effective antiviral drug for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing COVID-19. Therefore, the present study aims to explore the possibilities offered by naturally available and abundant flavonoid compounds, as a prospective antiviral drug to combat the virus. A library of 44 citrus flavonoids was screened against the highly conserved Main Protease (Mpro) of SARS-CoV-2 using molecular docking. The compounds which showed better CDocker energy than the co-crystal inhibitor of Mpro were further revalidated by flexible docking within the active site; followed by assessment of drug likeness and toxicity parameters. The non-toxic compounds were further subjected to molecular dynamics simulation and predicted activity (IC50) using 3D-QSAR analysis. Subsequently, hydrogen bonds and dehydration analysis of the best compound were performed to assess the binding affinity to Mpro. It was observed that out of the 44 citrus flavonoids, five compounds showed lower binding energy with Mpro than the co-crystal ligand. Moreover, these compounds also formed H-bonds with two important catalytic residues His41 and Cys145 of the active sites of Mpro. Three compounds which passed the drug likeness filter showed stable conformation during MD simulations. Among these, the lowest predicted IC50 value was observed for Taxifolin. Therefore, this study suggests that Taxifolin, could be a potential inhibitor against SARS-CoV-2 main protease and can be further analysed by in vitro and in vivo experiments for management of the ongoing pandemic.
Graphic abstract

Keywords: COVID-19, SARS-CoV-2, Flavonoids, Main protease, Molecular docking, Molecular dynamic
Introduction
The COVID-19 pandemic caused by SARS-CoV-2 has emerged as a global threat affecting more than 17 million people globally [1]. The rapid and dynamic spread of the disease has baffled healthcare officials across the world. With 668,910 global mortality till date (6th August 2020), and with no effective treatment available, the quest for discovering a drug or vaccine against SARS-CoV-2 has accelerated [1]. In this crisis, a renewed interest in traditional phytomedicinal plants or natural compounds has garnered significant attention due to its medicinal properties, low toxicity and adverse effects [2, 3]. At present, antagonistic approach by blocking the integral replication system of SARS-CoV-2 is being considered as one of the effective antiviral therapeutic strategies. SARS-CoV-2 is a positive sense single stranded RNA virus. Like any other virus, it uses the host cell machinery to translate its large polyprotein necessary to procreate. However, to be functional, the polyprotein has to be cleaved by viral main protease (Mpro) and other papain like proteases. As inhibition of Mpro would stop viral replication, this enzyme has become one of the best characterized and enticing drug targets for SARS-CoV-2 [4, 5].
Considering the urgent need for an effective therapeutic agent for SARS-CoV-2 virus, several researchers have emphasized the importance of natural compounds [2, 3, 6]. Natural plant products have been used for generations in Traditional Chinese as well as Indian Ayurvedic Medicines as antiviral treatments. Moreover, natural compounds are also a fundamental source for a large number of modern drugs. An important natural compound worth mentioning is chloroquine and hydroxychloroquine derived from secondary metabolites of Cinchona tree which is under clinical trial and has shown potential anti SARS-CoV-2 properties [7]. Among the most readily accessible secondary metabolites are flavonoids found abundantly in citrus fruits and studies have demonstrated their antiviral activities [8, 9]. In fact, a few studies have discussed the importance of flavonoid as an antiviral agent against other respiratory diseases including SARS-CoV-1 [10, 11]. Therefore, exploring the citrus flavonoids as inhibitors for SARS-CoV-2 can prove helpful in the search for an alternative first line treatment option.
The computational approaches under the urgent circumstances offer a great opportunity for testing the hypothesis of potential drug effect of the natural compounds. The present study intends to identify putative candidate compound as a potential therapeutic agent for COVID-19 by using computational approaches to screen natural citrus flavonoid compounds as potential inhibitors of the main protease (Mpro) of SARS-CoV-2.
Materials and methods
Selection and preparation of the compound library of flavonoid compounds
A total of 44 flavonoids were collected from already published articles to build the compound library [12]. The library of the flavonoids was selected based on the chemical skeleton C6-C3-C6.. Although the library of flavonoid compounds appears to be considerably small, the compounds are chemically similar to the co-crystal inhibitor of SARS-CoV-2 Mpro and thus increases the likelihood of finding potential inhibitor. The database of compound library was prepared and energy minimization was performed using the standard protocol of discovery studio 2018 (DS v 2018) [13].
Preparation of the target enzyme and selection of binding site
X-ray crystal structure of the enzyme main protease (Mpro), (PDB ID: 6M2N), co-crystalized with a flavonoid type (C6-C3-C6) inhibitor (3WL) was obtained from the Protein Data Bank websites [14, 15]. The target enzyme was cleaned, prepared and energy minimized using the standard protocol of DS v 2018 before the docking study [16]. The active binding site sphere with coordinates of X: − 33.0907, Y: − 63.8424, Z: 41.7832 and radius 9.2 Å was selected around the co-crystal inhibitor (3WL) present with the enzyme for docking.
Simulation-based docking of the compounds library
The entire compounds library was docked with the target using simulation-based docking protocol CDocker of the DS v 2018 which uses a CHARMm-based molecular dynamics (MD) algorithm to dock compounds into the active binding site of a receptor [17]. After docking, the binding positions of the compounds were analysed and compared with the binding poses of the co-crystal inhibitor (3WL) of the target.
Flexible docking and binding free energy calculation
In the CDocker protocol, the target protein or enzyme is taken as a rigid structure which may affect the accuracy in posing and scoring of ligands in the docking process. However, in nature, proteins or enzymes are not rigid and hence protein flexibility is a major factor which influences docking accuracy [18]. Therefore, we selected the best compounds from the preliminary simulation-based docking for further analysis using ‘Flexible Docking’ protocol of DS v 2018. This protocol allows for some receptor flexibility by the movement of side-chains of specified amino acids during docking which allows the receptor to adapt to different ligands in an induced-fit model [19]. The protocol uses a combination of components from other protocols like LibDock and CDocker to perform the docking, and is based on methods within CHARMm to sample side-chain and ligand conformations. From the final refined poses, the highest scored complex as defined by the calculated CDocker energy (kcal/mol) was considered for analysis and for the calculation of binding free energy. The binding free energy of the docked complexes was analysed using ‘Calculate Binding Energies’ protocol of DS v 2018.
Toxicity analysis
The compounds selected from the flexible docking were further analysed for different types of toxicities like tumorigenic, mutagenic, reproductive effective, irritant and for drug likeness using ORISIS Data Warrior 5.2.1 [20].
Molecular dynamics simulation study
The compounds which passed the toxicity analysis were then subjected to molecular dynamics simulation study to find their stability and to validate the docking study using DS v 2018. The receptor-ligand complexes of the compounds generated from the flexible docking study were taken for molecular dynamics study along with original X-ray crystal structure of the target with the co-crystal inhibitor. The complexes were initially prepared to remove any error in the structure. The CHARMm36 force field was used in the parametrization process of both the protein and ligands. The parametrization of the protein and ligands was carried out using protein–ligand complexes generated from flexible docking analysis using the default assign force field tool of DS v 2018. These complexes were then solvated using explicit periodical boundary condition in water cubic box of size 10 Å x 10 Å and 0.15 M NaCl was added to neutralize the system. Subsequently, energy minimization (5000 steps steepest descent and 5000 steps conjugate gradient), heating (20 ps) and equilibration (500 ps) were performed using ‘Standard Dynamic Cascade’ protocol of DS v 2018. Finally, the production was performed for 30 ns in NVT ensemble at 310 K for the whole protein–ligand complex where snapshots were saved every 2 ps. To assess the convergence of results after MD simulation, replicates of the analysis for 30 ns were performed. For the electrostatics calculations, the particle mesh ewald (PME) method was used. To constrain bonds containing hydrogen, the SHAKE algorithm was used and the time step was 2 fs. Taking into account the starting structure as reference for the entire protein–ligand complex, root-mean-square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (ROG) were computed to evaluate receptor-ligand conformation changes and their stability. The RMSD, RMSF and ROG values were calculated using ‘Analyze Trajectory’ protocol of simulation tool of DS v 2018. Over the course of the simulation, the distance of different hydrogen bonds formed were also analysed. Finally, different non-bond interactions were also analysed from the average interaction structure of the receptor-ligand complexes by comparing with the starting complexes [21, 22].
MM-PBSA based binding free energy calculation
The MM-PBSA based calculation of binding free energy (ΔG) is one of the important parameters to estimate the binding affinity of a compound to a target [23]. It also provides fast and accurate prediction of absolute binding affinity of a compound within the active binding site of a target protein in the form of binding free energy which is important for thermodynamic stability and particular potency of the compound in terms of inhibition or activation [24]. In this study, the binding free energies for each protein–ligand complex were calculated by using ‘Binding Free Energy–Single Trajectory’ protocol of DS v 2020 with the MM-PBSA method. The binding free energies of all the generated conformations were calculated, and finally, the average binding free energy (ΔG) was determined for each protein–ligand complexes.
Predicted activity determination
The predicted activity of the compounds was determined by 3D-QSAR analysis. 3D-QSAR analysis gives an idea on the activities of the compounds based on the similarities of their structural or physicochemical properties [25]. A total of 27 synthesized compounds reported as Mpro inhibitor were obtained from PostEra database along with their IC50 (µM) values to build the 3D-QSAR model [26]. The IC50 values of the compounds were converted in to pIC50 values by using an online tool before using in the study [27]. Initially, the 27 compounds were aligned using molecular overlay method (50% electrostatic and 50% steric fields), which was then divided into training set (16 compounds) and test set (11 compounds) based on molecular diversity in each group. The Grid BasedTemp model was generated using two probe types to calculate energy grids which indicates electrostatic and steric effects. The regression analysis was performed by cross validated Partial Least Square (PLS) method of LOO (Leave-one-out). The pIC50 values served as dependant variables to build the model which validates the test set for stability and predictability.
Analysis of atom wise contributions towards binding affinity
Different atoms of a compound have individual contributions towards the binding affinity and stability in the active binding site of target protein. These properties can provide valuable information to select a compound as a lead candidate for further drug design and discovery process. The role of the specific atoms in the overall binding affinity of the best poses for the selected compounds along with co-crystal inhibitor 3WL was calculated using SeeSAR bioinformatics tool [28].
Results and discussion:
In the preliminary docking study, CDocker energy as well as the CDocker interaction energy were calculated and considered for the screening of the compounds. The CDocker energy of Discovery Studio (DS) v 2018 provides comparatively accurate information regarding the binding affinity of the compounds in the active site of the target proteins [17]. On the other hand, CDocker interaction energy provides the different non-bonded interactions within the binding site of the target site of the protein [29]. In this study, since the screening compounds were the flavonoid compounds from citrus species, the flavonoid like co-crystal ligand 3WL of Mpro was used as control for the entire study. Molecular docking revealed that 5 compounds out of the 44 selected Citrus species flavonoid compounds showed better CDocker energy and CDocker interaction energy than the co-crystal ligand 3WL (Table 1).
Table 1.
Preliminary simulation-based docking results of the top five flavonoid compounds
| Compound code | Compound name | CDocker Energy (kcal/mol) | CDocker Interaction Energy (kcal/mol) |
|---|---|---|---|
| 3WL | - | − 30.6785 | − 31.338 |
| CF3 | Taxifolin | − 30.8173 | − 36.7392 |
| CF5 | Eriodictyol | − 32.5776 | − 37.1241 |
| CF7 | Isoscutellarein | − 33.0279 | − 36.7776 |
| CF8 | Luteolin | − 32.9575 | − 36.1835 |
| CF10 | Quercetin | − 33.8277 | − 37.0433 |
Considering the dynamic nature of the physiological conditions, the best 5 compounds were allowed to redock with the target protein in a flexible mode. During the flexible docking, the residues of the binding site of the target protein were kept flexible. The flexible docking analysis of the best 5 compounds also showed better CDocker energy as well as CDocker interaction energy (Table 2). The binding energy of the compounds to the target protein Mpro was calculated to understand the spontaneity of formation of drug-target/ligand–receptor complex suggesting the stable drug-target complex. The calculated binding energy of the compounds showed lower binding energy in comparison to the control co-crystal inhibitor 3WL shown in Table 2.
Table 2.
Flexible docking binding energy of the best five flavonoid compounds as compared to co-crystal inhibitor
| Compound | CDocker energy (kcal/mol) | CDocker interaction energy (kcal/mol) | LibDock Score | No of H-Bond | Binding energy (kcal/mol) |
|---|---|---|---|---|---|
| 3WL | − 34.8212 | − 35.2188 | 102.348 | 2 | − 24.9866 |
| Quercetin | − 39.6003 | − 42.9988 | 102.055 | 4 | − 31.5229 |
| Taxifolin | − 38.3342 | − 45.9748 | 109.909 | 5 | − 38.6097 |
| Isoscutellarein | − 37.2348 | − 38.4106 | 92.9587 | 2 | − 25.8374 |
| Eriodictyol | − 37.1527 | − 42.5989 | 97.6221 | 4 | − 39.0123 |
| Luteolin | − 36.3285 | − 41.5773 | 104.515 | 6 | − 35.0685 |
In the present study, the top 5 compounds formed higher number of H-bonds with the target protein than the co-crystal inhibitor 3WL. This may suggest that the test compounds have a higher tolerability against target protein putative mutations than 3WL [30]. The interaction and the number of H-bonds formed for the 5 screened compounds with Mpro are shown in Fig. 1.
Fig. 1.

Docking interaction of a 3WL, b Taxifolin (CF3), c Eriodictyol (CF5), d Isoscutellarein (CF7), e Luteolin (CF8) and F Quercetin (CF10) with Mpro. The green dashed line indicates the H-bonds between the ligands and the interacting residues of Mpro
From Fig. 1, it was observed that the compounds Taxifolin, Eriodictyol, Luteolin and Quercetin formed 4 or more H-bonds with the active site of SARS-CoV-2 Mpro, whereas compound Isoscutellarein and the co-crystal inhibitor 3WL formed 2 H-bonds with the target protein. Analysing the residues involved in interactions between the compounds and Mpro, showed that all the compounds interacted with the two important catalytic residues His41 and Cys145 of the active sites of Mpro. These two residues are present in the catalytic domain of SARS-CoV-2 Mpro and actively participate in the catalytic activities of Mpro. Hence, binding of the flavonoid compounds to these residues may reduce the catalytic activities of Mpro which eventually will lead to the reduction of viral replication.
The 5 compounds showing the best results in terms of CDocker energy, CDocker interaction energy, calculated binding energy and number of H-bonds were then subjected for the assessment of drug likeness and assessment of different toxicity parameters. It was observed that 3 compounds, namely Taxifolin, Eriodictyol and Luteolin did not show any toxicity against the toxicity parameters used in the study. On the other hand, the compounds Isoscutellarein and Quercetin showed the presence of mutagenic properties. Quercetin also showed the presence of tumorigenic property. The results of toxicity prediction and the drug likeness property analysis are shown in Table 3. Among all the screened compounds, Taxifolin possessed the highest drug likeliness property followed by Isoscutellarein and Luteolin. Based on the training dataset used by the ORISIS Data Warrior, the compounds with higher or positive drug likeliness values are considered as good drug candidates. Since the compounds Isoscutellarein and Quercetin showed the presence of toxic effects, we did not consider these compounds for further analysis. The non-toxic compounds, namely Taxifolin, Eriodictyol and Luteolin were further subjected to molecular dynamics simulation studies.
Table 3.
Toxicity and drug likeness analysis
| Compound | Drug likeness | Mutagenic | Tumorigenic | Reproductive Effective | Irritant |
|---|---|---|---|---|---|
| 3WL | 0.28194 | None | None | None | None |
| Taxifolin | 0.44477 | None | None | None | None |
| Eriodictyol | − 0.22006 | None | None | None | None |
| Isoscutellarein | 0.28194 | High | None | None | None |
| Luteolin | 0.28194 | None | None | None | None |
| Quercetin | − 0.082832 | High | High | None | None |
Molecular dynamics simulation study was performed to understand how the ligands bind to the receptor by mimicking in vitro and in vivo experiments [4]. Thereby, the RMSD, RMSF and ROG of the Mpro-ligand complexes were calculated over the simulation period of 30 ns and compared with the control (Mpro-3WL complex) to observe the stability of the complexes. To calculate the RMSD, RMSF and ROG of the complexes, the whole protein–ligand complexes were used. After completion of simulation, the RMSD plots for all the compounds were analyzed where it was observed that Mpro-3WL and Mpro-CF5 (Eriodictyol) had almost similar deviations with control complex (Mpro-3WL) within the simulation period. However, Mpro-CF3 (Taxifolin) and Mpro-CF8 (Luteolin) had comparatively higher deviation than Mpro-3WL; where Mpro-CF8 took more time (almost 10 ns) to reach plateau state (Fig. 2A). The fluctuations of the individual residues within the simulation period were plotted where the RMSF of the residues for the Mpro-3WL and Mpro-CF5 had almost similar pattern with minimum deviations from each other. For the complex Mpro-CF3 and Mpro-CF8, there were significant deviations which may indicate that the presence of ligands influenced the stability of the enzyme Mpro and changed its dynamic behaviour. Notably, the replicate analysis of the protein–ligand complexes showed similar RMSD deviations indicating a convergence of results. Comparing the RMSD of the complexes, Mpro-CF8 showed fluctuations all over the regions. Mpro-CF3 showed considerable fluctuations within residue 248 to 256 (Fig. 2B). These findings were also supported by calculated radius of gyration (ROG) for the Mpro-ligand complexes (Fig. 2C).
Fig. 2.

a Root mean square deviation of the receptor-ligand complexes; b Root mean square fluctuations of the residues of receptor-ligand complexes and c Radius of gyrations of the receptor-ligand complexes within the 30 ns simulation. In all the cases the complex Mpro-3WL is represented by blue line, Mpro-CF3 is represented by green line, Mpro-CF5 is represented by yellow line and Mpro-CF8 is represented by red line
In this study, we further analysed the interaction between the compounds with Mpro and checked the formation of H-bonds after 30 ns MD simulation. The interaction pattern of the compounds after 30 ns MD simulation is shown in Fig. 3. After 30 ns MD simulation, the co-crystal ligand 3WL formed 4 H-bonds, whereas compounds Taxifolin and Eriodictyol formed 7 and 4 H-bonds respectively with the target protein. Luteolin formed 3 H-bonds with the residues of the active site of SARS-CoV-2 Mpro after MD simulation for 30 ns. Although all compounds formed H-bonds with the catalytic residues i.e. His41 and Cys145 as observed during molecular docking, but after 30 ns simulation only Taxifolin interacted with these residues forming H-bonds. The co-crystal ligand 3WL formed weak interaction with Cys145 after 30 ns of MD simulation.
Fig. 3.

Interaction of a 3WL, b Taxifolin, c Eriodictyol and d Luteolin with Mpro after MD simulation for 30 ns. The green dashed line indicates the H-bonds between the ligands and the interacting residues of Mpro
The structural conformations of the protein–ligand complexes before and after MD simulation were also observed by superpositiong the complexes and are depicted in Fig. 4.
Fig. 4.

Superposition of the protein ligand complex of a Mpro-3WL, b Mpro-CF3, c Mpro-CF5 and d Mpro-CF8. The green colour complexes represent the protein–ligand complexes before MD simulation and red colour complexes represent the protein–ligand complexes after 30 ns MD simulation
In the MD simulation analysis, we analysed the different H-bonds formed within the active site of the target proteins with the flavonoid compounds during the process of simulation upto 30 ns. The number of H-bonds formed and their distances within the simulation period for each conformation were generated (Fig. 5). In Mpro-3WL complex total 5 hydrogen bonds were found, where 1 H-bond with Glu166 residue showed almost consistent average distance with low deviation. However, the other 4 H-bonds with Thr190, Asn142 and Glu166 initially had very large average distance but after approximately 18 ns it was reduced and stabilized. In Mpro-CF3 complex 7 H-bonds were observed where the H-bond with Leu50 residue showed substantial deviations around 10–20 ns and subsequently stabilized by the end of the simulation. In Mpro-CF5 complex 4 H-bonds were observed among which 2 H-bonds with Asp48 residue remained stable throughout the simulation. The H-bond with Ser46 residue stabilized within 4 ns of simulation, while the H-bond with Glu166 residue remained highly unstable with considerable fluctuation of distances throughout the simulation. In Mpro-CF8 complex 3 H-bonds were formed and the distance of bond with Asn119 varied throughout the simulation. The bond with Asp48 showed deviation around 18–26 ns and then reverted to its previous state. The distance for the H-bond with Ser46 gradually decreased to less than 5 Å. The 2D interaction generated for the complexes before and after simulation showed that the number of H-bonds formed increased for Mpro-3WL and Mpro-CF3. For Mpro-CF5, the number of H-bonds remained same but the interacting residues had changed. But for Mpro-CF8 the number of H-bonds decreased after simulation.
Fig. 5.

Distance of different hydrogen bonds formed within the simulation period for a 3WL; b Taxifolin, c Eriodictyol and d Luteolin
During MD simulation analysis, the binding free energies (ΔG) of the protein–ligand complexes were calculated upto 30 ns using MM-PBSA based method. From the result, the average ΔG of the Mpro-3WL complex was found to be − 51.1666 kcal/mol. The average ΔG of the Mpro-CF3, Mpro-CF5 and Mpro-CF8 were found to be − 60.3367 kcal/mol, − 68.3025 kcal/mol and − 55.7587 kcal/mol, respectively. It has been observed from the MM-PBSA analysis that the complexes formed between the flavonoids and the target, possessed lower ΔG than the complex of receptor-co crystal ligand. This indicates the formation of stable complexes with spontaneous interaction by the test ligands in the active binding pocket. The binding free energies (ΔG) of protein–ligand complexes during the MD simulation period are shown in Fig. 6.
Fig. 6.

Flucutation of binding free energies (ΔG) of protein–ligand complexes during the MD simulation period. The blue line represents the complex Mpro-3WL, brown line represents Mpro-CF3, grey line represents Mpro-CF5 and yellow line represents Mpro-CF8 complexes
The predicted activity (IC50) of the compounds was determined with the help of 3D-QSAR analysis. As the IC50 value of 3WL has not yet been reported, the IC50 value of 3WL was predicted by generating 3D-QSAR model from the inhibitor deposited in PostEra website [26]. To calculate the energy potential in 3D-QSAR method, 3 dimensional structures of a set of compounds were used. The calculated potential energies were then used as descriptors to build the 3D-QSAR model to corelate the 3D-structures and their biological activities. The generated QSAR model gives the information on correlation between the molecular field and the biological activities of the compounds [31]. In this study, the predicted activity i.e. IC50 of the compounds as well as control were determined by using the following linear equation.
where NEP: the number of descriptors of electrostatic potential (EP); CEP(i): model coefficient for electrostatic potential descriptor i; VEP: value of electrostatic potential on a grid point; NVDW: number of descriptors of van der Waals (VDW) interaction: CVDW(i): model coefficient for VDW descriptor i; VVDW: van der Waals interaction energy on a grid point.
The linear plot of the training set and the test set are depicted in Fig. 7. The determined R2 value for training set was found to be 0.912 and for test set was found to be 0.846 during validation. From the 3D-QSAR analysis, the predicted IC50 value of 3WL was observed to be 5.98 μM, whereas the compound Taxifolin was observed to be 9.63 μM followed by Luteolin (14.47 μM) and Eriodictyol (16.08 μM). As the actual IC50 value of 3WL has not been reported yet, the predicted IC50 value will not give the actual idea of its minimum inhibitory concentration. Thus, the complexes were considered for further SeeSAR analysis to assess the role of individual atoms towards the binding affinity.
Fig. 7:
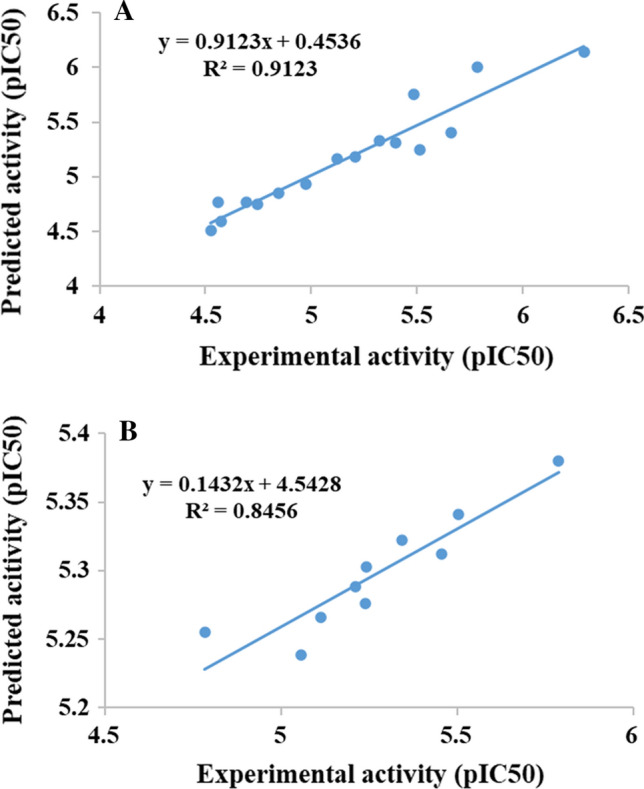
3D-QSAR plot of a training set and b test set
To further assess the binding affinity of 3WL and Taxifolin with Mpro before and after 30 ns MD simulation, HYDE (Hydrogen bonds and Dehydration) analysis was performed using SeeSAR of BiosolveIT [28]. HYDE analysis consistently designates hydrogen bonding between ligand and receptor, hydrophobic effect as well as desolvation. HYDE also helps in predicting the particular region of the complex which undergoes favourable and unfavourable binding ligand receptor. The HYDE scoring determined the Gibb’s free energy by calculating the difference between bonded and unbonded states of the complex [32]. The specific atoms which were favourable for good binding affinity (dark green sphere) and their individual HYDE values for the best compound Taxifolin and the co-crystal inhibitor 3WL in both before and after MD simulation, are shown in Fig. 8. Identification of the role of atoms present in the ligands is crucial in predicting overall binding affinity or interactions with the target sites of the protein. From Fig. 8a, it was observed that in case of 3WL (5,6,7‐trihydroxy‐2‐phenyl‐4H‐chromen‐4‐one), before MD simulation, the phenyl ring at 2nd position had major contributions towards the overall HYDE score (kcal/mol). But mainly the oxygen atom of the 6-hydroxyl group, carbon atom of the carbonyl group of 4 position and oxygen atom of the 1 position of the bicyclic ring system (with red coronas) had negative impact on the overall binding affinity of 3WL. Similarly, in case of Taxifolin ((2R,3S)‐2‐(3,4‐dihydroxyphenyl)‐3,5,7‐trihydroxy‐3,4‐dihydro‐2H‐1‐benzopyran‐4‐one), before MD simulation, the oxygen atom at the 1 position and the oxygen atom of the hydroxyl group at 3 position had negative impact towards the binding affinity of the compound. Moreover, the carbon atom at 7 position and the oxygen atom of the hydroxyl group at 7 position also demonstrated negative impact on the binding affinity. On the other hand, the atoms of the 3,4 dihydroxyphenyl group at 2 position of the bicyclic ring showed positive contributions towards the overall binding affinity of the molecule. In the bicyclic ring, the oxygen atom of the ketone group and carbon atom at the 6 and 8 position had positive effect towards the binding affinity of the molecule (Fig. 8b).
Fig. 8.
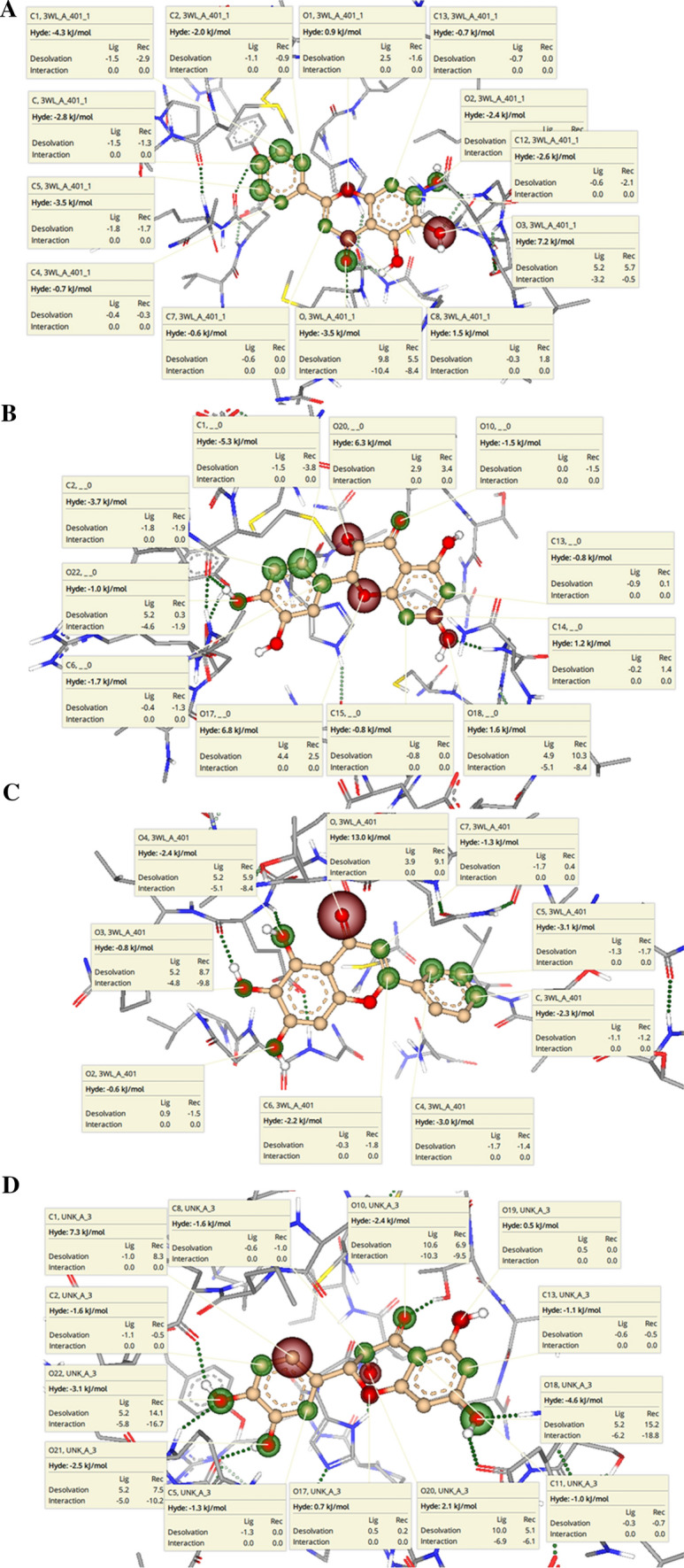
Visualization of binding of Mpro with a 3WL and b Taxifolin before MD simulation and c 3WL and d Taxifolin after MD simulation in SeeSAR with quantification of HYDE of the important non-hydrogen atoms which contribute in binding affinities
HYDE analysis was performed for the complexes after MD simulation also and represented in (Fig. 8c, d). From Fig. 8c, it was observed that in case of 3WL (5,6,7‐trihydroxy‐2‐phenyl‐4H‐chromen‐4‐one), the phenyl ring at second position had major contributions towards the overall HYDE score (kcal/mol). Further, the carbon atoms at 2 and 3 positions, and oxygen atoms of the hydroxyl groups at 5, 6 and 7 position had also showed positive contributions towards the overall binding affinity. However, the oxygen atom of the carbonyl group at 4 position (with red corona) was seen to have negative impact on the overall binding affinity of 3WL. Similarly, in case of Taxifolin ((2R,3S)‐2‐(3,4‐dihydroxyphenyl)‐3,5,7‐trihydroxy‐3,4‐dihydro‐2H‐1‐benzopyran‐4‐one), after MD simulation, the oxygen atom at the 1 position and the oxygen atom of the hydroxyl group at 3 and 5 position had negative impact towards the binding affinity of the compound. The carbon atom at 6 position of the 3,4-dihydroxyphenyl cyclic ring system present at 2 position, also had a major negative impact on the binding affinity. On the other hand, the carbon atoms of the 3,4-dihydroxyphenyl group at 2 and 5 position, and oxygen atoms of the hydroxyl groups at 3 and 4 positions showed positive contributions towards the overall binding affinity of the molecule. From the bicyclic ring, the oxygen atom of the ketone group and carbon atom at the 3, 4a and 6 position, and oxygen atom of the hydroxyl group at 7 position had positive effect towards the binding affinity of the molecule (Fig. 8d).
The ranges of binding affinity of Taxifolin and co-crystal inhibitor 3WL were also calculated before and after MD simulation and shown in Table 4. From the results, it was found that before MD simulation, Taxifolin had less binding affinity towards the target protein Mpro due to the major negative impact governed by the orientation of oxygen atom at 1 position and oxygen atom of the hydrpxyl group at 3 position. However, in case of complexes after MD simulation, Taxifolin showed better binding affinity towards the target protein Mpro than the co-crystal ligand 3WL. This suggests that during the course of reaction, Taxifolin possesses better binding affinity towards Mpro of SARS-CoV-2.
Table 4.
Predicted binding affinity ranges of 3WL and Taxifolin with Mpro before and after MD simulation
| Before MD simulation | After MD simulation | |||
|---|---|---|---|---|
| Compound | Lower boundary for estimated affinity | Upper boundary for estimated affinity | Lower boundary for estimated affinity | Upper boundary for estimated affinity |
| 3WL | 0.344957 mM | 34.273 mM | 20.240 mM | 2010.969 mM |
| Taxifolin | 68.357 mM | 6791.721 mM | 1.838 mM | 182.666 mM |
Taxifolin is a widely distributed natural flavonoid and waste material of forest industry offers an economically viable source for its extraction. Taxifolin has earlier been reported for its antiviral effects against coxsackievirus B and antiradical activities [33, 34]. We believe that no study has been undertaken concerning taxifolin’s potential inhibitory activities against respiratory viruses. It is worth mentioning that our study corroborates a recently published report of potential activity of taxifolin against the main protease of SARS-CoV-2 [35].
Conclusion
In this study, we screened flavonoid compounds of citrus species for their activity against SARS-CoV-2 targeting Mpro of the virus. From the computational analysis, we conclude that Taxifolin is the best druglike compound among all the selected flavonoids of citrus species without toxicity. Taxifolin binds to the target protein with comparatively better binding affinity than the co-crystal flavonoid like inhibitor 3WL. It forms H-bonds with two important catalytic residues of SARS-CoV-2 Mpro after molecular docking and remains stable till the completion of MD simulation for 30 ns. The results of the study suggest its potential inhibitory activity against SARS-CoV-2 Mpro with IC50 value 9.63 μM. The substantial effect of Taxifolin against the virus observed during the in silico study may be further validated with invitro and invivo experiments for clinical use of the compound. The present study will help in future endeavours for discovering a potential and effective treatment for COVID-19.
Acknowledgements
We acknowledge the support of the Drug Design Software facility of the Department of Pharmaceutical Sciences, Dibrugarh University established by the support of University Grants Commission, New Delhi (India) under UGC-SAP (DRS-I) [F.3-13/2016/DRS-I (SAP-II)]. Neelutpal Gogoi also acknowledge the supports from BioSolveIT GmbH, Sankt Augustin, Germany, for providing the special licence of SeeSAR software to carry out a part of the computational analysis.
Author contributions
NG, BG, PC & AKG developed the idea of the research work. NG & BG performed the software-based experimental part. NG, BG analysed the results and PC verified them. NG, BG, PC and AKG prepared the manusript. AD & DC provided input in the development stage of the manuscript. All the authors finalized and approved the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Compliance with ethical standards
Conflict of Interest
The authors declare that they have are no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO Coronavirus Disease (COVID-19) Dashboard WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/. Accessed 6 Aug 2020
- 2.Antonio ADS, Wiedemann LSM, Veiga-Junior VF. Natural products’ role against COVID-19. RSC Adv. 2020;10:23379–23393. doi: 10.1039/d0ra03774e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Islam MT, Sarkar C, El-Kersh DM, et al. Natural products and their derivatives against coronavirus: a review of the non-clinical and pre-clinical data. Phyther Res. 2020 doi: 10.1002/ptr.6700. [DOI] [PubMed] [Google Scholar]
- 4.Mahanta S, Chowdhury P, Gogoi N, et al. Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: an in silico based approach. J Biomol Struct Dyn. 2020;0:1–10. doi: 10.1080/07391102.2020.1768902. [DOI] [PubMed] [Google Scholar]
- 5.Gildenhuys S. Expanding our understanding of the role polyprotein conformation plays in the coronavirus life cycle. Biochem J. 2020;477:1479–1482. doi: 10.1042/BCJ20200223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Yang L. Turning the tide: natural products and natural-product-inspired chemicals as potential counters to SARS-CoV-2 Infection. Front Pharmacol. 2020;11:1013. doi: 10.3389/fphar.2020.01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Wang Y, Agostinis P et al 2020 Is hydroxychloroquine beneicial for COVID-19 patients? Cell Death Dis 11:512. 10.1038/s41419-020-2721-8 [DOI] [PMC free article] [PubMed]
- 8.Zakaryan H, Arabyan E, Oo A, Zandi K. Flavonoids: promising natural compounds against viral infections. Arch Virol. 2017;162:2539–2551. doi: 10.1007/s00705-017-3417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cataneo AHD, Kuczera D, Koishi AC, et al. The citrus flavonoid naringenin impairs the in vitro infection of human cells by Zika virus. Sci Rep. 2019;9:1–15. doi: 10.1038/s41598-019-52626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jo S, Kim S, Shin DH, Kim MS. Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem. 2020;35:145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meneguzzo F, Ciriminna R, Zabini F, Pagliaro M. Review of evidence available on hesperidin-rich products as potential tools against COVID-19 and hydrodynamic cavitation-based extraction as a method of increasing their production. Processes. 2020;8:1–18. doi: 10.3390/PR8050549. [DOI] [Google Scholar]
- 12.Gattuso G, Barreca D, Gargiulli C, et al. Flavonoid composition of citrus juices. Molecules. 2007;12:1641–1673. doi: 10.3390/12081641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.N Gogoi D Chetia B Gogoi A Das 2019 Multiple-targets Directed Screening of Flavonoid Compounds from Citrus Species to find out Antimalarial Lead with Predicted Mode of Action: An In Silico and Whole Cell-based In vitro Approach. Curr Comput Aided Drug Des 16:. https://doi.org/10.2174/1573409916666191226103000 [DOI] [PubMed]
- 14.RCSB PDB - 6M2N: SARS-CoV-2 3CL protease (3CL pro) in complex with a novel inhibitor. https://www.rcsb.org/structure/6m2n. Accessed 1 Aug 2020
- 15.RCSB PDB: Homepage. https://www.rcsb.org/. Accessed 1 Aug 2020
- 16.AK Goswami N Gogoi BJ Gogoi HK Sharma 2019 Network-pharmacology and DFT based approach towards identification of leads from Homalomena aromatica for multi-target in-silico screening on Entamoeba histolytica proteins. Curr Drug ther 14:. https://doi.org/10.2174/1574885514666190801102336
- 17.Wu G, Robertson DH, Brooks CL, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-a CHARMm-based MD docking algorithm. J Comput Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- 18.Lexa KW, Carlson HA. Protein flexibility in docking and surface mapping. Q Rev Biophys. 2012;45:301–343. doi: 10.1017/S0033583512000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koska J, Spassov VZ, Maynard AJ, et al. Fully automated molecular mechanics based induced fit protein-Ligand docking method. J Chem Inf Model. 2008;48:1965–1973. doi: 10.1021/ci800081s. [DOI] [PubMed] [Google Scholar]
- 20.Sander T, Freyss J, Von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 21.Lin CH, Chang TT, Sun MF, et al. Potent inhibitor design against h1n1 swine influenza: structure-based and molecular dynamics analysis for m2 inhibitors from traditional chinese medicine database. J Biomol Struct Dyn. 2011;28:471–482. doi: 10.1080/07391102.2011.10508589. [DOI] [PubMed] [Google Scholar]
- 22.Noha SM, Schmidhammer H, Spetea M. Molecular docking, molecular dynamics, and structure-activity relationship explorations of 14-Oxygenated N-Methylmorphinan-6-ones as potent μ-Opioid receptor agonists. ACS Chem Neurosci. 2017;8:1327–1337. doi: 10.1021/acschemneuro.6b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rastelli G, Del Rio A, Gianluca Degliesposti MS. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem. 2010;31:797–810. doi: 10.1002/jcc.21372. [DOI] [PubMed] [Google Scholar]
- 25.Vora J, Patel S, Sinha S, et al. Structure based virtual screening, 3D-QSAR, molecular dynamics and ADMET studies for selection of natural inhibitors against structural and non-structural targets of Chikungunya. J Biomol Struct Dyn. 2019;37:3150–3161. doi: 10.1080/07391102.2018.1509732. [DOI] [PubMed] [Google Scholar]
- 26.PostEra | COVID-19. https://covid.postera.ai/covid. Accessed 1 Aug 2020
- 27.Chandrabose Selvaraj, Sunil Kumar Tripathi KKR and SKS (2011) Tool development for Prediction of pIC50 values from the IC50 values - A pIC50 value calculator Association of Biotechnology and Pharmacy. In: Curr. Trends Biotechnol. Pharm. https://www.abap.co.in/tool-development-prediction-pic50-values-ic50-values-pic50-value-calculator. Accessed 1 Aug 2020
- 28.BioSolveIT-SeeSAR. https://www.biosolveit.de/SeeSAR/. Accessed 1 Aug 2020
- 29.S Rampogu M Rampogu Lemuel 2016 Network Based Approach in the Establishment of the Relationship between Type 2 Diabetes Mellitus and Its Complications at the Molecular Level Coupled with Molecular Docking Mechanism. Biomed Res Int 2016:. https://doi.org/10.1155/2016/6068437 [DOI] [PMC free article] [PubMed]
- 30.Liu X, Wang XJ. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J Genet Genomics. 2020;47:119–121. doi: 10.1016/j.jgg.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed N, Anwar S, Thet Htar T. Docking based 3D-QSAR study of tricyclic guanidine analogues of batzelladine K As anti-malarial agents. Front Chem. 2017;5:36. doi: 10.3389/fchem.2017.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider N, Hindle S, Lange G, et al. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J Comput Aided Mol Des. 2012;26:701–723. doi: 10.1007/s10822-011-9531-0. [DOI] [PubMed] [Google Scholar]
- 33.Galochkina AV, Anikin VB, Babkin VA, et al. Virus-inhibiting activity of dihydroquercetin, a flavonoid from Larix sibirica, against coxsackievirus B4 in a model of viral pancreatitis. Arch Virol. 2016;161:929–938. doi: 10.1007/s00705-016-2749-3. [DOI] [PubMed] [Google Scholar]
- 34.Roubalová L, Purchartová K, Papoušková B, et al. Sulfation modulates the cell uptake, antiradical activity and biological effects of flavonoids in vitro: an examination of quercetin, isoquercitrin and taxifolin. Bioorganic Med Chem. 2015;23:5402–5409. doi: 10.1016/j.bmc.2015.07.055. [DOI] [PubMed] [Google Scholar]
- 35.Fischer A, Sellner M, Neranjan S, et al. Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. Int J Mol Sci. 2020;21:3626. doi: 10.3390/ijms21103626. [DOI] [PMC free article] [PubMed] [Google Scholar]