

Abstract
Background:
A systemic pro-inflammatory state has been hypothesized to mediate the association between comorbidities and abnormal cardiac structure/function in heart failure with preserved ejection fraction (HFpEF). We conducted a proteomic analysis to investigate this paradigm.
Methods:
In 228 HFpEF patients from the multicenter PROMIS-HFpEF study, 248 unique circulating proteins were quantified by a multiplex immunoassay (Olink) and used to recapitulate systemic inflammation. In a deductive approach, we performed principal component (PC) analysis to summarize 47 proteins known a priori to be involved in inflammation. In an inductive approach, we performed unbiased weighted co-expression network analyses of all 248 proteins to identify clusters of proteins that overrepresented inflammatory pathways. We defined comorbidity burden as the sum of 8 common HFpEF comorbidities. We used multivariable linear regression and statistical mediation analyses to determine whether and to what extent inflammation mediates the association of comorbidity burden with abnormal cardiac structure/function in HFpEF. We also externally validated our findings in an independent cohort of 117 HFpEF cases and 30 comorbidity controls without HF.
Results:
Comorbidity burden was associated with abnormal cardiac structure/function and with PCs/clusters of inflammation proteins. Systemic inflammation was also associated with increased mitral E velocity, E/e’ ratio, and tricuspid regurgitation (TR) velocity; and worse right ventricular function (tricuspid annular plane systolic excursion [TAPSE] and right ventricular. [RV] free wall strain). Inflammation mediated the association between comorbidity burden and mitral E velocity (proportion mediated 19–35%), E/e’ ratio (18–29%), TR velocity (27–41%), and tricuspid annular plane systolic excursion (13%) (P<0.05 for all) but not RV free wall strain. TNF-R1, UPAR, IGFBP-7 and GDF-15 were the top individual proteins that mediated the relationship between comorbidity burden and echocardiographic parameters. In the validation cohort, inflammation was upregulated in HFpEF cases versus controls, and the most prominent inflammation protein cluster identified in PROMIS-HFpEF was also present in HFpEF cases (but not controls) in the validation cohort.
Conclusion:
Proteins involved in inflammation form a conserved network in HFpEF across 2 independent cohorts and may mediate the association between comorbidity burden and echocardiographic indicators of worse hemodynamics and RV dysfunction. These findings support the comorbidity-inflammation paradigm in HFpEF.
Keywords: heart failure with preserved ejection fraction, biomarkers, inflammation, echocardiography, comorbidities, mediation analysis
INTRODUCTION
Heart failure with preserved ejection fraction (HFpEF) is a systemic, multi-organ disorder that results in loss of cardiac, vascular, and skeletal muscle reserve, culminating in dyspnea and exercise intolerance. Several mechanisms have been proposed. The comorbidity-inflammation paradigm explains HFpEF as a result of a comorbidity-induced systemic pro-inflammatory state, which leads to endothelial dysfunction, coronary microvascular dysfunction, abnormal cardiac structure and function, and HFpEF.1 Indeed, comorbidities are prevalent in HFpEF and are known risk factors for cardiac dysfunction, the development of HFpEF, and adverse clinical outcomes.2–4 However, the evidence for the relationship between comorbidities, systemic inflammation, and HFpEF is based primarily on animal studies and small human studies that included cardiac biopsies.
Circulating inflammatory protein biomarkers appear to support the HFpEF comorbidity-inflammation paradigm. Several studies have found associations between inflammation biomarkers (e.g., TNFα, IL-6, hs-CRP, GDF-15, ST2, C-C motif chemokine 20, and TNF-receptor-1 [TNFR1]) and incident HFpEF, disease severity, and outcomes.5–8 However, these previous studies used a selective approach—investigating a single or limited number of proteins. High-throughput technologies including novel multiplexed protein assays enable the investigation of a large set of proteins in a single sample. The resultant biomarker profiles are potentially useful for characterizing underlying pathophysiological pathways.5, 9 Furthermore, previous studies did not evaluate the role of inflammation as a mediator of the association between comorbidity burden and cardiac dysfunction in HFpEF.
We therefore conducted a comprehensive proteomic evaluation of the comorbidity-inflammation paradigm in HFpEF. The main aims of our study were to: (1) evaluate the associations of comorbidity burden with cardiac structure/function; comorbidity burden with inflammation (defined by circulating inflammation biomarkers); and inflammation with cardiac structure/function; and (2) to perform a statistical mediation analysis to determine whether inflammation may mediate the association between comorbidity burden and abnormal cardiac structure/function in HFpEF.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study design
We analyzed blood samples and data from the PROMIS-HFpEF study, the design of which has been described previously.10 Briefly, PROMIS-HFpEF was a multicenter, observational study to evaluate the prevalence and correlates of coronary microvascular dysfunction in patients with HFpEF from Europe, the United States, and Southeast Asia. Patients were symptomatic, had a left ventricular ejection fraction (LVEF) ≥ 40%, and at least one of the following: (1) elevated natriuretic peptides; (2) prior HF hospitalization with evidence of either left ventricular (LV) hypertrophy or left atrial enlargement; (3) elevated pulmonary capillary wedge pressure at rest or with exercise; or (4) E/e’ ratio >15.
In the primary PROMIS-HFpEF study results paper (which focused on the prevalence and correlates of coronary microvascular dysfunction in HFpEF),10 only patients with analyzable coronary flow reserve (CFR) images and echocardiographic data (n=202) were included. For the present analysis, 228 of the total 263 enrolled patients (87%) with valid proteomic measurements were included.
We also analyzed data from a validation cohort of patients from Northwestern University. In this study, 117 out of 125 patients (94%) and 30 out of 31 comorbidity controls (97%) with available proteomic measurements were included. Patients were enrolled in this prospective observational study between November 2013 and May 2017. HFpEF patients were enrolled from the outpatient HFpEF clinic; control patients were enrolled from the outpatient general cardiology clinic or cardiac catheterization laboratory (elective, stable cases only). The diagnosis of HFpEF was made clinically by a cardiologist with expertise in HFpEF. Besides the presence of symptomatic HF and LVEF ≥50%, all patients had a prior history of HF hospitalization, evidence of elevated LV filling pressures (E/e’ > 15 or elevated LV filling pressures on invasive hemodynamic testing), or BNP > 100 pg/ml with evidence of LV hypertrophy or left atrial enlargement. None of the HFpEF patients in the Northwestern validation cohort were enrolled in the PROMIS-HFpEF study; thus, it represented an independent sample. Control patients were deemed eligible if they had 1 or more risk factors for HFpEF (e.g., hypertension, obesity, diabetes, chronic kidney disease) and only if there was no history of heart failure (any type, including those with recovered LVEF). For both the HFpEF and control patients, patients with the following conditions were excluded: dilated, hypertrophic, or infiltrative cardiomyopathy; greater than moderate left-sided valvular disease; congenital heart disease; constrictive pericarditis; or a history of heart transplantation.
All study participants in both the PROMIS-HFpEF study and in the Northwestern validation cohort gave written informed consent, and the institutional review board at each site in both studies approved the study. Both studies complied with the Declaration of Helsinki.
Definition of comorbidity burden
Clinical characteristics, including comorbidity status, were obtained in all patients in both cohorts. Comorbidities were defined based on physician documentation, review of the medical records, and patient self-report. Comorbidity burden was described by a simple score assigning 1 point to each of the following comorbidities: hypertension, diabetes mellitus, obesity (body mass index [BMI] ≥30 kg/m2), dyslipidemia, coronary artery disease (defined as previous myocardial infarction, percutaneous coronary intervention, coronary artery bypass grafting, or a significant [> 50%] stenosis in 1 or more coronary arteries), stroke, anemia, and chronic pulmonary disease. This score is in line with a recently published comorbidity score that was associated with abnormal cardiac structure and function.11 We intentionally excluded chronic kidney disease (CKD) and atrial fibrillation (AF) from the comorbidity score. CKD is known to be an important confounder in biomarker studies, due to the dominant renal clearance of many circulating biomarkers. Both CKD and AF can also be the consequence of HFpEF rather than the cause, which could bias the mediation analyses that were performed (see Statistical Analysis section below).
Echocardiography, coronary flow reserve, and peripheral arterial tonometry
All study participants in both cohorts underwent comprehensive 2-dimensional echocardiography with Doppler and tissue Doppler imaging using commercially available ultrasound systems, as described in detail previously.10, 12 Cardiac structure and function were quantified as recommended by the American Society of Echocardiography/European Association of Cardiovascular Imaging.13–15 Speckle-tracking analysis of the echocardiograms in the PROMIS-HFpEF study and in the Northwestern HFpEF cohort was performed as described previously.10, 16 CFR was measured during adenosine transthoracic Doppler echocardiography, and peripheral arterial tonometry was performed using an EndoPAT device as described previously.13
Proteomic measurements
In both the PROMIS-HFpEF study and the Northwestern cohort, circulating protein biomarkers were quantified by a multiplex immunoassay (92 analytes plus internal controls per panel, Proseek Multiplex96×96 Olink Bioscience, Uppsala, Sweden) as described previously,5 using the commercially available panels cardiovascular disease II, III and inflammation. Together these 3 panels measure 266 unique protein biomarkers (see Supplementary Table I for a full list of proteins and abbreviations). Further information about the Olink assay, including its reproducibility and internal validation, is available at http://www.olink.com and in the Supplemental Materials. When levels of any biomarker fell below the limit of detection (LOD) in ≥50% of the study population, the biomarker was excluded from further analysis (n=18 proteins, Supplementary Table I), leaving a total of 248 proteins that were included in the present analysis.
Statistical analysis
PROMIS-HFpEF patients were stratified by comorbidity burden (0–2, 3–4, or 5+ comorbidities). Baseline characteristics were compared between groups using one-way ANOVA (or Kruskal-Wallis tests, when appropriate) for continuous variables, or Fisher’s exact test for categorical variables. Figure 1 displays the multi-step analytic plan that we used to test the hypothesis that systemic inflammation mediates the association between comorbidities and abnormal cardiac structure/function in HFpEF.
Figure 1. Study Design Overview in the PROMIS-HFpEF Derivation Cohort.
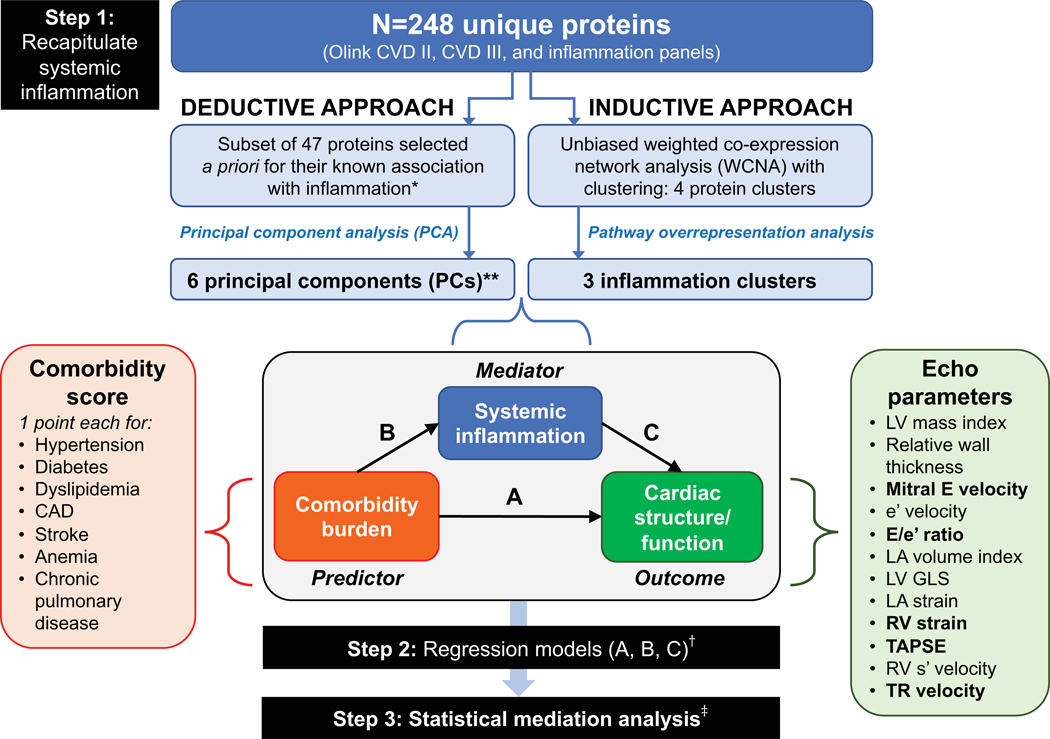
The analytic approach in the PROMIS-HFpEF derivation cohort started with recapitulating (summarizing) systemic inflammation using both inductive and deductive approaches in step 1. The deductive approach used a priori knowledge of proteins associated with inflammation based on functional annotation in the GO, KEGG, and Reactome databases, followed by principal components analysis. The inductive approach used an unbiased weight co-expression network analysis (WCNA) with clustering, followed by pathway overrepresentation analysis. The resulting principal components and inflammation protein clusters were then used as markers of systemic inflammation in subsequent analyses. In step 2, we used 1-way regression models to determine the association between (A) comorbidity burden with cardiac structure/function; (B) comorbidity burden and systemic inflammation; and (C) systemic inflammation and cardiac structure/function. Finally, in step 3, we performed formal statistical mediation analysis to determine whether markers of systemic inflammation (PCs and protein clusters) mediated the association between comorbidity burden and specific indices and cardiac structure/function. *Selected if annotated as inflammation-related in 2 or more of the databases (GO, KEGG, and Reactome).**These 6 PCs captured 57% of the variation in the 47 selected inflammation-related proteins. †All regression models were adjusted for age, sex, GFR, and study site; LA volume index, LA reservoir strain, e’ velocity, and E/e’ ratio were also further adjusted for atrial fibrillation. ‡ Mediation analysis was only performed for protein PCs/clusters and echocardiographic parameters that fulfilled a valid 3-way association in step 2 (i.e., statistically significant associations with a coherent direction had to be present in regression models corresponding to arrows A, B, and C). Echocardiographic parameters that met these criteria for mediation analysis are represented in bold font. Inflammation protein parameters that met these criteria were PC1, PC5, PC6, the turquoise cluster, and the yellow cluster. Abbreviations: CAD = coronary artery disease; Echo = echocardiographic; LV = left ventricle; LA = left atrial; RV = right ventricular; TAPSE = tricuspid annular plane systolic excursion; TR = tricuspid regurgitation; GO = gene ontology; KEGG = Kyoto Encyclopedia of Genes ad Genomes.
In step 1, we summarized ‘systemic inflammation’ based on the measured circulating proteins using 2 complementary approaches. In the deductive approach (step 1A), proteins were selected a priori based on their functional annotation in publicly available databases if these databases indicated that the protein was previously known to be associated with inflammation (Supplementary Table II). The resulting 47 proteins (see Supplementary Table I) were summarized using principal component (PC) analysis. PC analysis is a way to reduce the dimensionality of data that is correlated (i.e., instead of analyzing all 47 proteins individually, PC analysis can be used to summarize the information represented by the proteins into a much smaller number of variables while maintaining the informativeness of all 47 proteins).
In the inductive (unbiased) approach (step 1B), we used a weighted co-expression network analysis (WCNA) to cluster all 266 measured proteins given the possibility that some proteins may be involved in inflammation in HFpEF but may not be known a priori to be associated with inflammation. WCNA is a method often used to study biological networks; it defines clusters (modules) of proteins based on their relationship (weighted correlation) to each other. Although WCNA is able to collapse a large number of proteins into a smaller number of clusters, further analysis is required to determine which of the resultant protein clusters represents systemic inflammation. Thus, we used pathway overrepresentation analysis on the resulting clusters to select those representative of inflammatory pathways, which we defined as 2 or more inflammatory pathways represented in each protein cluster.
Once circulating inflammation was summarized by the deductive (protein PCs) and inductive (WCNA clusters) approaches, each patient is given a quantitative score (eigenvalue) that serves as a quantitative variable that summarizes the PCs or clusters. These eigenvalues, which summarize the variation in the proteins using the 2 analytic techniques (PC analysis and WCNA) were subsequently used in regression and mediation analyses.
In step 2, using unadjusted and multivariable-adjusted linear regression analyses, we investigated the 3-way associations underlying the inflammation paradigm in HFpEF, depicted by arrows A, B and C in Figure 1: (A) association of comorbidity burden with individual cardiac structure/function parameters; (B) association of comorbidity burden with individual inflammation PCs and clusters; and (C) association of inflammation PCs and clusters with cardiac structure/function parameters.
Step 3 involved statistical mediation analysis to investigate how inflammation mediates the association between comorbidity burden and cardiac structure/function. Only variables that fulfilled the 3-way associations in step 2 (i.e., all 3 associations were significant at P<0.05) were included in these analyses as this is a prerequisite of mediation analysis.17 In sensitivity analyses, we investigated interactions of the mediation effect using moderated mediation analysis, stratifying by gender and by history of inflammatory (autoimmune) disease and/or cancer. We performed these analyses to determine whether the results differed by gender or presence of conditions known to increase systemic inflammation.
We also explored several single proteins as mediators in mediation analyses using similar methods to those outlined above (3-way association analyses, followed by mediation analyses) by replacing the inflammation PCs/modules with individual proteins. We selected the proteins for these analyses based on our results from both the deductive and inductive approaches (i.e., main hubs of the PCs or WCNA modules, respectively) combined with the existing literature on the relationship of these proteins with inflammation. We also tested the association of inflammation PCs and WCNA modules with 6-minute walk distance and with CFR and reactive hyperemia index—reflective of coronary and systemic microvascular function, respectively.
Finally, we performed external validation analyses in the Northwestern cohort. Student t-tests, nonparametric Wilcoxon rank-sum tests, and Fisher’s exact tests were used to compare PROMIS-HFpEF with Northwestern-HFpEF patients, and Northwestern-HFpEF with Northwestern-control patients. Steps 1 and 2 of the analytic plan were repeated in the validation cohort. We tested whether the inflammation clusters identified in PROMIS-HFpEF cohort were conserved (replicated) in the Northwestern validation cohort as a way to determine whether the pattern of inflammation in the measured proteins was the same in the PROMIS-HFpEF and the Northwestern-HFpEF patients. Additionally, we evaluated whether inflammation was upregulated in HFpEF cases versus controls by volcano plot analysis (adjusted for estimated glomerular filtration rate [GFR]). Mediation analysis was precluded in the validation cohort because of the relatively small sample size.
All regression and mediation models were adjusted for age, sex, GFR, and study site. Models using e’, E/e’ or left atrial (LA) parameters as endpoints were additionally adjusted for AF at the time of echocardiography because these parameters were found to be confounded by AF. Regression and mediation analyses involving systemic inflammation were additionally adjusted for history of autoimmune disease and cancer. We also performed sensitivity analyses by excluding PROMIS-HFpEF patients with LVEF < 50% (n=23).
A two-sided P-value < 0.05 was considered statistically significant. Multiple hypothesis testing was accounted for using the false discovery rate (FDR) method by Benjamini & Hochberg. Analyses were performed using IBM SPSS Statistics, version 24.0 (IBM Corporation [Armonk, NY]) and R version 3.5.2 (The R Foundation for Statistical Computing, packages WGCNA and mediation). Protein biomarker networks were visualized using Cytoscape,18 and their potential biological roles were analyzed using Metascape.19 Additional details on the statistical analyses are included in the Supplementary Materials.
RESULTS
Baseline characteristics in PROMIS-HFpEF
The median comorbidity score was 3 (25th-75th percentile 2–4). Compared to HFpEF patients with a low comorbidity burden, patients with a higher comorbidity burden were of similar age but more often male, more symptomatic, had a higher prevalence of AF, worse renal function, and higher uric acid, high-sensitivity CRP, and white blood cell count values (Table 1). Echocardiographic characteristics stratified by comorbidity burden are shown in Table 2. Measures of cardiac structure, indexed to body size, were similar among the groups. Multiple measures of LV systolic and diastolic function, left atrial function, and RV function were increasingly abnormal with increasing comorbidity burden (Table 2).
Table 1.
Clinical and Laboratory Characteristics Stratified by Comorbidity Burden in PROMIS-HFpEF
| Characteristic | Comorbidity burden (number of comorbidities) |
P-value | ||
|---|---|---|---|---|
| 0–2 (n=67) | 3–4 (n=110) | 5+ (n=51) | ||
| Age, years | 78 (70–82) | 75 (70–81) | 73 (68–79) | 0.24 |
| Female, n (%) | 19 (28.4) | 53 (47.7) | 28 (54.9) | 0.007 |
| Race | 0.86 | |||
| -White | 60 (89.6) | 95 (85.6) | 45 (88.2) | |
| -Black | 2 (3.0) | 5 (4.5) | 3 (5.9) | |
| -Other | 5 (7.5) | 11 (9.9) | 3 (5.9) | |
| Body mass index, kg/m2 | 25.9 (23.0–29.0) | 27.6 (24.0–32.3) | 33.4 (29.7–39.5) | <0.001 |
| Heart rate, bpm | 69 (61–79) | 71 (60–80) | 67 (62–76) | 0.65 |
| Systolic BP, mmHg | 140 (121–156) | 140 (125–152) | 134 (126–150) | 0.84 |
| Diastolic BP, mmHg | 76 (70–83) | 79 (70–89) | 75 (66–81) | 0.11 |
| NYHA class | 0.001 | |||
| -I | 3 (4.5) | 1 (0.9) | 0 (0) | |
| -II | 7 (10.4) | 26 (23.4) | 21 (41.2) | |
| -III | 7 (10.4) | 26 (23.4) | 21 (41.2) | |
| Medical history, n (%) | ||||
| -Hypertension* | 39 (58.2) | 101 (91.0) | 50 (98.0) | <0.001 |
| -Obesity (BMI ≥ 30 kg/m2)* | 8 (11.9) | 39 (35.1) | 38 (74.5) | <0.001 |
| -Hyperlipidemia* | 9 (13.4) | 66 (59.5) | 46 (90.2) | <0.001 |
| -Diabetes* | 1 (1.5) | 25 (22.5) | 37 (72.5) | <0.001 |
| -Coronary artery disease* | 4 (6.0) | 19 (17.1) | 25 (49.0) | <0.001 |
| -Stroke* | 1 (1.5) | 10 (9.0) | 15 (29.4) | <0.001 |
| -Anemia* | 13 (20.0) | 39 (35.5) | 27 (52.9) | 0.001 |
| -Chronic pulmonary disease* | 5 (7.5) | 13 (11.7) | 16 (31.4) | 0.001 |
| -Atrial fibrillation | 26 (38.8) | 68 (61.3) | 37 (72.5) | 0.001 |
| -Chronic kidney disease | 30 (44.8) | 57 (51.4) | 35 (68.6) | 0.03 |
| -Valvular disease | 15 (22.4) | 37 (33.3) | 10 (19.6) | 0.12 |
| -Pulmonary hypertension | 14 (20.9) | 19 (17.1) | 10 (19.6) | 0.82 |
| -Malignancy | 17 (25.8) | 19 (17.1) | 5 (9.8) | 0.09 |
| -Inflammatory disease | 8 (11.9) | 11 (9.9) | 5 (9.8) | 0.92 |
| Laboratory data | ||||
| -Sodium, mmol/L | 140 (139–142) | 140 (138–142) | 141 (138–142) | 0.98 |
| -Potassium, mmol/L | 4.2 (3.9–4.4) | 4.3 (4.0–4.5) | 4.3 (4.1–4.6) | 0.13 |
| -BUN, mg/dL | 21.9 (16.0–26.1) | 22.1 (17.9–26.9) | 30.0 (17.9–44.5) | 0.007 |
| -Creatinine, mg/dL | 0.95 (0.81–1.15) | 1.07 (0.86–1.38) | 1.18 (1.02–1.63) | <0.001 |
| -GFR, mL/min/1.73 m2 | 63 (50–75) | 59 (45–76) | 52 (39–64) | 0.01 |
| -Cystatin-C, mg/L | 1.19 (0.96–1.61) | 1.35 (1.06–1.62) | 1.44 (1.18–1.96) | 0.008 |
| -NT-proBNP, pg/mL | 1180 (646–1882) | 1680 (831–2620) | 1503 (987–3378) | 0.07 |
| -hsCRP, mg/L | 2.1 (0.9–4.2) | 2.3 (1.15–5.5) | 3.9 (1.9–6.6) | 0.03 |
| -hsTnT, ng/L | 12 (10–18) | 14 (10–25) | 17 (10–33) | 0.02 |
| -Hemoglobin A1c, % | 5.6 (5.4–5.8) | 5.8 (5.6–6.7) | 6.9 (6.1–7.6) | <0.001 |
| -Albumin, g/dL | 3.7 (3.4–3.9) | 3.7 (3.4–4.0) | 3.7 (3.5–4.0) | 0.82 |
| -Uric acid, mg/dL | 6.22 (5.38–7.39) | 6.89 (5.71–8.57) | 7.23 (6.22–8.91) | 0.01 |
| -Hemoglobin, g/dL | 13.2 (12.4–14.1) | 13.0 (11.8–14.2) | 12.7 (11.2–13.6) | 0.11 |
| -Platelet count, K/mm3 | 225 (186–257) | 219 (180–246) | 208 (180–251) | 0.36 |
| -WBC count, K/mm3 | 5.8 (4.8–6.9) | 6.9 (5.7–8.0) | 7.1 (6.0–8.4) | <0.001 |
| -UACR, mg/g | 12.4 (6.2–38.9) | 37.2 (13.3–106.2) | 59.3 (21.2–184.1) | 0.001 |
| Medications, n (%) | ||||
| -Loop diuretic | 24 (35.8) | 60 (54.1) | 41 (80.4) | <0.001 |
| -Thiazide diuretic | 13 (19.4) | 10 (9.0) | 3 (5.9) | 0.05 |
| -ACE-inhibitor | 25 (37.3) | 28 (25.2) | 19 (37.3) | 0.15 |
| -ARB | 28 (41.8) | 50 (45.0) | 22 (43.1) | 0.91 |
| -Beta-blocker | 44 (65.7) | 86 (77.5) | 46 (90.2) | 0.006 |
| -MRA | 13 (19.4) | 33 (29.7) | 17 (33.3) | 0.18 |
| -CCB | 18 (26.9) | 35 (31.5) | 21 (41.2) | 0.26 |
| -Antiplatelet agent | 19 (28.4) | 25 (22.5) | 15 (29.4) | 0.55 |
| -Anticoagulant | 47 (70.1) | 95 (85.6) | 48 (94.1) | 0.002 |
| -Statin | 18 (26.9) | 67 (60.4) | 46 (90.2) | <0.001 |
| -Total number of medications | 7 (6–11) | 9 (6–12) | 11 (9–14) | <0.001 |
Values represent median (25th-75th percentile) for continuous variables and frequency (percent) for categorical variables.
Comorbidities included in the comorbidity score
BMI = body mass index; BP = blood pressure; HF = heart failure; BUN = blood urea nitrogen; NT-proBNP = N-terminal pro-B-type natriuretic peptide; GFR = glomerular filtration rate; hsCRP = high-sensitivity C-reactive protein; hsTnT= high-sensitivity troponin T; ACE = angiotensinogen converting enzyme; ARB = angiotensin II receptor blocker; MRA = mineralocorticoid receptor antagonist; CCB = calcium channel blocker.
Table 2.
Echocardiographic Characteristics Stratified by Comorbidity Burden in PROMIS-HFpEF
| Characteristic | Comorbidity burden (number of comorbidities) | P-value | ||
|---|---|---|---|---|
| 0–2 (n=67) |
3–4 (n=110) |
5+ (n=51) |
||
| LVEF, % | 59 (55–65) | 59 (55–63) | 61 (56–65) | 0.35 |
| LVEDVi, ml/m2 | 41.8 (32.6–52.7) | 41.7 (34.3–49.0) | 38.9 (32.9–50.2) | 0.79 |
| LVESVi, ml/m2 | 16.7 (11.6–22.0) | 16.8 (13.5–20.6) | 15.9 (12.3–19.3) | 0.65 |
| LVMi, g/m2 | 103 (81–119) | 104 (83–125) | 108 (85–133) | 0.37 |
| Relative wall thickness | 0.43 (0.38–0.51) | 0.44 (0.39–0.52) | 0.49 (0.43–0.56) | 0.02 |
| LAVi, ml/m2 | 35.9 (29.9–43.6) | 39.7 (30.9–47.7) | 39.0 (31.5–42.9) | 0.23 |
| Mitral E, cm/s | 89 (75–105) | 95 (81–114) | 108 (80–129) | 0.04 |
| e’ (average), cm/s | 8.2 (6.5–9.8) | 8.2 (6.9–10.5) | 8.0 (6.2–9.9) | 0.43 |
| E/e’ ratio (average) | 11.7 (9.1–16.1) | 12.1 (8.9–16.5) | 13.8 (10.8–16.7) | 0.28 |
| RVEDAi, mm2/m2 | 94.5 (79.1–107.8) | 101.6 (84.0–117.8) | 98.3 (81.8–119.7) | 0.07 |
| RVESAi, mm2/m2 | 50.3 (44.5–63.8) | 58.4 (47.8–75.1) | 53.8 (46.5–74.5) | 0.14 |
| RV wall thickness, cm | 0.46 (0.43–0.50) | 0.47 (0.43–0.53) | 0.51 (0.47–0.56) | <0.001 |
| TAPSE, cm | 1.9 (1.6–2.2) | 1.7 (1.5–2.1) | 1.7 (1.5–1.9) | 0.005 |
| RV s’ velocity, cm/s | 12.5 (9.7–14.4) | 11.2 (9.9–12.9) | 10.1 (9.1–12.4) | 0.01 |
| TR velocity, cm/s | 297 (274–325) | 293 (263–329) | 313 (271–362) | 0.18 |
| LV GLS, %-unit | 17.5 (14.1–19.4) | 16.2 (13.1–18.4) | 14.6 (12.4–17.1) | 0.008 |
| RV free wall strain, %-unit | 23.9 (20.5–28.0) | 21.0 (17.8–24.7) | 20.0 (15.5–24.6) | <0.001 |
| LA reservoir strain, %-unit | 25.2 (16.0–36.2) | 17.5 (12.5–31.6) | 18.0 (12.2–27.3) | 0.007 |
Values represent median (25th-75th percentile). Strain values are shown as absolute values for ease of interpretation. E/e’ ratio and e’ velocity represent average of septal and lateral mitral annulus values.
LVEDVi = left ventricular end diastolic volume index; LVESVi = left ventricular end systolic volume index; LVEF = left ventricular ejection fraction; LVMi = left ventricular mass index; RWT = relative wall thickness; LAVi =left atrial volume index; RVEDAi = right ventricular end diastolic area index; RVESAi = right ventricular end systolic area index; RV = right ventricular; TAPSE = tricuspid annular plane systolic excursion; TR = tricuspid regurgitation; GLS = global longitudinal strain of the left ventricle; LA = left atrial.
Inflammation proteomics
In the deductive approach, principal component analysis of 47 inflammation proteins, selected a priori, resulted in 6 principal components. The top 10 proteins that correlated most closely with each principal component are shown in Supplementary Table III. In the inductive approach, unbiased network-based clustering extracted 4 mutually exclusive protein clusters (Figure 2A), of which 3 were reflective of systemic inflammation on pathway overrepresentation analysis (Figures 2B–D): the turquoise cluster (main hub: TNFR1), yellow (main hub: insulin-like growth factor binding protein 7 [IGFBP 7]), and red (main hub: TNF-related apoptosis-inducing ligand receptor 2 [TRAILR2]) modules were reflective of ≥ 2 inflammatory pathways on pathway overrepresentation analysis (Figure 2B–E). Ninety-four proteins were not assigned to any cluster due to lack of correlation to other proteins (see Supplementary Table I for a list of all proteins and the assigned cluster for each protein).
Figure 2. Protein Clusters Identified by Weighted Co-expression Network Analyses in PROMIS-HFpEF Derivation Cohort and the Northwestern Validation Cohort.
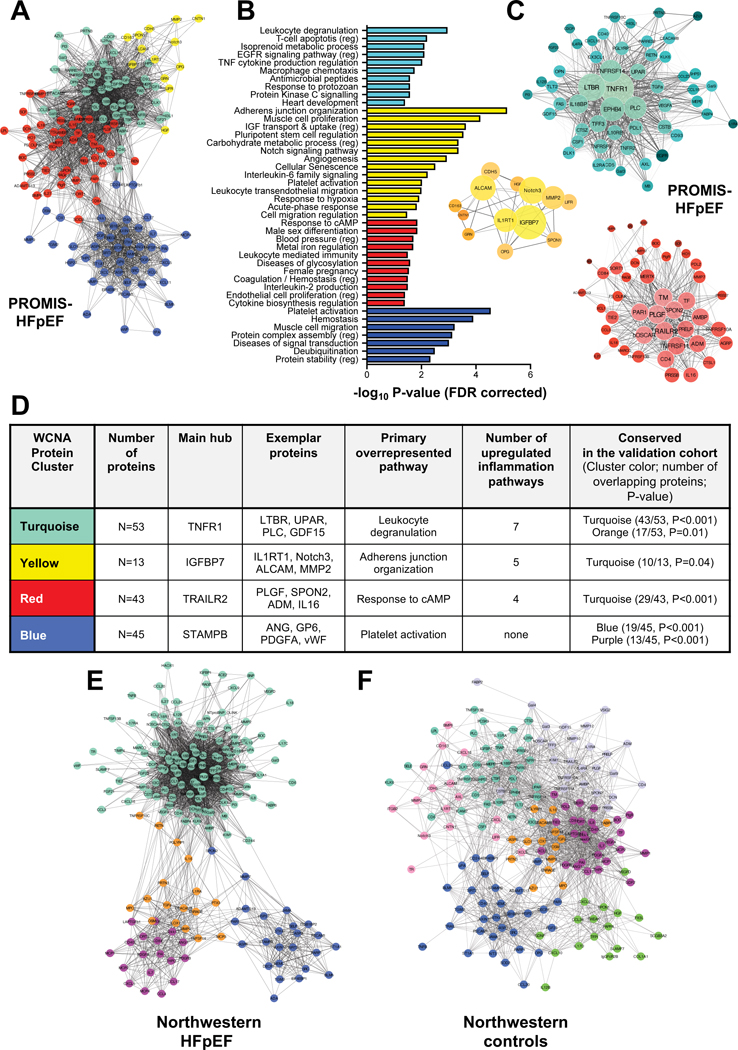
(A) Adjacency network-map of circulating proteins color-coded by cluster assignment by hierarchical clustering-based nearness or co-expression of proteins. For clarity of presentation only nodes (proteins) that were assigned to a cluster are shown (N=159/248); the remaining proteins lie on the outer edges of the network-map. (B) Overrepresented, non-redundant pathways in each cluster with false discovery rate corrected P-values. (C) Detailed network-maps of proteins in the 3 clusters that were representative of inflammation (i.e., overrepresentation of ≥2 inflammatory pathways). Node size reflects intra-cluster connectivity (i.e., the sum of weighted edges [correlations] with all other proteins in the cluster). Node color density reflects the strength of cluster membership. Edge thickness and transparency reflect the adjacency of proteins according to weighted co-expressions. (D) Summary of extracted clusters including (1) the number of assigned proteins per cluster, (2) the main hub (based on intra-cluster connectivity), (3) other exemplar proteins, the primary (most significant) overrepresented pathway; (4) the number of significantly upregulated inflammatory pathway; and (5) the cross-cohort conservation in the HFpEF patients in the Northwestern validation cohort, reflected by the number of overlapping proteins and corresponding P-value (tested under a hypergeometric distribution). (E) Adjacency network-map of circulating proteins in the Northwestern HFpEF patients in the validation cohort. Clusters with most significant overlap were assigned the same color as the corresponding cluster in PROMIS-HFpEF cohort. (F) Adjacency network-map of circulating proteins in the Northwestern control patients in the validation cohort. Cluster preservation was tested against the Northwestern HFpEF patients; clusters with significant overlap were assigned the same color as the corresponding cluster in the Northwestern HFpEF patients.
Combining the deductive and inductive approaches, a total of 9 parameters were extracted that summarize circulating protein markers of inflammation: 6 PCs based on inflammation proteins selected a priori and 3 unbiased protein clusters that reflect inflammation pathways.
Associations between comorbidity burden, inflammation, and cardiac structure/function
We confirmed the 3-way associations between comorbidity burden, inflammation, and cardiac structure/function. Comorbidity burden was independently associated with several echocardiographic parameters, except for LA volume index and LV mass index (Figure 3 and Supplementary Table IV). Comorbidity burden was also associated with multiple summary parameters of inflammation (Figure 4; PC1, PC3, PC5, PC6, and the turquoise and yellow inflammation clusters), independent of age, sex, and GFR and remained significant when additionally adjusting for a history of autoimmune disease and/or malignancy (Supplementary Table V). Finally, inflammation PCs and clusters were independently associated with increased mitral E velocity, E/e’ ratio, TR velocity, and with reduced RV function (TAPSE and RV free wall strain), but not with LV GLS, e’ velocity, LA volume, LA reservoir strain, or LV mass index (Figure 3 and Supplementary Table VI). Additional adjustment for a history of autoimmune disease and/or malignancy did not attenuate the overall associations between inflammation and cardiac function (Supplementary Table VI). As shown in the figures and tables, the majority of the identified associations remained significant after correcting for multiple comparisons using the FDR method.
Figure 3. Heatmap of Associations of Comorbidity Burden and Inflammation with Indices of Cardiac Structure and Function in PROMIS-HFpEF.
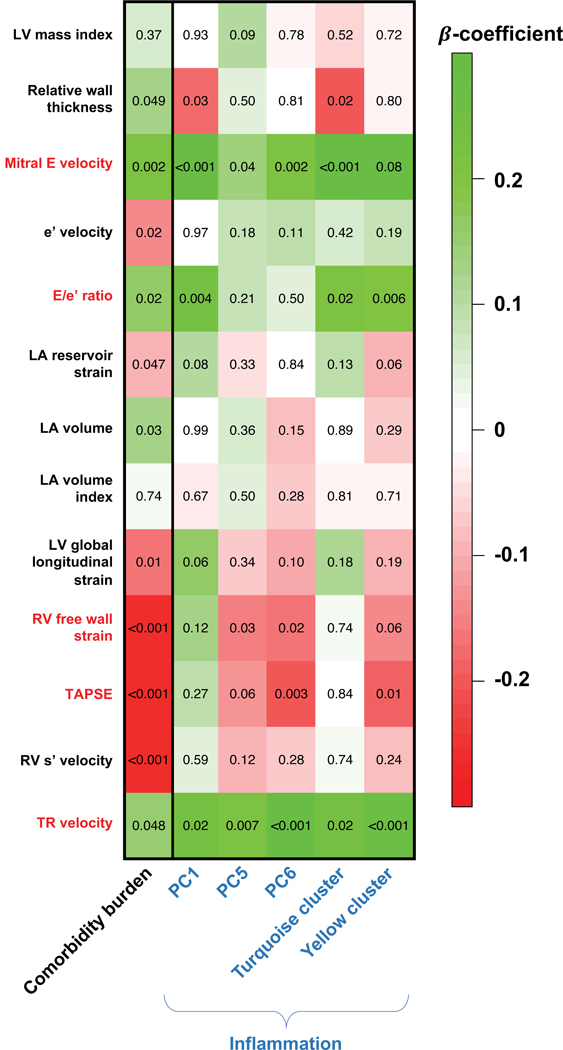
The values in the cells represent p-values for the associations. The color and intensity of each cell depicts the standardized β-coefficients from linear regression models adjusted for age, gender, glomerular filtration rate, and study site (and atrial fibrillation, for LA indices and E/e’ ratio). Echocardiographic indices that were significantly associated with both comorbidity burden and inflammation (and thus fulfilling the assumptions for mediation analysis) are marked by red font. PC2, PC4, and the red cluster are not shown because of their lack of association with comorbidity burden; PC3 is not shown because all associations had P-values >0.20. Of the statistically significant associations (P<0.05), the yellow cluster was no longer associated with TAPSE after adjustment for inflammatory (autoimmune disease) and/or malignancy. Of the statistically significant associations (P<0.05), the following were no longer significant after correction for multiple testing (false discovery rate): PC1 and relative wall thickness; PC5 and mitral E velocity; PC5 and RV free wall strain; PC6 and RV free wall strain; turquoise cluster and relative wall thickness; turquoise cluster and E/e’ ratio; and turquoise cluster and TR velocity. LV = left ventricular; LA = left atrial; RV = right ventricular; TAPSE = tricuspid annular plane systolic excursion; TR = tricuspid regurgitation; PC = principal component.
Figure 4. Mediation Analysis of Inflammation Principal Components/Clusters as Mediators of the Association Between Comorbidity Burden and Mitral E Velocity in PROMIS-HFpEF.
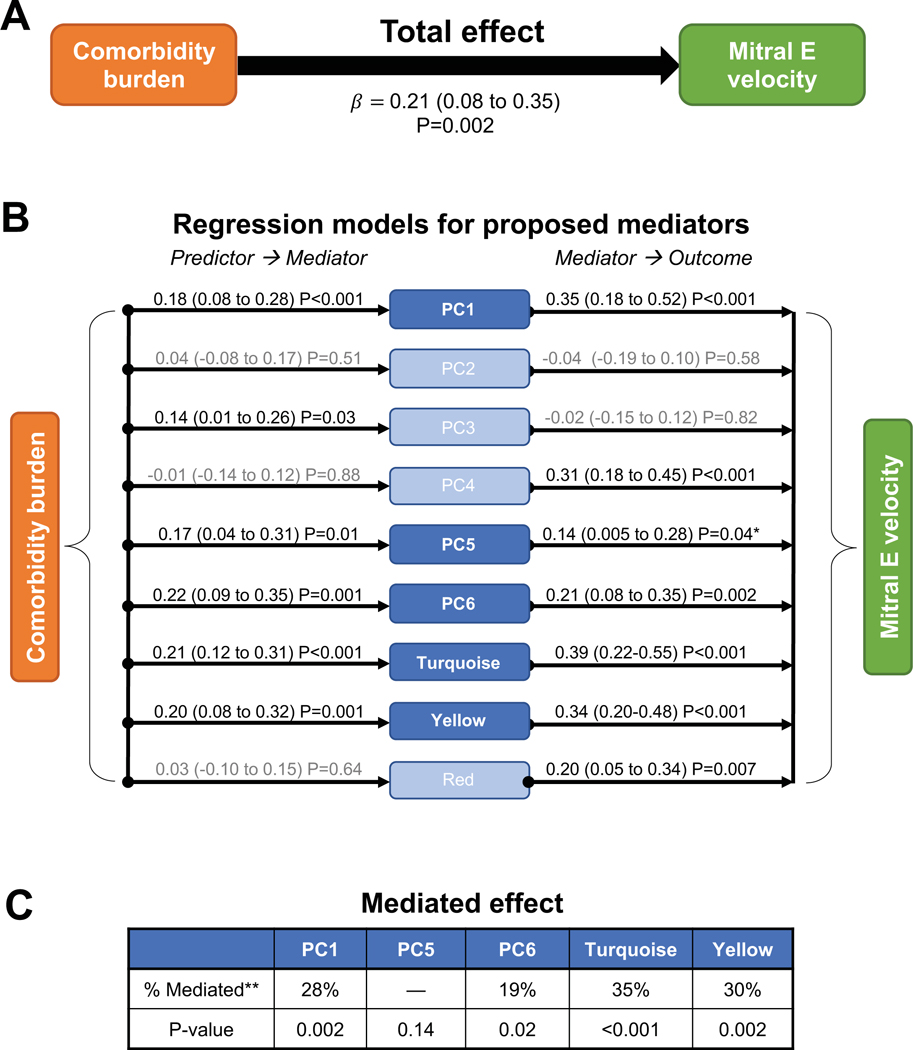
Mediation modeling including testing of underlying assumptions. Mitral E velocity was used as an example outcome measure. Values adjacent to the arrows in (A) and (B) depict standardized β-coefficients (95% CIs) and P-values from linear regression models adjusted for age, gender, glomerular filtration rate, study site, and atrial fibrillation. (A) Total effect of the association of comorbidity burden with mitral E velocity on linear regression analysis, a prerequisite for mediation analysis. (B) Investigating the assumptions that comorbidity burden is associated with increased inflammation (reflected by protein PCs/clusters) and that inflammation is associated with increased mitral E velocity in HFpEF. Mediators in light blue do not fulfill the assumptions for mediation analysis because there is no statistically significant effect between predictor and mediator and/or between mediator and outcome. (C) Mediated effect in mediation analysis of parameters fulfilling the underlying assumptions, indicating that 19–35% of the association of comorbidity burden with increased mitral E velocity is mediated by systemic inflammation. *P>0.05 after false discovery rate correction. **Percent mediated = mediated effect / total effect × 100.
Inflammation as mediator between comorbidities and cardiac structure/function
All variables that fulfilled the assumption of a 3-way association between comorbidities, inflammation, and cardiac structure/function (mitral E velocity, E/e’ ratio, TAPSE, TR velocity and RV free wall strain) were entered into formal mediation analyses. Inflammation mediated the associations between comorbidity burden and each of these echocardiographic indices except for RV free wall strain. For example, inflammation mediated the association between comorbidity burden and mitral E velocity (Figure 4 and Supplementary Table VII): 19–35% of the association between comorbidity burden and mitral E velocity was explained by inflammation PCs/modules. As shown in Supplementary Table VII, mediation results for the other echocardiographic indices were as follows: E/e’ (18–29%), TAPSE (13%), and TR velocity (27–41%). The most consistent inflammation mediators were PC1, PC6, and the turquoise and yellow clusters. Mediation analysis results did not differ by sex or history of autoimmune disease and/or malignancy (P>0.90 for all interactions). Results were similar after correcting for multiple comparisons using the FDR method (Supplementary Table VII) and after exclusion of patients who had HF with mid-range EF (LVEF 40–49%; n=23) (Supplementary Table VIII).
As an exploratory analysis, 15 individual proteins that were highly representative of the inflammation PCs/clusters were tested individually as mediators between comorbidities and cardiac structure/function. TNFR1, UPAR, IGFBP7 and GDF15 were identified as mediators between comorbidity burden and mitral E velocity, E/e’ ratio, and TR velocity; LTBR and IL1-RT1 were mediators for mitral E velocity and TR velocity, and PLC was a mediator for TR velocity only (Supplementary Table IX).
In additional exploratory analyses, we found that in the HFpEF patients, inflammation PCs and clusters were independently associated with reduced CFR and lower 6MWT distance, but not with reactive hyperemia index. Additional adjustment for coronary artery disease, smoking, and E/e’ ratio in the regression models for CFR did not attenuate most of these associations (Supplementary Table X).
External validation
The Northwestern HFpEF patients (median comorbidity score 4, 25th-75th percentile 3–5) had a similar comorbidity burden compared to PROMIS-HFpEF patients (P=0.83 for the comparison between cohorts). However, Northwestern HFpEF patients were younger, more often obese, and had less structural remodeling (lower LV mass index, LA volume index, and RV size) but a lower average e’ velocity (Supplementary Table X). Compared to comorbidity controls, HFpEF patients were more often female; more frequently had obesity and anemia; had less coronary artery disease; and had worse renal function and cardiac structure/function (Supplementary Table XI).
Northwestern HFpEF patients exhibited significantly more systemic inflammation compared to controls. As shown in Figure 5A (volcano plot), 25 individual proteins were overexpressed in HFpEF patients versus controls. Together, they were highly representative of inflammatory pathways including cellular response to TNF, cytokine-cytokine receptor interaction, leukocyte chemotaxis, acute inflammatory response, and macrophage differentiation (Figure 5B).19. Four of the 6 inflammation PCs were significantly higher in HFpEF patients compared to controls, even after adjustment for age, sex, and GFR (Figure 5C).
Figure 5. Inflammation in HFpEF Patients versus Comorbidity Controls in the Northwestern Validation Cohort.

(A) Volcano plot showing the upregulated proteins in HFpEF versus comorbidity control patients in the Northwestern validation cohort. Dashed lines show the cut-off used for defining upregulation (false discovery rate-corrected P<0.01 and a > 1.5 fold-change). (B) Significantly overrepresented pathways in HFpEF patients versus comorbidity controls based on the upregulated proteins, indicating upregulation of several inflammatory pathways. (C) Principal component scores summarizing circulating inflammation proteins in HFpEF patients (red) versus comorbidity controls (turquoise). Boxes depict 25th-75th percentiles, horizontal lines through the box depict the median (50th percentile), whiskers depict the upper and lower extremes, and dots depict outliers.
The cluster with the most inflammation pathways that was identified in the PROMIS-HFpEF on WCNA analysis (turquoise cluster) was the most conserved cluster in the Northwestern HFpEF patients (P<0.001, Figure 2A, 2D, 2E, and 2F), whereas it was the least conserved cluster in Northwestern control patients. The overall coexpression network of circulating proteins was very different in Northwestern HFpEF patients versus controls (Figure 2E and 2F).
In the Northwestern cohort, comorbidity burden was associated with increased inflammation (turquoise cluster) after adjustment for age, sex and GFR (Supplementary Table XII). Inflammation PCs/clusters were associated with multiple indices of cardiac structure/function (increased LA volume index, mitral E velocity, E/e’ ratio, and TR velocity, and reduced TAPSE), but adjustment for age, sex and GFR largely attenuated these associations (Supplementary Tables XIIIA and XIIIB).
DISCUSSION
In a comprehensive proteomic investigation of the comorbidity-inflammation paradigm in HFpEF, we found that (1) comorbidity burden is associated with worse cardiac structure and function; (2) comorbidity burden is associated with heightened systemic inflammation; (3) systemic inflammation is associated with worse cardiac function; and importantly, (4) systemic inflammation appears to mediate the association between comorbidity burden worse cardiac hemodynamic stress (elevated intracardiac pressures) and its consequences (worse RV function).
Comorbidity burden and cardiac structure/function in HFpEF
In the first step of our analyses, we found associations between comorbidity burden and altered cardiac structure and function in HFpEF. A similar association between comorbidity burden and worse LV longitudinal strain, e’ velocity, and E/e’ ratio prior to the onset of HF was previously shown in the Hypertension Genetic Epidemiology Network (HyperGEN) study.11 In patients with prevalent HF, a higher comorbidity burden was found to be associated with increased hospitalizations and mortality, with a stronger association for hospitalizations in HFpEF compared to heart failure with reduced ejection fraction (HFrEF).4 Together this evidence supports the idea that comorbidities are drivers of HFpEF development and progression.
Inflammation as a mediator between comorbidities and cardiac structure/function in HFpEF
The second step of our analysis was assessment of systemic inflammation as a potential mediator between comorbidities and cardiac alterations seen in HFpEF—a well-recognized hypothesis1, 20 that is based on pre-clinical and relatively small human biopsy studies of HFpEF.5–7, 9, 21–27 Previous biomarker studies in HFpEF have also supported the prominent role of inflammation in HFpEF. In the KaRen study, a panel of 98 biomarkers was investigated. Inflammation-related biomarkers were the strongest associates of both HFpEF severity and outcome.5 Two other studies compared HFpEF versus HFrEF and found specific biomarker profiles in HFpEF to be enriched with inflammation pathways,7 one of which was also externally validated.9
Our study is unique in that it is the first study to our knowledge to: (1) evaluate inflammation comprehensively in HFpEF, using both deductive and inductive approaches, along with external validation; and (2) investigate the role of inflammation as a mediator between comorbidities and abnormal cardiac structure/function in HFpEF. Mediation analysis is a statistical technique that provides causal inference within observational studies such as ours. We found that systemic inflammation is more prominent in HFpEF compared to controls with comorbidities who do not have HF; and systemic inflammation is a significant and relevant mediator of the association between comorbidity burden and echocardiographic indices of cardiac filling pressures and RV dysfunction in the setting of HFpEF.
In HFpEF, systemic inflammation has been related to increased cardiomyocyte passive tension and increased myocardial collagen deposition, both of which would result in reduced LV compliance.21, 25, 26 In the absence of a widely accepted non-invasive method to measure LV compliance,28, 29 echocardiographic markers of LV filling pressures (e.g., E velocity, E/e’ ratio, TR velocity) can be used to indicate increased LV diastolic stiffness. Thus, our finding that inflammation PCs and modules primarily mediated the association between comorbidity burden and markers of LV filling pressures supports the hypothesis that comorbidity-induced systemic inflammation is associated with increased myocardial stiffening. Another potential explanation for our findings is that comorbidity-induced systemic inflammation is an important trigger for congestion.30
Our results are further supported by a similar associative trend in the validation cohort, although the smaller study size prohibited mediation analysis, and statistical power was limited (especially for multivariable analyses). Still, we found that inflammation was upregulated in HFpEF compared to controls and that inflammatory proteins are coexpressed differently in cases versus comorbidity controls, which supports the notion that systemic inflammation is related to HF symptomatology (i.e., HFpEF) in patients with comorbidities. These analyses suggest that proteins involved in T cell-mediated toxicity and other inflammatory pathways are connected differently in HFpEF versus controls. Finally, the turquoise module (with the central hub of TNFR1) was the most conserved module across the two independent HFpEF cohorts, validating its biological importance in HFpEF.
Specific inflammation pathways in HFpEF
Several representative inflammation-related proteins in our study show potential for further investigation, either as a biomarker for screening or as a potential therapeutic target to further investigate in pre-clinical studies. These proteins included the main hubs (most strongly associated with) of the inflammation summary markers (PC1, PC6, turquoise cluster, yellow cluster) that appeared to mediate the association between comorbidity burden and cardiac structure/function in our analyses. Soluble TNFR1 represents the main hub of the most important inflammation cluster in our study (turquoise cluster) and PC1 and also individually mediated the association between comorbidities and cardiac structure/function. Soluble TNFR1 is released in response to pro-inflammatory stimuli31 and was associated with incident HF in 2 independent Swedish cohorts32 and in the US-based Health ABC study.33 Interestingly, it was more closely associated with risk of HFpEF than HFrEF. In the KaRen biomarker study, soluble TNFR1 was associated with both clinical outcome and with E/e’ ratio.5 Although prior trials of TNFα antagonism in HFrEF did not improve outcomes,34 divergent effects attributed to stimulation of TNFR1 versus TNFR235 in combination with our results, and the possibility that TNFα pathways are different in HFpEF compared to HFrEF could evoke new interest in the TNF pathway, particularly in HFpEF.
Additional individual proteins implicated in systemic inflammation that appeared to mediate the association between comorbidities and cardiac structure and function in our study included: (1) growth differentiation factor-15 (GDF15)—a member of the transforming growth factor-beta superfamily stimulated by inflammation, oxidative stress, and tissue hypoxia or injury—is associated with incident HF and prognosis in both HFpEF and HFrEF;5, 32, 36 (2) urokinase plasminogen activator receptor (UPAR)—whose ligand uPA is involved in cardiac macrophage accumulation, macrophage oxidative stress, and subsequent cardiac fibrosis37, 38—was found to be associated with incident HF32 and worse outcome in prevalent HF;39 (3) lymphotoxin β receptor (LTBR)—a known orchestrator of immune cell trafficking which binds 2 immune-cell specific ligands and is expressed on endothelial cells and smooth muscle cells40–42—is associated with HF, atherosclerosis, endothelial cell inflammation, and diabetes; (4) interleukin-1 receptor type-1 (IL-1RT1, most correlated protein with PC6) is involved in the IL-1 signaling cascade, and the IL-1 blocker anakinra has been studied in HFpEF in the phase 2 D-HART trial in HFpEF where it did not improve cardiorespiratory fitness but did lower hsCRP and NT-proBNP;43 and (5) perlecan (PLC), a large basement membrane proteoglycan which regulates inflammation, cardiac development, and angiogenesis.44 An additional protein identified in our study, IGFBP7 (the main hub of the yellow cluster)—which is localized to endothelial cells and related to cellular senescence and fibrosis, may link it to the pro-inflammatory cascade in HFpEF—is associated with both adverse outcome and worse diastolic function in HFpEF.45
Clinical impact and future research
Inflammation biomarkers may have potential in screening, diagnostic, and prognostic purposes for HFpEF.5–7, 9, 24, 32, 46 Considering that comorbidities are thought to be precursors of HFpEF by increasing systemic inflammation—which is supported by our results—it would be worthwhile to investigate whether we can select high-risk patients based on their level of circulating inflammation (or enrichment of specific inflammation modules/proteins in the blood) for early diagnosis and prevention efforts. Moreover, our results support the pursuit of inflammation as a therapeutic target for prevalent HFpEF. Treating in an early stage may improve efficacy of therapy in HFpEF considering that inflammation precedes cardiac stiffening, which may be very difficult to reverse. Broad anti-inflammatory or immunomodulatory therapies including anti-TNF therapy (RENEWAL program), immuno-modulation (ACCLAIM trial), and intravenous immunoglobulin therapy did not show benefit in prior HFrEF trials.47 Our study, coupled with epidemiological studies showing a greater role of inflammation in preceding incident HFpEF compared to HFrEF, provide rationale to reexamine targeting inflammation in HFpEF, the clinical trials for which have thus far been limited.43, 48 Thus, both non-specific anti-inflammatory therapies (e.g., colchicine, statins, n3-PUFA) as well as more targeted therapies (e.g., anti-MCP1, recombinant IL-10, ICAM/VCAM-antagonists) still hold promise in HFpEF.47, 49 Furthermore, HFpEF is known to be heterogeneous, and inflammation may be more or less dominant in specific subtypes; thus future studies and trials may benefit from: (1) characterization of the circulating inflammation profile in HFpEF patients in order to provide appropriate and targeted therapy; (2) confirmation of causality of inflammation in the pathogenesis of HFpEF; and (3) investigation of specific inflammation targets.
Strengths and limitations
Strengths of our study include the prospective, multi-center, multi-national nature of the PROMIS-HFpEF study and validation of our results in an independent cohort of HFpEF patients and comorbidity controls. We utilized a comprehensive inflammation proteomic evaluation using both deductive and inductive approaches with a large set of protein biomarkers. Finally, we performed mediation analysis as a method for causal inference. Although mediation does not prove causality, it goes beyond simple, unidirectional association analyses.
Several limitations should be considered when interpreting the results of our study. Our results may be biased because the proteins we measured—despite the relatively large number studied here—are still only a small subset of all the proteins in the circulation, and do not necessarily reflect intracellular or tissue-specific proteins. We attempted to mitigate this limitation using pathway analysis which can broaden the inference obtained from the smaller subset of proteins measured in our study. In addition, our investigation was not exploratory but based on the specific, published hypothesis that inflammation in an important pathogenic stimulus in HFpEF based on prior pre-clinical and clinical studies. Moreover, we intentionally used both deductive (PC analysis) and inductive (WCNA) approaches to identify related inflammation biomarkers. Interestingly, both approaches returned analogous inflammation protein profiles based on the top 10 most correlated proteins with PC1 versus the top 10 proteins of the turquoise cluster, providing a form of internal validation of our findings. This reduces the potential for bias, plus we additionally provide external validation of our results. Mediation analysis in a cross-sectional study should be interpreted with caution because of the potential for associations to be bidirectional. We cannot exclude the possibility that elevated cardiac filling pressures and worse RV function (reflective of worse HF) is the trigger for increased inflammation and elevated biomarkers such as TNFR1. However, prior studies that have found biomarkers such as TNFR1 are associated with incident HF, coupled with pre-clinical studies showing a role for dysregulated TNFR1 signaling in the pathogenesis of HF50 argue against inflammation as solely a consequence (and not a cause) of HFpEF. Nevertheless, population-based studies of incident HFpEF, mechanistic studies in animal models, and replication of our results with alternative proteomic assays or platforms should be performed to validate our findings.
Conclusions
In a comprehensive proteomic investigation of the comorbidity-inflammation paradigm in HFpEF, we found that (1) comorbidity burden is associated with abnormal cardiac structure and function; (2) comorbidity burden is associated with heightened systemic inflammation; (3) circulating proteins reflective of systemic inflammation are associated with worse cardiac function and are upregulated in HFpEF compared to non-HF controls with comorbidities; and importantly, (4) inflammation appears to mediate the association between comorbidity burden and worse cardiac hemodynamics and RV function.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new?
To investigate the hypothesis that HFpEF is the result of comorbidity-induced systemic inflammation, we performed a comprehensive proteomic analysis including 248 unique circulating proteins in the PROMIS-HFpEF study and validated our results externally.
We found that circulating proteins involved in inflammation mediate the association between comorbidity burden and echocardiographic parameters of worse hemodynamics and right ventricular function in HFpEF, supporting the comorbidity-inflammation paradigm.
Inflammatory proteins formed a conserved network across 2 independent HFpEF cohorts, and inflammatory pathways were upregulated in HFpEF cases versus controls, providing additional supportive evidence for the role of inflammation in HFpEF.
What are the clinical implications?
If validated in future studies, our findings could support reconsidering inflammation as a therapeutic target in patients with HFpEF and could lead to the investigation of inflammation targets in HFpEF.
Comprehensive characterization of the circulating inflammation profile may assist with the diagnosis of HFpEF and the identification of a HFpEF subgroup that has increased burden of inflammation due to comorbidities.
ACKNOWLEDGEMENTS
None.
FUNDING SOURCES
The PROMIS-HFpEF study was funded by AstraZeneca. The Northwestern University HFpEF proteomic validation study was funded by the American Heart Association (#15CVGPSD27260148). S.S-vW is supported by a fellowship grant from the Netherlands Heart Institute. A.S. is supported by grants from the Finnish Cardiovascular Foundation and Academy of Finland. L.H.L. is supported by Karolinska Institutet, the Swedish Research Council [grant 523-2014-2336], the Swedish Heart Lung Foundation [grants 20150557, 20190310], and the Stockholm County Council [grants 20170112, 20190525]. C.S.L. is supported by a Clinician Scientist Award from the National Medical Research Council of Singapore. S.J.S. is supported by grants from the National Institutes of Health (R01 HL140731, R01 HL120728, R01 HL107577, and R01 HL149423) and the American Heart Association (#16SFRN28780016).
DISCLOSURES
S.S-vW has received research grants and speaker fees from Roche Diagnostics.
A.S. has received consulting fees from Astra Zeneca; and has received speaker fees from Astra Zeneca, Abbott, Amgen, Bayer, and Novartis.
R.S.T. has served on an advisory board/speaker’s panel for Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Johnson & Johnson, Merck, Novo Nordisk, and Pfizer; and has served on a speaker’s panel for Roche Diagnostics.
M.L.F. and L-M.G. are employees of AstraZeneca.
L.H.L. has received research grants from AstraZeneca, Novartis, Boehringer Ingelheim, Boston Scientific, Vifor Pharma, Relypsa, Pharmacosmos and consulting/speaker honoraria from AstraZeneca, Novartis, Bayer, Vifor Pharma, Sanofi, Lexicon, Myokardia, Orion Pharma, Merck/MSD, Respicardia, and Medscape.
C.S.L. has received research support from Boston Scientific, Bayer, Roche Diagnostics, AstraZeneca, Medtronic, and Vifor Pharma; has served as consultant or on the advisory board/ steering committee/executive committee for Abbott Diagnostics, Amgen, Applied Therapeutics, AstraZeneca, Bayer, Biofourmis, Boehringer Ingelheim, Boston Scientific, Corvia Medical, Cytokinetics, Darma Inc., Eko.ai Pte Ltd, JanaCare, Janssen Research & Development LLC, Medtronic, Menarini Group, Merck, MyoKardia, Novartis, Novo Nordisk, Radcliffe Group Ltd., Roche Diagnostics, Stealth BioTherapeutics, The Corpus, Vifor Pharma and WebMD Global LLC; and serves as co-founder and non-executive director of EKo.ai Pte Ltd.
S.J.S. has received research grants from Actelion, AstraZeneca, Corvia, Pfizer, and Novartis; and has served as consultant or on the advisory board/steering committee/executive committee for Actelion, Amgen, AstraZeneca, Aria, Axon, Bayer, Bristol Myers Squib, Boehringer-Ingelheim, Cardiora, Cyclerion, Cytokinetics, Eisai, GSK, Imara, Ionis, Ironwood, Lilly, Merck, MyoKardia, Novartis, Novo Nordisk, Pfizer, Regeneron, Sanofi, Shifamed, and United Therapeutics.
Non-standard Abbreviations and Acronyms
- CFR
coronary flow reserve
- FDR
false discovery rate
- GDF-15
growth differentiation factor-15
- GLS
global longitudinal strain
- HFpEF
heart failure with preserved ejection fraction
- hs-CRP
high-sensitivity C-reactive protein
- IGFBP7
insulin Like Growth Factor Binding Protein 7
- IL-6
interleukin-6
- IL1-RT1
interleukin-1 receptor type 1
- LA
left atrial
- LOD
limit of detection
- LTBR
lymphotoxin beta receptor
- LV
left ventricular
- PCA
principal component analysis
- PLC
perlecan
- PROMIS-HFpEF
PRevalence Of MIcrovascular dySfunction in Heart Failure with Preserved Ejection Fraction
- RV
right ventricular
- TAPSE
tricuspid annular plane systolic excursion
- TNFR1
tumor necrosis factor receptor 1
- TNFα
tumor necrosis factor-alpha
- TR
tricuspid regurgitation
- TRARILR2
TNF-related apoptosis-inducing ligand receptor 2
- UPAR
urokinase plasminogen activator receptor
- WCNA
weighted coexpression network analysis
Footnotes
All other authors report no relevant disclosures.
SUPPLEMENTAL MATERIALS
REFERENCES
- 1.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 2.Komajda M, Carson PE, Hetzel S, McKelvie R, McMurray J, Ptaszynska A, Zile MR, Demets D, Massie BM. Factors associated with outcome in heart failure with preserved ejection fraction: findings from the Irbesartan in Heart Failure with Preserved Ejection Fraction Study (I-PRESERVE). Circ Heart Fail. 2011;4:27–35. [DOI] [PubMed] [Google Scholar]
- 3.Kristensen SL, Mogensen UM, Jhund PS, Petrie MC, Preiss D, Win S, Kober L, McKelvie RS, Zile MR, Anand IS, Komajda M, Gottdiener JS, Carson PE, McMurray JJ. Clinical and Echocardiographic Characteristics and Cardiovascular Outcomes According to Diabetes Status in Patients With Heart Failure and Preserved Ejection Fraction: A Report From the I-Preserve Trial (Irbesartan in Heart Failure With Preserved Ejection Fraction). Circulation. 2017;135:724–735. [DOI] [PubMed] [Google Scholar]
- 4.Ather S, Chan W, Bozkurt B, Aguilar D, Ramasubbu K, Zachariah AA, Wehrens XH, Deswal A. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol. 2012;59:998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hage C, Michaelsson E, Linde C, Donal E, Daubert JC, Gan LM, Lund LH. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients With Heart Failure With Preserved Ejection Fraction: A Holistic Proteomic Approach. Circ Cardiovasc Genet. 2017;10:e001633. [DOI] [PubMed] [Google Scholar]
- 6.Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, Liu Y, Hoffmann U, Bauer DC, Newman AB, Kritchevsky SB, Harris TB, Butler J. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol. 2010;55:2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanders-van Wijk S, van Empel V, Davarzani N, Maeder MT, Handschin R, Pfisterer ME, Brunner-La Rocca HP. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail. 2015;17:1006–1014. [DOI] [PubMed] [Google Scholar]
- 8.AbouEzzeddine OF, McKie PM, Dunlay SM, Stevens SR, Felker GM, Borlaug BA, Chen HH, Tracy RP, Braunwald E, Redfield MM. Suppression of Tumorigenicity 2 in Heart Failure With Preserved Ejection Fraction. J Am Heart Assoc. 2017;6:e004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, Metra M, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, Lang CC, Ng LL, Zannad F, Zwinderman AH, Hillege HL, van der Meer P, Voors AA. Identifying Pathophysiological Mechanisms in Heart Failure With Reduced Versus Preserved Ejection Fraction. J Am Coll Cardiol. 2018;72:1081–1090. [DOI] [PubMed] [Google Scholar]
- 10.Shah SJ, Lam CSP, Svedlund S, Saraste A, Hage C, Tan RS, Beussink-Nelson L, Fermer ML, Broberg MA, Gan LM, Lund LH. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. 2018;39:3439–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvaraj S, Aguilar FG, Martinez EE, Beussink L, Kim KY, Peng J, Rasmussen-Torvik L, Sha J, Irvin MR, Gu CC, Lewis CE, Hunt SC, Arnett DK, Shah SJ. Association of comorbidity burden with abnormal cardiac mechanics: findings from the HyperGEN study. J Am Heart Assoc. 2014;3:e000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC, Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2015;131:269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28:1–39 e14. [DOI] [PubMed] [Google Scholar]
- 14.Nagueh SF, Smiseth OA, Appleton CP, Byrd BF 3rd, Dokainish H, Edvardsen T, Flachskampf FA, Gillebert TC, Klein AL, Lancellotti P, Marino P, Oh JK, Alexandru Popescu B, Waggoner AD, Houston T, Oslo N, Phoenix A, Nashville T, Hamilton OC, Uppsala S, Ghent Liege B, Cleveland O, Novara I, Rochester M, Bucharest R, Louis M. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2016;17:1321–1360. [DOI] [PubMed] [Google Scholar]
- 15.Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K, Solomon SD, Louie EK, Schiller NB. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23:685–713. [DOI] [PubMed] [Google Scholar]
- 16.Freed BH, Daruwalla V, Cheng JY, Aguilar FG, Beussink L, Choi A, Klein DA, Dixon D, Baldridge A, Rasmussen-Torvik LJ, Maganti K, Shah SJ. Prognostic Utility and Clinical Significance of Cardiac Mechanics in Heart Failure With Preserved Ejection Fraction: Importance of Left Atrial Strain. Circ Cardiovasc Imaging. 2016;9:e003754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee H, Herbert RD, McAuley JH. Mediation Analysis. JAMA. 2019;321:697–698. [DOI] [PubMed] [Google Scholar]
- 18.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franssen C, Chen S, Hamdani N, Paulus WJ. From comorbidities to heart failure with preserved ejection fraction: a story of oxidative stress. Heart. 2016;102:320–330. [DOI] [PubMed] [Google Scholar]
- 21.Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016;4:312–324. [DOI] [PubMed] [Google Scholar]
- 22.van der Pol A, Gil A, Tromp J, Sillje HHW, van Veldhuisen DJ, Voors AA, Hoendermis ES, Grote Beverborg N, Schouten EM, de Boer RA, Bischoff R, van der Meer P. OPLAH ablation leads to accumulation of 5-oxoproline, oxidative stress, fibrosis, and elevated fillings pressures: a murine model for heart failure with a preserved ejection fraction. Cardiovasc Res. 2018;114:1871–1882. [DOI] [PubMed] [Google Scholar]
- 23.van Dijk CG, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van Heerebeek L, Duncker DJ, Verhaar MC, Fontoura D, Lourenco AP, Leite-Moreira AF, Falcao-Pires I, Joles JA, Cheng C. Distinct Endothelial Cell Responses in the Heart and Kidney Microvasculature Characterize the Progression of Heart Failure With Preserved Ejection Fraction in the Obese ZSF1 Rat With Cardiorenal Metabolic Syndrome. Circ Heart Fail. 2016;9:e002760. [DOI] [PubMed] [Google Scholar]
- 24.van Empel V, Brunner-La Rocca HP. Inflammation in HFpEF: Key or circumstantial? Int J Cardiol. 2015;189:259–263. [DOI] [PubMed] [Google Scholar]
- 25.van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–839. [DOI] [PubMed] [Google Scholar]
- 26.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44–52. [DOI] [PubMed] [Google Scholar]
- 27.Hamdani N, Franssen C, Lourenco A, Falcao-Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, Gonzalez A, Tschope C, Diez J, Linke WA, Leite-Moreira AF, Paulus WJ. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–1249. [DOI] [PubMed] [Google Scholar]
- 28.Flachskampf FA, Biering-Sorensen T, Solomon SD, Duvernoy O, Bjerner T, Smiseth OA. Cardiac Imaging to Evaluate Left Ventricular Diastolic Function. JACC Cardiovasc Imaging. 2015;8:1071–1093. [DOI] [PubMed] [Google Scholar]
- 29.Mitter SS, Shah SJ, Thomas JD. A Test in Context: E/A and E/e’ to Assess Diastolic Dysfunction and LV Filling Pressure. J Am Coll Cardiol. 2017;69:1451–1464. [DOI] [PubMed] [Google Scholar]
- 30.Valentova M, von Haehling S, Bauditz J, Doehner W, Ebner N, Bekfani T, Elsner S, Sliziuk V, Scherbakov N, Murin J, Anker SD, Sandek A. Intestinal congestion and right ventricular dysfunction: a link with appetite loss, inflammation, and cachexia in chronic heart failure. Eur Heart J. 2016;37:1684–1691. [DOI] [PubMed] [Google Scholar]
- 31.Ueland T, Kjekshus J, Froland SS, Omland T, Squire IB, Gullestad L, Dickstein K, Aukrust P. Plasma levels of soluble tumor necrosis factor receptor type I during the acute phase following complicated myocardial infarction predicts survival in high-risk patients. J Am Coll Cardiol. 2005;46:2018–2021. [DOI] [PubMed] [Google Scholar]
- 32.Stenemo M, Nowak C, Byberg L, Sundstrom J, Giedraitis V, Lind L, Ingelsson E, Fall T, Arnlov J. Circulating proteins as predictors of incident heart failure in the elderly. Eur J Heart Fail. 2018;20:55–62. [DOI] [PubMed] [Google Scholar]
- 33.Marti CN, Khan H, Mann DL, Georgiopoulou VV, Bibbins-Domingo K, Harris T, Koster A, Newman A, Kritchevsky SB, Kalogeropoulos AP, Butler J. Soluble tumor necrosis factor receptors and heart failure risk in older adults: Health, Aging, and Body Composition (Health ABC) Study. Circ Heart Fail. 2014;7:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anker SD, Coats AJ. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol. 2002;86:123–130. [DOI] [PubMed] [Google Scholar]
- 35.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119:1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouabdallaoui N, Claggett B, Zile MR, McMurray JJV, O’Meara E, Packer M, Prescott MF, Swedberg K, Solomon SD, Rouleau JL. Growth differentiation factor-15 is not modified by sacubitril/valsartan and is an independent marker of risk in patients with heart failure and reduced ejection fraction: the PARADIGM-HF trial. Eur J Heart Fail. 2018;20:1701–1709. [DOI] [PubMed] [Google Scholar]
- 37.Fuhrman B, Khateeb J, Shiner M, Nitzan O, Karry R, Volkova N, Aviram M. Urokinase plasminogen activator upregulates paraoxonase 2 expression in macrophages via an NADPH oxidase-dependent mechanism. Arterioscler Thromb Vasc Biol. 2008;28:1361–1367. [DOI] [PubMed] [Google Scholar]
- 38.Moriwaki H, Stempien-Otero A, Kremen M, Cozen AE, Dichek DA. Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice. Circ Res. 2004;95:637–644. [DOI] [PubMed] [Google Scholar]
- 39.Koller L, Stojkovic S, Richter B, Sulzgruber P, Potolidis C, Liebhart F, Mortl D, Berger R, Goliasch G, Wojta J, Hulsmann M, Niessner A. Soluble Urokinase-Type Plasminogen Activator Receptor Improves Risk Prediction in Patients With Chronic Heart Failure. JACC Heart Fail. 2017;5:268–277. [DOI] [PubMed] [Google Scholar]
- 40.Dahl CP, Gullestad L, Fevang B, Holm AM, Landro L, Vinge LE, Fiane AE, Sandberg WJ, Otterdal K, Froland SS, Damas JK, Halvorsen B, Aukrust P, Oie E, Yndestad A. Increased expression of LIGHT/TNFSF14 and its receptors in experimental and clinical heart failure. Eur J Heart Fail. 2008;10:352–359. [DOI] [PubMed] [Google Scholar]
- 41.Halvorsen B, Santilli F, Scholz H, Sahraoui A, Gulseth HL, Wium C, Lattanzio S, Formoso G, Di Fulvio P, Otterdal K, Retterstol K, Holven KB, Gregersen I, Stavik B, Bjerkeli V, Michelsen AE, Ueland T, Liani R, Davi G, Aukrust P. LIGHT/TNFSF14 is increased in patients with type 2 diabetes mellitus and promotes islet cell dysfunction and endothelial cell inflammation in vitro. Diabetologia. 2016;59:2134–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grandoch M, Feldmann K, Gothert JR, Dick LS, Homann S, Klatt C, Bayer JK, Waldheim JN, Rabausch B, Nagy N, Oberhuber A, Deenen R, Kohrer K, Lehr S, Homey B, Pfeffer K, Fischer JW. Deficiency in lymphotoxin beta receptor protects from atherosclerosis in apoE-deficient mice. Circ Res. 2015;116:e57–68. [DOI] [PubMed] [Google Scholar]
- 43.Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, Del Buono MG, Billingsley H, Wohlford G, Viscusi M, Oddi-Erdle C, Abouzaki NA, Dixon D, Biondi-Zoccai G, Arena R, Abbate A. IL-1 Blockade in Patients With Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2018;11:e005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gubbiotti MA, Neill T, Iozzo RV. A current view of perlecan in physiology and pathology: A mosaic of functions. Matrix Biol. 2017;57–58:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gandhi PU, Gaggin HK, Redfield MM, Chen HH, Stevens SR, Anstrom KJ, Semigran MJ, Liu P, Januzzi JL Jr. Insulin-Like Growth Factor-Binding Protein-7 as a Biomarker of Diastolic Dysfunction and Functional Capacity in Heart Failure With Preserved Ejection Fraction: Results From the RELAX Trial. JACC Heart Fail. 2016;4:860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng JM, Akkerhuis KM, Battes LC, van Vark LC, Hillege HL, Paulus WJ, Boersma E, Kardys I. Biomarkers of heart failure with normal ejection fraction: a systematic review. Eur J Heart Fail. 2013;15:1350–1362. [DOI] [PubMed] [Google Scholar]
- 47.Glezeva N, Baugh JA. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail Rev. 2014;19:681–694. [DOI] [PubMed] [Google Scholar]
- 48.Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, Jahangiri A, Falcao RA, Kontos MC, Shah KB, Voelkel NF, Dinarello CA, Abbate A. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol. 2014;113:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tschope C, Van Linthout S, Kherad B. Heart Failure with Preserved Ejection Fraction and Future Pharmacological Strategies: a Glance in the Crystal Ball. Curr Cardiol Rep. 2017;19:70. [DOI] [PubMed] [Google Scholar]
- 50.Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD. Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy. Circulation. 2019;139:206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.