

Abstract
Objectives
To identify predictors of portal hypertension, liver transplantation, and death in North American youth with alpha-1-antitrypsin (AAT) deficiency, and compare with AAT deficiency patients elsewhere.
Study design
The Childhood Liver Disease Research Network Longitudinal Observational Study of Genetic Causes of Intrahepatic Cholestasis is a prospective, cohort study of pediatric cholestatic liver diseases, including AAT deficiency, enrolling PIZZ and PISZ subjects 0-25 years of age seen since November 2007 at 17 tertiary care centers in the US and Canada. Data from standard-of-care baseline and annual follow-up visits were recorded from medical records, history, physical examination, and laboratory studies. Participants with portal hypertension were identified based on data collected.
Results
We enrolled 350 participants (60% male) with a native liver; 278 (79%) entered the cohort without portal hypertension and 18 developed portal hypertension during follow-up. Thirty participants required liver transplantation; 2 patients died during 1077 person-years of follow-up. There was no difference in participants with or without preceding neonatal cholestasis progressing to transplantation or death during the study (12% vs 7%; P = .09), or in experiencing portal hypertension (28% vs 21%; P = .16); the hazard ratio for neonatal cholestasis leading to portal hypertension was P = .04. Development of portal hypertension was associated with a reduced height Z-score.
Conclusions
Portal hypertension in youth with AAT deficiency impacts growth measures. Progression to liver transplantation is slow and death is rare, but the risk of complications and severe liver disease progression persists throughout childhood. A history of neonatal cholestasis is a weak predictor of severe disease.
Alpha-1-antitrypsin (AAT) deficiency occurs in 1 in 2000-3500 births in North American and European populations.1,2 It is associated with chronic liver disease, cirrhosis, and hepatocellular carcinoma. The Z mutant allele of the AAT gene is associated with the vast majority of AAT liver disease, either as the classical form of homozygous ZZ AAT deficiency, or as the SZ compound heterozygous form, which also has an increased risk of liver disease.1
The most common childhood presentation of AAT deficiency is neonatal cholestasis1,2 Although the cholestasis often resolves spontaneously, a minority of these children go on to develop cirrhosis and portal hypertension in the first few years of life. Children with AAT less commonly present at later ages with signs or symptoms of liver disease or are diagnosed during family testing. A minority of children with AAT progress to liver failure and liver transplantation or death. The factors influencing liver disease are not known, and it is not possible to predict which patients will progress to severe liver disease.
The mechanism of the liver injury in AAT deficiency involves the accumulation within hepatocytes of the AAT mutant Z protein, which triggers hepatocellular death, compensatory regeneration and, in some patients, hepatic fibrosis.1,3–5 However, given the wide variability in clinical course, an important role for genetic and environmental modifiers is likely.1,2,6,7 In the early 1970s, a cohort of 127 ZZ AAT-deficient children was identified in Sweden by newborn screening and followed longitudinally.8–10 This unbiased, population-based cohort had a low rate (<5%) of life-threatening liver disease in childhood, and all patients who developed severe disease had preceding neonatal cholestasis. There was no evidence of liver injury or complications of liver disease from the second through the fourth decades of life. This finding is in contrast with other single-center retrospective reports from more genetically mixed groups of patients in North America and Western Europe that report a wider range of liver complications with progression throughout childhood, sometimes without preceding neonatal cholestasis.1,2,11–14 It maybe that some complications of liver disease, or progressive disease in childhood, are too infrequent to be detected in the Swedish cohort of 127 individuals. However, it is also possible that a different set of disease modifiers, associated with more severe disease, are found outside of Sweden.
The Childhood Liver Disease Research Network is a National Institutes of Health-supported consortium of pediatric tertiary care centers in North America, 17 of which collected data during this study period, focused on the study of rare pediatric liver diseases, as described.15–19 The Longitudinal Observational Study of Genetic Causes of Intrahepatic Cholestasis (NCT00571272) is a study conducted by this network with the goal of describing the natural history and genetic and environmental modifiers of a group of metabolic-cholestatic liver diseases, including AAT deficiency. We hypothesized that the prospective study of a cohort of children and young adults with AAT deficiency would provide insight into the natural history and clinical variability. The goal of this analysis was to evaluate undefined predictors of portal hypertension, liver transplantation, and death in North American youth with AAT deficiency, and determine how they differ from other AAT deficiency patients around the world.
Methods
Population and Assessments
This report includes participants with AAT deficiency with their native livers enrolled into Longitudinal Observational Study of Genetic Causes of Intrahepatic Cholestasis from November 30, 2007, through November 30, 2017.15–18 Institutional review board approvals and consents were obtained at each site. Eligibility for enrollment of AAT participants in the Longitudinal Observational Study of Genetic Causes of Intrahepatic Cholestasis study include PIZZ or PISZ serum protein phenotype or genotype, with a corresponding low serum level of AAT protein (defined as less than the laboratory wild-type reference range), age birth to 25 years, and evidence of liver disease as defined by documentation of one of the following: neonatal cholestasis (conjugated hyperbilirubinemia and jaundice within the first 3 months of life); 1.25 or more times the upper limit of normal alanine transaminase, aspartate transaminase (AST), or gamma-glutamyl transpeptidase (GGTP); chronic hepatomegaly; clinical findings or complications of portal hypertension or cirrhosis; impaired liver synthetic function; or abnormal liver biopsy histology, other than globular inclusions of AAT, showing liver injury (inflammation, fibrosis, or necrosis), as described.19
At enrollment, medical history and physical examination were obtained, including review of available medical records and standard of care laboratories. Updates in medical history and physical examination were documented at annual follow-up visits, and other data collected as previously described.19
Outcomes
Primary outcomes were the onset of either definite, or possible, clinically evident portal hypertension and liver transplantation or death. Definite clinically evident portal hypertension was defined as either ascites (treatment with diuretics for a history of or currently present ascites) or endoscopic evidence of esophageal or gastric varices or clinical findings consistent with portal hypertension. The clinical findings indicating portal hypertension were the presence of both splenomegaly (spleen >2 cm below the costal margin) and thrombocytopenia (platelet count <150 000/mm3). Possible clinically evident portal hypertension was used to designate participants with splenomegaly or thrombocytopenia, but not both (definition adapted from18). For this analysis, these groups were considered together as definite/possible portal hypertension to increase the power of our conclusions owing to low numbers of severe event outcomes.18
Other outcomes assessed were growth and anthropomorphic outcomes, such as weight, height, and weight-for-height Z-scores, body mass index, head circumference, right mid-arm circumference, and right triceps skinfold thickness.
Statistical Analyses
Descriptive statistics of patient characteristics, follow-up, and longitudinal patterns for outcomes were used to describe the study population and its natural history. Kaplan-Meier methods were used to characterize the time-to-definite/possible portal hypertension and to liver transplantation or death. Last follow-up for those without an event was defined as their last completed clinical visit; thus, participants were censored at the time of liver transplantation, death, drop-out, or last visit for time to definite/possible portal hypertension and were censored at drop-out or last visit for time-to-transplantation or death. (Note that no participants were censored for liver transplantation in this cohort because all transplanted participants experienced definite/possible portal hypertension before transplantation and death.)
To identify potential predictors Cox proportional hazards models were used with either fixed or time-dependent covariates. Time-dependent predictors are those assessed longitudinally at repeated visits (eg, laboratory measures). We examined the relationship between a history of neonatal cholestasis and definite/possible portal hypertension (including those with portal hypertension at study entry and those who developed portal hypertension during the course of the study) in participants with native liver using a χ2 test, and assessed time-to-portal hypertension and time-to-liver transplantation, relative to a personal history of neonatal cholestasis, in a Cox model. We also explored the impact of changes in laboratory measures of liver function (albumin, total bilirubin, direct bilirubin, AST, alanine transaminase, GGTP, platelets, AST/platelets ratio, and international normalized ratio) and changes in growth measures (head circumference Z-score, right mid-arm circumference, right triceps skinfold thickness, body mass index, body mass index Z-score, weight Z-score, and height Z-score) on outcomes. Because of the limited number of events, it was not possible to develop multivariable models to define risk scores for these important outcomes. A P value of less than .05 is considered statistically significant.
We used linear mixed models for longitudinal data, adjusted for age, to assess the effect of definite/possible portal hypertension on changes in growth measures. These models were used to predict the average change in anthropometric measures for a participant of average age at baseline with and without portal hypertension; predicted values and 95% CIs are presented graphically for measures that are statistically significant.
Results
Demographics and Course of the AAT Liver Disease Cohort
Of 350 participants with their native liver, 60% were male; the median age at baseline enrollment was 4.2 years (IQR, 7.9 years; range, 0.1-24.9 years). The majority of participants were non-Hispanic and white (Table I); 286 participants (90%) were homozygous ZZ and 32 (10%) were SZ.1,2,8 The median length of follow-up was 2.5 years (range, 0-9.7). Overall, 112 participants (32%) withdrew, were discontinued owing to being followed at noncontinuing centers (grants not renewed), were lost to follow-up, or had other reasons for withdrawal from the study (Table III and Table IV; available atwww.jpeds.com).
Table I.
Patient characteristics at study entry and during follow-up
| Characteristic | Statistic | Baseline | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 | Year 6 | Year 7 | Year 8 | Year 9 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Participants | N | 350 | 229 | 179 | 151 | 102 | 73 | 56 | 47 | 37 | 18 |
| Visits per participant | Mean (SD) | 3.55 (2.48) | – | – | – | – | – | – | – | – | – |
| Median (IQR) | 3 (4) | – | – | – | – | – | – | – | – | – | |
| Min, max | 1,11 | – | – | – | – | – | – | – | – | – | |
| Phenotype | SZ | 32 (10.1%) | – | – | – | – | – | – | – | – | – |
| ZZ | 286 (89.9%) | – | – | – | – | – | – | – | – | – | |
| Age (years) | N | 350 | 229 | 179 | 151 | 102 | 73 | 56 | 47 | 37 | 18 |
| Mean (SD) | 6.05 (5.49) | 6.82 (5.09) | 7.31 (4.87) | 8.91 (4.99) | 9.83 (4.85) | 9.99 (4.21) | 10.86 (4.04) | 11.52 (3.75) | 12.96 (3.97) | 13.24 (4.24) | |
| Median (IQR) | 4.23 (7.98) | 5.59 (7.38) | 5.93 (6.38) | 7.7 (7.07) | 8.64 (7.27) | 9.04 (5.3) | 9.94 (5.58) | 10.72 (4.49) | 12.79 (4.76) | 12.13 (4.25) | |
| Min, max | 0.12, 24.87 | 0.96, 22.48 | 1.77, 22.85 | 2.78, 23.9 | 4.05, 24.96 | 4.95, 25.97 | 6.06, 24.54 | 7.35, 25.79 | 8.25, 26.88 | 9.77, 28.07 | |
| Age group (years) | 0-<5 | 191 (54.6%) | 107 (46.7%) | 77 (43%) | 42 (27.8%) | 13 (12.7%) | 1 (1.4%) | – | – | – | – |
| 5-<10 | 83 (23.7%) | 70 (30.6%) | 60 (33.5%) | 56 (37.1%) | 47 (46.1%) | 43 (58.9%) | 28 (50%) | 20 (42.6%) | 11 (29.7%) | 2 (11.1%) | |
| 10-<15 | 43 (12.3%) | 31 (13.5%) | 25 (14%) | 31 (20.5%) | 26 (25.5%) | 23 (31.5%) | 21 (37.5%) | 20 (42.6%) | 18 (48.6%) | 13 (72.2%) | |
| 15-<20 | 28 (8%) | 17 (7.4%) | 13 (7.3%) | 17 (11.3%) | 12 (11.8%) | 4 (5.5%) | 5 (8.9%) | 5 (10.6%) | 6 (16.2%) | 2 (11.1%) | |
| 20-<25 | 5 (1.4%) | 4 (1.7%) | 4 (2.2%) | 5 (3.3%) | 4 (3.9%) | 1 (1.4%) | 2 (3.6%) | 1 (2.1%) | 1 (2.7%) | – | |
| ≥25 | – | – | – | – | – | 1 (1.4%) | – | 1 (2.1%) | 1 (2.7%) | 1 (5.6%) | |
| Ethnicity | Hispanic | 21 (6.1%) | 15 (6.6%) | 13 (7.3%) | 9 (6%) | 8 (7.8%) | 5 (6.8%) | 3 (5.4%) | 4 (8.5%) | 3 (8.1%) | 3 (16.7%) |
| Non-Hispanic | 324 (93.9%) | 212 (93.4%) | 165 (92.%) | 141 (94%) | 94 (92.2%) | 68 (93.2%) | 53 (94.6%) | 43 (91.5%) | 34 (91.9%) | 15 (83.3%) | |
| Sex | Male | 211 (60.5%) | 144 (62.9%) | 112 (62.6%) | 86 (57%) | 61 (59.8%) | 39 (53.4%) | 32 (57.1%) | 22 (46.8%) | 20 (54.1%) | 11 (61.1%) |
| Female | 138 (39.5%) | 85 (37.1%) | 67 (37.4%) | 65 (43%) | 41 (40.2%) | 34 (46.6%) | 24 (42.9%) | 25 (53.2%) | 17 (45.9%) | 7 (38.9%) | |
| Race | White | 322 (93.6%) | 210 (92.5%) | 167 (93.3%) | 140 (92.7%) | 96 (94.1%) | 70 (95.9%) | 52 (92.9%) | 43 (91.5%) | 34 (91.9%) | 17 (94.4%) |
| Black | 3 (0.9%) | 3 (1.3%) | 1 (0.6%) | 1 (0.7%) | – | – | – | – | – | – | |
| Other | 19 (5.5%) | 14 (6.2%) | 11 (6.1%) | 10 (6.6%) | 6 (5.9%) | 3 (4.1%) | 4 (7.1%) | 4 (8.5%) | 3 (8.1%) | 1 (5.6%) |
Seventy-two participants (21%) had definite/possible clinically evident portal hypertension at entry into the study. Of the 278 participants (79%) who entered the study without portal hypertension, 18 developed definite/possible portal hypertension during follow-up; the incidence rate is 2.1 per 100 person-years of follow-up. Thirty-two participants underwent liver transplantation or died during 1077 person-years of follow-up (annual incidence rate of 3.0 transplants per 100 person-years of follow-up; Table V and Table VI [available at www.jpeds.com]). One patient died with their native liver with definite/possible portal hypertension and one after transplant. Twenty-nine of the 30 transplanted participants entered the study with definite/possible portal hypertension, and all transplanted participants developed portal hypertension before transplant.
Kaplan-Meier curves for portal hypertension-free survival (Figure 1, A) and liver transplant-free survival (Figure 2) are shown for participants by presence or absence of neonatal cholestasis. We chose to focus on this comparison because neonatal cholestasis, although it only affects a minority of ZZ infants, is by far the most common presentation for AAT deficiency in childhood. Our data show there is no statistically significant difference for either definite/possible portal hypertension-free survival or liver transplant-free survival based on neonatal cholestasis. Prevalent participants with definite/possible portal hypertension at entry into the study are reflected in the drop at time 0. For those without portal hypertension at entry, there is a slow but continued progression; Figure 1, B shows the portal hypertension-free Kaplan-Meier curve for participants without portal hypertension at the time of study entry. For all participants combined at 2 years, the probability of developing definite/possible portal hypertension is 5% (95% CI, 2%-9%). Similarly, the incidence of liver transplantation or death increases slowly over time, with the 2-year probability of liver transplant or death of all participants estimated to be 8% (95% CI, 5%-11%).
Figure 1.
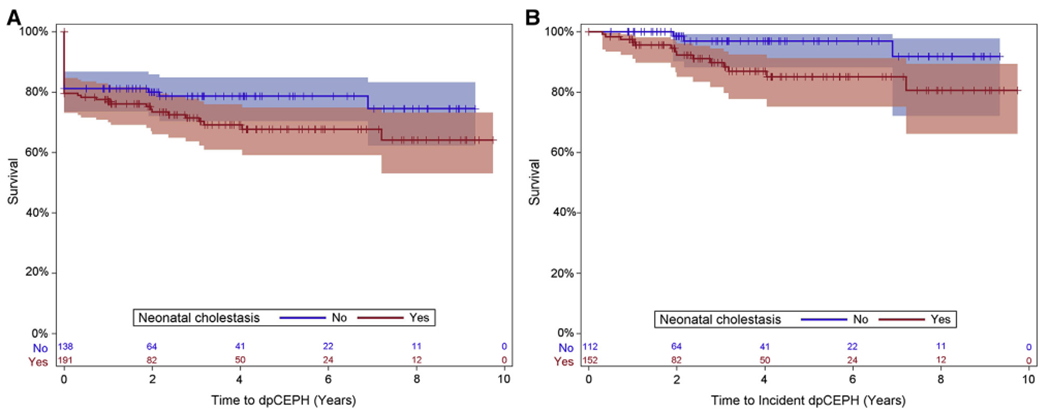
A, Definite/possible clinically evident portal hypertension (dpCEPH)-free Kaplan-Meier curve: all participants by neonatal cholestasis status. B, dpCEPH-free Kaplan-Meier curve: incident dpCEPH cases only.
Figure 2.
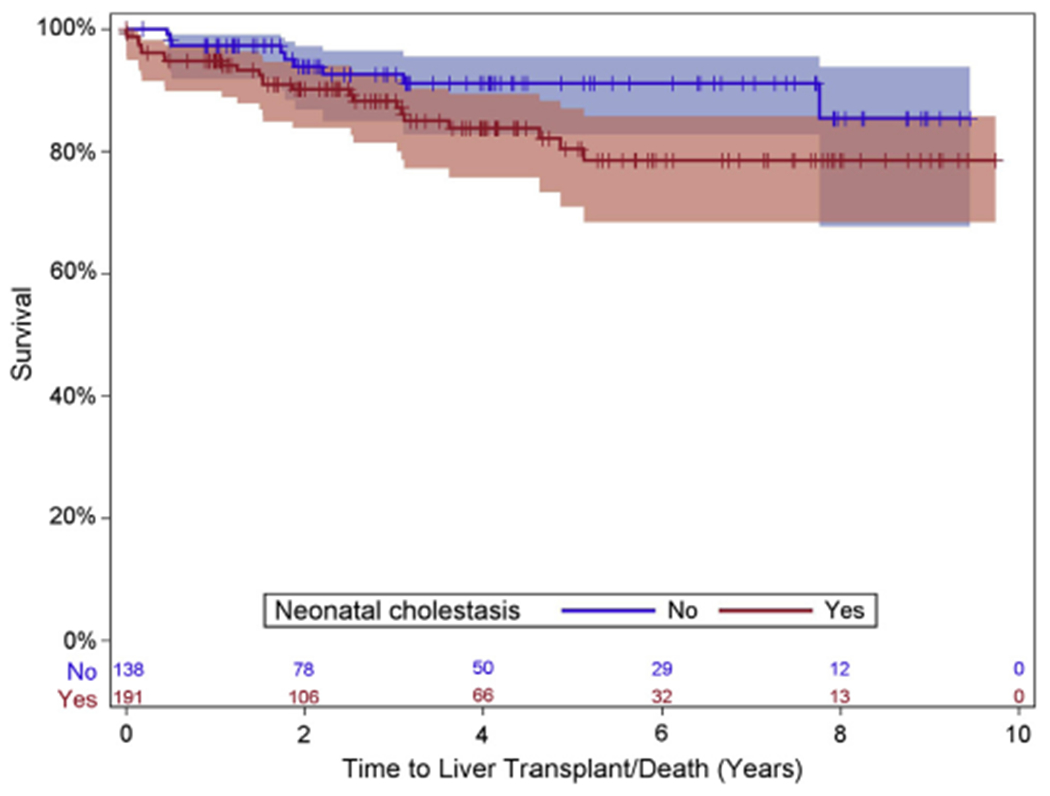
Liver transplant-free survival Kaplan-Meier curve. All participants by neonatal cholestasis status.
Predictors of Future Severe Disease and Disease Associations
Prospective standard of care physical examination and laboratory factors were analyzed for their ability to predict the development of portal hypertension (Table II) or liver transplantation or death (Table VI; available at www.jpeds.com). We found no statistically significant associations of anthropometric and growth measurements with risk of future portal hypertension. Over time, participants with increasing total bilirubin, AST, GGTP, and AST/platelet ratio, along with decreasing platelet count, were associated with increased risk of future portal hypertension. Owing to small numbers of events, neither a single measure nor a group of measures, could be found with useful predictive specificity for the development of portal hypertension. However, when a plot of absolute GGTP values vs age was constructed, many of the relatively few participants who developed definite/possible portal hypertension (Figure 3 [available at www.jpeds.com], red dots, n = 11 participants with GGTP values recorded) had higher GGTP values, especially among those 3 years of age or younger. Decreasing serum albumin and platelet count, and increasing total bilirubin, direct bilirubin, AST, and international normalized ratio were univariately associated with future liver transplant or death (Table VII).
Table II.
Univariate predictors of time-to-dpCEPH: Incident dpCEPH cases only
| Predictor | HR | 95% CI | P value |
|---|---|---|---|
| Head circumference Z-score | 0.65 | 0.42-1.00 | .051 |
| Right mid arm circumference | 0.93 | 0.2-1.07 | .32 |
| Right triceps skinfold thickness | 1.05 | 0.97-1.13 | .27 |
| BMI | 0.89 | 0.68-1.17 | .40 |
| BMI Z-score | 0.61 | 0.32-1.14 | .12 |
| Weight Z-score | 0.66 | 0.43-1.03 | .07 |
| Height Z-score | 0.67 | 0.42-1.07 | .09 |
| Albumin (per 1 g/dL increase) | 0.52 | 0.23-1.16 | .11 |
| Total bilirubin (per 1 mg/dL increase) | 1.97 | 1.33-2.90 | .0007 |
| Direct bilirubin (per 1 mg/dL increase) | 0.32 | 0.76-2.28 | .32 |
| AST (per 10 U/L increase) | 1.06 | 1.01-1.10 | .01 |
| ALT (per 10 U/L increase) | 1.00 | 1.00-1.01 | .25 |
| GGTP (per 100 U/L increase) | 1.20 | 1.10-1.31 | <.0001 |
| Platelets (103/mm3) | 0.99 | 0.98-1.00 | .04 |
| INR (per 0.1) | 1.21 | 0.95-1.54 | .12 |
| AST/platelets | 5.93 | 2.30-15.33 | .0002 |
| Neonatal cholestasis | 3.70 | 1.06-12.87 | .04 |
ALT, alanine transaminase; BMI, body mass index; dpCEPH, definite or possible clinically evident portal hypertension; HR, hazard ratio; INR, international normalized ratio; KM, Kaplan-Meier.
Model: Cox proportional hazards model with time-varying covariates (except for neonatal cholestasis, which is fixed).
We also examined whether development of definite/possible portal hypertension affected subsequent growth measures, after adjusting for age. Smaller changes in height Z-score (estimate = −0.21), triceps skinfold thickness (estimate = −0.17, log scale), and mid-arm circumference (estimate = −0.06, log scale) were found in children with, than in those without, definite/possible portal hypertension (P < .05 for each). Figure 4 (available at www.jpeds.com) represents the predicted average differences of these anthropometric measures for a representative AAT participants from our study (6 years old).
Twenty-eight percent of participants with a history of neonatal cholestasis had definite/possible portal hypertension (at entry or during follow-up) compared with 21% without a history of neonatal cholestasis (P = .16); the hazard ratio in those with neonatal cholestasis history relative to those without is 3.70 (95% CI, 1.06-12.87; P = .04). Twelve percent of participants with a history of neonatal cholestasis y were transplanted or died during the follow-up period compared with 7% without neonatal cholestasis history; the hazard ratio for liver transplantation or death for those with neonatal cholestasis history relative to those without is 1.93 (95% CI, 0.89-4.17; P = .09).
We found that 50 of 350 participants reported asthma or similar respiratory difficulties. There were no other clear associations in AAT participants between medical histories, medications, or other diagnoses reported in the cohort and the development of portal hypertension, death, or need for transplantation.
Discussion
We chose to focus on portal hypertension as one of our most important outcomes for children with AAT deficiency, because it is the most common severe liver disease outcome seen in children with this disease. Furthermore, it is common for AAT-deficient children with portal hypertension to remain stable for many years before decompensating to liver transplantation, which made it more likely we would capture a larger number of events than if we focused solely on the outcome of liver transplantation. The finding that the rate of definite/possible portal hypertension was 2.1 per 100 person-years is consistent with previous studies, suggesting that a large portion of patients with AAT deficiency are asymptomatic and well throughout childhood, and likely go unrecognized. This interpretation of the genetic prevalence data has been common. However, symptomatic North American children with AAT deficiency tend to be identified early in life during workup for signs or symptoms of liver disease, most commonly the neonatal cholestasis syndrome Therefore, our reported cohort is representative of the currently diagnosed body of young patients in the US and Canada. We followed a broad age range of individuals at multiple sites for long periods of time, including some for a decade. Our cohort’s characteristics are consistent with the known North-West European genetic origin of this disease, but are also likely to reflect the mixed gene pool of the US and Canada, which is hypothesized to have an important impact on outcomes.1,2,8
Our study highlights that most children with AAT-associated neonatal cholestasis do not develop portal hypertension within the current cohort follow-up period. Furthermore, most of our North American AAT participants did well, and the neonatal cholestasis resolved spontaneously, which is consistent with reports from other countries. We recognize that more patients left the study than we anticipated, which is a limitation; 16% were lost to follow-up, and another 13% exited the study because they were originally enrolled at study sites that were discontinued owing to grants not renewed. Although the existing follow-up data on these subjects was analyzed, there were likely events following discontinuation that were not captured.
From our analysis, it is evident that progression to portal hypertension or liver transplantation is not limited to those with a history of neonatal cholestasis and that the absence of neonatal cholestasis does not rule out the possibility of the future development of severe disease. This observation has been reported anecdotally, but has sometimes been questioned owing to a lack of systematic data collection.2,12,14 This finding is in contrast with the Swedish birth cohort, in which 100% of the severe disease participants had a history of neonatal cholestasis. It should be noted, however, that some of our potential participants might have had neonatal cholestasis and gone on to liver transplantation, before enrollment, and were therefore missed. In that case, this current report would underestimate the predictive value of neonatal cholestasis. Taken together, these data support the practice of continuing to monitor young patients with AAT for the development of portal hypertension, or other complications of liver disease, and to provide anticipatory guidance for health maintenance in the setting of increased risk of liver disease. For example, maintaining a healthy weight, administration of hepatitis A and B vaccines, counseling about the use of potentially hepatotoxic medications or supplements, counseling for strict avoidance of smoking or second-hand smoke, and nonsteroidal anti-inflammatory drug avoidance if portal hypertension is suspected.
Our cohort is selected for the presence of liver disease and identified a wider range of disease courses. The development of definite/possible portal hypertension was documented, and liver transplants occurred throughout childhood. This factor strengthens our study, in that nearly all reported outcomes in childhood of this disease are captured. However, the fact that this is not an unbiased birth cohort limits our ability to be specific about the frequency of these events in the population of asymptomatic, undiagnosed patients.
Modifier genes are likely to play an important role in progression to severe AAT disease, and it is possible that populations in different regions of the world have different sets of modifier genes. As our analysis continues, we have embarked on an ambitious genetic analysis of the cohort in an effort to identify disease modifiers whose physiologic relevance can be examined and possibly leveraged to find to new therapies.
Throughout the process of building and analyzing this cohort, we have sought to identify clinical markers obtained during routine care that would help physicians and families to identify patients at high risk of progression to severe liver disease. Although the development of an algorithm with useful specificity for predicting the hepatic prognosis of youth with AAT is not yet possible from these data, some features portending a poor prognosis were evident. First, very high GGTP levels in the first few years of life were commonly seen in participants with portal hypertension. In general, worsening liver chemistries tended to be seen as portal hypertension developed. However, no specific cut-off values with predictive significance have yet been found.
Our study does have limitations. First, it is a liver disease cohort, not an unbiased birth cohort, as noted, and the participants were mostly, but not exclusively, evaluated and managed at tertiary care centers. Also, this is a slowly progressive disease, and a large proportion of the cohort has been followed for only a few years. Therefore, conclusions about the disease risk or health of the pool of asymptomatic or undiagnosed AAT patients cannot be made.
Patient samples linked to the database are under study in an attempt to identify AAT disease-specific biomarkers, different from typical clinical tests that would be obtained during routine care, that might be used to predict progression to portal hypertension, liver transplant, and other complications. This type of new, powerful biomarker determination is only made possible by our combination of detailed, prospective data collection and continued long-term follow-up, with links to a biospecimen repository. ■
Acknowledgments
We thank Heather Van Doren, Senior Medical Editor with Arbor Research Collaborative for Health, who provided editorial assistance on this manuscript.
Glossary
- AAT
Alpha-1-antitrypsin
- AST
Aspartate transaminase
- GGTP
Gamma-glutamyl transpeptidase
Appendix
Members of ChiLDReN (As of 10/28/2019)
Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL (supported by NIDDK DK62436 and NCATS UL1TR001422): Estella Alonso, MD; Lee Bass, MD; Susan Kelly, RN, BSN; Mary Riordan, CCRP; Hector Melin-Aldana, MD
Cincinnati Children’s Hospital Medical Center, Cincinnati, OH (supported by NIDDK DK62497 and NCATS UL1TR000077): Jorge Bezerra, MD; Kevin Bove, MD; James Heubi, MD; Alexander Miethke, MD; Greg Tiao, MD; Julie Denlinger, BSN, RN; Erin Chapman
Children’s Hospital Colorado, Aurora, CO (supported by NIDDK DK62453 and NCATS UL1TR001082): Ronald Sokol, MD; Amy Feldman, MD; Cara Mack, MD; Michael Narkewicz, MD; Frederick Suchy, MD; Shikha Sundaram, MD; Johan Van Hove, MD; Benigno Garcia; Mikaela Kauma; Kendra Kocher, CCRP; Matthew Steinbeiss, MS, CCRP; Mark Lovell, MD
The Children’s Hospital of Philadelphia, Philadelphia, PA (supported by NIDDK DK62481): Kathleen Loomes, MD; David Piccoli, MD; Elizabeth Rand, MD; Pierre Russo, MD; Nancy Spinner, PhD; Jessi Erlichman, MPH; Samantha Stalford, MPH; Dina Pakstis; Sakya King
Children’s Hospital of Pittsburgh, Pittsburgh, PA (supported by NIDDK DK62466 and NCATS UL1TR000005): Robert Squires, MD; Rakesh Sindhi, MD; Veena Venkat, MD; Kathy Bukauskas, RN, CCRC; Patrick McKiernan, MD; Lori Haberstroh, RN, BSN; James Squires, MD, MS UCSF Children’s Hospital, San Francisco, CA (supported by NIDDK DK62500 and NCATS UL1TR000004): Philip Rosenthal, MD; Laura Bull, PhD; Joanna Curry; Camille Langlois, MS; Grace Kim, MD
Saint Louis University School of Medicine, St. Louis, MO (supported by NIDDK DK62453): Jeffery Teckman, MD; Vikki Kociela, BSN, CCRC; Rosemary Nagy, RDN, MBA; Shraddha Patel, PhD; Jacqueline Cerkoski, BSN
Riley Hospital for Children, Indiana University School of Medicine, Indianapolis, IN (supported by NIDDK DK84536 and NCATS UL1TR001108): Jean P. Molleston, MD; Molly Bozic, MD; Girish Subbarao, MD; Ann Klipsch, RN; Cindy Sawyers, BSRT; Oscar Cummings, MD
Seattle Children’s Hospital, Seattle WA (supported by NIDDK DK84575 and NCATS UL1TR000423): Simon Hor- slen, MB, ChB, FRCPCH; Karen Murray, MD; Evelyn Hsu, MD; Kara Cooper, CCRC; Melissa Young, CCRC; Laura Finn, MD
The Hospital for Sick Children, Toronto, Ontario, CANADA (supported by NIDDK DK103135): Binita Kamath, MD; Vicky Ng, MD; Claudia Quammie, CCRP; Juan Putra, MD; Deepika Sharma, MSc; Aishwarya Parmar, BSc
University of Utah, Salt Lake City, UT (supported by NIDDK DK103140): Stephen Guthery, MD; Kyle Jensen, MD; Ann Rutherford; Amy Lowichik, MD, PhD; Linda Book, MD; Rebecka Meyers, MD; Tyler Hall
Children’s Hospital Los Angeles, Los Angeles, CA (supported by NIDDK DK84538 and NCATS UL1TR000130): Kasper Wang, MD; Sonia Michail, MD; Danny Thomas, MD; Catherine Goodhue, CPNP; Rohit Kohli, MBBS, MS; Larry Wang, MD, PhD; Nisreen Soufi, MD; Daniel Thomas, MD
Children’s Healthcare of Atlanta, Atlanta, GA (supported by NIDDK DK062470 and NCATS UL1TR000454): Saul Karpen, MD, PhD; Nitika Gupta, MD, DCH, DNB, MRCPH; Rene Romero, Jr., MD; Miriam B. Vos, MD, MSPH; Rita Tory, MS, CCRP; John-Paul Berauer, MD; Carlos Abramowsky, MD; Jeanette McFall, MPH.
Texas Children’s Hospital, Houston, TX (supported by NIDDK DK103149): Benjamin Shneider, MD; Sanjiv Harpavat, MD; Paula Hertel, MD; Daniel Leung, MD; Mary Tessier, MD; Deborah Schady, MD; Laurel Cavallo; Diego Olvera; Christina Banks; Cynthia Tsai
King’s College Hospital, London, UK: Richard Thompson, BM, BCh, MRCP, MRCPCH
National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD: Edward Doo, MD; Jay Hoofnagle, MD; Averell Sherker, MD, FRCP; Rebecca Torrance, RN, MSN; Sherry Hall, MS
Scientific Data Coordinating Center, Ann Arbor, MI (supported by NIDDK DK62456): John Magee, MD; Robert Merion, MD, FACS; Cathie Spino, DSc; Wen Ye, PhD
Supported by funding from the Alpha-1 Foundation (University of Colorado Denver and Saint Louis University School of Medicine) and by U01 grants from the National Institute of Diabetes, Digestive and Kidney Diseases and UL1 grants from the National Center for Advancing Translational Sciences (NCATS): DK 62445 [Mt. Sinai School of Medicine], DK 62497 and UL1 TR000077 [Cincinnati Children’s Hospital Medical Center, University of Cincinnati], DK103149 [Texas Children’s Hospital], DK 62470 [Children’s Healthcare of Atlanta, Emory University], DK 62481 and UL1 TR000003 [The Children’s Hospital of Philadelphia, University of Pennsylvania], DK 62456 [University of Michigan], DK 84536 and UL1 TR000006 [Riley Hospital for Children, Indiana University], DK 84575 and UL1 TR000423 [Seattle Children’s Hospital, University of Washington], DK 62500 and UL1 TR000004 [UCSF Children’s Hospital, University of California, San Francisco], DK 62503 and UL1 TR000424 [Johns Hopkins School of Medicine], DK 62466 and UL1 TR000005 [Children’s Hospital of Pittsburgh, University of Pittsburgh], DK 62453 and UL1 TR002535 [University of Colorado Denver, Children’s Hospital Colorado], DK 62452 and UL1 TR000448 [Washington University School of Medicine, St. Louis, St. Louis Children’s Hospital], DK 84538 and UL1 TR000130 [Children’s Hospital Los Angeles, University of Southern California], DK 62436 and UL1 TR000150 [Ann and Robert H. Lurie Children’s Hospital of Chicago, Northwestern University], DK103140 [University of Utah], DK103135 [Hospital for Sick Children (Toronto). J.T. has grants from NIH, Arrowhead, Dicerna, Vertex, and provides consulting for Arrowhead, Dicerna, Vertex, KorroBio, Camp4, Takada, and the Alpha-1 Foundation. P.R. has grants from Gilead, Abbvie, Merck, Retrophin; and provides consulting for Gilead, Abbvie, Retrophin, Audentes, Albireo, and Mirum. C.S. worked as a statistical consultant for Albireo from 6 Feb 2018 to 14 Feb 2019. K.M. serves as a consultant for Albireo and Gilead. N.K. served on the High Tide advisory panel (one occasion) in 2019; she receives a royalty from Elsevier on the Wilson Disease book, which she co-edited with Eve Roberts. S.K. is a consultant for Albireo, Intercept, Mirum, Retrophin, and Spruce Bioscience. J.M. has funded research from Gillead, Abbvie, and Mirum. L.B. is on the speakers bureau of Mead Johnson Nutrition and is a consultant for Horizon Pharmaceuticals. B.K. is a consultant for Mirum, Shire, and Albireo; she has an unrestricted grant from Mirum. K.L. is a consultant for Mirum and Albireo. R.S. reports COIs with Retrophin, Albireo, and Mirum. J.H. is consultant to Mirum and Retrophin and has equity interest in Asklepion Pharmaceuticals, LLC. The remaining authors declare no conflicts of interest. A.S. is an employee of NIH. The study sponsors did not have a role in study design; collection, analysis, and interpretation of data; writing of the report; or the decision to submit the paper for publication.
Table III.
Patient disposition
| Disposition | No. (%) | Median length of follow-up,* years (range) |
|---|---|---|
| Active with native liver | 206 (58.9) | 3.45 (0.00-9.74) |
| Death or transplant | 32 (9.1) | 1.64 (0.00-7.76) |
| Withdrawn | 10 (2.9) | 2.03 (0.00-4.05) |
| Lost to follow-up | 57 (16.3) | 1.13 (0.00-5.20) |
| Other† | 45 (12.9) | 2.17 (0.00-6.11) |
| Total | 350 (100.0) | 2.47 (0.00-9.74) |
From baseline visit to either transplant or death (for those with an event), or last known follow-up.
Other reasons for study removal included: Completed study, noncontinuing site, transferred to adult care, or moved out of network.
Table IV.
Demographics and baseline characteristics by clinical outcome
| Characteristics | Survival with native liver (n = 208) | Died or underwent liver transplantation (n = 32) | Censored* (n = 110) |
|---|---|---|---|
| Age at visit (years) | |||
| N | 208 | 32 | 110 |
| Mean (SD) | 5.0 (4.7) | 5.8 (4.7) | 8.1 (6.5) |
| Median (Q1, Q3) | 3.2 (1.2, 7.5) | 4.6 (1.6, 9.1) | 6.9 (2.1, 13.0) |
| Min, Max | 0.1 (18.6) | 0.3 (15.2) | 0.1 (24.9) |
| Sex | |||
| Female | 89 (43%) | 10 (31.3%) | 39 (35.5%) |
| Male | 118 (57%) | 22 (68.8%) | 71 (64.5%) |
| Race | |||
| Black | 1 (0.5%) | 2 (6.5%) | 0 (0%) |
| Other | 11 (5.4%) | 1 (3.2%) | 7 (6.4%) |
| White | 191 (94.1%) | 28 (90.3%) | 103 (93.6%) |
| Ethnicity | |||
| Hispanic | 10 (4.9%) | 2 (6.5%) | 9 (8.3%) |
| Non-Hispanic | 196 (95.1%) | 29 (93.5%) | 99 (91.7%) |
| Neonatal cholestasis | |||
| No | 84 (44.2%) | 9 (28.1%) | 45 (42.1%) |
| Yes | 106 (55.8%) | 23 (71.9%) | 62 (57.9%) |
| Incident dpCEPH | 10 (4.8%) | 3 (9.4%) | 5 (4.5%) |
| Time to incident dpCEPH, median years (Q1, Q3) | 2.1 (1.9, 4.0) | 0.7 (0.4,1.1) | 2.4 (1.9, 2.8) |
ChiLDReN, Childhood Liver Disease Research Network; dpCEPH, definite, or possible, clinically evident portal hypertension.
Participants were censored if they were lost to follow-up, withdrew from the study, transferred care to a non-ChiLDReN hospital, or were at a discontinued ChiLDReN site.
Table V.
Longitudinal patterns for clinical outcomes
| Outcomes | Baseline | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 | Year 6 | Year 7 | Year 8 | Year 9 |
|---|---|---|---|---|---|---|---|---|---|---|
| dpCEPH | ||||||||||
| No. at risk | 350 | 196 | 151 | 121 | 93 | 65 | 48 | 39 | 23 | 10 |
| Cumulative no. of events | 72 | 76 | 82 | 85 | 87 | 88 | 88 | 89 | 90 | 90 |
| KM estimate (95% CI) | 1.00 | 0.78 (0.74-0.82) | 0.75 (0.70-0.80) | 0.74 (0.69-0.79) | 0.72 (0.67-0.77) | 0.71 (0.66-0.77) | 0.71 (0.66-0.77) | 0.70 (0.63-0.76) | 0.68 (0.61-0.75) | 0.68 (0.61-0.75) |
| Person-years (time on study) | 0 | 211 | 384 | 519 | 623 | 698 | 753 | 798 | 829 | 846 |
| Transplant or death | ||||||||||
| No. at risk | 350 | 249 | 193 | 155 | 120 | 83 | 63 | 51 | 25 | 11 |
| Cumulative no. of events | 0 | 11 | 20 | 23 | 28 | 30 | 31 | 31 | 32 | 32 |
| KM estimate (95% CI) | 1.00 | 0.96 (0.94-0.98) | 0.92 (0.89-0.95) | 0.91 (0.87-0.94) | 0.87 (0.83-0.92) | 0.86 (0.80-0.91) | 0.84 (0.79-0.90) | 0.84 (0.79-0.90) | 0.82 (0.75-0.89) | 0.82 (0.75-0.89) |
| Person-years (Time on study) | 0 | 266 | 485 | 658 | 793 | 888 | 959 | 1017 | 1058 | 1077 |
dpCEPH, definite, or possible, clinically evident portal hypertension; KM, Kaplan-Meier.
Table VI.
Longitudinal pattern for incident dpCEPH
| Variables | Baseline | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 | Year 6 | Year 7 | Year 8 | Year 9 |
|---|---|---|---|---|---|---|---|---|---|---|
| No. at risk | 278 | 196 | 151 | 121 | 93 | 65 | 48 | 39 | 23 | 10 |
| Cumulative no. of events | 0 | 4 | 10 | 13 | 15 | 16 | 16 | 17 | 18 | 18 |
| KM estimate (95% CI) | 1.00 | 0.98 (0.96-1.00) | 0.95 (0.91-0.98) | 0.93 (0.89-0.97) | 0.91 (0.87-0.95) | 0.90 (0.85-0.95) | 0.90 (0.85-0.95) | 0.88 (0.81-0.94) | 0.85 (0.77-0.93) | 0.85 (0.77-0.93) |
| Person-years (Time on study) | 0 | 211 | 384 | 519 | 623 | 698 | 753 | 798 | 829 | 846 |
dpCEPH, definite, or possible, clinically evident portal hypertension; KM, Kaplan-Meier.
Table VII.
Univariate predictors of time-to-transplant/death: All participants
| Predictor | HR | 95% CI | P Value |
|---|---|---|---|
| Head circumference Z-score | 0.76 | 0.15-3.74 | .73 |
| Right mid-arm circumference | 0.75 | 0.53-1.07 | .11 |
| Right triceps skinfold thickness | 0.81 | 0.56-1.18 | .27 |
| BMI | 1.00 | 0.93-1.08 | .91 |
| BMI Z-score | 1.49 | 0.45-4.94 | .51 |
| Weight Z-score | 0.58 | 0.28-1.21 | .15 |
| Height Z-score | 0.60 | 0.29-1.24 | .17 |
| Albumin (per 1 g/dL increase) | 0.09 | 0.03-0.29 | <.0001 |
| Total bilirubin (per 1 mg/dL increase) | 1.31 | 1.17-1.47 | <.0001 |
| Direct bilirubin (per 1 mg/dL increase) | 3.56 | 1.58-8.01 | .0021 |
| AST (per 10 U/L increase) | 1.18 | 1.09-1.26 | <.0001 |
| ALT (per 10 U/L increase) | 1.00 | 1.00-1.01 | .48 |
| Platelets (103/mm3) | 0.99 | 0.98-1.00 | .0128 |
| AST/platelets | 1.05 | 0.98-1.14 | .18 |
| INR (per 0.1) | 1.83 | 1.32-2.54 | .0003 |
| Neonatal cholestasis | 1.93 | 0.89-4.17 | .0941 |
ALT, alanine transaminase; BMI, body mass index; HR, hazard ratio; INR, international normalized ratio; KM, Kaplan-Meier.
Model: Cox proportional hazards model with time-varying covariates (except for Neonatal Cholestasis, which is fixed).
Figure 3.
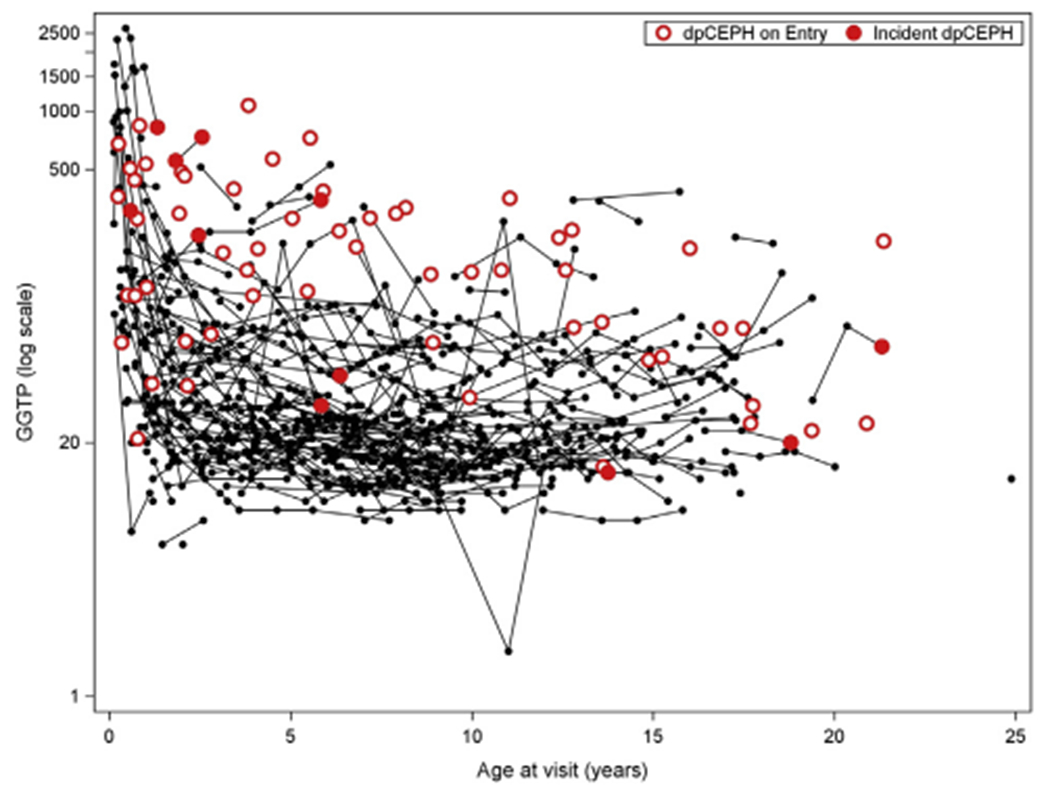
Relationship of GGTP, age, and definite/possible clinically evident portal hypertension (dpCEPH): all participants.
Figure 4.
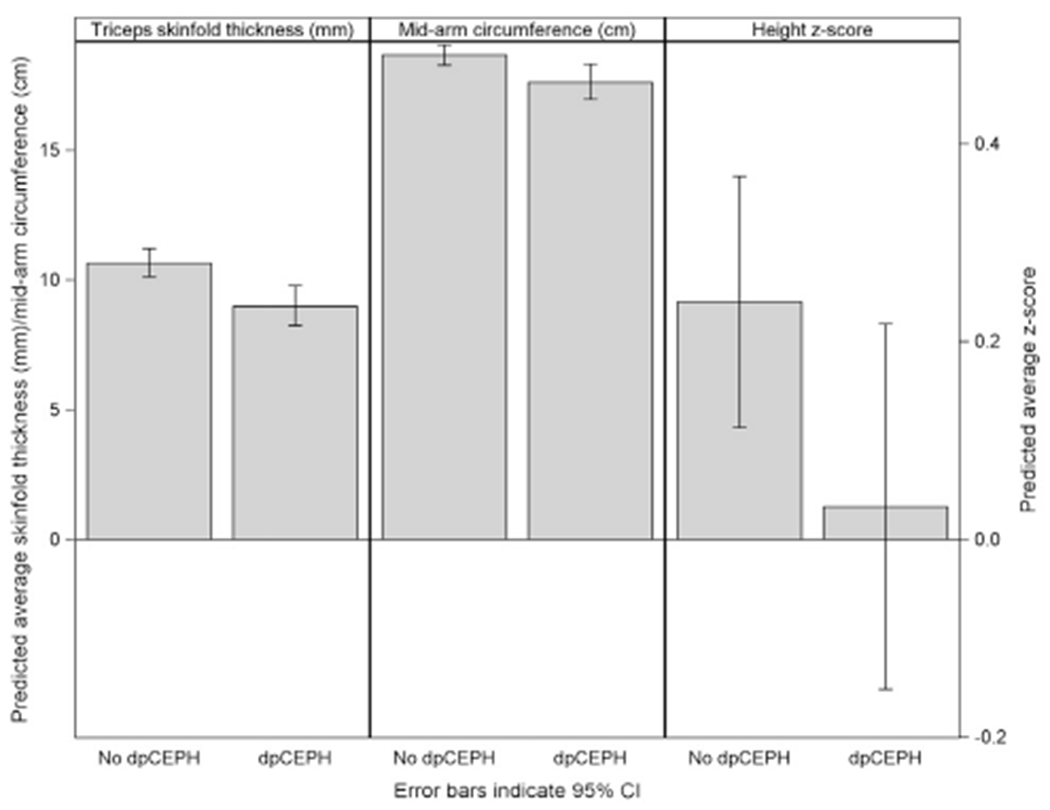
Predicted anthropometric outcomes at age 6 by definite/possible clinically evident portal hypertension (dpCEPH) vs no dpCEPH: all participants.
References
- 1.American Thoracic Society/European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003;168: 818–900. [DOI] [PubMed] [Google Scholar]
- 2.Teckman JH. Alpha1-antitrypsin deficiency in childhood. Semin Liver Dis 2007;27:274–81. [DOI] [PubMed] [Google Scholar]
- 3.Lindblad D, Blomenkamp K, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology 2007;46:1228–35. [DOI] [PubMed] [Google Scholar]
- 4.Marcus NY, Blomenkamp K, Ahmad M, Teckman JH. Oxidative stress contributes to liver damage in a murine model of alpha-1-antitrypsin deficiency. Exp Biol Med (Maywood) 2012;237:1163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlmutter DH, Silverman GA. Hepatic fibrosis and carcinogenesis in alpha1-antitrypsin deficiency: a prototype for chronic tissue damage in gain-of-function disorders. Cold Spring Harb Perspect Biol 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan S, Huang L, McPherson J, Muzny D, Rouhani F, Brantly M, et al. Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency. Hepatology 2009;50: 275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudnick DA, Shikapwashya O, Blomenkamp K, Teckman JH. Indomethacin increases liver damage in a murine model of liver injury from alpha-1-antitrypsin deficiency. Hepatology 2006;44:976–82. [DOI] [PubMed] [Google Scholar]
- 8.Sveger T Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976;294:1316–21. [DOI] [PubMed] [Google Scholar]
- 9.Sveger T The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr Scand 1988;77:847–51. [DOI] [PubMed] [Google Scholar]
- 10.Sveger T, Thelin T, McNeil TF. Young adults with alpha 1-antitrypsin deficiency identified neonatally: their health, knowledge about and adaptation to the high-risk condition. Acta Paediatr 1997;86:37–40. [DOI] [PubMed] [Google Scholar]
- 11.Mowat AP. Alpha 1-antitrypsin deficiency (PiZZ): features of liver involvement in childhood. Acta Paediatr Suppl 1994;393:13–7. [DOI] [PubMed] [Google Scholar]
- 12.Volpert D, Molleston JP, Perlmutter DH. Alpha1-antitrypsin deficiency-associated liver disease progresses slowly in some children. J Pediatr Gastroenterol Nutr 2000;31:258–63. [DOI] [PubMed] [Google Scholar]
- 13.Wall M, Moe E, Eisenberg J, Powers M, Buist N, Buist AS. Long-term follow-up of a cohort of children with alpha-1-antitrypsin deficiency. J Pediatr 1990;116:248–51. [DOI] [PubMed] [Google Scholar]
- 14.Ibarguen E, Gross CR, Savik SK, Sharp HL. Liver disease in alpha-1-antitrypsin deficiency: prognostic indicators. J Pediatr 1990;117:864–70. [DOI] [PubMed] [Google Scholar]
- 15.Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol 2010;53:170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sokol RJ. Reloading against rare liver diseases. J Pediatr Gastroenterol Nutr 2010;50:9–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamath BM, Piccoli DA, Magee JC, Sokol RJ. Childhood Liver Disease Research and Education Network. Pancreatic insufficiency is not a prevalent problem in Alagille syndrome. J Pediatr Gastroenterol Nutr 2012;55:612–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shneider BL, Abel B, Haber B, Karpen SJ, Magee JC, Romero R, et al. Portal hypertension in children and young adults with biliary atresia. J Pediatr Gastroenterol Nutr 2012;55:567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teckman JH, Rosenthal P, Abel R, Bass LM, Michail S, Murray KF, et al. Baseline analysis of a young alpha-1-antitrypsin deficiency liver disease cohort reveals frequent portal hypertension. J Pediatr Gastroenterol Nutr 2015;61:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]