

Abstract
Emerging evidence indicates that metabolic programs regulate B cell activation and antibody responses. However, the metabolic mediators that support the durability of the memory B cell and long-lived plasma cell populations are not fully elucidated. Adenosine monophosphate-activated protein kinase (AMPK) is an evolutionary conserved serine/threonine kinase that integrates cellular energy status and nutrient availability to intracellular signaling and metabolic pathways. Here we use genetic mouse models to show that loss of AMPKα1 in B cells led to a weakened recall antibody response associated with a decline in the population of memory-phenotype B cells. AMPKα1-deficient memory B lymphocytes exhibited aberrant mitochondrial activity, decreased mitophagy, and increased lipid peroxidation. Moreover, loss of AMPKα1 in B lymphoblasts was associated with decreased mitochondrial spare respiratory capacity. Of note, AMPKα1 in B cells was dispensable for stability of the bone marrow-resident long-lived plasma cell population, yet absence of this kinase led to increased rates of Ig production and elevated serum antibody concentrations elicited by primary immunization. Collectively, our findings fit a model in which AMPKα1 in B cells supports recall function of the memory B cell compartment by promoting mitochondrial homeostasis and longevity, but restrains rates of immunoglobulin production.
INTRODUCTION
B cells confer long-lasting protection against microbes or their exotoxins via the production of highly specific antibodies. Upon encounter with cognate antigen, naïve B cells rapidly proliferate and ultimately differentiate into antigen-specific memory B cells or antibody-secreting plasma cells, a subset of that persists for decades after clearance of the antigen (1). The persistence of memory B cell and long-lived plasma cell populations is critical for protection against subsequent antigen exposure and achieving long-lasting immunological memory.
After activated B cells undergo proliferation and differentiation, long-lived plasma cells and memory B cells arise from both germinal center (GC)-dependent and -independent pathways (2). After a phase as plasmablasts, long-lived plasma cells can reside in the bone marrow and continuously synthesize and release antigen-specific antibody into the blood stream to bind antigen (3). By contrast, antigen-specific memory B cells continue to circulate through the body and have the capacity to reenter germinal centers or rapidly differentiate into ASC upon subsequent exposure to antigen (4). Though both are long-lived cells in the B lineage, memory B cells appear to be metabolically quiescent while plasma cells continuously synthesize large amounts of secretory glycosylated antibody (5). Apart from diversity in function, long-lived B cells must also adapt to varied nutrient availability in the microenvironment (6,7). Mechanisms that support longevity in the face of metabolic stressors and demands are incompletely understood.
Bone marrow plasma cell longevity is attributable in part to the expression of Bcl6-related transcription factor ZBTB20 and the ability to shuttle pyruvate to the mitochondria (8–10). Emerging evidence implicates autophagy as critical for the long-term survival of both memory and plasma cells (11–13). However, the mode of autophagy that supports the longevity of the B lineage is unknown.
The energy sensor adenosine monophosphate-activated protein kinase (AMPK) is one upstream activator of canonical autophagy. AMPK is a conserved serine/threonine kinase that couples nutrient availability to cellular metabolism (14). During periods of bioenergetic insufficiency, AMPK can restore cellular energy homeostasis by targeting multiple downstream targets that trigger pathways that increase ATP generation while inhibiting those that consume ATP. Specifically, AMPK promotes fatty acid oxidation, autophagy, and mitochondrial biogenesis while inhibiting protein and fatty acid synthesis (14). Several studies link age-related diseases to the dysregulation of mitochondrial dynamics, suggesting that AMPK may support longevity of cells through regulation of mitochondrial homeostasis (15). Furthermore, AMPK negatively regulates another energy sensor, mechanistic target of rapamycin complex 1 (mTORC1), which has been reported to be critical for effective primary antibody responses and the generation of memory (16).
Several reports indicate that AMPK activity in T cells is critical for effective activation and survival (17). In one study, AMPK supported the survival of the CD8 memory population; others showed defects in primary CD8 T cell activation and effector functions with the loss of AMPK (18–20). In contrast to the T lineage, two studies have reported negative findings about AMPK in humoral responses (21,22). B and T cells from mice with unconditional null alleles encoding the isoform AMPKα1 were sensitized to cell death upon metabolic stressors, but no effect on primary or boosted antibody responses was observed (21). However, later work showed that this whole-body knockout leads to diffuse inflammation as a consequence of AMPKα1 deficiency in other cell populations including erythrocytes and antigen presenting cells, which may affect B cell function (23,24). More recently, work with a B cell-intrinsic knockout of AMPKα1 concluded that loss of AMPK down-regulates transcriptional expression of IgD (22). However, whether AMPK governs humoral memory remains unresolved.
Since AMPK is a master regulator that controls multiple aspects of metabolism, including mitochondrial homeostasis, mTORC1, and autophagy all of which play critical roles in B cells fate and function we sought to test if these downstream pathways were AMPK dependent in the B lineage. Here we show that AMPK promoted mitochondrial homeostasis and limited mTORC1 activity in this population while balancing effector functions and long-term maintenance of phenotypically defined memory B cells (MphenBC) as well as recall capacity after rechallenge. These findings provide evidence of a divergence between the cellular sources of two limbs in humoral memory –plasma cells and B cell memory – in their dependence on AMPK.
MATERIALS AND METHODS
Mice and immunizations
B cell-specific AMPKα1-deficient mice were generated by crossing Prkaa1 flox mice, purchased from the Jackson Laboratory (Bar Harbor, ME), with mice harboring Cre recombinase under three types of control element: mb1-Cre (25), huCD20-CreERT2, and Rosa26-CreERT2 (16). Age-matched Prkaa1 +/+ mice with the corresponding Cre transgene were used as wildtype controls and co-housed with Prkaa1 f/f mice. Mice were housed at Vanderbilt University under specified pathogen-free conditions and both males and females (6–10 wk old) were used according to approved mouse Institutional Animal Care and Use Committee protocols. Due to practical limitations on mobilization of matched mice for immunization experiments with otherwise unmanipulated mice, the Rosa26-CreERT2 knock-in mice were used for purification of B cells for in vitro or transfer experiments as described below. For immunizations in mice with B lineage-specific loss of AMPKα1, huCD20-CreERT2 and mb1-Cre lines were immunized with 4-hydroxy-3-nitrophenylacetyl hapten conjugated to keyhole limpet hemocyanin (NP-KLH, Biosearch Technologies, Novato, CA) emulsified in Imject Alum (50 μg in 50 μL) (Thermo Scientific, Waltham, MA). Recall responses were induced with 100 μg NP-KLH in alum (100 μL). To induce deletion of Prkaa1 in mature B cells prior to immunization, mice with huCD20-CreERT2 or Rosa26-CreERT2 transgenes received three doses (3 mg each) of tamoxifen from Cayman Chemical (Ann Arbor, MI) dissolved in safflower oil, injected intraperitoneally every other day (16).
For transfer experiments, CD45.1+ IgHa recipient mice were generated by crossing CD45.1+ IgHb and CD45.2+ IgHa mice, each purchased from Jackson Laboratory (stock # 002014 and 008341, respectively). CD45.2+ IgHb B cells (10 × 106) purified from Rosa26-CreERT2 ± Prkaa1 f/f mice, polyclonal wildtype CD4+ T cells (4 × 106), and CD4+ T cells from OT-II transgenic mice (1 × 106) were transferred into allotype-disparate CD45.1+ IgHa mice pre-conditioned with sub-lethal irradiation (split dose of 3.75 Gy × 2) two days prior to transfer. These chimeric mice were immunized with NP16-ovalbumin (Biosearch Technologies) emulsified Imject Alum (100 μg in 100 μL).
PCR
Genomic DNA was extracted from sorted cells using cell lysis buffer (1% SDS, 50 mM Tris Base, 10 mM EDTA pH 8) and 100 ng genomic DNA was used as a template for Prkaa1 and CreERT2 amplifications using primers previously described (26).
Flow cytometry
Unless otherwise specified, all mAbs were from BD Pharmingen (San Jose, CA), Life Technologies (Carlsbad, CA), eBiosciences (San Diego, CA), or Tonbo Biosciences (San Diego, CA). For detection of GC- and memory-phenotype B cells, splenocytes (3 × 106) were stained with anti-B220, -GL7, -Fas, -IgD, -CD38, NP-APC and a dump channel containing anti-CD11b, -CD11c, -F4/80, -Gr-1, and 7-AAD in 1% BSA and 0.05% sodium azide in PBS. To phenotype different memory B cell subsets, a second panel consisted of anti-B220, -CD38, -CD80, -PD-L2, -IgG1, NP-APC and the aforementioned dump channel with the addition of anti-IgD, -CD4, -CD8, -GL7. In analyses of transfer experiments, allotype-specific antibodies were used to distinguish donor (CD45.2) and recipient (CD45.1) B cells as described (27). For flow analyses of mitochondria, 1–3 × 106 cells were washed in PBS and stained with 200 nM MitoTracker Green (Invitrogen, Carlsbad, CA), 50 nM MitoTracker Deep Red (Invitrogen), and Ghost-780 in PBS for 20 min at 37°C, then washed again (1% BSA in PBS) and further stained with anti-B220, -CD138, or -CD38. MitoSox (5 μM), and Bodipy C-11 581/591 (1.25 μM) staining were performed similarly. For intracellular phospho-flow analysis, cells were fixed with 4% PFA followed by methanol permeabilization. Peptide-specific anti-phospho-S6 (Cell Signaling Technologies) was as described (16). Samples were analyzed using a FACSCanto flow cytometer driven by BD FACS Diva software and were processed using Flow-Jo software (FlowJo LLC, Ashland OR).
Preparative sorting
Starting from single-cell suspensions of splenocytes, memory-phenotype B cells (MphenBC) were enriched by depleting IgD+ and Thy1.2+ cells using biotinylated antibodies followed by BD IMag™ streptavidin particles on an IMag™ Cell Separation Magnet (BD Biosciences, San Jose, CA). Cells were stained as described for flow cytometry and dump negative, IgD− CD38+, B220+ cells were sorted into 10% FBS in PBS for downstream applications.
ELISA and ELISpot
For detection of circulating NP-binding antibody of all- and high-affinity after immunization with NP-KLH in alum adjuvant, serial dilutions of sera were added to NP24-BSA (Biosearch Technologies) coated (0.1 μg/well) 96-well plates (Costar, Washington D.C.) and incubated overnight at 4°C followed by incubation with either HRP-conjugated anti-IgM or -IgG1 (Southern Biotech), or biotinylated anti-IgG1a or anti-IgG1b (BD Biosciences) for transfer experiments, followed by Streptavidin-HRP (R & D Systems, Minneapolis, MN). The plates were developed using Ultra TMB Substrate (Thermo Scientific) and optical densities at 450 nm were measured. To compile results across biologically independent experiments, optical densities within the linear range of serially diluted sera were combined. Using OD values generated from a dilution in the linear range of the curve, “recall effects” were calculated as described (28), using ELISA data from each mouse and subtracting the OD value from its serum collected the day before re-challenge from the OD value of the serum one week post re-challenge. For detection of secreted antibody after in vitro studies, supernatants from cultured cells were added to anti-Ig(H+L) (Southern Biotech, Birmingham, AL)-coated 96 well plates before detection with HRP-conjugated antibodies. Normal mouse IgG (Thermo Scientific) was used as a standard to interpolate concentrations of IgG for tissue culture supernatants.
To detect antigen-specific antibody secreting cells (ASCs) after immunization, 96-well high protein binding membrane plates (Millipore, Burlington MA) were coated with 1 μg/well NP24-BSA. Splenocytes or bone marrow cells (5 – 20 × 105) were added and plates were incubated at 37° overnight followed by incubation with biotinylated anti-IgM, -IgG1, -IgG1a, or -IgG1b antibodies prior to incubation with VectaStain ABC kit (Vector Laboratories, Burlingame, CA) and development using 3-amino-9-ethylcarbzole (Sigma, St. Louis, MO). ASCs were quantified using an ImmunoSpot Analyzer (Cellular Technology, Shaker Heights, OH).
In vitro culture
Splenic B cells were purified from Rosa26-CreERT2 mice (Prkaa1 +/+ and Prkaa1 f/f) by negative selection using biotinylated anti-CD43, -Thy1.2, and -F4/80 (>85% CD19+) followed by streptavidin particles and IMag™ Cell Separation Magnet (BD Biosciences). To induce plasma cell differentiation, B cells were seeded at 5 × 105 per mL and treated with 5 μg/mL LPS (Sigma), 10 ng/mL BAFF (AdipoGen, San Diego CA), 10 ng/mL IL-4 (Peprotech, Rocky Hill, NJ), 5 ng/mL IL-5 (Peprotech), and 50 nM 4-hydroxy-tamoxifen (4-OHT) (Sigma). Cells were cultured for 2–8 days in RPMI-1640 supplemented with 10% FBS (Peak, Denver, CO), 100 U/mL penicillin (Invitrogen), 100 μg/mL streptomycin (Invitrogen), 3 mM L-glutamine (Invitrogen), and 0.1 mM 2-mercaptoethanol (Sigma). B cells were also expanded on the NB21 feeder line as previously described (, 30). Every three days, supernatants were frozen for further analysis and the expanded B cells were reseeded on fresh NB21 feeder cells in new media and 4-OHT. For spontaneous antibody secretion, day 8 LPS cultures or day 9 NB21.2D9 cultures were subjected to Ficoll spin (Invitrogen) to eliminate dead cells and debris. Cells were then washed and 5 × 104 cells were seeded in 100 μL of fresh media in a 96 well plate for 8 hours. Supernatants were frozen and levels of secreted IgG1 were determined by ELISA.
Immunoblots
Unless otherwise indicated, all immunoblots depict relative protein from whole B cell extracts derived from splenocytes of Rosa26-CreERT2 mice (Prkaa1 +/+ or Prkaa1 f/f) followed by a 2-day culture with LPS, BAFF, and 4-OHT at 5 × 106 per mL. For immunoblotting for AMPK targets after glucose starvation, 2-day LPS blasts were washed and reseeded in glucose-free RPMI (Invitrogen) supplemented with 10% dialyzed HyClone FBS (Thermo Scientific) for specified amount of time. Cells were washed twice in cold PBS and lysed in RIPA buffer (Sigma catalog # 0278) in the presence of phosphatase inhibitor (Thermo Scientific) and a protease inhibitor cocktail (Sigma). Twenty to one hundred micrograms of cell lysates were resolved by SDS-PAGE, transferred to PVDF membranes, and immunoblotted for phospho-AMPKα1 (T172), AMPKα, phospho-S6 (S235/236), total S6, phospho-ACC (S79), total ACC, phospho-ULK1 (S317), total ULK1, phospho-4E-BP1, total 4E-BP1, and/or LC3 using monoclonal rabbit antibodies purchased from Cell Signaling Technology (Danvers, MA). Actin (Santa Cruz Biotechnology, Dallas, TX) was detected on all blots as a loading control. Immunoblots were visualized using Odyssey Imaging system (Li-Cor) after incubation with secondary reagents, anti-rabbit IgG-680, or anti-mouse IgG-800 (Invitrogen).
Seahorse assays
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using an XFe96 extracellular flux analyzer (Seahorse Bioscience). Briefly, 5 × 105 two-day LPS and BAFF activated B cells were seeded per well of a Cell-Tak (5 μg/mL; Corning) coated plate. Glycolytic and mitochondrial stress tests were performed as previously described (16, 31). Maximum respiration and spare respiratory capacity were calculated using formulas derived from the Seahorse platform.
Immunocytochemistry
LPS blasts (5 × 105 cells from 48 hr cultures) in 0.5 mL were seeded on poly-D-lysine coated coverslips in a 24 well plate and stained with 100 nM MitoTracker Deep Red for 20 min at 37° before centrifugation to ensure cellular adherence to coverslips. Coverslips were incubated overnight with anti-B220 or CD138-PE and either anti-LC3, anti-ULK1 or anti-Lamp1 using rabbit monoclonal antibodies (Cell Signaling Technology) after methanol fixation and blocking in 1% BSA in PBST. Coverslips then were mounted onto slides using ProLong Gold anti-fade reagent (Invitrogen) after incubation with secondary antibody anti-rabbit IgG 488 (Invitrogen) to visualize rabbit antibodies using an Olympus FV-1000 fluorescent confocal microscope. LC3-puncta were assessed using ImageJ. Co-localization of lysosomes and mitochondria as an indicator of mitophagy was determined using Just Another Co-localization Plugin (JACoP) in ImageJ. Manders’ coefficient represents the percentage of mitochondrial pixels (blue channel) that overlay Lamp1 or ULK1 pixels (green channel) where 0 = no co-localization; 1 = 100% co-localization.
Pulse-chase analyses of antibody production
After 7 days of culture with LPS, BAFF, IL-5, IL-4, and 4-OHT as above, viable cells were recovered after Ficoll step gradient centrifugation, rinsed, and recounted. Equal numbers of cells were then pulsed for 1 hour with 2 μCi 3[H]-leucine (Moravek, 60 Ci/mmol, Brea, CA) per 2 × 106 cells per mL of arginine-, lysine-, and leucine-deficient RPMI (Sigma) supplemented back with complete RPMI levels of L-arginine (1.149 mM; Sigma), L-lysine (0.219 mM; Sigma), and 10% of complete RPMI levels of L-leucine (0.038 mM; Sigma). Media also contained 10% dialyzed HyClone FBS, 25 mM HEPES (Invitrogen) and phenol red (Sigma). After pulsing cells, 2 × 106 cells were lysed in RIPA buffer in the presence of protease and phosphatase inhibitors and supernatants were frozen for the zero chase time point. Remaining cells were washed in PBS and resuspended in the aforementioned media but with 100% complete RPMI levels of L-leucine (0.382 mM) at 1 × 106 cells per mL. At each specified chase time point, 2 × 106 cells were lysed and the supernatant collected. Samples were stored at −20°. Secreted and intracellular IgG antibodies were purified from supernatant and lysate samples respectively using protein G agarose beads (Santa Cruz Biotechnology). Precipitates were subjected to SDS-PAGE in reducing conditions, transferred to PVDF membranes, and rocked for 30 minutes in 2 M sodium salicylate (Sigma) prior to fluorography at −80° for 1 – 14 days as described previously (32). Molar amounts of 3[H]-leucine incorporated into antibody were calculated after membranes were subjected to liquid scintillation counting using a Beckman Coulter LS 6500.
RESULTS
AMPK in B cells dampens primary antibody responses and initial memory B cell formation in vivo.
We set out to test the role of AMPK on B cell function over the course of an immune response. Previous studies have provided evidence that AMPKα1, encoded by Prkaa1, is the only isoform of the essential catalytic subunit of AMPK expressed in B cells (21,22,33). Thus, to generate mice with a conditional B cell-specific deletion of AMPK, we crossed Prkaa1 floxed mice to transgenic animals expressing a hydroxytamoxifen-inducible Cre recombinase under the control of the huCD20 promoter (huCD20-CreERT2). This B lineage-restricted promoter is active from the pre-B to mature B cell stage (34). AMPKα expression was undetectable in LPS-activated B lymphoblasts from tamoxifen-injected Prkaa1 f/f huCD20-CreERT2 mice when compared to Prkaa1 +/+ huCD20-CreERT2 controls (Fig. 1A). LPS-activated Prkaa1 Δ/Δ B cells failed to phosphorylate AMPKα target ACC S79 even in the absence of glucose indicating a loss of AMPK function in Prkaa1-deleted cells (Supplemental Fig 1). Since there was no detectable band in Prkaa1-deleted cells with the AMPKα antibody which detects both AMPKα1 and α2 catalytic isoforms, we conclude that only AMPKα1 is substantially expressed in B cells and that loss AMPKα1 did not lead to compensatory induction of AMPKα2.
Figure 1. Inactivation of Prkaa1 in the B lineage leads to elevated primary antibody responses and initial MphenBC population.
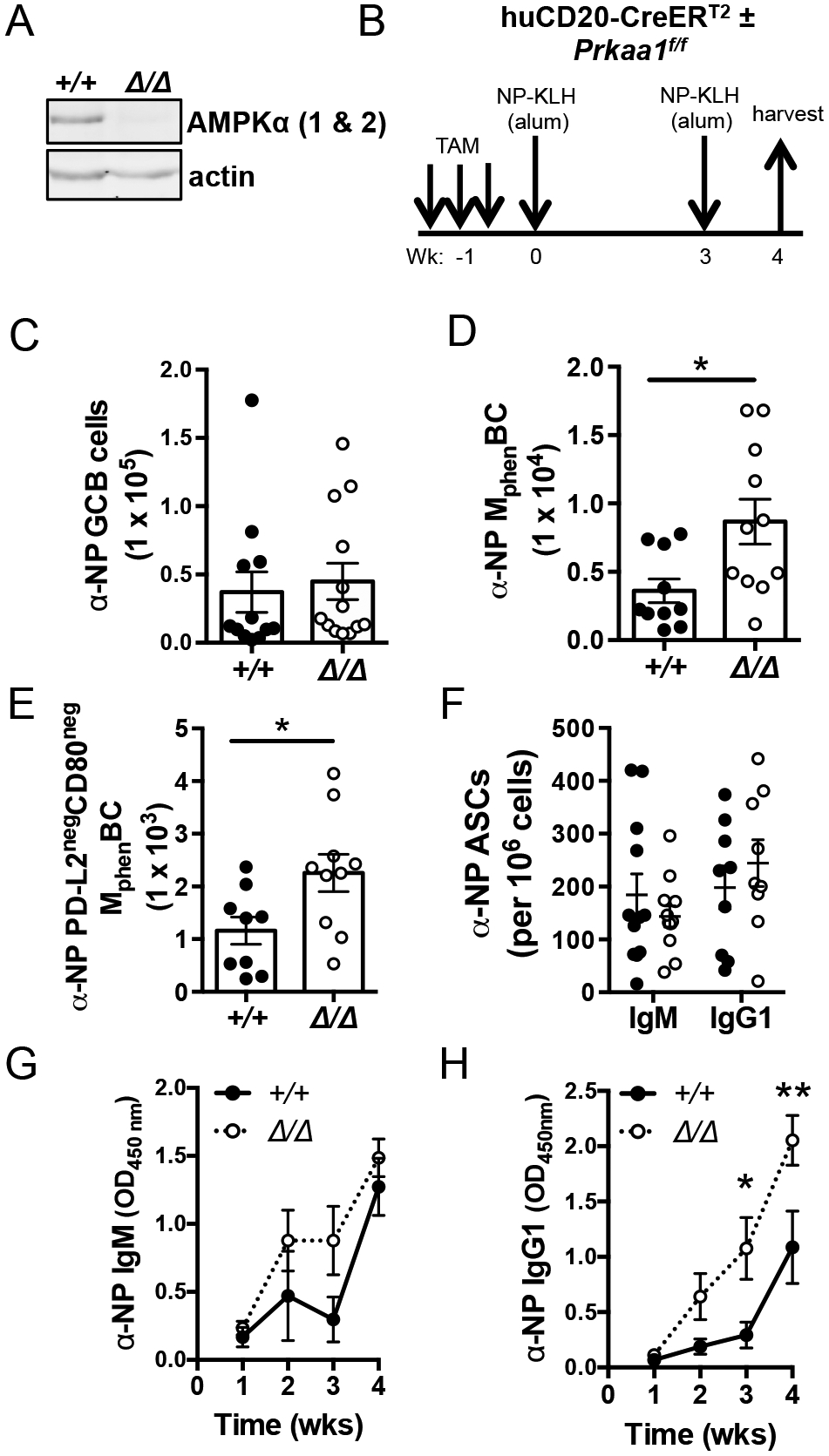
(A) Immunoblot for AMPKα (1 and 2) and actin expressed in LPS-activated B cells purified after in vivo tamoxifen regimen. Data representative of immunoblots from n = 3 vs. 3 mice. (B) Schematic of immunization strategy using mice harboring a tamoxifen-inducible Cre expressed specifically in the B lineage. (C) Total number of splenic NP+ germinal center B cells (B220+ IgD− Fas+ GL7+ NP+). (D, E) Total and CD80neg PD-L2neg splenic NP+ MphenBC (B220+ IgD− GL7− CD38+ NP+). (F) Total splenic NP-specific IgM- and IgG1-secreting cells detected per 106 plated by ELISpot. (C-F) Each circle (controls, filled ●; Prkaa1 Δ/Δ, open ○) represents sample from one mouse with mean ± SEM also displayed. (G, H) Circulating NP-specific IgM and IgG1 after immunization strategy as described in (A). Data represent mean ± SE from four independent experiments with at least n = 9 Prkaa1 +/+ vs. n = 9 Prkaa1 Δ/Δ. P values determined by Mann-Whitney U non-parametric t-test or ANOVA where appropriate. * indicates p < 0.05, ** indicates p < 0.01.
A prior study provided evidence that a B cell-specific deletion of AMPKα1 driven by CD19-Cre had no effect on GC and no substantial change in early plasma cell generation or initial levels of circulating antibody two weeks after NP-CGG immunization (22). However, the impact of AMPKα1 on other outcomes of GC, including the generation of memory and long-lived plasma cells remained unexamined. We assessed the effect of B cell intrinsic AMPKα1 on the generation of B cells with memory phenotype (MphenBC) and antibody secreting cells (ASCs), the latter of which peak in formation during late germinal centers at least three weeks after exposure to antigen (35). To do so, we immunized tamoxifen-treated huCD20-CreERT2 mice (Prkaa1 f/f or Prkaa1 +/+) with NP-KLH followed by boosting after three weeks and analyses of NP-specific humoral responses one week after the booster immunization (Fig. 1B). Consistent with a prior study (22), we observed no difference in NP-specific germinal center B cells (B220+ NP+ IgDneg GL7+ Fas+) for AMPKα1-deficient B cells (Fig. 1C). However, induced loss of AMPKα1 from mature B cells led to almost a two-fold increase in NP-specific MphenBC (B220hi NP+ IgDneg GL7neg CD38+) (Fig. 1D). We further examined whether AMPKα1 differentially supported the generation of different MphenBC subsets distinguished by PD-L2 and CD80 expression (36–38). The double-negative (CD80neg PD-L2neg ) MphenBC population, which makes up >85% of total MphenBC, are >90% are IgM+ (38). Hapten-binding CD80neg PD-L2neg MphenBC were increased in the absence of AMPKα1 to a similar extent as NP-specific MphenBC (Fig. 1E, Supplemental Fig. 1B). Double-positive antigen-specific MphenBC which make up ~5% of total MphenBC, were unaffected in AMPKα1 null mice (Supplemental Fig. 1C). Although we cannot exclude a contribution of renewed proliferation of MphenBC after the boost, we infer that AMPK dampened the initial MphenBC population size, particularly CD80neg PD-L2neg MphenBC, after a short-term prime/boost immunization strategy. Additionally, the frequencies of splenic (Fig. 1F) and bone marrow (Supplemental Fig. 2A) anti-NP IgM- and IgG1-secreting ASCs were unaffected by the loss of AMPKα1 from the B lineage. Despite similar numbers antigen-specific IgM and IgG1 ASCs, there was a substantial increase in circulating anti-NP IgM and IgG1 as early as two weeks after the initial immunization in the absence of AMPK (Fig. 1G, H). The elevated antigen-specific IgG1 concentrations in the sera were maintained several weeks after immunization despite the apparently normal numbers of anti-NP IgG1 ASCs in the marrow (Supplemental Fig. 2B). Collectively these data indicate that AMPK restrained both the initial CD80neg PD-L2neg MphenBC population and primary antibody production in vivo without an observable increase in the antigen-specific ASCs or GC B cells derived from AMPKα1-deficient B cells.
AMPK attenuates antibody synthesis in plasma cells.
Several models, none mutually exclusive of another, could account for the apparent paradox of persistently higher antibody concentrations in the absence of any increase in ASCs derived from AMPKα1-deficient B cells. First, loss of AMPKα1 in B cells may lead to increased rates of antibody production per ASC. Second, loss of AMPKα1 from B cells might lead to more formation of plasma cells that generate higher circulating antibody but die before time of analysis. To explore these possibilities, we used in vitro cultures to test the effect of AMPKα1 on plasma cell differentiation and antibody production. For these in vitro studies, B cells purified from tamoxifen-injected Rosa26-CreERT2 mice (Prkaa1 f/f or +/+) were cultured with IL-4 on NB-21.2D9 feeder cells described (29, 30). When CD138 and B220 expression were analyzed every three days for nine days of co-culture with the BAFF, CD40L, and IL-21-expressing feeder cells, no differences were observed in the number of plasma cells (B220lo CD138+) (Fig. 2A), suggesting that AMPKα1 is dispensable for plasma cell differentiation. To test the effect of B cell AMPKα1 on antibody production throughout the co-culture, IgG1 was measured in supernatants. Despite similar numbers of plasma cells, cultures with AMPKα1-deficient B cells accumulated three-fold higher concentrations of antibody in their supernatants from day six to nine (Fig. 2B). Thus, consistent with the in vivo serologies, loss of AMPKα1 from B cells led to an increase in antibody in the supernatant despite comparable plasma cell numbers. These in vitro results indicated that the increase in antibody in the supernatant observed from AMPKα1-deficient B cells was due to an increase in antibody production on a per cell basis.
Figure 2. Loss of AMPKα1 has no defect on plasma cell differentiation but leads to increased antibody synthesis per cell.

(A) Representative flow plots of CD138 vs. B220 expression during plasma cell differentiation after B cells from tamoxifen-treated Rosa26-ERT2Cre mice (Prkaa1 +/+ or Prkaa1 f/f ) were co-cultured on NB-21.2D9 feeder cells in the presence of 4-OHT and IL-4 (left panel). Total number (mean ± SEM) of plasma cells (CD138+B220lo) throughout co-culture (right panel). (B) Mean (± SEM) concentrations of IgG1 detected in supernatants collected from days 3, 6, and 9 of co-culture. (C) Representative wells and quantification of ELISpot analysis depicting numbers of IgG1 secreting cells per 500 plated cells. (D) Mean spot size of IgG1 ASCs after nine days of co-culture. (E) Relative levels of IgG1 detected in the supernatant eight hours after plating 5 × 104 / 100 μL of day 9 co-cultured cells. Data normalized to wildtype controls. Data represent mean ± SE from at least two independent experiments with n = 6 Prkaa1 +/+ vs. n = 4 or 5 Prkaa1 Δ/Δ mice. (F) Protein G precipitation of intracellular and secreted IgG1 from 7 day-LPS cultures after one hour of labeling with [3H]-leucine and the indicated chase times. (G) [3H]-leucine incorporation into IgG1 collected from lysates and supernatants at indicated chase times. (H) Ratio of [3H]-leucine incorporation in the supernatant to incorporation in the lysate, derived from (G), as an indicator of secretion efficiency. Data are representative of three independent experiments using n = 3 Prkaa1 +/+ vs. n = 3 Prkaa1 Δ/Δ mice. (C-H) Each circle (controls, filled ●; Prkaa1 Δ/Δ, open ○) represents sample from one mouse with mean ± SEM also displayed. P values determined by Mann-Whitney U non-parametric t-test or ANOVA where appropriate. ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001.
We next characterized differentiated cells at d 9 of co-culture, when the greatest difference in antibody production with the loss of AMPKα1 was observed. To test if similar frequencies of B220lo CD138+ cells translated to similar numbers of functional ASCs, we performed ELISpot analyses. These assays detected ~40 IgG1-secreting cells per 500 plated differentiated cells for both AMPKα1-deficient and -sufficient B cells (Fig. 2C). Despite similar numbers of IgG1-secreting cells after nine days of co-culture with the loss of AMPKα1, the mean spot size for AMPKα1-deficient plasma cells was increased (Fig. 2D). This finding suggested that the amount of antibody secreted per cell was greater with the loss of AMPKα1. To test this rigorously, we measured the effect of AMPKα1 on short-term (8 hr) antibody secretion after re-plating equal numbers of in vitro differentiated cells in fresh media. IgG1 detected in the supernatant from AMPKα1-deficient cells was ~1.5-fold greater than from controls (Fig 2E). Collectively, data indicate that AMPKα1 in B cells neither hinders nor promotes plasma cell generation but attenuates the amount of antibody produced per plasma cell.
We next examined whether the increase in antibody production per cell from AMPKα1-deficient B cells was due to an increase in immunoglobulin synthesis and/or enhanced activity of the secretory pathway for immunoglobulin. To obtain enough plasma cells for downstream biochemical assays, we activated and cultured wild-type and AMPKα1-deficient B cells using LPS. Similar to the co-culture system, the frequency of plasma cells generated by LPS, BAFF, IL-4, and IL-5 were unaffected with the loss of AMPKα1 (Supplemental Fig. 2C) but IgG1 detected in the supernatant from AMPKα1-deficient cells were enhanced two-fold (Supplemental Fig. 2D).
To distinguish the role of AMPKα1 on rates of antibody synthesis and/or immunoglobulin secretion, we performed [3H]-leucine pulse-chase analyses after re-plating equal numbers of LPS-differentiated cells. AMPKα1-deficient cells exhibited increased intracellular and secreted [3H]-leucine-labeled immunoglobulin (Fig. 2F,G). Specifically, AMPKα1-deficient cultures had ~6.5 fmol [3H]-leucine incorporated in antibody after 1h of labeling compared to ~3.5 fmol of wild-type controls (Fig. 2G). The similar ratio of secreted to intracellular [3H]-antibody after labeling was most consistent with increased synthesis as the basis for higher IgG1 production (Fig. 2H). Together, these data demonstrate that AMPKα1 in B cells restrains rates of antibody synthesis in plasma cells.
AMPKα1 suppresses mTORC1 activity in the B lineage.
AMPKα1 inhibits mechanistic target of rapamycin complex 1 (mTORC1) activity both indirectly through the activation of TSC2 and directly by an inhibitory phosphorylation of Raptor, an essential component of mTORC1 (39). Loss of Raptor in B cells led to poor antibody responses and inefficient generation of memory B cells in vivo (16). To evaluate mTORC1 activity in AMPKα1-deficient B cells, we performed immunoblots for target phospho-proteins of mTORC1 using extracts of B lymphoblasts. Consistent with the canonical model, loss of AMPKα1 led to elevated expression of downstream mTORC1 targets, phospho-S6S235/236 and phospho-4E-BP1T37/46 (Fig. 3A, B). Similarly, splenic MphenBC from Prkaa1 f/f huCD20- CreERT2 mice had elevated levels of phospho-S6 compared to wildtype controls after undergoing the immunization strategy illustrated in Fig. 1B (Fig. 3C, top panel). Enhanced phospho-S6 in AMPKα1-null plasma cells was also observed in the bone marrow (Fig. 3C, bottom panel). Because mTORC1 promotes protein synthesis, the elevated levels of antibody production observed with AMPKα1-deficient plasma cells are consistent with the increase in mTORC1 activity.
Figure 3. Increased mTORC1 activity in the absence of AMPKα1 in the B lineage.

(A, B) Immunoblot of mTORC1 targets pS6 S235/236 (A) and p4E-BP1 T37/46 (B) after 2 days of LPS and BAFF activation. Data represent three independent experiments or quantified in the right panels as the ratio of the indicated phospho-protein to signal for the holoprotein. P values were determined by paired t-test. (C) Representative plot and quantification of the mean fluorescence intensity of phospho-S6 in splenic MphenBC (B220+IgD−GL7−CD38+) and bone marrow plasma cells (CD138+TACI+). Data represent two independent experiments with n = 6 vs. 6 mice. P values determined by Mann-Whitney U non-parametric t-test.
AMPKα1 promotes the long-term persistence and function of memory B cells.
Early after a prime/boost primary antibody response, B lineage-intrinsic AMPKα1 limited the MphenBC population (Fig. 1D, E) and antibody synthesis (Fig. 1G, H). Because mTORC1 hyperactivity in TSC1-deficient T lineage cells undermined their longevity (40), we assessed if AMPKα1 affects the persistence of MphenBC in vivo, bearing in mind that mTORC1 activity in MphenBC is decreased compared to the overall pool of GC B cells as indicated by flow cytometric measurements of phospho-S6 in these two populations (Supplemental Fig. 2E). Because of technical issues relating to new B cell production after tamoxifen treatments in the huCD20-CreERT2 mice, we used the potent B lineage-specific mb1-Cre transgene to drive excision of Prkaa1 conditional alleles (25). This mode of excision, which starts as early as the pro-B cell stage, is constitutive but tamoxifen-independent. Lack of AMPKα1 throughout B lymphoid ontogeny had no discernable effect on the pre-immune B cell populations of mb1-Cre Prkaa1 f/f mice (Supplemental Fig. 3A–B). Similar to primary response data observed in huCD20-CreERT2 mice, Prkaa1 f/f mb1-Cre mice exhibited normal frequencies of NP-specific germinal center B cells one week after immunization compared to wildtype controls (Supplemental Fig. 3C). To test the effect of AMPK on the longevity of the MphenBC population, we immunized mb1-Cre mice (Prkaa1 f/f and Prkaa1+/+) with NP-KLH, analogous to Fig. 1B–F except mice were harvested eight weeks after the boost (Fig. 4A) instead of one (Fig. 1). In sharp contrast to the increase in total and CD80neg PD-L2neg antigen-specific MphenBC populations one week post boost (Fig. 1D), the frequencies of these MphenBC populations were reduced eight weeks post boost when B lineage were AMPKα1-deficient (Fig. 4B, C). Together with our data at the early time-point after immunization (Fig. 1D), we infer that although AMPKα1 limits MphenBC generation/expansion during an early phase of a primary response, while GC are active, it enhances the long-term persistence of memory B cells defined by standard phenotypic criteria (i.e., MphenBC).
Figure 4. AMPKα1 supports the long-term persistence of antigen-specific MphenBC and recall humoral response.
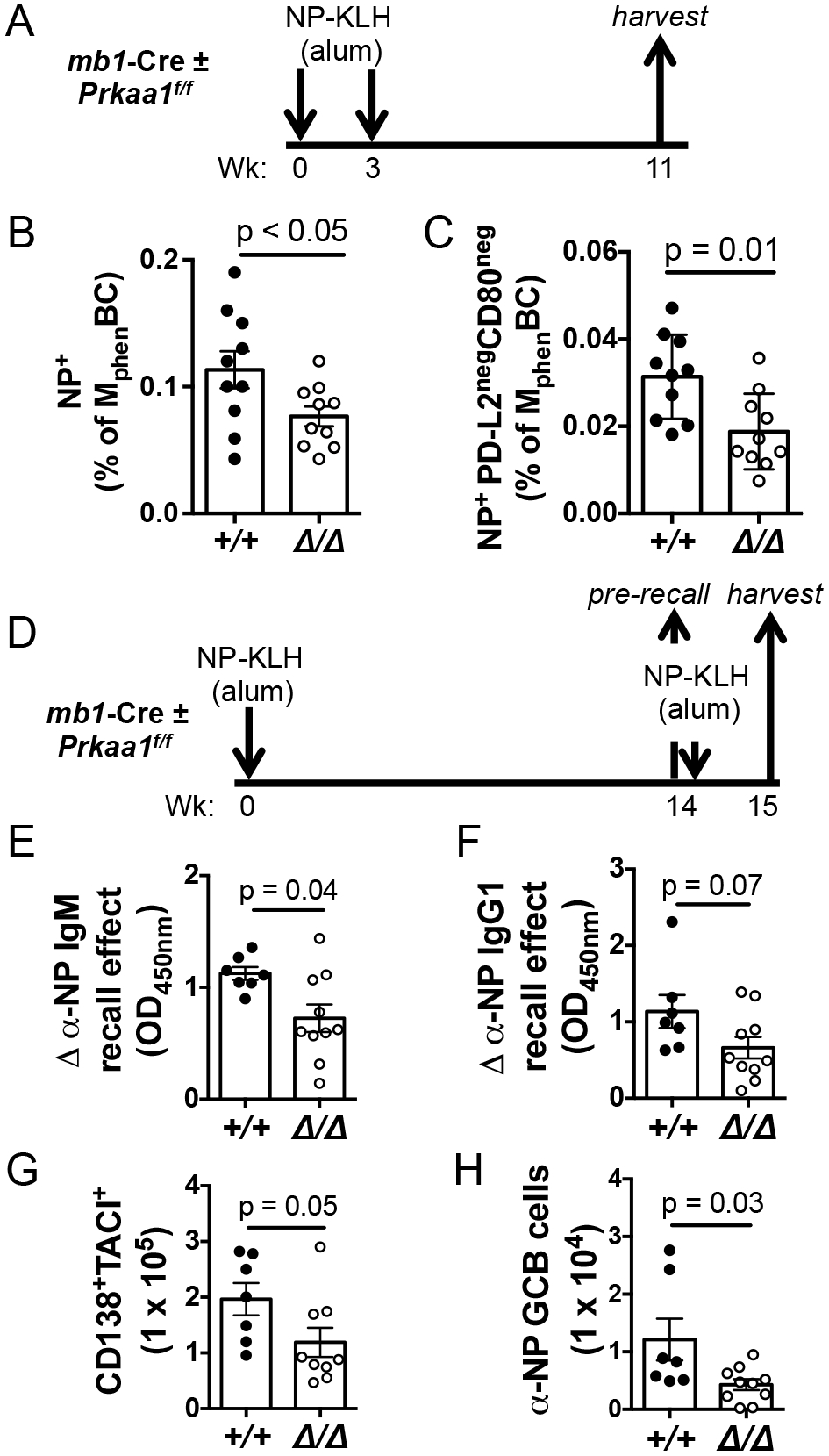
(A) Schematic of immunization strategy with a hapten-carrier using mice harboring mb1-Cre, a tamoxifen-independent cre recombinase expressed specifically in the B lineage. (B) Frequency of MphenBC that are NP-specific. (C) Frequency of NP-specific PD-L2negCD80neg within the memory phenotype B cell gate. Data represent 3 independent experiments with 10 vs. 10 mice. P values determined by Mann-Whitney U non-parametric t-test. (D) Schematic of immunization strategy to assess humoral recall. (E) Quantitation of the magnitudes of increase in anti-NP IgM and (F) anti-NP IgG1 in sera one week after recall immunization relative to pre-recall concentrations. Shown are the calculated ‘recall effects’ as defined in (28) and the Methods (G) Total numbers of splenic CD138+TACI+ cells and (H) NP-specific germinal center B cells one week after recall challenge.
We next sought to test whether the decline in MphenBC in the absence of AMPKα1 had consequences on humoral recall, i.e., memory function. Upon subsequent encounter with antigen, memory B cells rapidly differentiate into ASCs or reenter germinal centers to generate faster and more robust humoral responses than at the initial encounter. Accordingly, we re-challenged mb1-Cre mice (Prkaa1 f/f vs Prkaa1 +/+) 14 wk after a primary immunization (Fig. 4D). To assess the strength of recall responses Prkaa1-null MphenBC, we measured circulating anti-NP IgM and anti-NP IgG1 in sera collected both immediately prior to and a week after re-challenge, thereby allowing for the possible persistence of higher Ab levels in the absence of AMPKα1 (Fig. 1H). For each mouse, then, a “recall effect” (mnestic induction of increased Ab concentration) was calculated (28). Anti-NP IgM and IgG1 increased substantially less in response to the recall immunization of Prkaa1 f/f mb1-Cre mice than mb1-Cre controls (Fig. 4E, F). These data indicate that, in contrast to the primary response (Fig. 1), recall Ab responses were weaker when B cells lacked AMPKα1. Consistent with the observed decrease in recall-induced Ab, the CD138+ TACI+ splenic plasmablast/plasma cell populations (41) also were attenuated in mice harboring AMPKα1-deficient B cells (Fig. 4G). In contrast to the primary response, antigen-specific GC B cells, a fate of reactivated CD80neg PD-L2neg MphenBC (36, 37), were also diminished in re-challenged AMPKα1-deficient mice. We infer from these data that AMPKα1 not only supports the longevity of MphenBC but also maintains the functional capacity of recall humoral responses.
Mice in the previous model (Prkaa1 f/f mb1-Cre) harbor B cells that are deficient in AMPKα1 from very early in B lineage development, so that a defect of programming during the primary response might contribute to the finding of weaker recall. To test the role of AMPKα1 after primary immunity, we used adoptive transfers with a tamoxifen-inducible system in which Prkaa1 can be deleted after a normal primary response (Fig. 5A). Naïve B cells (CD45.2+ IgHb allotype) from Rosa26-CreERT2 mice (Prkaa1 f/f or Prkaa1 +/+) were transferred into sub-lethally irradiated CD45.1+ IgHa allotype-disparate recipients along with helper T cells (a mix of CD4+ T cells from wild-type and OTII transgenic mice). Recipients were then immunized with NP-ovalbumin and boosted (as in Fig. 1A) to generate normal primary responses. Tamoxifen injections to induce deletion of Prkaa1 in the donor B cell population, including MphenBC, were deferred to week nine. To assess recall responses, mice were re-challenged with NP-ovalbumin several months later,h and harvested one week after the recall immunization (Fig. 5A). PCR products from flow-sorted donor CD45.2+ MphenBC at the time of harvest documented effective deletion of Prkaa1 in the cells from mice that received Prkaa1 f/f Rosa26-CreERT2 donor B cells (Fig. 5B). Using allotype-specific detection reagents, donor and recipient anti-NP IgG1 were measured in the sera before and after recall challenge. Compared to mice receiving wild-type CreERT2 B cells, mice that received CreERT2 Prkaa1 f/f B cells had weaker anti-NP IgG1b recall responses after re-challenge (Fig. 5C). In contrast, an internal control of recipient-derived a-allotype Ab found that levels of anti-NP IgG1a were similar regardless of the donor cell source. Consistent with these serologies after re-challenge, anti-NP IgG1b-secreting cells and donor-derived CD45.2+ CD138+ TACI+ plasmablasts and plasma cells in the spleen were diminished in mice that received B cells from which AMPKα1 was depleted (Fig. 5D, E). AMPKα1-deficient donor-derived germinal center B cells were also decreased compared to wildtype donor germinal center B cells (Fig. 5F). These data support the conclusion that after a normal primary humoral response is established, the recall capacity of B cells is enhanced by AMPKα1.
Figure 5. Loss of AMPKα1 after primary immunity impairs subsequent recall function.
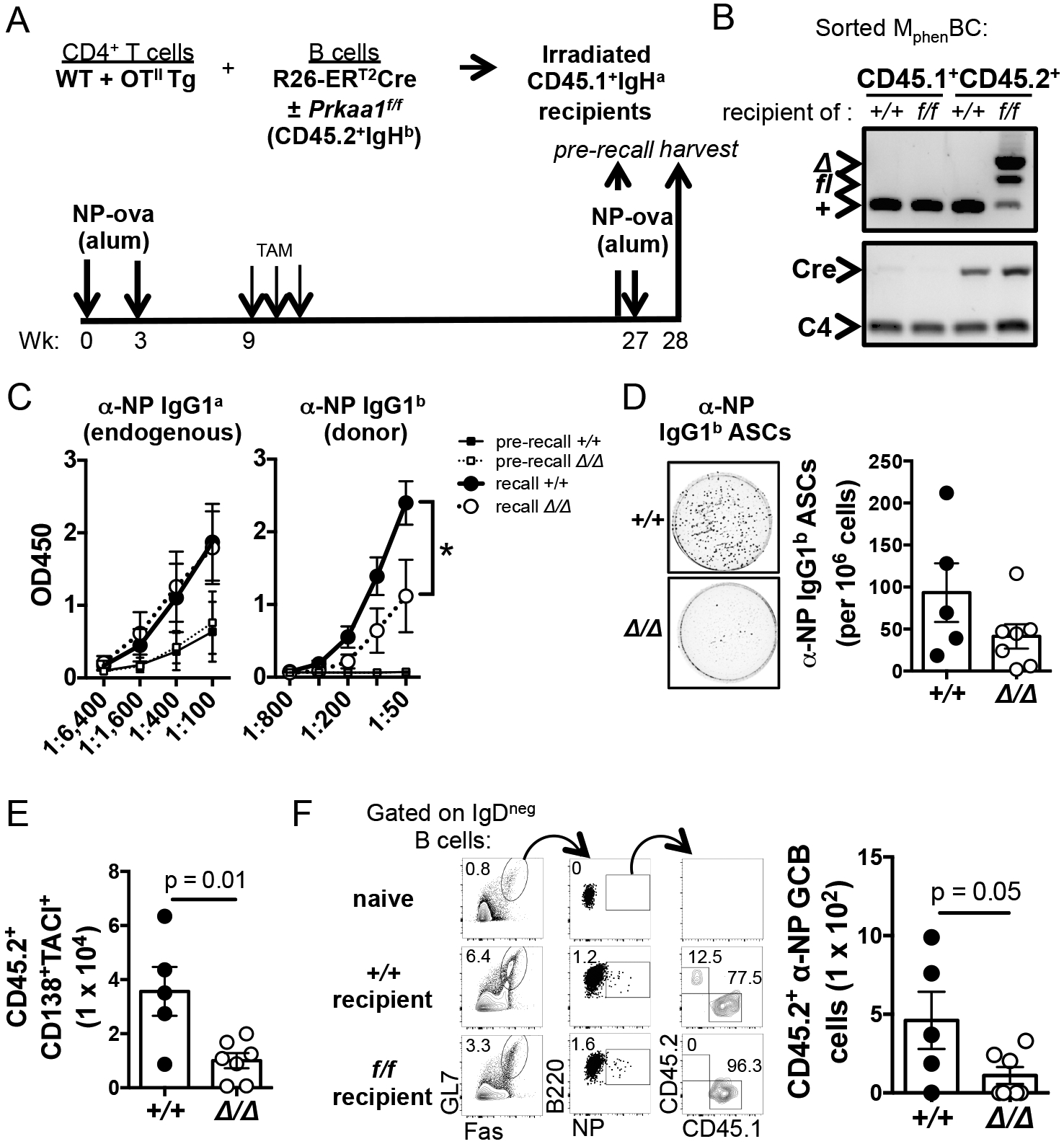
(A) Experimental design to evaluate the effect of AMPKα1 in MphenBC after a normal primary response. CD4+ T cells and CD45.2+ IgHb B cells from Rosa26- CreERT2 (Prkaa1 f/f or Prkaa1 +/+) mice were transferred into naïve wild-type allotype-disparate CD45.1+ IgHa mice prior to immunization. Deletion of Prkaa1 in MphenBC was induced by tamoxifen injections nine weeks after the initial immunization. Mice were rechallenged 18 wk thereafter and harvested 1 wk after the recall immunization. (B) Prkaa1 and Cre PCR of DNA from purified splenic CD45.1+ or CD45.2+ MphenBC (B220+ IgD− GL7− CD38+). (C) Circulating anti-NP IgG1a (recipient-derived) and anti-NP IgG1b (donor-derived) immediately before and one week post rechallenge. (D) Representative wells and quantification of ELISpot analyses determining numbers of splenic NP-specific IgG1b ASCs at harvest. (E) Numbers of donor CD45.2+ CD138+ TACI+ plasma cells in the spleen after rechallenge. (F) (Left panel) Representative flow plots depicting frequency of splenic NP-specific germinal center B cells that are recipient-derived (CD45.1+) vs. donor-derived (CD45.2+) after rechallenge. (Right panel) Total number of donor-derived NP-specific germinal center B cells in the spleen after rechallenge.
AMPKα1 is dispensable for autophagy in B cells.
AMPK regulates many aspects of intracellular metabolism including supporting autophagy, the degradation and recycling of cellular components (42). Autophagy appears to support the survival of memory B cells and limits antibody production in favor of sustaining a long-lived plasma cell population (11–13). We hypothesized that the decline in the MphenBC population and/or increased antibody synthesis observed in B cell-specific AMPKα1-deficient mice was due to a defect in autophagy in the B lineage. Surprisingly, the ability of LPS-activated B cells to form LC3-puncta indicative of autophagosome formation after glucose starvation was no different in AMPKα1-deficient B cells compared to controls (Supplemental Fig. 4A,B). Furthermore, LC3-I conversion into the faster migrating LC3-II upon glucose starvation was independent of AMPKα1 expression (Supplemental Fig. 4C). Thus, LPS-activated B cells appear able to undergo glucose starvation-induced autophagy by an AMPKα1-independent mechanism. Finally, normal frequencies and sizes of LC3-puncta were visualized in freshly purified bone marrow plasma cells of tamoxifen-treated Prkaa1 f/f, Rosa26-CreERT2 mice (Supplemental Fig. 4D). This result further supports that autophagy occurs in the B lineage even in the absence of AMPKα1. Collectively, these data suggest that B lineage cells can induce AMPKα1-independent autophagy, potentially by non-canonical pathways (43).
Altered mitochondrial quality control and function in AMPKα1-deficient B cells.
In light of the role of AMPK as a regulator of intermediary metabolism and mitochondrial function, we next tested metabolic performance of activated B cells that were AMPKα1-sufficient or -deficient. Extracellular flux analyses revealed no changes in any aspect of the extracellular acidification rate (ECAR) with the loss of AMPKα1 (Fig. 6A). In contrast, analyses of the oxygen consumption rate (OCR), before and after treatment with different mitochondrial stressors, determined that mitochondrial oxidative phosphorylation was impaired in AMPKα1-deficient B cells (Fig. 6B). Basal respiration, represented by the OCR values before the addition of ATP synthase V inhibitor oligomycin, was not altered by the loss of AMPKα1 (Fig. 6B left panel). However, loss of AMPKα1 led to defects in maximal and spare respiratory capacity (Fig. 6B middle, and right panels). The oxygen consumption rate was specific to the electron transport chain as the OCR levels were dependent on complex I and III inhibitors rotenone and antimycin A. Taken together, these data indicate that AMPKα1 in activated B cells promotes the establishment or maintenance of optimal respiratory function of mitochondria.
Figure 6. Loss of AMPKα1 in B cells leads to decreased mitochondrial function and defects in mitophagy.
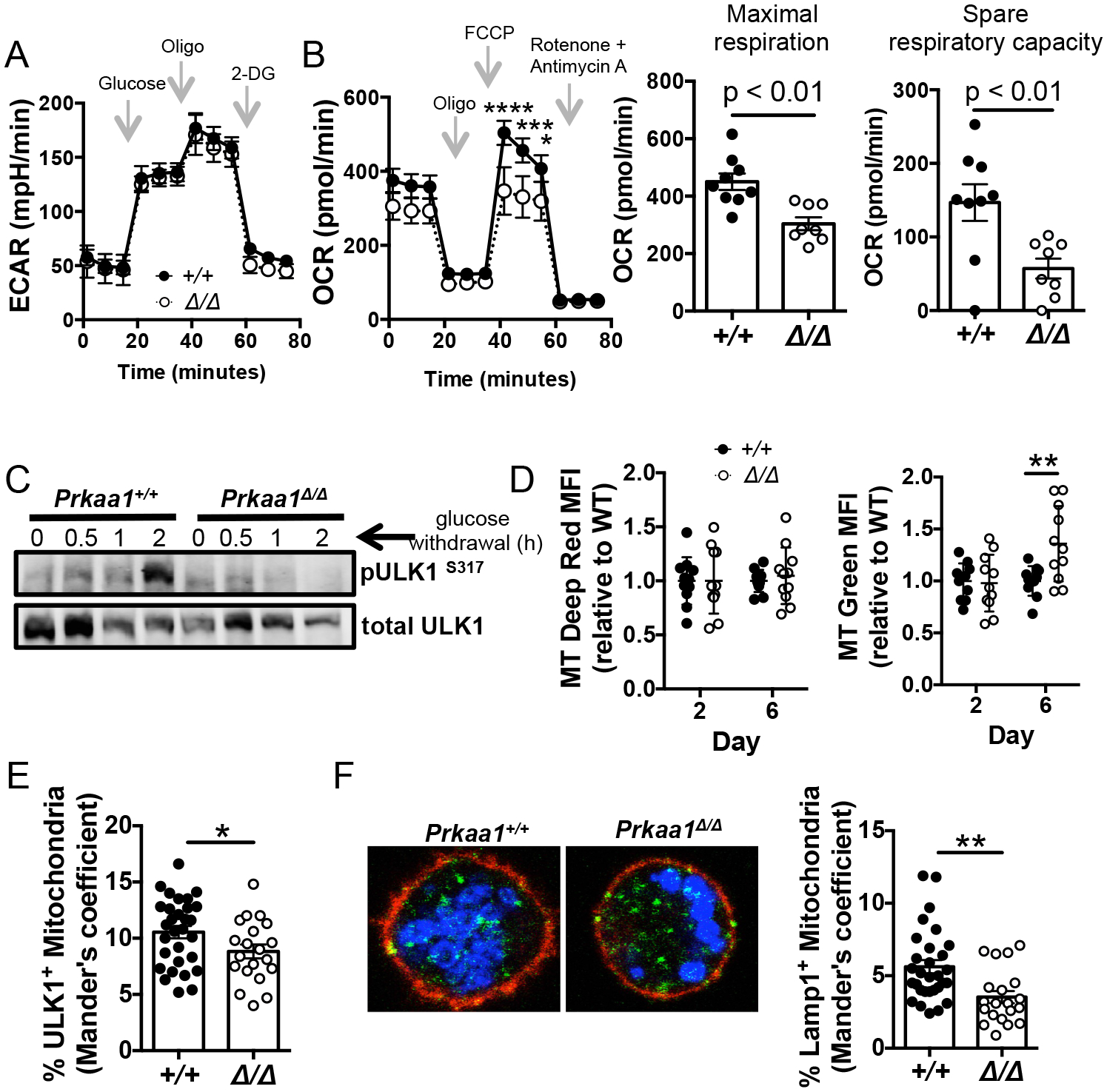
(A) Glycolytic stress test after B cells from tamoxifen-treated Rosa26-ERT2Cre mice (Prkaa1 +/+ or Prkaa1 f/f ) were activated for 2 days with BAFF and LPS in the presence of 4-OHT. (B) Mitochondrial stress test after B cells from tamoxifen-treated Rosa26-ERT2Cre mice ± Prkaa1 f/f were activated for 2 days with BAFF and LPS in the presence of 4-OHT (left panel). Maximal respiration and spare respiratory capacity calculated by Seahorse report generator (right panels). Data represent three independent experiments with n = 9 vs. 8 mice. (C) Expression of pULK1S317, an AMPK target that initiates mitophagy, after glucose deprivation in day 2 LPS-activated cells. Data is representative of three independent experiments. (D) Relative MFI values for MitoTracker Deep Red (left panel) and MitoTracker Green (right panel) in the B220+ gate after activation with LPS, BAFF, IL-4, and IL-5 in the presence of 4-OHT. Data represent 4 independent experiments with 10 vs. 11 mice. (E) Quantification of ULK1 and MitoTracker Deep Red co-localization after 2 day activativation with LPS and BAFF. (F) Representative immunofluorescence of MitoTracker Deep Red and Lampl co-localization on day 2 LPS-activated cells. (Blue = MitoTracker Deep Red; green = Lamp1; red = B220). Quantification of Lamp1 and MitoTracker Deep Red co-localization (right panel). Data are representative of two independent experiments with n = 4 vs. 3 mice and 10 fields/mouse. P values determined by Mann-Whitney U non-parametric t-test or ANOVA where appropriate. * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001.
AMPK phosphorylates multiple downstream targets that support different aspects of mitochondrial homeostasis and quality, including mitochondrial biogenesis and mitophagy, i.e., the selective degradation of mitochondria (44). As we observed no defect in overall autophagy in the mutant B cells, we explored whether AMPK was essential for mitophagy in LPS-activated B cells. Unc-51 like kinase (ULK1), a substrate of AMPK, is involved in the initiation of canonical autophagy and is essential for mitophagy (45–48). LPS-activated wildtype B cells increased phosphorylation of the S317 site of ULK1, a target of AMPK, after 2 h of glucose starvation (Fig. 6C). Induction of ULK1 (S317) phosphorylation due to glucose withdrawal was dependent on AMPKα1 expression. Mitophagy is critical for the removal of damaged or superfluous mitochondria. We reasoned that a defect in mitophagy would lead to the accumulation of dysfunctional mitochondria over time. To assess the effect of AMPKα1 on mitochondria accumulation after in vitro activation, wild-type and mutant B cells were stained with MitoTracker Green and MitoTracker Deep Red as their fluorescence reports total mitochondrial mass and actively respiring mitochondria respectively. Mitochondrial mass was comparable regardless of AMPKα1 expression on day 2, but by day 6 AMPKα1-deficient B cells had increased mitochondrial mass in the B220+ gate compared to controls (Fig. 6D right panel). The increased mitochondrial mass over time was not accompanied with increased mitochondrial activity (Fig. 6D left panel) suggesting that the relative increased MitoTracker Green staining (d2 – d6) represented nonfunctional mitochondria. The evidence is consistent with a model where AMPKα1 in B cells is important for the clearance of ineffective mitochondria, as characterized for other cell types (49).
Once activated in a manner dependent in part on AMPK-mediated phosphorylation, ULK1 migrates to the mitochondria and triggers a signaling cascade that leads to the recruitment of LC3 machinery and fusion with the lysosome (46,47). To test if AMPKα1 enhances mitophagy in B cells, we assessed in LPS-activated B cells the association of mitochondria with ULK1 and Lamp1, a lysosomal marker by confocal imaging. Co-localization analyses with wild-type and AMPKα1-deficient B cells revealed that the percentage of ULK1+ mitochondria was decreased in the absence of AMPKα1 (Fig. 5E), suggesting that AMPK plays a role in ULK1 recruitment to mitochondria. As further evidence of a defect in mitophagy, the frequencies of Lamp1+ mitochondria indicative of mitochondrial fusion to the lysosome were diminished by loss of AMPK (Fig. 6F). Collectively, these findings support a model in which AMPKα1 promotes mitochondrial clearance and quality control in B cells, likely due to a non-redundant requirement for ULK1 phosphorylation at S317, and supporting mitochondrial function.
AMPKα1 maintains mitochondria and protects memory B cells against lipid peroxidation.
We next tested if the defect in mitochondrial homeostasis applied to the phenotypically-defined memory B cell population, i.e., MphenBC. To obtain sufficient cells, mitochondrial parameters were analyzed by flow cytometry using total rather than NP-specific MphenBC (B220+ GL7neg IgDneg CD38+) 7–10 wk after immunizations of mb1-Cre mice (Prkaa1 f/f vs Prkaa1 +/+) with NP-KLH (Fig. 7A, left panel). Actively respiring mitochondria were modestly decreased in the AMPKα1-deficient MphenBC, whereas total mitochondrial mass was unchanged compared to mb1-Cre controls (Fig. 7A, right panel). The ratio of mitochondrial membrane potential to total mitochondrial mass (Fig. 7A), indicative of mitochondrial quality, was diminished in AMPKα1-deficient MphenBC (Fig. 7B) (50). These data indicate that AMPKα1 supports mitochondrial quality maintenance in phenotypically defined memory B cells. We next tested if the decrease in mitophagy observed in LPS blasts (Fig. 6F) was also observed MphenBC immediately ex vivo from mb1-Cre mice (Prkaa1 f/f vs Prkaa1 +/+). Co-localization analysis revealed that MphenBC from mb1-Cre, Prkaa1 f/f mice harbored fewer Lamp1+ mitochondria, consistent decreased levels of mitophagy in memory B cells lacking AMPKα1 (Fig 7C). Levels of mitochondrial derived reactive oxygen species (mtROS) play a key role in many cellular functions including regulating B cell fate and function (51,52). Consistent with the defect in quality control, AMPKα1-deficient MphenBC exhibited increased mtROS compared to controls (Fig. 7D). To test if increased mtROS observed in AMPKα1-deficient MphenBC was accompanied with changes in lipid peroxidation, we used Bodipy 581/591 C11 and found it to be elevated in AMPKα1-deficient MphenBC (Fig 7E). Collectively, these results suggest that AMPKα1 protects memory B cells against excessive oxidative stress and cell death involving lipid peroxidation by contributing to mitophagic quality control.
Figure 7. AMPKα1 supports mitochondrial quality and limits lipid peroxidation in MphenBC.
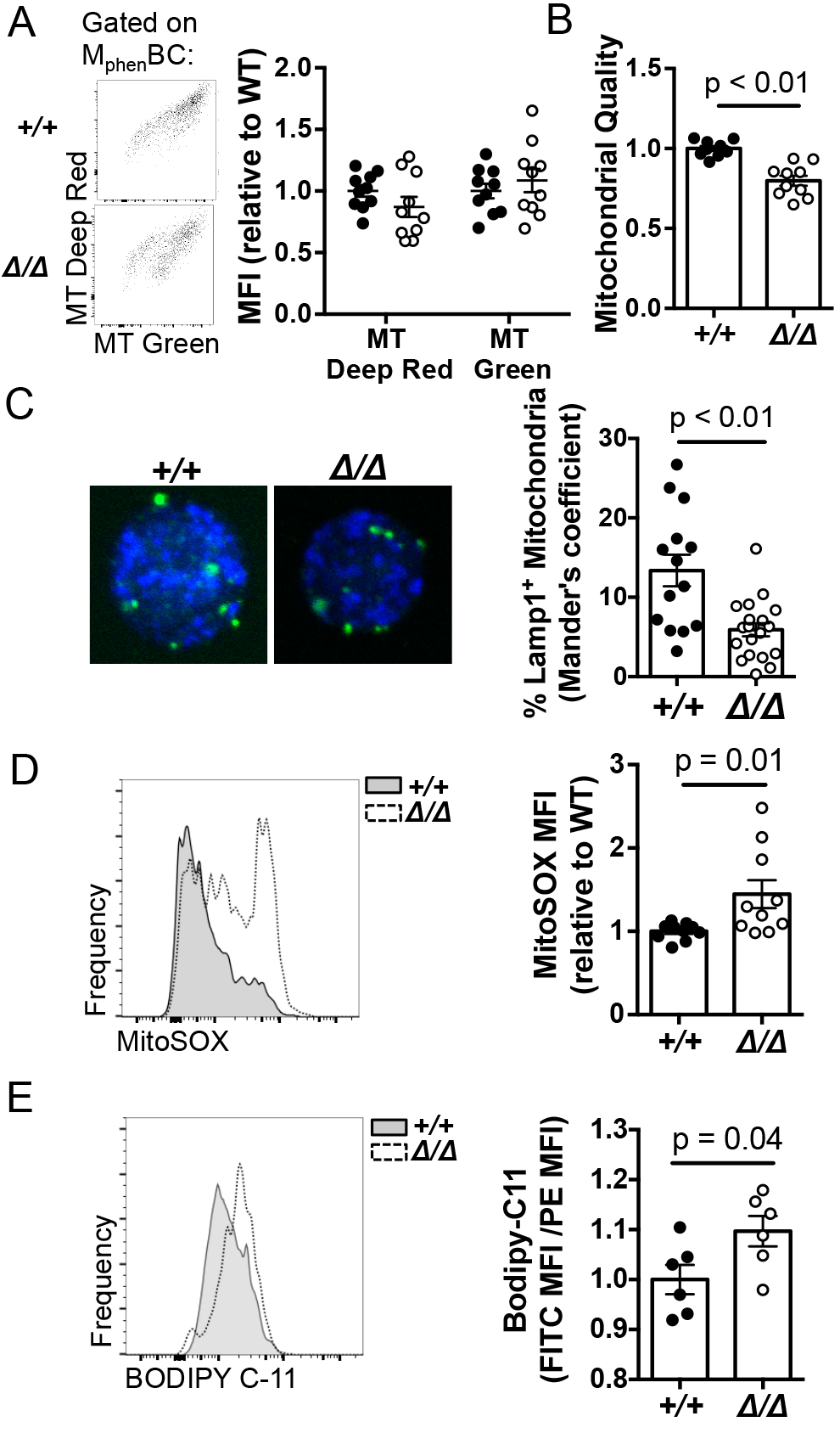
(A) (Left panel) Representative flow plots of MitoTracker Deep Red vs. MitoTracker Green of MphenBC (B220+CD38+ after IgD depletion) from mb1-Cre (Prkaa1 f/f vs Prkaa1 +/+) mice 11 wks after immunization with a hapten-carrier. (Right panel) Quantification of MitoTracker Deep Red and MitoTracker Green MFI values normalized to wildtype. (B) Mitochondrial quality, i.e. the normalized ratio of MitoTracker Deep Red to MitoTracker Green, as an indicator of the proportion of functional mitochondria to the total. (C) Representative images and quantification of the co-localization of MitoTracker Deep Red (blue) and Lamp1 (green) of flow sorted MphenBC (B220+ GL7− IgD− CD38+) from mb1-Cre mice (Prkaa1 +/+ or Prkaa1 f/f ). (D) Representative MitoSOX histogram of MphenBC from mb1-Cre mice 11 wks after immunization with a hapten carrier (left panel). Quantification of normalized MitoSOX values (right panel). (E) Representative histogram of BODIPY C-11 (which fluoresces in the FL-1 channel upon lipid oxidation) of MphenBC from mb1-Cre mice 11 wks after immunization (left panel). Normalized ratio of FITC (FL-1) MFI to PE (FL-2) MFI of MphenBC after staining with Bodipy 581/591 C11 as a measure of lipid peroxidation. Data are representative of three independent experiments with n=10 vs. 10 mice where the average of wildtype values in each experiment was taken and all mice were normalized to that value. P values determined by Mann-Whitney U non-parametric t-test.
DISCUSSION
The orchestration of intracellular metabolic pathways varies to support developmental and functional needs throughout the lifespan of a B lineage cell (6,7,53). Furthermore, as B cells and their progeny migrate through or take residence in distinct tissues, they likely have the capacity to persist in distinct microenvironments with differing nutrient availability (6,7). Relatively little is known about the metabolic pathways that support the persistence and function of memory B cells and long-lived plasma cells, each of which confers one facet of durable humoral immunity. The energy sensor AMPK regulates multiple aspects of intracellular metabolism. Its overall program favors energy conservation and tilts balance toward catabolism while restraining anabolism in response to declining levels of ATP (14). Here we have shown that AMPK supports the longevity of memory B cells essential for robust recall humoral responses and regulates the antibody production of plasma cells both in short- and long-term times after immunization. In particular, the lack of AMPK in B cells led to increased increased mTORC1 activity as well as initial and long-term immunoglobulin synthesis in response to immunization with haptenated carrier. After the initial population increase, AMPK deficiency resulted in declines in the memory-phenotype B cell population along with the accumulation of dysfunctional mitochondria, decreased mitophagy, increased mitochondrial ROS, and increased lipid peroxidation. The functional impact of these changes was a reduced strength of recall antibody response after rechallenge. These findings indicate that AMPK protects the maintenance of a memory B cell population and suggest that this function is achieved at least in part through the regulation of mitochondrial turnover. In contrast, AMPK was dispensable for the persistence of bone marrow resident long-lived plasma cells but played a critical role in regulating their rate of antibody synthesis.
Immunological memory is a hallmark component of the adaptive immune system whereby long-lived cells of the T and B lineages confer long-lasting protective immunity against re-infection. Though there are some distinctions in the balance of oxidative phosphorylation and glycolysis employed by B and T cells during activation (31), such that B lymphoblasts better couple pyruvate generation to the mitochondria than their T cell counterparts, both subsets undergo clonal expansion and generate long-lived quiescent memory populations (54,55). Thus, there may be parallels in the metabolic programming between the two lineages. Akin to the role of AMPK in maintaining CD8 memory T cells (18), our data indicate that AMPK promotes the persistence of memory-phenotype B cells and their function in humoral recall responses. Analogous to reports in the T lineage (18,19,56), we found that AMPK antagonizes mTORC1 activity in B cells, a cross-talk that is likely to play a role in regulating rates of protein synthesis. The finding that AMPK promotes longevity and catabolism in both the B and T cell lineages suggests a conserved function of AMPK in these long-lived memory subsets. However, the long-lived plasma cell population provides a second limb of humoral memory (57) and it is not clear if any T cell subset is analogous to this population of terminally differentiated cells that erased most central features of B cell identity (58). Unlike quiescent memory populations, long-lived plasma cells require high rates of protein and glucose metabolism to support the synthesis and glycosylation of antibodies (5). Surprisingly, our data indicate that unlike in memory lymphocytes, AMPK is dispensable for maintaining the marrow-resident long-lived plasma cell population. This finding highlights a stark molecular distinction between memory B cells and bone marrow plasma cells in their metabolic requirements.
In primary humoral responses, mTORC1 is critical for memory B cell generation and antibody production (16,59). In the present study, we observed enhanced mTORC1 activity in AMPK-deficient ex vivo memory B cells and plasma cells. This measurement corresponded to an increase in the initial number of MphenBC observed one week after a booster immunization as well as enhanced antibody production from plasma cells. Thus, our data suggest that AMPK moderates mTORC1 activity in a manner that maintains a biologically appropriate range of mTORC1 signaling that ultimately leads to appropriate levels of antibody and memory B cell generation. Such “Goldilocks mTOR” may be evolutionally conserved for purposes of maintaining controlled primary humoral responses as it does in T cell differentiation (60). We have uncovered that AMPK plays a part in the fine-tuning of mTORC1 during B cell activation. Elevated mTORC1 signaling and enhanced circulating class-switched circulating antibody were maintained in AMPK-deficient long-lived bone marrow plasma cells. In light of evidence indicating that rapamycin treatment hindered antibody secretion from long-lived plasma cells (59), our data suggest that in addition to immunoglobulin synthesis rates, the regulation of mTORC1 activity in long-lived plasma cells is governed at least in part by AMPK.
AMPK and mTORC1 reciprocally regulate autophagy, a conserved self-degrading cellular process (45). Autophagy is essential for the long-term persistence of both memory B and CD4 T cell populations (11,12,61). Loss of either two autophagy-essential genes, Atg5 or Atg7, was tied to mitochondrial dysfunction and enhanced lipid peroxidation in the B and T cell lineages (11,61,62). In a manner seeming analogous to our findings with AMPKα1, Atg5 in B cells was critical for limiting excessive immunoglobulin production by plasma cells (13), which was attributed to a function in restraining endoplasmic reticulum stress signaling. It was surprising, therefore, to observe that autophagy after glucose withdrawal was normal for AMPK-deficient B cells despite the expected absence of activating phosphorylation of ULK1. These findings suggest that activated B cells can also induce autophagy via non-canonical, AMPK-independent pathways (43). Despite normal formation of LC3 puncta, the defect in ULK1 (S317) phosphorylation was associated with a defect in ULK1 recruitment to the mitochondria, and failure to induce mitophagy in AMPK-deficient B cells. In support of our findings, the AMPK-ULK1 axis is reported to be essential for hypoxia- or exercise-induced mitophagy in mouse embryonic fibroblasts and skeletal muscle cells respectively (47,48). Interestingly, previous studies demonstrating the importance of autophagy in lymphocyte persistence and mitochondrial homeostasis use genetic models by conditional inactivation of Atg5 or Atg7, both of which may be involved in multiple autophagy pathways including mitophagy (43). Accordingly, the reason memory B cell persistence is promoted by Atg7 may be attributable in part to mitochondrial-specific autophagy activated via AMPKα1.
Our data show that AMPK promotes both the long-term persistence of memory B lymphocytes and mitophagy in B lineage cells. There are substantial antecedents that connect mitochondria to memory B cell persistence (63). Memory B cell longevity is linked to enhanced expression of mitochondrial pro-survival proteins in the Bcl2 superfamily, both Bcl2 itself (64) and BH3-only Puma (65). Furthermore, mitochondrial homeostasis is associated with the persistence of memory B cells (11,63). Mitophagy has also been associated with maintaining memory in a natural killer cell population (66). In that setting, it likely functioned through clearance of dysfunctional mitochondria, which protected cells from the accumulation of excessive mitochondrially-derived ROS, lipid peroxidation, and cell death. Our findings with AMPK-deficient B cells align with this previous work, in that we observed increased mitochondrial ROS and abnormal function in mitochondrial testing during metabolic flux analyses. Spare oxidative phosphorylation, or the reserved capacity to amplify respiration in response to increased demand, was impaired in B cells lacking AMPK, similar to observations on memory CD8 T cell longevity (67). The types of memory lymphocytes assayed in prior work and ours are known to circulate through blood, tissue, and lymphatics (68). Accordingly, we speculate that spare respiratory capacity may be critical for memory cells to adapt to substantial differences in these microenvironments and the attendant metabolic stresses as they survey and pass through distinct tissues.
One observation in this work is that memory B cells and long-lived plasma cells exhibited distinct metabolic profiles and dependence on AMPK. Though AMPK supported the persistence of the memory B cell population, it dampened long-lived plasma cell antibody synthesis function without hindering survival. Activation of the AMPK pathway by metformin has been associated with increased memory B cells and improved antibody responses to influenza vaccine in type II diabetic patients (69). Our data may provide insight on the efficacy of drugs that target AMPK to achieve longer lasting humoral responses and improve vaccine design.
Supplementary Material
KEY POINTS.
AMPK dampens mTORC1 activity and the synthesis of antibody in plasma cells.
AMPK promotes mitochondrial homeostasis in B lymphoblasts and memory B cells.
AMPK supports the persistence of the memory B cell population and humoral recall.
ACKNOWLEDGEMENTS
We thank Ariel Raybuck for sharing mice, techniques, and ideas throughout the duration of the work as well as careful readings of the manuscript. We thank Majan Rafat for assistance in quantification of confocal images, Kevin Chen for performing some ELISAs, Lan Wu for managing and troubleshooting FACSCanto equipment, and Linda Sealy and Janice Blum for guidance and support. We also thank the High-Throughput Screening, Cell Imaging Shared Resource, Flow Cytometry Shared Resource, and Cell and Developmental Biology Cores at Vanderbilt University and Vanderbilt University Medical Center for their equipment, expertise, and assistance.
Experimental work was supported by NIH grants AI113292 and HL106812 (to M.R.B.). Additional support for S.M. was provided by NIH grants R25-GM062459 and T32 CA009592-29, followed by a supplement to AI113292 and P. M. & I. Departmental funds, and by Vanderbilt University’s Provost Graduate Fellowship. Additional support for P.J.B. was provided by FAPESP 2018/08563-8. NIH Shared Instrumentation Grant 1S10OD018015 as well as scholarships via the Cancer Center Support Grant (CA068485) and Diabetes Research Center (DK0205930) helped defray costs of Vanderbilt Cores.
Abbreviations Used
- AMPK
adenosine monophosphate activated protein kinase
- ULK1
Unc-51-like kinase
- 7-AAD
7-aminoactinomycin D
- ASC
Ab-secreting cell
- 2DG
2-deoxyglucose
- FCCP
Trifluoromethoxy carbonylcyanide phenylhydrazone
- ROS
reactive oxygen species
- Ab
antibody
- GC
germinal center
- mTORC1
mechanistic target of rapamycin complex 1
- MT Green
MitoTracker Green
- MT Deep Red
MitoTracker Deep Red
- mtROS
mitochondrial ROS
- ECAR
extracellular acidification rate
- OCR
oxygen consumption rate
- NP
nitrophenol
- NP-KLH
NP conjugated to keyhole limpet hemacyanin
- MphenBC
memory-phenotype B cells
- 4-OHT
4-hydroxytamoxifen
REFERENCES
- 1.Slifka MK, Antia R, Whitmire JK, and Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity. 8: 363–372. [DOI] [PubMed] [Google Scholar]
- 2.Victora GD and Nussenzweig MC. 2012. Germinal centers. Annu. Rev. Immunol 30: 429–457. [DOI] [PubMed] [Google Scholar]
- 3.Brynjolfsson SF, Berg LP, Ekerhult TO, Rimkute I, Wick M, Mårtensson I, and Grimsholm O. 2018. Long-lived plasma cells in mice and men. Frontiers in Immunol. 9: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weisel F and Shlomchik M. 2017. Memory B cells of mice and humans. Annu. Rev. Immunol 35: 255–284. [DOI] [PubMed] [Google Scholar]
- 5.D’Souza L and Bhattacharya D. 2019. Plasma cells: you are what you eat. Immunological Reviews. 288: 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boothby M and Rickert RC. 2017. Metabolic regulation of the immune humoral response. Immunity. 46: 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jellusova J and Rickert RC. 2017. A brake for B cell proliferation: appropriate responses to metabolic stress are crucial to maintain B cell viability and prevent malignant outgrowth. Bioessays. doi: 10.1002/bies.201700079. [DOI] [PubMed] [Google Scholar]
- 8.Chevrier S, Emslie D, Shi W, Kratina T, Wellard C, Karnowski A, Erikci E, Smyth GK, Chowdhury K, Tarlinton D, and Corcorna LM. 2014. The BTB-ZF transcription factor Zbtb20 is driven by Irf4 to promote plasma cell differentiation and longevity. J. Exp. Med 211: 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y and Bhattacharya D. 2014. Adjuvant-specific regulation of long-term antibody responses by ZBTB20. J. Exp. Med 211: 841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, McCommis KS, Fahrmann J, Pizzato HA, Nunley RM, Lee J, Wolfgang MJ, Patti GJ, Finck BN, Pearce EL, and Bhattacharya D. 2016. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. 45: 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, Corry DB, Kheradmand F, and Wang J. 2014. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nature Medicine. 20: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M, Kodali S, Jang A, Kuai L, and Wang J. 2015. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol. 194: 2607–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, Fagioli C, Merlini A, Mariani E, Pasqualetto E, Orfanelli U, Ponzoni M, Sitia R, Casola S, and Cenci S. 2013. Plasma cells require autophagy for sustainable immunoglobulin production. Nature Immunology. 14: 298–305. [DOI] [PubMed] [Google Scholar]
- 14.Garcia D and Shaw RJ. 2017. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Molecular Cell. 66: 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srivastava S 2017. The mitochondrial basis of aging and age-related disorders. Genes. 398: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raybuck AL, Cho SH, Li J, Rogers MC, Lee K, Williams CL, Shlomchik M, Thomas JW, Chen J, Williams JV, and Boothby MR. 2018. B cell-intrinsic mTORC1 promotes germinal center-defining transcription factor gene expression, somatic hypermutation, and memory B cell generation in humoral immunity. J Immunol. 200: 2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma EH, Poffenberger MC, Wong AH, and Jones RG. 2017. The role of AMPK in T cell metabolism and function. Current Opinion in Immunology. 46: 45–52. [DOI] [PubMed] [Google Scholar]
- 18.Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, and Cantrell DA. 2013. AMPKα1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol. 43: 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, Pelletier J, Piccirillo CA, Krawczyk CM, Divangahi M, and Jones RG. 2015. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 42: 41–54. [DOI] [PubMed] [Google Scholar]
- 20.Rao E, Zhang T, Zhu G, Hao J, Persson XT, Egilmez NK, Suttles J, and Li B. 2015. Deficiency of AMPK in CD8+ T cells suppresses their anti-tumor function by inducing protein phosphatase-mediated cell death. Oncotarget. 6: 7944–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer AS Denanglaire B Viollet O Leo, and Andris F. 2008. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol. 38: 948–956. [DOI] [PubMed] [Google Scholar]
- 22.Waters LR, Ahsan FM, Hoeve J, Hong JS, Kim DN, Minasyan A, Braas D, Graeber TG, Zangle TA, and Teitell MA. 2019. Ampk regulates IgD expression but not energy stress with B cell activation. Scientific Reports. 9: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S, Dale GL, Song P, Viollet B, and Zou M. 2010. AMPKα1 deletion shortens erythrocyte life span in mice. J of Biological Chemistry. 285: 19976–19985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carroll KC, Viollet B, and Suttles J. 2013. AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J Leukoc Biol. 94: 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M. 2006. Testing gene function early in the B cell lineage in mb1-cre mice. PNAS. 103: 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakada D, Saunders TL, and Morrison SJ. 2010. Lkb1 reguates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 468: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee K, Heffington L, Jellusova J, Nam KT, Raybuck A, Cho SH, Thomas JW, Rickert RC, and Boothby M. 2013. Requirement for Rictor in homeostasis and function of mature B lymphoid cells. Blood. 122: 2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho SH, Raybuck A, Wei M, Erickson J, Nam KT, Cox RG, Trochtenberg A, Thomas JW, Williams J, and Boothby M. 2013. B cell-intrinsic and -extrinsic regulation of antibody responses by PARP14, an intracellular (ADP-ribosyl) transferase. J Immunol. 191: 3169–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, and Kelsoe G. 2016. Complex antigens drive permissive clonal selection in germinal centers. Immunity. 44: 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nojima T, Haniuda K, Moutai T, Matsudaira M, Mizokawa S, Shiratori I, Azuma T, and Kitamura D. 2011. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nature Communications. 465: 1–11. [DOI] [PubMed] [Google Scholar]
- 31.Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, Gavin AL, Abel ED, Kelsoe G, Green DR, Rathmell JC. 2014. Metabolic reprogramming is required for the antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 192: 3626–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucher LA and Lego T. 1988. Use of water soluble-fluor sodium salicylate for fluorographic detection of tritium in thin-layer chromatograms and nitrocellulose blots. Analytical Biochemistry. 178: 327–330. [DOI] [PubMed] [Google Scholar]
- 33.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, Mamer OA, Avizonis D, DeBerardinis RJ, Siegel PM, and Jones RG. 2013. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 17: 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, and Shlomchik MJ. 2007. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 179: 3351–3361. [DOI] [PubMed] [Google Scholar]
- 35.Weisel FJ, Zuccarino-Catania G, Chikina M, Shlomchik MJ. 2016. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity. 44: 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, and Shlomchik MJ. 2014. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunology. 15: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weisel F and Shlomchik M. 2017. Memory B cells of mice and humans. Annu Rev Immunol. 35: 255–84. [DOI] [PubMed] [Google Scholar]
- 38.Tomayko MM, Steinel NC, Anderson SM, and Shlomchik MJ. 2010. Cutting edge: hierarchy of maturity of murine memory B cell subsets. J. Immunol 181: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 30: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shrestha S, Yang K, Wei J. Karmaus PWF, Neale G, and Chi H. 2014. Tsc1 promotes the differentiation of memory CD8+ cells via orchestrating the transcriptional and metabolic programs. PNAS. 111: 14858–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, Roth E, Mielenz D, Berek C, Côrte-Real J, Jäck H, and Schuh W. 2017. A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur J Immunol. 47: 1389–1392. [DOI] [PubMed] [Google Scholar]
- 42.Tamargo-Gómez I and Mariño G. 2018. AMPK: regulation of metabolic dynamics in the context of autophagy. Int J of Mol Sci. 19: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Codogno P, Mehrpour M, and Proikas-Cezanne T. 2012. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Molecular Cell Biology. 13: 7–12. [DOI] [PubMed] [Google Scholar]
- 44.Herzig S and Shaw RJ. 2017. AMPK: guardian of metabolism and mitochondrial homeostasis. Molecular Cell Biology. 19: 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Kundu M, Viollet B, and Guan K. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 13: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu W, Tian W, Hu Z, Chen G, L Huang W Li, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Zhang X, Li L, Zhang L, Sui S, Zhau B, and Feng D. 2014. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Reports. 15: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, Zhong W, Chen Y, Wu W, Lin C, Chen H, Hou X, Zhang L, Sui S, Zhao B, Hu Z, Li L, and Feng D. 2015. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Letters. 589: 1847–1854. [DOI] [PubMed] [Google Scholar]
- 48.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, and Yan Z. 2017. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. doi: 10.1038/s41467-01700520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu H, Foretz M, Xie Z, Zhang M, Zhu Z, Xing J, Leclerc J, Gaudry M, Viollet B, and Zou M. 2014. PRKAA1/AMPKα1 is required for autophagy-dependent mitochondrial clearance during erythrocyte maturation. Autophagy. 10: 1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsui C, Martinez-Martin N, Gaya M, Maldonado P, Llorian M, Legrave NM, Rossi M, MacRae JI, Cameron AJ, Parker PJ, Leitges M, Bruckerbauer A, and Batista FD. 2018. Protein kinase C-β dictates B cell fate by regulating mitochondrial remodeling, metabolic reprogramming, and heme biosynthesis. Immunity. 48: 1144–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sena LA, and Chandel NS. 2012. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 48: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jang K, Mano H, Aoki K, Hayashi T, Muto A, Nambu Y, Takahashi K, Itoh K, Taketani S, Nutt SL, Igarashi K, Shimizu A, and Sugai M. 2015. Mitochondrial function provides instructive signals for activation-induced B-cell fates. Nature Communications. doi: 10.1038/ncomms7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Egawa T and Bhattacharya D. 2019. Regulation of metabolic supply and demand during B cell activation and subsequent differentiation. Curr Opin Immunology. 57: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones DD, Wilmore JR, and Allman D. 2015. Cellular dynamics of memory B cell populations: IgM+ and IgG+ memory B cells persist indefinitely as quiescent cells. J Immunol. 195: 4753–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konjar Š and Veldhoen M. 2019. Dynamic metabolic state of tissue resident CD8 T cells. Front Immunol. doi: 10.3389/fimmu.2019.01683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, and Jones RG. 2011. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. 187: 4187–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manz RA, Thiel A, and Radbruch A. 1997. Lifetime of plasma cells in the bone marrow. Nature. 388: 133–134. [DOI] [PubMed] [Google Scholar]
- 58.Nutt SL, Hodgkin PD, Tarlinton DM, and Corcoran LM. 2015. The generation of antibody-secreting plasma cells. Nature Reviews Immunology. 15: 160–171. [DOI] [PubMed] [Google Scholar]
- 59.Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, and Allman D. 2016. mTOR has distinct functions in generation versus sustaining humoral immunity. J Clin Investigation. 126: 4250–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeng H and Chi H. 2017. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol. 46: 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murera D, Arbogast F, Arnold J, Bouis D, Muller S, and Gros F. 2018. CD4 T cell autophagy is integral to memory maintenance. Sci Rep. doi: 10.1038/s41598-018-23993-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pua HH, Guo J, Komatsu M, and He YW. 2009. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 182: 4046–4055. [DOI] [PubMed] [Google Scholar]
- 63.Sandoval H, Kodali S, and Wang J. 2018. Regulation of B cell fate, survival, and function by mitochondria and autophagy. Mitochondrion. 41: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nuñez G, Hockenbery D, McDonnell TJ, Sorensen CM, and Korsmeyer SJ. 1991. Bcl-2 maintains B cell memory. Nature. 353: 71–73. [DOI] [PubMed] [Google Scholar]
- 65.Clybouw C, Fischer S, Auffredou MT, Hugues P, Alexia C, Bouillet P, Raphael M, Leca G, Strausser A, Tarlinton DM, Vazquez A. 2011. Regulation of memory B-cell survival by the BH3-only protein Puma. Blood. 118: 4120–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Sullivan TE, Johnson LR, Kang HH, and Sun JC. 2015. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 43: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van der Windt GJ, Everts B, Chang C, Curtis JD, Freitas TC, Amiel E, Pearce EJ, and Pearce EL. 2012. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 36: 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tangye SG and Tarlinton DM. 2009. Memory B cells: effectors of long-lived immune responses. Eur J Immunol. 39: 2065–2075. [DOI] [PubMed] [Google Scholar]
- 69.Diaz A, Romero M, Vazquez T, Lechner S, Blomberg BB, Frasca D. 2017. Metformin improves in vivo and in vitro B cell function in individuals with obesity and Type-2 Diabetes. Vaccine. 35: 2694–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.