

Abstract
Amphetamine-based (Amph) drugs are metabolized in humans to their hydroxylamine (AmphNHOH) and nitroso (AmphNO) derivatives. The latter metabolites are known to bind to the Fe centers of cytochrome P450 and other heme enzymes to inhibit their activities. Although these AmphNHOH/AmphNO metabolites are present in vivo, their interactions with the blood protein hemoglobin (Hb) and the muscle protein (Mb) have been largely discounted due to a perception that the relatively small heme active sites of Hb and Mb will not be able to accommodate the large AmphNO group. We report the 2.15 Å resolution X-ray crystal structure of the AmphNO adduct of adult human hemoglobin as the Hb[α-FeIII(H2O)][β-FeII(AmphNO)] derivative. We show that the binding of AmphNO to the β subunit is enabled by an E helix movement and stabilization of ligand binding by H-bonding with the distal His63 residue. We also observe an AmphNHOH group in the Xe2 pocket in close proximity to the α heme site in this derivative. Additionally, UV-vis spectroscopy was used to characterize this and related wt and mutant Mb adducts. Importantly, our X-ray crystal structure of this Hb-nitrosoamphetamine complex represents the first crystal structure of a wild-type heme protein adduct of any amphetamine metabolite. Our results provide a framework for further studies of AmphNHOH/AmphNO interactions with Hb and Mb as viable processes that potentially contribute to the overall biological inorganic chemistry of amphetamine drugs.
Keywords: iron, amphetamine, X-ray structure, hemoglobin, myoglobin, nitroso
Graphical Abstract
1. Introduction
Amphetamine (Amph) and methamphetamine (“meth”) are Schedule II drugs that although have selected approved medical use, have a high potential for abuse [1]. For example, a methamphetamine drug, namely Desoxyn®, has limited use for the treatment of obesity and attention-deficit hyperactivity disorder (ADHD). The d-isomer of methamphetamine has a higher abuse potential due to its more potent physiological and behavioral effects [2]. Amphetamine drugs including Adderall®, have been prescribed for the treatment of ADHD and narcolepsy. The metabolism of methamphetamine and amphetamine in humans and other mammals has been studied extensively [2-4]. In humans, methamphetamine is metabolized by cytochrome P450 2D6 to generate Amph, N-hydroxymethamphetamine, and p-hydroxymethamphetamine [2]. Amph metabolism in mammals also generates its hydroxylamine derivative, namely N-hydroxyamphetamine (AmphNHOH; Fig. 1) [2, 3]. Curiously, the metabolism of Amph to AmphNHOH in humans has also been shown to be carried out by a flavin-containing monooxygenase FMO3 [5], complementing the more recognized cytochrome P450-mediated monooxygenation reaction.
Fig. 1.
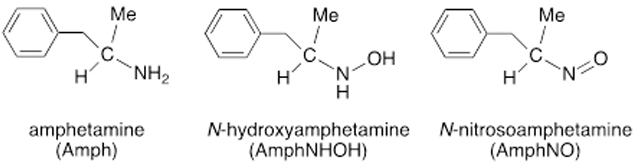
Amphetamine (Amph) and its oxidative products AmphNHOH and AmphNO.
The P450-mediated metabolism of amphetamine and its analogues to their monooxygenated N-hydroxylamines has been known for quite a long time [6-12]. Interestingly, the generated AmphNHOH and analogues interact with heme proteins to form ferrous nitroso FeII–N(O)Amph derivatives that inhibit the function of the proteins. In the case of cytochrome P450, the FeII–N(O)Amph derivatives are characterized by their 455 nm spectral signatures [8, 13-20]. The AmphNO metabolite (right of Fig. 1) also binds, as judged by UV-vis spectroscopy, to the heme sites of microperoxidase 8 [21, 22], prostaglandin H synthase [23], and the inducible isoform of nitric oxide synthase [24].
Curiously, although administration of Amph and methamphetamine are known to result in their metabolism to the AmphNHOH derivatives, the interactions of the latter with the blood protein hemoglobin (Hb) and muscle protein myoglobin (Mb) have not been pursued to any significant extent. We previously reported our preliminary UV-vis spectral investigations of these systems and described the X-ray crystal structure of the FeII–N(O)Amph derivative of a distal pocket Mb mutant in which the bulky ligand was oriented towards the exterior solvent region [25], perhaps avoiding steric clashes with the interior distal pocket residues. Interestingly, and in the absence of structural data, there has been some speculation that the active site distal pockets of Hb and Mb might be too small to accommodate the large AmphNO ligand [21-24]. Here, we adopt a structural biology approach to prepare and determine the 2.15 Å-resolution crystal structure of an AmphNO adduct of the human blood protein hemoglobin. This structure represents the first three-dimensional structure of any wild-type protein–AmphNO adduct, and only the second heme–AmphNO (protein or heme model) crystal structure to be reported to date.
2. Materials and Methods
The ferric human HbIII–H2O [26, 27] and ferric sperm whale (sw) MbIII–H2O (wild-type (wt), and mutant H64Q and H64V) [28] proteins used in this study were purified as reported previously [25]. The recombinant wt swMb plasmid was a kind gift from Dr. Mario Rivera. N-hydroxyamphetamine (AmphNHOH) was synthesized following a published method and its purity was verified by 1H NMR spectroscopy [29].
2.1. UV-vis spectroscopic studies
Unless otherwise stated, UV-vis spectroscopic monitoring of the reactions were carried out aerobically using quartz cuvettes (Starna) equipped with screw caps, and the measurements were recorded using a Hewlett-Packard 8453 spectrophotometer.
The Hb reaction with AmphNHOH was performed as reported previously [25]. To a sample of ferric HbIII–H2O (10 μL of a 30 mg/mL solution) in 2.5 mL of 0.1 M sodium phosphate buffer at pH 7.4, was added AmphNHOH (10 μL of a 200 mM solution), and the reaction was monitored spectroscopically. The comparative anaerobic reaction of ferrous deoxyHbII with AmphNHOH was similarly performed using degassed reagents (by bubbling with nitrogen gas for ~30 min prior to use) in an MBraun anaerobic glove box.
The analogous reactions of AmphNHOH with the ferric swMbIIIs were performed in a similar fashion as described above for the ferric HbIII–H2O reaction.
The oxidation of the heme-AmphNO products by ferricyanide back to their ferric forms was also probed spectroscopically. For example, the ferric MbIII–H2O (wt and mutant) proteins in 0.1 M sodium phosphate buffer at pH 7.4 were mixed with solid AmphNHOH until the ferrous MbII–AmphNO complexes formed. Aliquots (5 μL) of these product solutions were added to 2.5 mL of 0.1 M sodium phosphate buffer at pH 7.4. Finally, potassium ferricyanide (5 μL of a 100 mM solution) was added to the cuvette and the reoxidation reaction monitored by UV-vis spectroscopy.
2.2. Crystallization of the product from the ferric HbIII–H2O reaction with AmphNHOH
The co-crystallization method was utilized to obtain crystals of the product resulting from the reaction of ferric HbIII–H2O with AmphNHOH. A 2 mL solution of ferric HbIII–H2O (30 mg/mL in 50 mM sodium phosphate, pH 7.4) was mixed with 0.5 mL AmphNHOH (90 mM in methanol) and allowed to react for one week prior to setting up crystallization trays; due to the different protein:ligand ratio used for crystallography purposes, the completion of the reaction was verified by UV-vis spectroscopy. Crystals were grown using the batch method using conditions described by Safo and Abraham for ferrous Hbs [27]. Monoject blood collection tubes (10.25×64 mm, COVIDIEN) were used for the aerobic batch crystallizations [27], and they contained 3.2 M Na/K phosphate buffer at pH 6.47, 5 μL toluene, and varying ratios of buffer to protein reaction solutions. Crystals formed within a few days, but were harvested after ~1 week. Crystals were harvested and cryoprotected using well solution with the addition of 10% glycerol, and flash frozen in liquid nitrogen until needed for the diffraction experiment.
2.3. X-ray data collection and processing
Diffraction data was collected at the University of Oklahoma Macromolecular Crystallography Lab (MCL) using a Rigaku MicroMax 007HF microfocus X-ray generator coupled to a PILATUS 200K detector. The data was collected at 100 K with Cu Kα radiation (λ = 1.54178 Å) from the generator operated at 40 kV/30 mA. The X-ray diffraction data was processed using HKL3000 [30] and the resulting sca files were converted to mtz using Scalepack2mtz in the CCP4 program suite [31]. PHASER MR (CCP4) [32] was used to determine the initial phases. The model used for molecular replacement was ferric R-state human aquomethemoglobin (ferric HbIII–H2O) at 2.0 Å resolution (PDB ID: 3P5Q) with the heme, water molecules, and ligands removed from the structure. Refinement was performed using Refmac5 (CCP4) [33]. Models were rebuilt using COOT [34] and validated using MolProbity [35] to check for unusual residue conformations and contacts.
Ten initial cycles of restrained refinement were run with Refmac5, and the R factor decreased from 0.353 to 0.273. Ligands and water were added to the model based on the Fo–Fc electron density maps in the subsequent refinement cycles. Two AmphNO ligands (ligand id: 3QM), one AmphNHOH molecule (ligand id: K7M) and one glycerol molecule were added to the model. The model for AmphNHOH was built in Phenix using REEL [36]. The coordinates in a new pdb file and the topologies in a monomer library cif file were produced using eLBOW [37]. The C-terminal residues R141 in α1, 140-141 in α2 and 146 in β2, as well as the N-terminal residues 1 in β1, and 1–2 in β2, were omitted due to lack of electron density. The final R factor and Rfree were 0.216 and 0.287, respectively. Selected data collection and refinement statistics are collected in Table 1. The final structure was deposited in the PDB with accession number 7JJQ.
Table 1.
Data collection and refinement statistics
| Data collectiona | Refinement statistics | ||
|---|---|---|---|
| Space group | P41 | No. of protein atoms | 4335 |
| Cell dimensions (a, b, c (in Å); α, β, γ (in °) | 53.34, 53.34, 190.65 90.00, 90.00, 90.00 | R factorc | 0.216 |
| Resolution (Å) | 40.85-2.15 | Rfreed | 0.287 |
| I/σ[I] | 14.22 (4.60) | RMSD Bond length (Å) | 0.021 |
| No. of observed reflns | 37504 | RMSD Bond angles (°) | 1.530 |
| No. of unique reflns | 26737 (2436) | Overall Mean B Factor (Å3) | 27.05 |
| Multiplicity | 1.4 (1.0) | Ramachandran plot (%)e | |
| Completeness (%) | 93.0 (85.0) | most favored residues | 94.0 |
| Rmergeb | 0.043 (0.025) | outliers | 1.1 |
| CC1/2 | 0.982 |
Values in parentheses correspond to the highest resolution shells.
Rmerge = Σ∣I–<I>∣/Σ(I), where I is the individual intensity observation and <I> is the mean of all measurements of I.
R = Σ∥Fo∣–∣Fc∣∣/Σ∣Fo∣, where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree was calculated by using 5% of the randomly selected diffraction data which were excluded from the refinement.
As calculated using MolProbity.
The figures were generated using Pymol [38]. 2Fo–Fc electron density maps were calculated by Fast Fourier Transform (FFT; CCP4) [39]. Fo–Fc electron density maps were generated by first deleting all ligands from the final pdb file of the structure. This pdb file was used to create a new Fc mtz file using SFall (CCP4). The Fc mtz file was used to generate the Fo–Fc electron density map using FFT. The resulting FFT map files were converted to ccp4 files and displayed in Pymol.
2.4. UV-vis of the Hb-AmphNO crystal
In order to characterize the redox state of the crystal used for X-ray diffraction data collection and to determine if X-ray exposure altered the redox state, the UV-vis spectrum of this crystal was obtained after data collection. After diffraction data collection was complete, the crystal was removed from the goniometer and placed into a drop of 2–5 μL of 0.1 M sodium phosphate buffer at pH 7.4. Once the crystal was fully dissolved, the aerobic UV-vis spectrum of the solution was obtained using a Synergy HTX (Biotek) instrument.
3. Results and Discussion
3.1. UV-vis spectroscopic monitoring of the reactions of AmphNHOH with the swMb and Hb proteins
UV-vis spectroscopy was used to monitor the reaction between ferric wt swMbIII–H2O and AmphNHOH. As noted previously for this reaction [25], the Soret band of the ferric wt MbIII–H2O at λmax 409 nm shifted slowly to 423 nm over a 24 h period, and new peaks in the Q band region at 543 and 576 nm also appeared. These shifts are indicative of, and consistent with, the formation of a ferrous nitrosoalkane MbII–RNO (R = alkyl) derivative [40]. Once formed, this ferrous wt swMb–nitrosoalkane derivative is fairly stable in aerobic solution, showing only mild decomposition over a one week period as judged by UV-vis spectroscopy [25].
Two distal pocket mutants, namely H64Q (with distal H-bonding capacity) and H64V (without distal H-bonding capacity), were also examined for this reaction. Addition of excess AmphNHOH to solutions of the ferric H64Q and H64V swMbIII mutants in phosphate buffer at pH 7.4 resulted in similar spectral changes in their UV-vis spectra (Figs. 2B and 2C). For the H64Q MbIII–H2O mutant, the Soret band shifted from 409 nm to 424 nm and the peak in the Q band region at 503 nm was replaced with two peaks at 543 nm and 571 nm (Fig. 2B). Similarly, for the ferric H64V MbIII mutant, the Soret band shifted from 395 nm to 425 nm, and the peak in the Q band region at 504 nm was replaced with two peaks at 541 nm and 572 nm appeared (Fig. 2C). The reaction was essentially complete after ~30 min for the H64Q mutant, and after ~60 min for the H64V mutant. These shifts are similarly indicative of the formation of ferrous MbII–RNO derivatives [41, 42], and solutions of the products were likewise quite stable in air for days at ambient temperature, suggesting strong interactions of the AmphNO ligands with the heme centers that were not disrupted by dioxygen displacement of the ligands.
Fig. 2.
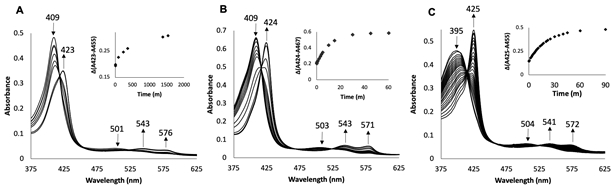
UV-vis spectral changes during the reaction of AmphNHOH with ferric (A) wt swMb and the (B) H64Q and (C) H64V Mb mutants. A plot of ΔAbs(423–455) against time for wt (A), ΔAbs(424-467) against time for the H64Q mutant (B) and ΔAbs(425–455) against time for the H64V mutant (C). Conditions: 0.1 M phosphate buffer at pH 7.4, [Mb] = 2 μM, final [AmphNHOH] = 80 μM.
The mechanism of formation of heme-N(O)R (R = alkyl/aryl) products from the reactions of the ferric precursors and organohydroxylamines has been described previously; organo-RNHOH reduces the ferric center to the ferrous state [43] presumably via initial coordination of the organo-RNHOH followed by a sequence of proton and electron transfers [21-23, 44]. We note that an alternate process in the cytochrome P450 case has been reported, in which the NADPH-enabled reduction of the ferric center occurs [45].
To further verify the oxidation state of the metal centers in our wt and mutant Mb–AmphNO products, the compounds were treated with the oxidant potassium ferricyanide. In each case, the spectra of the Mb–AmphNO products were replaced by the original peaks of their respective ferric MbIII–H2O precursors (Fig. 3). These results are consistent with our formulation of the Mb–AmphNO products as ferrous derivatives.
Fig. 3.
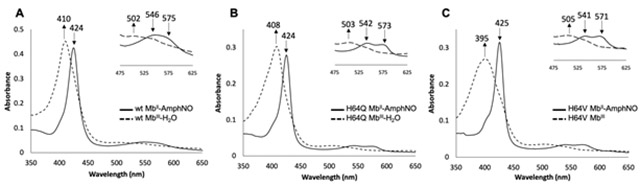
UV-vis spectroscopy of the reactions of ferrous (A) wt MbII–AmphNO, (B) H64Q MbII–AmphNO, and (C) H64V MbII–AmphNO with ferricyanide. Conditions: 100 mM sodium phosphate at pH 7.4, [Mb–AmphNO] = 2.4 μM, [ferricyanide] = 200 μM
The time it took for the products to revert to their ferric precursors varied. For wt Mb, the reaction was complete in ~40 min, whereas these reactions were much quicker for the H64Q mutant (10 min) and for the H64V mutant (5 min).
In the Hb case, the reaction of excess AmphNHOH with ferric HbIII–H2O in phosphate buffer at pH 7.4 results in a spectral shift of the Soret band from 406 nm to 421 nm, with formation of new bands at 541 and 559 nm (Fig. 4) [25]. Once formed, the product is fairly stable in aerobic solution, showing no observable decomposition for the Hb derivative after ~3 d at ambient temperature as judged by UV-vis spectroscopy.
Fig. 4.
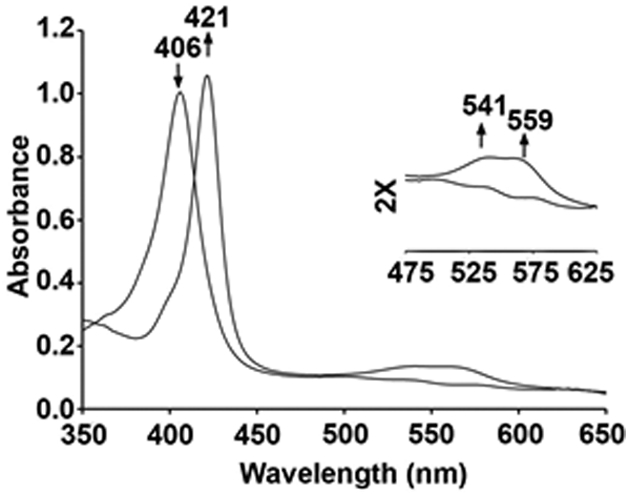
UV-vis spectral changes during the reaction of ferric HbIII–H2O with AmphNHOH. Conditions: 0.1 M phosphate buffer at pH 7.4, [Hb] = 12.5 μM, final [AmphNHOH] = 100 μM.
3.2. Molecular structure of the Hb–nitrosoamphetamine adduct
There have been long-standing discussions regarding whether bulky ligands can be accommodated within the “relatively small” active sites of Hb and Mb. Our UV-vis spectroscopy results indicate that Fe–N(O)R derivatives are formed, but the spectral data do not provide information on exactly how these AmphNO ligands are oriented within the heme protein active sites. We previously reported the structure of the H64A swMbII–AmphNO compound, but this structure showed that the organic “Amph” fragment was oriented away from the interior of the distal pocket and towards the solvent exterior [25], thus essentially avoiding steric clashes within the distal pocket. Consequently, we devoted considerable effort to crystallization attempts of these products in order to determine their crystal structures, especially for the wt proteins that retained their distal His ligands. We were, after several crystallization attempts, able to obtain suitable crystals of the product from the reaction of AmphNHOH with hemoglobin, but not of the wt or mutant MbII derivatives reported here. We describe the 2.15 Å resolution X-ray crystal structure of the Hb derivative below.
3.2.1. The overall quaternary structure
The overall structure of the tetrameric α2β2 Hb product is shown in Figure 5A. In order to determine the quaternary state of this product, a comparison of the β2 FG corner and α1 C interface of representative structures of known quaternary states was made (Fig. 5B) [46]. Based on the comparisons of this signature “switch” region [46], we conclude that the structure of the product from the Hb/AmphNHOH reaction (green in Fig. 5) matches that of an R-state Hb (magenta in Fig. 5B).
Fig. 5.

(A) The overall tetrameric structure. (B) Comparison of the α1β2 interface of representative Hb structures [Green: this work. Cyan: T-state deoxyHb (PDB id 1B86; RMSD = 2.500 Å), Magenta: R-state Hb(CO) (PDB id 1AJ9; RMSD = 0.396 Å), Yellow: R2-state Hb(CO) (PDB id 1BBB; RMSD = 1.611 Å), Light pink: R3-state Hb(CO) (PDB id 1YZI; RMSD = 0.580 Å)]
3.2.2. The heme ligands
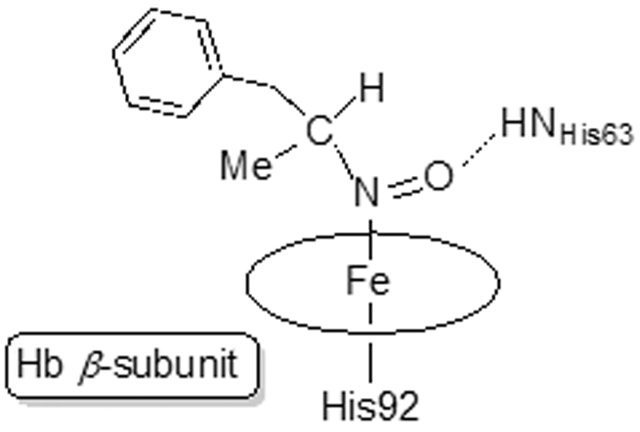
Our crystallization conditions yielded a product in which only half of the Hb heme sites, namely the hemes of the β subunits, were bound to AmphNO. Figure 6 shows both the α and β heme sites. A water molecule bound directly to heme Fe was modeled at full occupancy into the α site based on the electron density map (Fig. 6A). The electron density in the β subunit and the resulting Fo–Fc map matched that of a modeled AmphNO (or AmphNHOH) bound directly to Fe; we modeled the ligand as the N-bound nitroso AmphNO derivative (Fig. 6B; rather than AmphNHOH) based on the spectral similarity with other Fe–nitrosoalkane derivatives of Hb and Mb [40-42]. The d-isomer of AmphNO best matched the electron density above the heme. What was somewhat striking was that the hydrophobic portion of the bulky AmphNO ligand was unexpectedly oriented towards the interior of the active site (see later), with the phenyl plane of the AmphNO ligand roughly perpendicular (~74°) to the heme plane. A hydrogen bond was evident between the nitroso O-atom of the AmphNO and the distal His63 residue (Fig. 6B), with the nitroso N–O bond nearly eclipsing an Fe–N(heme) bond (with a N(por)–Fe–N–O torsion angle of ~2°). As we note in the Introduction, nitrosoalkane binding to the Fe centers of heme proteins are known to inhibit the functions of these proteins [8, 13-24].
Fig. 6.

Final model of the active site and Fo-Fc omit maps of (A) the α1 subunit (contoured at 3 σ) and (B) the β1 subunit (contoured at 2 σ).
To verify the Fe oxidation states extant in the crystal obtained from the Hb/AmphNHOH reaction, we retrieved the crystal used in the X-ray diffraction data collection and obtained its UV-vis spectrum (after dissolving the used crystal in buffer). The resulting UV-vis spectrum is shown in Fig. 7, and displays two peaks in the Soret region at λ 405 and 421 nm. The former band corresponds to that of authentic ferric metHbIII–H2O, and the latter to the ferrous HbII–AmphNO product. This UV-vis spectral result was consistent with our X-ray crystallography results. We thus formulate the identity of the crystalline product we obtained from the Hb/AmphNHOH reaction as the Hb[α-FeIII(H2O)][β-FeII(AmphNO)] derivative. We do not discount the possibility that the lack of AmphNO in the α subunit was due to the partial decomposition of the fully occupied HbII–AmphNO (obtained in solution) under our crystallization conditions.
Fig. 7.
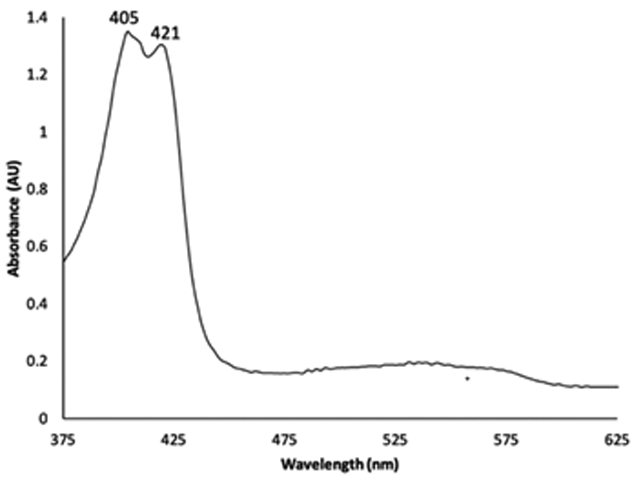
UV-vis spectroscopy of the dissolved crystal of the product after X-ray diffraction data collection.
3.2.3. E-helix movement allows for AmphNO binding
To better understand how Hb was in fact able to accommodate the bulky AmphNO ligand, the Hb[α-FeIII(H2O)][β-FeII(AmphNO)] structure was aligned with that of ferric HbIII-H2O (PDB id: 3P5Q) as shown in Fig. 8. The overall RMSD of the two structures is 0.283 Å. Not surprisingly, significant differences are observed between the structures in the β subunits, more specifically in the E helix (Fig. 8A) and EF corner regions, but not in the α subunits. Some notable subtle differences are observed, however, within the active sites; the largest difference in the β active site, other than ligand identity, lies in the position of the Val67 residue which was displaced by 3.5 Å (Fig. 8B). The Val67 residue is part of the E helix and is one of several residues to undergo positional changes upon AmphNO binding to Fe (Fig. 8A). This is coincident with the observation that the E helix partially unravels, specifically between residues 65–76, inducing large shifts (~1.5 and up to 8.0 Å in Cα; e.g., Ala70 and Asp73), thus allowing for the accommodation of the phenyl moiety of AmphNO which would otherwise clash with the Val67 residue.
Fig. 8.

Overlay of the (A) β1 E helix and (B) the β1 active sites of Hb[α-FeIII(H2O)][β-FeII(AmphNO)] (green, this work) and aquometHb (cyan, PDB id: 3P5Q).
3.2.4. The E helix movement determines the orientational preference for the AmphNO ligand in Hb[α-FeIII(H2O)][β-FeII(AmphNO)]
The crystal structure the Hb[α-FeIII(H2O)][β-FeII(AmphNO)] product is only the second such structure of any heme–AmphNO derivative. In our previously published crystal structure of H64A Mb–AmphNO [25], the bulky "Amph" moiety was oriented towards the exterior of the protein, whereas in Hb[α-FeIII(H2O)][β-FeII(AmphNO)] it was oriented towards the interior of the protein. Given that the previously reported H64A Mb–AmphNO structure does not contain the distal H64 residue (in the E helix), we were interested in determining if the presence of the His64 residue and/or this E helix movement was responsible for the orientational preferences in these derivatives.
The Cα alignment of the β subunit of Hb[α-FeIII(H2O)][β-FeII(AmphNO)] with both ferrous H64A swMbII–AmphNO (RMSD = 1.368 Å, PDB id: 5KD1) and ferric wt swMbIII–H2O (RMSD = 1.183 Å, PDB id: 2MBW) allowed us to further analyze the binding of AmphNO to Fe at the active sites of these heme proteins (Fig. 9). A close look at the E helix region (Fig. 9A) reveals that partial unraveling only occurred in the β1 subunit of the Hb structure, but not in the swMbII–AmphNO structure. This lack of E helix movement in the swMbII–AmphNO structure enables the distal Val68 (analogous to Val67 in Hb) to remain in its original location (e.g., as with swMbIII–H2O) to create a steric hindrance that disfavors an AmphNO orientation towards the protein interior. In contrast, the Val67 residue in the Hb structure is shifted ~2.6 Å (Cα) away from its original position to create space for accommodating a bulky heme ligand. Further, the presence of the distal H-bonding residue His63 in the Hb structure (analogous to His64 in swMb) allows for H-bonding stabilization of the bound AmphNO ligand (Fig. 7B), a feature that is coincident with the Val67 (and E helix) movement that allows an orientational preference of the AmphNO ligand towards the hydrophobic interior of the protein.
Fig. 9.

Overlay of the (A) E helix region, and (B) heme active sites of ferric wt swMbIII-H2O (magenta, PDB id: 2MBW), ferrous H64A swMbII–AmphNO (cyan, PDB id: 5KD1) and the β1 subunit of Hb[α-FeIII(H2O)][β-FeII(AmphNO)] (green, this work). Residue identifiers are labeled black for Hb and grey for Mb.
Given that similar heme–AmphNO products are formed with the wt, H64Q and H64V Mbs (e.g., the UV-vis spectral data in Fig. 2), we speculate that due to the presence of H-bonding residues in wt and H64Q, that similar orientations of AmphNO towards the protein interior occurs. Conversely, in the H64V derivative, which more closely resembles the previously reported H64A Mb–AmphNO structure, the AmphNO ligand is oriented towards the protein exterior. Crystallization attempts are ongoing for other heme–AmphNO derivatives to further probe this speculation.
3.2.5. An AmphNHOH molecule in the Xe2 pocket of the α2 subunit
The presence of internal cavities within globular proteins such as Mb and Hb have been identified by various methods including photocrystallography, time-resolved spectroscopy, and in silico molecular dynamics [47-52]. Crystal structures of these proteins soaked in Xe allows a direct detection of such internal cavities. For example, four such Xe sites (labeled Xe1–Xe4) have been identified in swMb [48, 50], and twelve such sites have been identified in tetrameric T-state human deoxyHbII [47]. Such “Xe pockets” have been implic ated in transient ligand binding, and are frequently associated with modulation of ligand migration and kinetics.
In our Hb[α-FeIII(H2O)][β-FeII(AmphNO)] crystal structure, we determined the presence of new electron density in the Xe2 sites of the α subunits as shown in Fig. 10. The electron density in the α1 subunit was best modeled as a glycerol molecule (Fig. 10A-C; glycerol from the crystal cryoprotectant). The glycerol interacts with a nearby fixed water and both the sidechain and mainchain of the Trp14 residue (Fig. 10C).
Fig. 10.

The crystal structure of the α1 subunit (A-C) and α2 subunit (D-F) from the Hb[α-FeIII(H2O)][β-FeII(AmphNO)] derivative. (A) The α1 chain displaying glycerol in the Xe2 pocket, (B) the Fo-Fc omit map (contoured at 2σ) and (C) the nearest interactions in the α1 Xe2 pocket. (D) The α2 chain displaying AmphNHOH in the Xe2 pocket, (E) the Fo-Fc omit map (contoured at 2σ) and (F) the nearest interactions in the α2 Xe2 pocket.
In the α2 subunit (Fig. 10D-F), the electron density was modeled as an AmphNHOH molecule at this site. We note that our UV-vis studies on the interaction of Hb with AmphNHOH imply that the AmphNO ligand binds to both the α and β subunits, however only binding to the β subunit was observed in our crystal structure. Consequently, an alternate formulation of this molecule could well be the oxime derivative PhCH2C(Me)=NOH that would result from dissociation of the AmphNO ligand and its tautomerization to the oxime (e.g., nitromethane CH3NO with an α-H atom next to the-NO group readily tautomerizes to formaldoxime CH2=NOH [53]). However, the sum of angles around the α-C in the AmphNHOH/AmphNO molecule in the Xe2 was ~330° (i.e., not planar), disfavoring the oxime formulation. The AmphNO formulation is also reasonable, given that this Xe2 site occupation is present only in the α subunit where the expected AmphNO ligand was not observed. In any event, this molecule in the α2 Xe2 site is stabilized in this pocket through H-bonding with a nearby water which further H-bonds with the backbone C=O of the Gly18 and Trp14 residues (Fig. 10F). To the best of our knowledge, this structure represents the first demonstration of bulky ligand occupation of a Xe site in Hb.
4. Conclusions
AmphNHOH and AmphNO are metabolites that form in humans and other mammals upon administration of amphetamine and methamphetamine based drugs. However, their interactions with the blood protein hemoglobin and the muscle protein myoglobin have been largely ignored due, in large part, to a perception that the active site distal pockets in these proteins are not large enough to accommodate the bulky AmphNO heme ligand. We have successfully demonstrated, using X-ray crystallography, that the bulky AmphNO ligand is readily accommodated in the Hb active site, and that E-helix movement and distal H-bonding of the nitroso –NO group with the His63 residue helps direct the Fe–N(O)Amph orientation. Our results serve as an entry point into more detailed studies of AmphNHOH/AmphNO interactions with Hb and Mb as viable processes that potentially contribute to their harmful effects in humans.
Synopsis:
A nitrosoamphetamine (AmphNO) adduct of human adult hemoglobin (Hb), namely Hb[α-FeIII(H2O)][β-FeII(AmphNO)], displaying direct binding of an amphetamine metabolite to the β heme Fe center, has been characterized by X-ray crystallography to 2.15 Å resolution. This represents the first structural determination of an amphetamine derivative bound to a wild-type protein.
Acknowledgements
This material is based upon work supported by (while GBR-A was serving at) the National Science Foundation (NSF; Grants CHE-1900181 and CHE-1566509). Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF. We thank Dr. Erwin G. Abucayon for assistance with the synthesis of the AmphNHOH reagent. This paper reports data obtained in the University of Oklahoma Macromolecular Crystallography Laboratory which is supported, in part, by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103640, and by a Major Research Instrumentation award from the National Science Foundation under award number 092269.
References:
- [1].U.S.Department of Justice, A DEA Resource Guide, 2015. Website: http://www.dea.gov/druginfo/factsheets.shtml, pp. 9, 46, 50-51. [Google Scholar]
- [2].Li L, Everhart T, Jacob III P, Jones R, Mendelson J, Stereoselectivity in the Human Metabolism of Metamphetamine, Br. J. Clin. Pharmacol, 69 (2010) 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Florence VM, Di Stefano EW, Sum CY, Cho AK, The Metabolism of (R)-(−)-Amphetamine by Rabbit Liver Microsomes: Initial Products, Drug Metab. Disp, 10 (1982) 312–315. [PubMed] [Google Scholar]
- [4].Reynolds GP, Elsworth JD, Blau K, Sandler M, Lees AJ, Stern GM, Deprenyl is Metabolized to Methamphetamine and Amphetamine in Man, Br. J. Clin. Pharmacol, 6 (1978) 542–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cashman JR, Xiong YN, Xu L, Janowsky A, N-Oxygenation of Amphetamine and Methamphetamine by the Human Flavin-Containing Monooxygenase (Form 3): Role in Bioactivation and Detoxification, J. Pharmacol. Exp. Ther, 288 (1999) 1251–1260. [PubMed] [Google Scholar]
- [6].Beckett AH, Belanger PM, Metabolic Incorporation of Oxygen into Primary and Secondary Aliphatic-Amines and Consequences in Carbon-Nitrogen Bond-Cleavage, J. Pharm. Pharmacol, 27 (1975) 547–552. [DOI] [PubMed] [Google Scholar]
- [7].Wright J, Cho AK, Gal J, The Role of N-Hydroxylamphetamine in the Metabolic Deamination of Amphetamine, Life Sci., 20 (1977) 467–474. [DOI] [PubMed] [Google Scholar]
- [8].Bensoussan C, Delaforge M, Mansuy D, Particular Ability of Cytochromes P450 3A to Form Inhibitory P450-Iron-Metabolite Complexes Upon Metabolic Oxidation of Aminodrugs, Biochem. Pharmacol, 49 (1995) 591–602. [DOI] [PubMed] [Google Scholar]
- [9].Fukuto JM, Di Stefano EW, Burstyn JN, Valentine JS, Cho AK, Mechanism of Oxidation of N-Hydroxyphentermine by Superoxide, Biochemistry, 24 (1985) 4161–4167. [DOI] [PubMed] [Google Scholar]
- [10].Lin LY, Di Stefano EW, Schitz DA, Hsu L, Ellis SW, Lennard MS, Tucker GT, Cho AK, Oxidation of Methamphetamine and Methylenedioxymethamphetamine by CYP2D6, Drug Metab. Disp, 25 (1997) 1059–1064. [PubMed] [Google Scholar]
- [11].Burstyn JN, Iskandar M, Brady JF, Fukuto JM, Cho AK, Comparative Studies of N-Hydroxylation and N-Demethylation by Cytochrome P-450, Chem. Res. Toxicol, 4 (1991) 70–76. [DOI] [PubMed] [Google Scholar]
- [12].Hirata M, Lindeke B, Orrenius S, Cytochrome P450 Product Complexes and Glutathione Consumption Produced in Isolated Hepatocytes by Norbenzphetamine and Its N-Oxidized Congeners, Biochem. Pharmacol, 28 (1979) 479–484. [DOI] [PubMed] [Google Scholar]
- [13].Mansuy D, Rouer E, Bacot C, Gans P, Chottard JC, Leroux JP, Interaction of Aliphatic N-Hydroxylamines with Microsomal Cytochrome P450: Nature of The Different Derived Complexes and Inhibitory Effects on Monooxygenases Activities, Biochem. Pharmacol, 27 (1978) 1229–1237. [DOI] [PubMed] [Google Scholar]
- [14].Mansuy D, Beaune P, Chottard JC, Bartoli JF, Gans P, The Nature of the “455 nm Absorbing Complex” Formed During the Cytochrome P450 Dependent Oxidative Metabolism of Amphetamine, Biochem. Pharmacol, 25 (1976) 609–612. [DOI] [PubMed] [Google Scholar]
- [15].Jonsson J, Lindeke B, On the Formation of Cytochrome P-450 Product Complexes During the Metabolism of Phenylalkylamines, Acta Pharm. Suec, 13 (1976) 313–320. [PubMed] [Google Scholar]
- [16].Franklin MR, The Formation of a 455 nm Complex During Cytochrome P-450-Dependent N-Hydroxyamphetamine Metabolism, Mol. Pharmacol, 10 (1974) 975–985. [Google Scholar]
- [17].Franklin MR, Complexes of Metabolites of Amphetamines with Hepatic Cytochrome P-450, Xenobiotica, 4 (1974) 133–142. Chem. Abs. 182:118715h. [Google Scholar]
- [18].James RC, Franklin MR, Comparisons of the Formation of Cytochrome P-450 Complexes Absorbing at 455 nm In Rabbit and Rat Microsomes, Biochem. Pharmacol, 24 (1975) 835–838. [DOI] [PubMed] [Google Scholar]
- [19].Lindeke B, Paulsen U, von Bahr C, Groth C-G, Cytochrome P-455 Complex Formation in the Metabolism of Phenylalkylamines. V. Complex Formation in Human Liver Microsomes from Various Sources, Biochem. Pharmacol, 30 (1981) 388–390. [DOI] [PubMed] [Google Scholar]
- [20].Mansuy D, Gans P, Chottard J-C, Bartoli J-F, Nitrosoalkanes as Fe(II) Ligands in the 455-nm-Absorbing Cytochrome P-450 Complexes Formed from Nitroalkanes in Reducing Conditions, Eur. J. Biochem, 76 (1977) 607–615. [DOI] [PubMed] [Google Scholar]
- [21].Ricoux R, Ludowska E, Pezzotti F, Mahy J-P, New Activities of a Catalytic Antibody with a Peroxidase Activity: Formation of Fe(II)-RNO Complexes and Stereoselective Oxidation of Sulfides, Eur. J. Biochem, 271 (2004) 1277–1283. [DOI] [PubMed] [Google Scholar]
- [22].Ricoux R, Boucher J-L, Mansuy D, Mahy J-P, Formation of Iron(II) Nitrosoalkane Complexes: A New Activity of Microperoxidase 8, Biochem. Biophys. Res. Commun, 278 (2000) 217–223. [DOI] [PubMed] [Google Scholar]
- [23].Mahy JP, Mansuy D, Formation of Prostaglandin Synthase-Iron-Nitrosoalkane Inhibitory Complexes upon in Situ Oxidation of N-Substituted Hydroxylamines, Biochemistry, 30 (1991) 4165–4172. [DOI] [PubMed] [Google Scholar]
- [24].Renodon A, Boucher J-L, Wu C, Gachhui R, Sari M-A, Mansuy D, Stuehr D, Formation of Nitric Oxide Synthase-Iron(II) Nitrosoalkane Complexes: Severe Restriction of Access to the Iron(II) Site in the Presence of Tetrahydrobiopterin, Biochemistry, 37 (1998) 6367–6374. [DOI] [PubMed] [Google Scholar]
- [25].Wang B, Powell SM, Guan Y, Xu N, Thomas LM, Richter-Addo GB, Nitrosoamphetamine binding to Myoglobin and Hemoglobin: Crystal Structure of the H64A Myoglobin-Nitrosoamphetamine Adduct, Nitric Oxide, 67 (2017) 26–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Antonini E, Brunori M, Hemoglobin and Myoglobin In Their Reactions With Ligands, North-Holland, Amsterdam, 1971. [Google Scholar]
- [27].Safo MK, Abraham DJ, X-ray Crystallography of Hemoglobins, Methods Mol. Med, 82 (2003) 1–19. [DOI] [PubMed] [Google Scholar]
- [28].Springer BA, Sligar SG, High-Level Expression of Sperm Whale Myoglobin in Escherichia coli, Proc. Natl. Acad. Sci. USA, 84 (1987) 8961–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mourad MS, Varma RS, Kabalka GW, Reduction of α,β-Unsaturated Nitro-Compounds with Boron Hydrides: A New Route to N-Substituted Hydroxylamines, J. Org. Chem, 50 (1985) 133–135. [Google Scholar]
- [30].Otwinowski Z, Minor W, Processing of X-Ray Diffraction Data Collected in Oscillation Mode, Methods Enzymol., 276 (1997) 307–326. [DOI] [PubMed] [Google Scholar]
- [31].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Overview of the CCP4 Suite and Current Developments, Acta Crystallogr., D67 (2011) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, PHASER Crystallographic Software, J. Appl. Cryst, 40 (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Murshudov GN, Vagin AA, Dodson EJ, Refinement of Macromolecular Structures by the Maximum-Likelihood Method, Acta Cryst., D53 (1997) 240–255. [DOI] [PubMed] [Google Scholar]
- [34].Emsley P, Cowtan K, COOT: Model-Building Tools for Molecular Graphics, Acta Cryst., D60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [35].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: All-atom Structure Validation for Macromolecular Crystallography, Acta Cryst., D66 (2010) 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Moriaty NW, Draizen EJ, Adams PD, An Editor for the Generation and Customization of Geometry Restraints, Acta Cryst., D73 (2017) 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Moriaty NW, Grosse-Kunstleve RW, Adams PD, Electronic Ligand Builder and Optimization Workbench (eLBOW): A Tool for Ligand Coordinate and Restraint Generation, Acta Cryst., D65 (2009) 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schrödinger-LLC, The PYMOL Molecular Graphics System New York, NY, 2018. [Google Scholar]
- [39].Read RJ, Schierbeek AJ, A Phased Translation Function, J. Appl. Crystallogr, 21 (1988) 490–495. [Google Scholar]
- [40].Mansuy D, Chottard JC, Chottard G, Nitrosoalkanes as Fe(II) Ligands in the Hemoglobin and Myoglobin Complexes Formed from Nitroalkanes in Reducing Conditions, Eur. J. Biochem, 76 (1977) 617–623. [DOI] [PubMed] [Google Scholar]
- [41].Copeland DM, West AH, Richter-Addo GB, Crystal Structures of Ferrous Horse Heart Myoglobin Complexed with Nitric Oxide and Nitrosoethane, Proteins: Struct., Func., Genet, 53 (2003) 182–192. [DOI] [PubMed] [Google Scholar]
- [42].Yi J, Ye G, Thomas LM, Richter-Addo GB, Degradation of Human Hemoglobin by Organic C-Nitroso Compounds, Chem. Commun, 49 (2013) 11179–11181. [DOI] [PubMed] [Google Scholar]
- [43].Mulvey D, Waters WA, A Mechanistic Study of the Decomposition of Phenylhydroxylamine to Azoxybenzene and Aniline and its Catalysis by Iron(II) and Iron(III) Ions Stabilized by Ethylenediaminetratraacetic Acid, J. Chem. Soc., Perkin Trans 2, (1977) 1868–1876. [Google Scholar]
- [44].Mansuy D, Battioni P, Chottard J-C, Riche C, Chiaroni A, Nitrosoalkane Complexes of Iron-Porphyrins: Analogy between the Bonding Properties of Nitrosoalkanes and Dioxygen, J. Am. Chem. Soc, 105 (1983) 455–463. [Google Scholar]
- [45].Fukuto JM, Brady JF, Burstyn JN, VanAtta RB, Valentine JS, Cho AK, Direct Formation of Complexes between Cytochrome P-450 and Nitrosoarenes, Biochemistry, 25 (1986) 2714–2719. [DOI] [PubMed] [Google Scholar]
- [46].Safo MK, Ahmed MH, Ghatge MS, Boyiri T, Hemoglobin-Ligand Binding: Understanding Hb Function and Allostery on Atomic Level, Biochim. Biophys. Acta, 1814 (2011) 797–809. [DOI] [PubMed] [Google Scholar]
- [47].Savino C, Miele AE, Draghi F, Johnson KA, Sciara G, Brunori M, Vallone B, Pattern of Cavities in Globins: The Case of Human Hemoglobin, Biopolymers, 91 (2009) 1097–1107. [DOI] [PubMed] [Google Scholar]
- [48].Brunori M, Bourgeois D, Vallone B, The Structural Dynamics of Myoglobin, J. Struct. Biol, 147 (2004) 223–234. [DOI] [PubMed] [Google Scholar]
- [49].Tetreau C, Blouquit Y, Novikov E, Quiniou E, Lavalette D, Competition with Xenon Elicits Ligand Migration and Escape Pathways in Myoglobin, Biophys J, 86 (2004) 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tilton RF, Kuntz ID, Petsko GA, Cavities in Proteins: Structure of a Metmyoglobin-Xenon Complex Solved to 1.9 Å, Biochemistry, 23 (1984) 2849–2857. [DOI] [PubMed] [Google Scholar]
- [51].Knapp JE, Pahl R, Cohen J, Nichols JC, Schulten K, Gibson QH, Srajer V, Royer WE, Ligand Migration and Cavities within Scapharca Dimeric Hbl: Studies by Time-Resolved Crystallography, Xe Binding, and Computational Analysis, Structure, 17 (2009) 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mouawad L, Maréchal J-D, Perehia D, Internal Cavities and Ligand Passageways in Human Hemoglobin Characterized by Molecular Dynamics Simulations, Biochim. Biophys. Acta, 1724 (2005) 385–393. [DOI] [PubMed] [Google Scholar]
- [53].Gowenlock BG, Batt L, The Isomerization of Nitrosomethane to Formaldoxime, J. Mol. Struct. (THEOCHEM), 454 (1998) 103–104. [Google Scholar]