

Abstract
Background
Recent studies suggested that crosstalk between ERα and EGFR/HER2 pathways plays a critical role in mediating endocrine therapy resistance. Several inhibitors targeting EGFR/HER2 signaling, including FDA-approved lapatinib and gefitinib as well as a novel dual tyrosine kinase inhibitor (TKI) sapitinib, showed greater therapeutic efficacies. However, how 3D chromatin landscape responds to the inhibition of EGFR/HER2 pathway remains to be elucidated.
Methods
In this study, we conducted in situ Hi-C and RNA-seq in two ERα+ breast cancer cell systems, 1) parental MCF7 cells and its associated tamoxifen-resistant MCF7TR cells; and 2) parental T47D cells and its associated tamoxifen-resistant T47DTR cells, before and after the treatment of sapitinib.
Results
We identified differential responses in topologically associated domains (TADs), looping genes and expressed genes. Interestingly, we found that many differential TADs and looping genes are reversible after sapitinib treatment, indicating that EGFR/HER2 signaling may play a role in reshaping and rewiring the high order genome organization. We further examined and recapitulated the reversible looping genes in 3D spheroids of breast cancer cells, demonstrating that 3D cell culture spheroid of breast cancer cells could be a potential preclinical breast cancer model for studying 3D chromatin regulation.
Conclusions
Our study has provided significant insights into our understanding of 3D genomic landscape changes in response to EGFR/HER2 Inhibition in endocrine-resistant breast cancer cells. Our data provides a rich resource for further evaluating chromatin structural responses to EGFR/HER2 targeted therapies in endocrine-resistant breast cancer cells. Our analyses suggest that these alterations of chromatin structures and transcriptional programs may provide new avenues for intervention or designing of patient selection for targeted endocrine treatment.
Keywords: Altered domains, reversible gene looping, EGFR/HER2 inhibitor, tamoxifen resistant breast cancer
Introduction
The regulatory mechanisms in breast cancer cells are complex, as reflected by differential responses to adjuvant endocrine therapy including relapse. ~50% of ERα-positive (ERα+) breast cancer patients treated with anti-estrogens such as tamoxifen (Tam) subsequently exhibit intrinsic or develop acquired resistance to this hormonal therapy [1,2]. Many studies including ours have demonstrated that estrogen (E2)-induced transcriptional program is rewired or altered in Tam-resistant breast cancer cells [3–8]. In a recent study, we used the Hi-C technique to investigate dynamic changes of three-dimensional (3D) chromatin architectures in Tam-sensitive (TS) and Tam-resistant (TR) breast cancer cells and found that E2-induced highly dynamic domains in TS cells are predominantly associated with active open chromatin, enhanced ERα but decreased CTCF binding, and significantly altered in TR cells [9]. We further revealed that ERα-bound promoter-enhancer looping genes enclosed within altered domains are enriched with genes with functions and pathways associated with cancer aggressiveness, glycolysis and metabolism, and focal adhesion. Our results suggested that these ERα-associated, dynamically reorganized active domains in regulating gene looping events may result in higher susceptibility to alterations in TR cells. This prompted us to speculate that these genome domains and looping genes may be responsible for driving acquired tamoxifen resistance.
Although molecular mechanisms underlying acquired tamoxifen resistance are not fully understood, several mechanisms have been proposed. For instance, loss of ERα expression and ERα mutations may lead to the transcriptional repression of the ERα gene and overpopulation of ERα-cells from heterogeneous ERα+ tumors [10–12]; differential metabolic activation of tamoxifen may affect the therapeutic outcomes [13,14]; the presence of ERα-cancer stem cells has the potential to differentiate to luminal cancer cells and thus to seed relapses and metastasis [15–17]; and gene amplification or alteration of certain transcription factors or regulators could also contribute to endocrine resistance by altering the ERα-dependent transcriptome [18–22]. Recent studies suggested that other signaling pathways such as EGFR/HER2, IGF-IR, and AKT/PTEN could play critical roles in mediating endocrine therapy resistance through modulating ERα activity [23–27]. Our previous study has further demonstrated that targeting EGFR/HER2 signaling pathway by a dual tyrosine kinase inhibitor (TKI), sapitinib (Sap), significantly inhibits TR cell growth in vitro and delays TR tumor growth in vivo. It also showed a greater inhibitory efficacy than other FDA approved anti-EGFR drugs such as lapatinib and gefitinib, indicating a therapeutic potential of this inhibitor [28]. However, how high order chromatin landscape responds to inhibition of EGFR/HER2 pathways in endocrine-resistant breast cancer cells remains to be elucidated.
In this study, we set out to conduct genome-wide 3D genomic and transcriptomic profiling of differential responses to EGFR/HER2 inhibition in tamoxifen-sensitive vs -resistant breast cancer cells. We performed in situ Hi-C and RNA-seq in two ERα+ breast cancer cell systems, 1) parental MCF7 cells and its associated tamoxifen-resistant MCF7TR cells; and 2) parental T47D cells and its associated tamoxifen-resistant T47DTR cells, before and after the treatment of AZD8931 (Sap), a dual TKI of EGFR/HER2. We identified differential responses of topologically associating domains (TADs), looping genes as well as expressed genes in TR cells upon the treatment of Sap. We further examined selected differential looping genes in 3D cell culture spheroids. Our data provides a rich resource for further evaluating chromatin structural responses to anti-EGFR/HER2 therapies in endocrine-resistant breast cancer.
Results
Identification of differential topologically associating domains
We have conducted in situ Hi-C in two ERα+ breast cancer cell systems, MCF7 and MCF7TR as well as T47D and T47DTR, before and after Sap treatment, each with biological replicates (Supplementary Table S1). We first evaluated the reproducibility of two biological replicates (Supplementary Figure S1) and then used TopDom [29] to identify TADs for the combined replicates for each condition and found a majority of sizes of TADs are less than 2Mb whose distribution of the number of different sizes of TADs is very similar among all six conditions (Supplementary Figure S2). We then compared the changes of TADs between MCF7 and MCF7TR, T47D and T47DTR, MCF7TR and MCF7TR+Sap, and T47DTR and T47DTR+Sap, respectively. We were able to define eight types of TAD changes: a) No-change; b) Conserve-expand; c) Conserve-shrink; d) Shift; e) Split; f) Fuse; g) Neo (from a border boundary or GAP to a new TAD); h) Del (from a TAD to GAP or border boundary), where types a-c are relatively conserved (RC) and types d-h are drastically changed (DC) (Figure 1A,B and Supplementary Figure S3). Interestingly, we observed that the percentages of the numbers of RC and DC TADs are very similar in two cell systems for all of the comparisons, 61% RC vs 39% DC between MCF7 and MCF7TR, 70% RC vs 30% DC between MCF7TR and MCF7TR+Sap, 58% RC vs 42% DC between T47D and T47DTR, and 66% RC vs 34% DC between T47DTR and T47DTR+Sap, respectively. When closely examining the four types of DC TADs, we found that a significant number of TAD changes were reversible upon Sap treatment. In other words, 32% or 23% of Neo-, Del-, Split- or Fuse-TADs in MCF7TR vs MCF7, or T47DTR vs T47D respectively, were reversed to the original TADs, i.e., Del-, Neo-, Fuse- or Split-TADs in MCF7 or T47D, upon Sap treatment of TR cells, respectively (Figure 1C, Supplementary Files S1–4). Interestingly, the number of reversible DC TADs is quite similar between MCF7 and T47D cell lines, except for the Neo-Del switch, which is more in MCF7 cell system than in T47D cell system. We further compared the reversible DC TADs between the two cell systems and found that there are only 2% common reversible DC TADs, suggesting that chromatin structural responses to EGFR/HER2 inhibition may be in a cell-type-specific manner (Figure 1D). Screenshots of two examples of reversible DC TADs in MCF7 cell system, Del-Neo and Split-Fuse, are shown in Figure 1E.
Figure. 1.
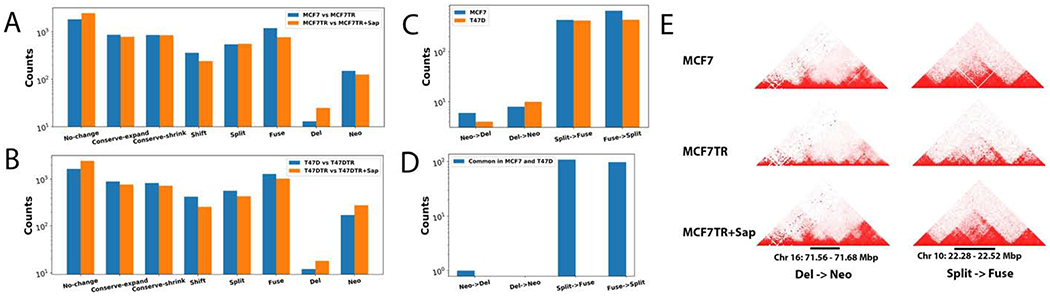
Identifying differential TADs by in situ Hi-C in MCF7, MCF7TR, T47D, T47DTR cells before and after Sap treatment. A. A histogram showing the number of differential TADs in each of eight types of TAD changes between MCF7 and MCF7TR cells, and between MCF7TR and MCF7TR+Sap conditions, respectively. B. A histogram showing the number of differential TADs in each of eight types of TAD changes between T47D and T47DTR cells, and between T47DTR and T47DTR+Sap conditions, respectively. C. A histogram showing the number of reversible DC TADs in MCF7 and T47D cell lines, respectively. D. A histogram showing the common reversible DC TADs between MCF7 and T47D cell lines. E. Screenshots of two examples of reversible DC TADs, Del->Neo and Split->Fuse, in MCF7 cell line.
Identification of differential significant Hi-C interactions
To link Hi-C interactions to looping genes, we first used HOMER [30] to identify significant interacting fragments (SIFs) from Hi-C data and then associated each end of SIFs to either a promoter (P) or a distal region (D). We thus obtained a total number of 153,875, 154,223, 291,737, 128,960, 112,981, and 201,895 SIFs for MCF7, MCF7TR, MCF7TR+Sap, T47D, T47DTR, and T47DTR+Sap, respectively (Figure 2A,B). Consequently, we found that a significant number of SIFs could be assigned to a promoter-distal (PD) looping gene either as P1D1 (if both P and D are associated with the same gene), or P1D2 (if associated with two different genes) (Figure 2A,B), where SIFs were annotated based on the definition used in our previous studies [9,31]. We further compared the SIFs and P1D1/2 looping genes between MCF7 and MCF7TR, MCF7TR and MCF7TR+Sap, T47D and T47DTR, T47DTR and T47DTR+Sap, and identified a significant number of differential SIFs and P1D1/2 looping genes (DLGs) such as gained loops (Gain) in MCF7TR/T47DTR, MCF7TR+Sap/T47DTR+Sap, or lost loops (Loss) in MCF7/T47D, MCF7TR/T47DTR (Figure 2C,D, Upper panel). Our results showed that there were much more Gain than Loss DLGs in both MCF7 and T47D cell systems. Interestingly, we found that many P1D1 DLGs were reversible such that genes with gained loops in TR cells were lost again or genes with lost loops were regained after Sap treatment (Figure 2C,D, Lower panel), indicating that EGFR/HER2 signaling may play a role in reshaping and rewiring the high order genome organization. Surprisingly, we identified very few common reversible looping genes (Gain then Loss or Loss then Gain) between two breast cancer cell systems (Figure 2E), suggesting looping-mediated response to the inhibition of EGFR/HER2 signaling might be tightly dependent on the expression levels of EGFR and HER2 genes in TS and TR cells.
Figure 2.
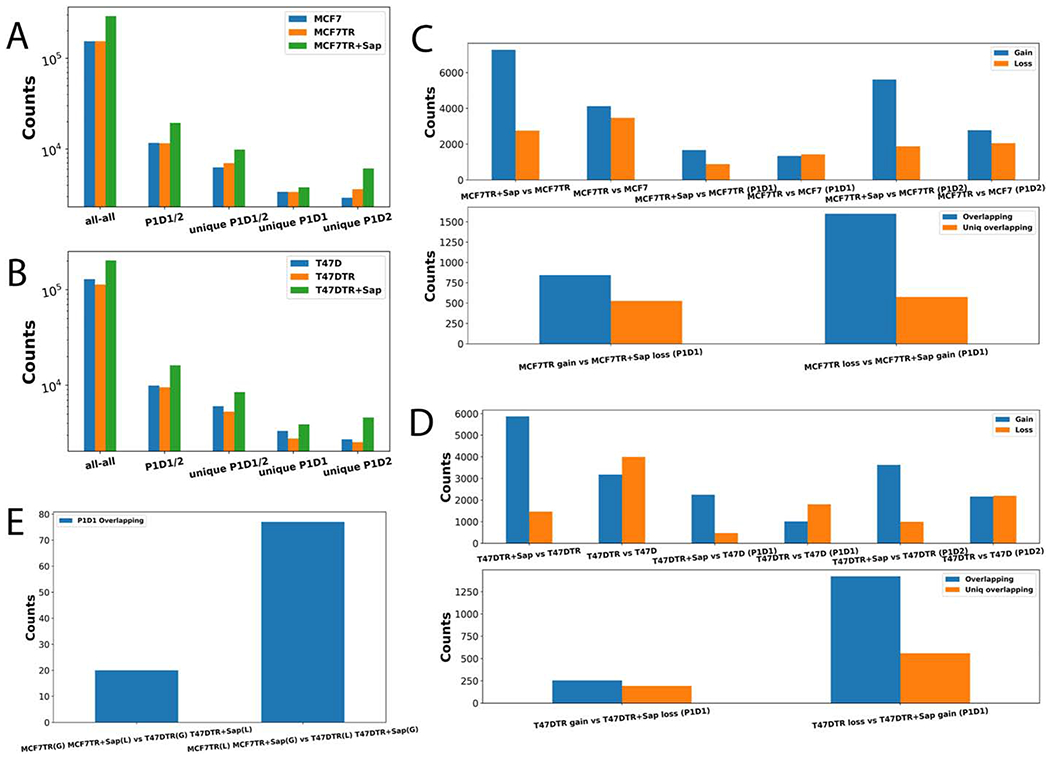
Identifying differential significant interacting fragments (DSIFs). A. A histogram showing all-all SIFs, P1D1/2 loops and unique P1D1/2 loops in MCF7, MCF7TR, and MCF7TR+Sap conditions, respectively. B. A histogram showing all-all SIFs, P1D1/2 loops and unique P1D1/2 loops in T47D, T47DTR, and T47DTR+Sap conditions, respectively. C. Upper: A histogram showing differential SIFs and differential P1D1/2 looping genes (DLGs) between MCF7 vs MCF7TR, and between MCF7TR vs MCF7TR+Sap, respectively; Lower: an overlapping P1D1 DLGs for Gain in MCF7TR then Loss in MCF7TR+Sap, and an overlapping P1D1 DLGs for Loss in MCF7TR then Gain in MCF7TR+Sap. D. Upper: A histogram showing differential SIFs and differential P1D1/2 looping genes (DLGs) between T47D and T47DTR, and between T47DTR and T47TR+Sap. Lower: an overlapping P1D1 DLGs for Gain in T47DTR then Loss in T47DTR+Sap, and an overlapping P1D1 DLGs for Loss in T47DTR then Gain in T47DTR+Sap. E. The number of common rDLGs between MCF7 and T47D cell systems.
Pathway enrichment and gene ontology analyses on reversible differential looping genes.
We used DAVID [32] to perform pathway enrichment and gene ontology (GO) analyses on four sets of significant reversible P1D1 DLGs (rDLGs) with a Fisher’s exact test. Sets A and B include 576 genes with Loss in MCF7 vs MCF7TR then Gain in MCF7TR vs MCF7TR+Sap, and 527 genes with Gain in MCF7 vs MCF7TR then Loss in MCF7TR vs MCF7TR+Sap at P < 3e-308 (Supplementary File S5). Sets C and D include 560 genes with Loss in T47D vs T47DTR then Gain in T47DTR vs T47DTR+Sap, and 192 genes with Gain in T47D vs T47DTR then Loss in T47DTR vs T47DTR+Sap at P < 9e-185 (Supplementary File S6). We identified common and distinct top ten enriched biological pathways and molecular functions among four sets of rDLGs (p-values <= 0.05 and Fold enrichments >= 1) (Figure 3). For example, metabolic processes were enriched in both Sets A and C, indicating metabolic alterations in TR breast cancer cells may be tightly associated with EGFR/HER2 signaling pathway that is positively regulated through looping events. The calcium signaling pathway was only enriched in MCF7 cell system, suggesting that looping-mediated calcium signaling alterations may be dependent more on HER2-expression than EGFR-expression, since HER2 is expressed at low level in parental T47D cells [27]. VEGF signaling pathway and regulation of actin cytoskeleton were enriched in Set D, suggesting that these two altered pathways responded positively to inhibition of EGFR/HER2 pathways through the reversible gene looping since both parental T47D and T47DTR are sensitive to both EGFR and HER2 expression.
Figure 3.
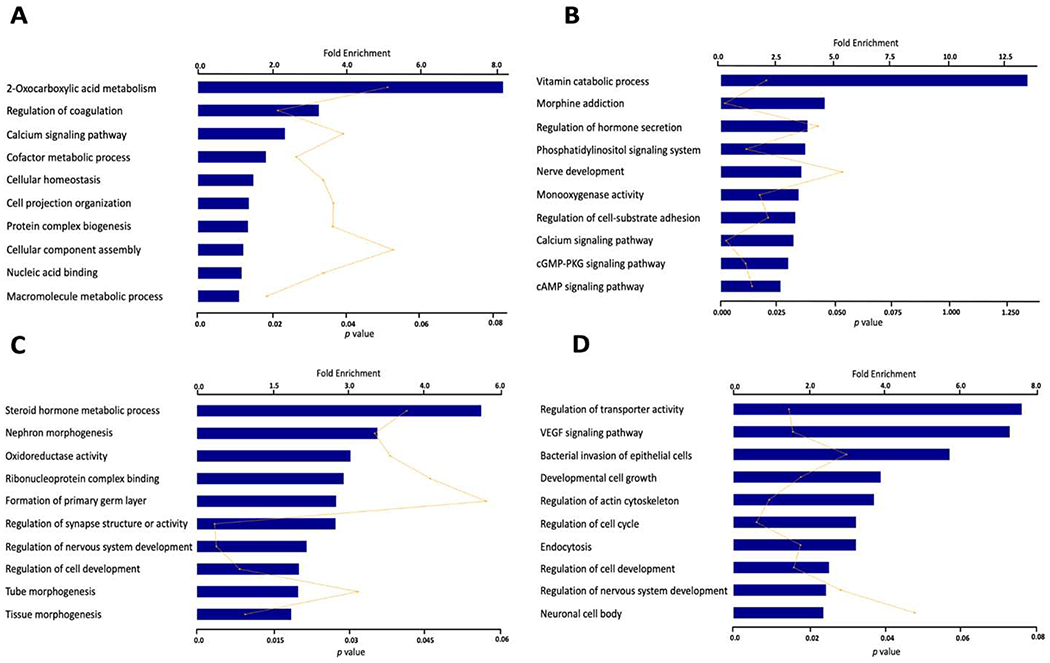
The top enriched biological pathways and molecular functions associated with four sets of reversible differentially looping genes (rDLGs). A. 576 looping changing genes which lost loops in MCF7 vs MCF7TR but gained loops in MCF7TR vs MCF7TR+Sap. B. 527 looping changing genes which gained loops in MCF7 vs MCF7TR but lost loops in MCF7TR vs MCF7TR+Sap. C. 560 looping changing genes which lost loops in T47D vs T47DTR but gained loops in T47DTR vs T47DTR+Sap. D. 192 looping changing genes which gained loops in T47D vs T47DTR but lost loops in T47DTR vs T47DTR+Sap.
Identification of differentially expressed looping genes
To examine gene expression levels for those DLGs, we conducted RNA-seq in MCF7 and T47D cells as well as MCF7TR and T47DTR cells, before and after Sap treatment, each with three biological replicates (Supplementary Table S2). We used DESeq2 [33] to identify differential expressed genes (DEGs) with p-adj <= 0.05 and Fold Change (FC) >= 1.5 or <=−1.5 between MCF7 and MCF7TR, between T47D and T47DTR, between MCF7TR and MCF7TR+Sap, and between T47DTR and T47DTR+Sap, respectively (Figure 4A,B, Supplementary Files S7–10). Interestingly, we found that a significant number of up- (Up) or down-regulated (Down) genes in both TR cells were reversed after Sap treatment (Figure 4C,D). We also identified a small proportion of up/down-regulated genes that showed even higher/lower expressed after Sap treatment respectively, suggesting that this group of genes may escape EGFR/HER2 signaling in response to other crosstalk pathways. When correlating reversible DEGs (rDEGs) with rDLGs, we identified only 46 reversible differentially expressed looping genes (rDELGs) between 821 rDEGs and 1,103 rDLGs in MCF7 cell system, including 11 genes Loss & Down in MCF7 vs MCF7TR then Gain & Up in MCF7TR vs MCF7TR+Sap, 14 genes Loss & Up in MCF7 vs MCF7TR then Gain & Down in MCF7TR vs MCF7TR+Sap, 5 genes Gain & Down in MCF7 vs MCF7TR then Loss & Up in MCF7TR vs MCF7TR+Sap, and 16 genes Gain & Up in MCF7 vs MCF7TR then Loss & Down in MCF7TR vs MCF7TR+Sap (Figure 4E). We also identified 48 rDELGs between 1,678 rDEGs and 752 rDLGs in T47D cell system, including 11 genes Loss & Down in T47D vs T47DTR then Gain & Up in T47DTR vs T47DTR+Sap, 22 genes Loss & Up in T47D vs T47DTR then Gain & Down in T47DTR vs T47DTR+Sap, 3 genes Gain & Down in T47D vs T47DTR then Loss & Up in T47DTR vs T47DTR+Sap, and 12 genes Gain & Up in T47D vs T47DTR then Loss & Down in T47DTR vs T47DTR+Sap (Figure 4F).
Figure 4.
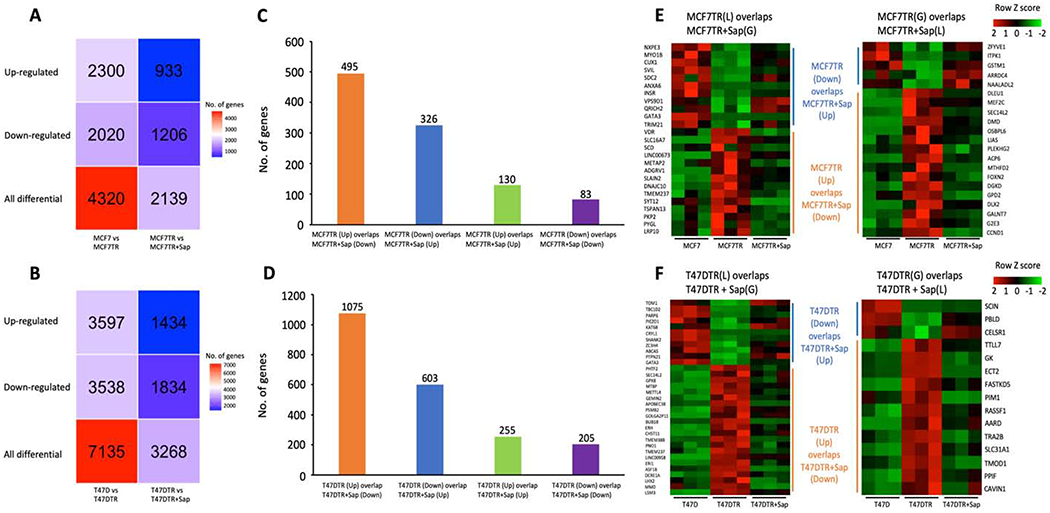
Differentially expressed genes (DEGs) and their relationship with differentially looping genes. A. The heatmap of DEGs in MCF7 vs MCF7TR and MCF7TR vs MCF7TR+Sap. B. The heatmap of DEGs in T47D vs T47DTR and T47DTR vs T47DTR+Sap. C. The bar graph of common DEGs that: 1). Up in MCF7 vs MCF7TR then Down in MCF7TR vs MCF7TR+Sap, 2). Down in MCF7 vs MCF7TR then Up in MCF7TR vs MCF7TR+Sap, 3). Up in MCF7 vs MCF7TR and Up in MCF7TR vs MCF7TR+Sap, and 4). Down in MCF7 vs MCF7TR and Down in MCF7TR vs MCF7TR+Sap. D. The bar graph of common DEGs that: 1). Up in T47D vs T47DTR then Down in T47DTR vs T47DTR+Sap, 2). Down in T47D vs T47DTR then Up in T47DTR vs T47DTR+Sap, 3). Up in T47D vs T47DTR and Up in T47DTR vs T47DTR+Sap, and 4). Down in T47D vs T47DTR and Down in T47DTR vs T47DTR+Sap. E. The heatmap of gene expression for 46 rDELGs in MCF7 cell system. F. The heatmap of gene expression for 48 rDELGs in T47D cell system.
Examination of differentially expressed looping genes in 3D cell culture spheroids
3D cell culture spheroid has lately been used to investigate how cancerous cells interact with other cells due to its higher degree of clinical and biological relevance. To examine if differential looping and expressed genes identified in 2D monolayer cells could be recapitulated in 3D spheroid of cells, we first grew 3D cell culture spheroids of MCF7 and MCF7TR with IHC evaluated ERα expression in spheroids (Figure 5A). We then tested the effect of Sap inhibition on spheroids growth by treating MCF7 and MCF7TR 3D spheroids with 10uM Sap for 6 days. As expected, although Sap inhibited the growth of both MCF7 and MCFTR spheroids, Sap was much more effective in inhibition of MCFTR spheroid growth (Figure 5B). We further performed 3C-qPCR on randomly selected two rDLGs and RT-qPCR on randomly selected three rDEGs. 3C-qPCR validations confirmed the differential looping intensity of four randomly selected pairs between MCF7 MCFTR and Sap treated spheroids. We observed significantly higher interactions of FOXN2 and SOLE in MCF7TR spheroids in comparison to MCF7, but lower interactions in MCF7TR spheroids upon Sap treatment (Figure 5C). We further confirmed the trend of the reversible expression of CDK14, FOXN2, and ARSJ between MCF7 vs MCF7TR spheroids and between MCF7TR and Sap-treated MCF7TR spheroids (Figure 5D). Despite of highly variability of spheroids, we were able to get a significant p-value of 0.01, 0.05 or 0.1 for all 3C-qPCR and RT-qPCR replicates between the comparisons. Taken together, our results demonstrate that 3D cell culture spheroid of breast cancer cells can not only recapitulate differential expressed looping genes identified in 2D cells but also could be a potential preclinical breast cancer model for studying 3D chromatin regulation.
Figure 5.
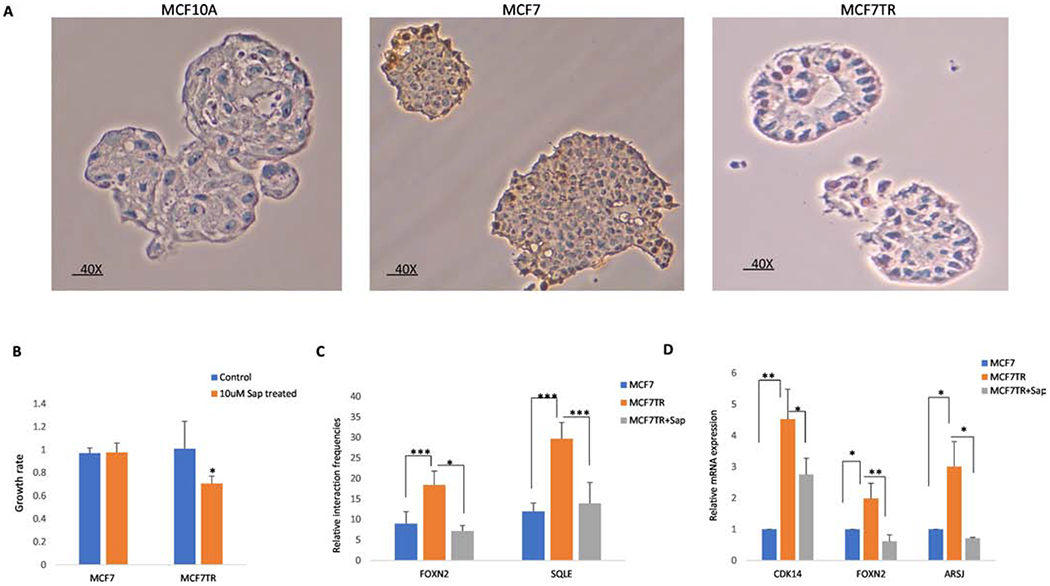
Validations of differentially expressed and looping genes in 3D cell culture spheroids. A. IHC staining of 3D spheroids of MCF7 and MCF7TR. Images were captured at a higher magnification (40X). B. Growth rate in 3D spheroids of MCF7 and MCF7TR after Sap treatment. *p <= 0.1 was considered statistically significant. C. Three biological replicates of 3C-qPCR were performed for each gene loop. One-tail paired t-test analysis was performed to compare the interaction frequencies among MCF7, MCF7TR and Sap treated spheroids. Error bars represent standard deviation with three experiments. ***p <= 0.01; **p <= 0.05; *p <= 0.1. D. RT-qPCR analysis in MCF7, MCF7TR, and Sap treated spheroids. **p <= 0.05; *p <= 0.1 was considered statistically significant.
Discussion
Studies have shown that crosstalk between ERα and EGFR/HER2 pathways functionally contributes to acquired endocrine-resistant breast cancer [25,34]. Several EGFR/HER2 signaling inhibitors showed greater inhibitory efficacies in tamoxifen-resistant settings than in a naive setting [28,35]. To explore the relationship between 3D chromatin structure and crosstalk of these two pathways, we have investigated 3D chromatin landscapes of differential responses to the perturbation of EGFR/HER2 signaling in two ERα+ tamoxifen resistant breast cancer cell lines. To the best of our knowledge, our study is the first to examine how 3D chromatin organization responds to drug inhibition of EGFR/HER2 signaling pathway. We identified thousands of differential TADs, differential looping genes and differential expressed genes between TS and TR cells, before and after Sap-treated TR cells. Furthermore, we found that many differential TADs and differential looping genes are reversible upon Sap treatment, indicating that EGFR/HER2 signaling may play a role in reshaping and rewiring the high order genome organization (Figures 1,2).
Interestingly, our pathway enrichment analyses reveal both common and distinct biological pathways or molecular functions among four groups of reversible differential looping genes. Although metabolic alterations in breast cancer and endocrine-resistance have been previously reported [13,14], our study further identifies a link between metabolic processes and the downregulation of EGFR/HER2 signaling that alters gene activities through dynamic, reversible loop formations associated with TR metabolic processes. Further functional or mechanistic experiments will be needed to substantiate our initial finding. For example, metabolism assays could be used to confirm if the levels of metabolites and cellular energy intakes are affected in TR cells treated with Sap. In addition, metabolism-associated cellular functions that confer phenotypical reprogramming upon inhibition to EGFR/HER2 pathway could be examined. Results from these approaches could potentially elucidate the role of metabolism and EGFR/HER2 crosstalk in re-sensitizing tamoxifen resistant cells. It has been shown that VEGF signaling pathway and regulation of actin cytoskeleton were altered in TR breast cancer cells [36,37]. Our study suggests that these two altered pathways are more specifically associated with resistant cells expressing higher level of both EGFR and HER2, and a reversible gene looping may be a possible mechanism for driving this crosstalk.
Remarkably, we were able to demonstrate that differential looping and expressed genes identified in 2D monolayer cells were recapitulated in 3D spheroids of breast cancer cells. This result is very significant due to spheroids’ bio-similarity to in vivo morphology, cell connectivity, polarity, and tissue architecture, and more physiologically relevant. However, we recognize several limitations in this validation. For example, we only tested a few selected genes and thus need to expand to a larger list of genes particularly associated with relevant biological signaling pathways. Using 3D organoid of patient tissues may be a better strategy for further translational studies including drug screening, cancer modeling, and toxicity testing, since it serves as a better in vitro model as compared to 2D or 3D co-culture systems. In addition, it is necessary to establish an in vivo model of TS and TR xenografts to re-examine the role of altered looping genes in this crosstalk [38,39].
In summary, our study has provided significant insights into our understanding of 3D genomic landscape changes in response to EGFR/HER2 Inhibition in endocrine-resistant breast cancer cells. The high quality and large 3D chromatin data will provide a rich resource for further evaluating chromatin structural responses to anti-EGFR/HER2 therapies in endocrine-resistant breast cancer. Our analyses suggest that these alterations of chromatin structures and transcriptional programs may provide new avenues for intervention or designing of patient selection for targeted endocrine treatment.
Material and Methods
Cell lines and reagents
Human breast carcinoma cell lines MCF7 and T47D and their tamoxifen resistant (TR) cell lines were originated from Osborne et al [40]. MCF7 or T47D cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin (pen/strep) until 90% confluent. MCF7TR or T47DTR cells were cultured in phenol red free RPMI-1640 containing 10% charcoal-stripped FBS, 1% pen/strep and 100nM Tamoxifen (Sigma-Aldrich). Tamoxifen was replaced every 48 hours. Cells were kept in a cell culture incubator with 37 °C and 5% CO2 until they reach 90% confluence. The cells were treated with Sapitinib (AZD8931) at the concentrations at 0uM to 30uM, the gradient concentration is 5uM. Cell absorbance was recorded at day 1 and day 7.
In situ Hi-C
In situ Hi-C experiments were performed as described previously [9,41]. The breast cancer cells were crosslinked with 1% formaldehyde and lysed with ice cold lysis buffer (10mM Tris-Hcl pH 8.0, 10mM NaCl, 0.2 % NP-40, 1mM DTT) to collect nuclei. The pelleted nuclei were digested with 200 units of HindIII (NEB, R3104L) at 37°C for overnight. The HindIII digested fragment overhangs were filled with biotin-labelled dATP (Life Technologies,19524-016) in a Klenow end-filling reaction. Four hundred units of T4 DNA Ligase (NEB, M0202) was added for ligation and samples were incubated for 4 h at room temperature with slow rotation. The ligation products were purified, and the chromatin was sheared to a size of 300-500bp using Covaris sonicator (Covaris Woburn, MA). Dynabeads MyOne Streptavidin T1 beads (Life technologies, 65601) were used to pull down the Biotin-labelled DNA. The end repair, dA tailing was performed and ligated with Illumina TruSeq adapters to form final Hi-C ligation products. Each Hi-C library was amplified with 12 cycles of PCR using Illumina primers. The Hi-C library was purified and then sequenced with lllumina HiSeq3000.
RNA-seq
Total RNA was extracted by ZYMO Research Quick-RNA MiniPrep kit from lysed 10 million of breast cancer cells in RNA Lysis Buffer, then removed most of gDNA with Spin-Away Filter. After that, the mixture of RNA was transferred with ethanol to Zymo-Spin IIICG column to remove trace DNA by DNase I on the column, then washed twice with RNA wash buffer followed by elution with 50 μl DNase/RNase-free water. RNA-seq library was prepared with NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEB #E7490). The Oligo dT Beads were washed with RNA binding buffer and incubated with total 1ug RNA to purify mRNA, followed with more washing by beads washing buffer. Then mRNA was eluted with elution buffer and reverse transcribed. After that, the first and the second strand cDNA were synthesized. After purification of Double-stranded DNA, adaptor was added. Adaptor-ligated DNA was enriched by PCR followed by purification, then the DNA library was sequenced with Illumina HiSeq3000.
3D spheroid formation
3D spheroids were generated as described in [42]. Spheroids formed by MCF7, MCF7TR were grown on Matrigel. Briefly, each well on a six-well plate was coated with 300ul of Matrigel (Corning) and the plate was kept in a 37 °C incubator for 30 min for gel formation. The Matrigel was overlaid with 2ml of cell culture medium containing 5 X 104 cells per well using MCF7 or MCF7TR cells. For Sap treatment of MCFTR cells, spheroids were treated with 100nM Sap to each well, and isolated after for 48 hours.
Cell growth assay
For Sap treatment, 96-well plate was precoated with 35 ul Matrigel and then placed in a 37 °C incubator to solidify. MCF7 and MCF7TR 2D cells cultured in the RPMI 1640 medium were mixed with 5% Matrigel (4,000 cells per well) and the cells were transferred to a precoated 96 well plate. Sap with different concentrations ranging from 0uM to 30uM was tested, the gradient concentration is 10uM. Growth of the 3D spheroids was quantified at day 1 and day 6 by BioTek™ ELx800™ Absorbance Microplate Reader. The assay was carried out with six replicates for each experiment. The experiments were repeated and analyzed three times separately.
Immunohistochemistry staining
Immunohistochemistry (IHC) staining was performed to detect ERα in MCF7 and MCFTR, with MCF10A as a negative control. The arrays were sectioned in 3 μm thickness and placed on a poly-lysine coated slides to dry. The sections were dewaxed by baking the slides at 60 °C for 30 min followed by two washes of xylene, 5 min each at room temperature. Antigen retrieval was performed for 24 min with 10Mm sodium citrate solution [pH 6.0], kept boiling in microwave and 30 min cooling to room temperature. Sections were subject to endogenous peroxidase activity blocking with 3% hydrogen peroxidase for 20 min and rinsed with PBS for 9 min, blocked with 10% normal goat serum for 1 h at room temperature, and then incubated with Estrogen receptor alpha (ERα) antibody for overnight in cold room. Spheroids were then washed with PBS three times and incubated with Goat anti-rabbit Poly-HRP for 30-45 minutes. Diaminobenzidine (DAB)-based detection was performed to detect antibody binding and slides were counterstained with hematoxylin. Appropriate controls were used for all conditions.
3C-qPCR
3C-qPCR was performed as previously described [43]. MCF7, MCF7TR, and Sap-treated MCF7TR spheroids were cross-linked with 37% formaldehyde for 10 min at room temperature. The reaction was quenched by the addition of 1M glycine for 5 min at room temperature. The spheroids were lysed with 500 μL of cold lysis buffer (10 Mm Tris–HCl Ph 8.0, 10 Mm NaCI, 0.2% Igepal CA630) with protease inhibitors for at least 15 min on ice. After lysis, the cell nuclei were pelleted and the chromatin was digested using 200 units of Hindlll (NEB) at 37 °C overnight and then the digestion was stopped at 65 °C for 20 min. Digested DNA fragments were ligated using T4 DNA ligase (NEB) for 4 h at 16 °C. Samples were reverse cross-linked with Proteinase K at 65 °C overnight. 3C samples were then purified using phenol–chloroform extraction. The 3C template was dissolved in 10mM Tris-HCI and DNA concentrations were measured using Nanodrop. For 3C several primers are designed for a restriction fragment of interest. All 3C primers were designed by “Primer 3”. Firstly, we designed 4 different primers for each gene to detect the specific interaction frequency between genes and the regulatory elements. All pairs of primers will amplify ligation products that are the result of head-to-head ligation of the corresponding restriction fragments. We isolated DNA and quantified using nanodrop in order to take equal concentration for all the groups. The primer mix was done for all the primers with the anchor sequence of specific gene. Primers used are listed in Supplementary Table S3 Interactions were measured using a 3C-qPCR assay for ligation products between each anchor Hindlll fragment and each target Hindlll fragment. Results are presented as relative interaction frequencies compared with those GAPDH as an internal control [43]. The frequency of ligation events was estimated by measuring the intensity of a PCR product after gel electrophoresis.
RT-qPCR
The spheroids were treated and harvested with Cell Recovery Solution (Corning Incorporated, Corning, NY, USA) to remove the Matrigel. Total RNA was extracted from MCF7, MCF7TR, Sap-treated MCF7TR spheroids using Quick -RNATM Mini Prep (Zymo Research, USA) according to the manufacturer’s instructions. Each 10 μL reaction consists of Superscript III RT/Platinum Taq mix, 5ul of 2X SYBR green reaction mix, 1 ul of test primer, 300ng RNA and distilled water. Quantitative Real-time PCR was performed on Light Cycler® 480 Instrument II real-time PCR system (Roche Diagnostics, Penzberg, Germany) and Ct values were outputted for quantification. Initial enzyme activation was performed at 95C for 15 min, followed by 70 cycles of denaturation at 95C for 15 s and primer annealing/extension at 60C for 70 s. Melting curve analysis was performed at 95C for 5 s, 65C for 60 s, and 45C for 30 s. Primers used are listed in Supplementary Table S4. The expression levels of target genes were normalized against endogenous control ACTB. Data analysis was done using 2-ΔΔCt method. Each PCR reaction was performed in triplicate, and the data presented were the average of three independent experiment results for all PCR reactions.
Hi-C interaction matrices and significant interactions
All raw reads were mapped to HG19 reference genome by using HiCUP [45] and the biological replicates were merged together. HOMER [30] was used to obtain Hi-C interaction matrices with default parameters and 40Kb tiling window size. The significant interactions or SIFs of each data set were called by HOMER in 40Kb resolution with default parameter (P<=0.001), except for MCF7TR and T47D (P<=0.05) since the numbers of uniquely mapped reads were relatively low. SIFs were further mapped to promoter (P, from +5Kb to −1Kb of 5’TSS) and distal regions (D, +/−500Kb to 10Kb relative to 5’TSS) based on UCSC RefSeq HG19 where the mapping definition was used in our previous studies [9,29]. In the case of a 40Kb bin mapped to multiple genes, all genes are considered. SIFs and P1D1/2 looping genes were compared by using an in-house python code to obtain DLGs
Topologically associating domains (TAD)
TopDom [29] was used to identify TAD, boundary, and gap for intra-chromosomal interactions. All interaction matrices (40Kb resolution) were normalized by Hi-Corrector [46] before using TopDom. TADs were compared by using the in-house Python program to obtain the differential TADs (or TAD changes) between MCF7/T47D vs MCF7/T47DTR, and between MCF7/T47DTR vs MCF7/T47DTR+Sap, respectively. Eight types of TAD changes were defined as the following: a) No-change: no changed TAD size and length between two conditions, A and B; b) Conserve-expand: a TAD identified in both conditions A and B, with the length of the TAD increasing by at most 300 Kb in condition B; c) Conserve-shrink: a TAD identified in both conditions A and B, with the length of the TAD decreasing by at most 300 Kb in condition B; d) Shift: a TAD identified in condition B that overlaps with a TAD in condition A, with the position shifting by more than 300 Kb; e) Split: a TAD in condition A becoming multiple TADs in condition B; f) Fuse: multiple TADs in condition A becoming one TAD in condition B; g) Neo: a border boundary or GAP in condition A becoming a TAD in condition B; h) Del: a TAD in condition A becoming a GAP or border boundary in condition B. Types a-c) are relatively conserved (RC) and types d-h) are drastically changed (DC). The definition of TAD changes was similar to our previous studies [9,31].
A more detailed procedure of classifying these eight types is described in the following five steps. 1) If a TAD position remains the same between the two conditions then it is defined as no-change TAD. 2) Then the program tries to find Fuse TAD in which multiple TADs from one condition are merged into the same TAD in the other condition. Here, the maximum allowed TAD position difference is 100Kb between the first condition and the second one. 3) Subsequently, the rest of TAD change types are detected, where the maximum allowed TAD position difference is 300Kb for conserve-Shrink and conserve-Expand, respectively. If the maximum TAD position difference is greater than 300Kb between the two conditions, then it is defined as Shift TAD. 4) If a TAD in condition one is split to multiple TADs in the second condition, then it is called Split TAD. Here the maximum allowed TAD position difference is 100Kb between a TAD in condition one and all split TADs in the second condition. 5) If a gap/boundary element in condition one becomes TAD in the second condition, then it is called Neo-TAD. On the other hand, if a TAD of condition one becomes gap/boundary element in the other condition, then it is called del-TAD.
In order to evaluate the statistical significance of each type of TAD change, we calculate an expected P-value by a random sampling method. For example, we randomly select a TAD to evaluate its TAD change between two conditions, and counts how many times a TAD change from a random drawing meets with a pre-defined TAD type. The random selection is repeated 10,000 times. In this way, an expected P-value of each of eight types of TAD changes is thus calculated and shown in the Supplementary Table S5.
Supplementary Material
Highlights.
We identified differential responses in topologically associated domains (TADs), looping genes and expressed genes.
We found that many differential TADs and looping genes are reversible after sapitinib treatment, indicating that EGFR/HER2 signaling may play a role in reshaping and rewiring the high order genome organization.
We further examined and recapitulated the reversible looping genes in 3D spheroids of breast cancer cells, demonstrating that 3D cell culture spheroid of breast cancer cells could be a potential preclinical breast cancer model for studying 3D chromatin regulation.
Acknowledgements
We thank UTHSA Next Generation Sequencing Facility for services rendered for production of the in situ Hi-C and RNA-seq data.
Funding
This project was partially supported by grants from NIH R01GM114142 (VXJ), U54CA217297 (VXJ), R01CA220578 (RL, YH) and CPRIT RP170126 (VXJ, YH, RL), P50CA186784 (SPORE) (R.S.), Breast Cancer Research Foundation (BCRF) 16-142, 17-143, 18-145 (R.S.). JW is supported by South-Eastern Norway Regional Health Authority (HSØ 2017061 and HSØ 2018107), and the usage of high performance computer resource is supported by the Norwegian Research Council NOTUR project (nn4605k).
Abbreviations
- ERα+
Estrogen receptor α-positive
- TS
Tam-sensitive
- TR
Tam-resistant
- 3D
Three-dimensional
- TKI
Dual tyrosine kinase inhibitor
- Sap
Sapitinib or AZD8931
- TADs
Topologically associating domains
- SIFs
Significant interacting fragments
- DLGs
Differentially P1D1/2 looping genes
- DEGs
Differentially expressed genes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and materials
Raw and processed in situ Hi-C for MCF7, MCF7TR+Sap, T47DTR+Sap cells and Raw and processed RNA-seq data for MCF7, MCF7TR, MCF7TR+Sap, T47D, T47DTR and T47DTR+Sap cells are deposited in GEO under accession number GSE144380 and GSE128676.
Ethics approval and consent to participate
Not applicable
Competing interests
Rachel Schiff reported research funding from AstraZeneca, GlaxoSmithKline, Gilead Sciences, and PUMA Biotechnology, and is a consulting/advisory committee member for Macrogenics and Eli Lilly outside the submitted work. The remaining authors declare no competing interests.
Additional information
Supplementary Information includes four tables and one figure and ten files can be found with this article online.
Author Statement
N/A
References
- 1.Brodie A, Sabnis G. Adaptive changes result in activation of alternate signaling pathways and acquisition of resistance to aromatase inhibitors. Clin Cancer Res. 2011. July 1; doi: 10.1158/1078-0432.CCR-10-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004. December; doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 3.Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, Salisbury JD, Cheng AS, Li L, Abbosh PH, Huang TH, Nephew KP. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006. December 15; doi: 10.1158/0008-5472.CAN-06-1666. [DOI] [PubMed] [Google Scholar]
- 4.Frasor J, Chang EC, Komm B, Lin CY, Vega VB, Liu ET, Miller LD, Smeds J, Bergh J, Katzenellenbogen BS. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 2006. July 15; doi: 10.1158/0008-5472.CAN-05-4269. [DOI] [PubMed] [Google Scholar]
- 5.Gu F, Hsu HK, Hsu PY, Wu J, Ma Y, Parvin J, Huang TH, Jin VX. Inference of hierarchical regulatory network of estrogen-dependent breast cancer through ChIP-based data. BMC Syst Biol. 2010. December 17; doi: 10.1186/1752-0509-4-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu PY, Hsu HK, Lan X, Juan L, Yan PS, Labanowska J, Heerema N, Hsiao TH, Chiu YC, Chen Y, Liu Y, et al. Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer. Cancer Cell. 2013. August 12; doi: 10.1016/j.ccr.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnani L, Stoeck A, Zhang X, Lánczky A, Mirabella AC, Wang TL, Gyorffy B, Lupien M. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc Natl Acad Sci U S A. 2013. April 16; doi: 10.1073/pnas.1219992110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Dily F, Vidal E, Cuartero Y, Quilez J, Nacht AS, Vicent GP, Carbonell-Caballero J, Sharma P, Villanueva-Cañas JL, Ferrari R, De Llobet LI, Verde G, Wright RHG, Beato M. Hormone-control regions mediate steroid receptor-dependent genome organization. Genome Res. 2019. January; 29(1):29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y, Gerrard DL, Wang J, Li T, Yang Y, Fritz AJ, Rajendran M, Fu X, Schiff R, Lin S, Frietze S, Jin VX. Temporal dynamic reorganization of 3D chromatin architecture in hormone-induced breast cancer and endocrine resistance. Nat Commun. 2019. April 3; doi: 10.1038/s41467-019-09320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Release of methyl CpG binding proteins and histone deacetylase 1 from the Estrogen receptor alpha (ER) promoter upon reactivation in ER-negative human breast cancer cells. Mol Endocrinol. 2005. July; doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Q, Atadja P, Davidson NE. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol Ther. 2007. January; doi: 10.4161/cbt.6.1.3549. [DOI] [PubMed] [Google Scholar]
- 12.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, Smith A, Wilson J, Morrow C, Wong WL, De Stanchina E, Carlson KE, Martin TS, Uddin S, Li Z, Fanning S, Katzenellenbogen JA, Greene G, Baselga J, Chandarlapaty S. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017. March;7(3):277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, Hayes DF, Desta Z, Flockhart DA. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst. 2003. December 3; doi: 10.1093/jnci/djg108. [DOI] [PubMed] [Google Scholar]
- 14.Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, Skaar T, Storniolo AM, Li L, Araba A, Blanchard R, Nguyen A,Ullmer L, Hayden J, Lemler S, Weinshilboum RM, Rae JM, Hayes DF, Flockhart DA. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005. January 5; doi: 10.1093/jnci/dji005. [DOI] [PubMed] [Google Scholar]
- 15.Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H. CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006; doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ouhtit A, Abd Elmageed ZY, Abdraboh ME, Lioe TF, Raj MH. In vivo evidence for the role of CD44s in promoting breast cancer metastasis to the liver. Am J Pathol. 2007. December; doi: 10.2353/ajpath.2007.070535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, Allan AL. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009. August; doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu J, Zhu Y, Liu Y, Sun L, Lv X, Wu Y, Hu P, Su F, Gong C, Song E, Liu B, Liu Q. E2F7 overexpression leads to tamoxifen resistance in breast cancer cells by competing with E2F1 at miR-15a/16 promoter. Oncotarget. 2015. October 13; doi: 10.18632/oncotarget.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, Shea M, Nardone A, De Angelis C, Heiser LM, Anur P, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci U S A. 2016. October 25; doi: 10.1073/pnas.1612835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeselsohn R, Cornwell M, Pun M, Buchwalter G, Nguyen M, Bango C, Huang Y, Kuang Y, Paweletz C, Fu X, Nardone A, De Angelis C, Detre S, Dodson A, Mohammed H, Carroll JS, Bowden M, Rao P, Long HW, Li F, Dowsett M, Schiff R, Brown M. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc Natl Acad Sci U S A. 2017. May 30; doi: 10.1073/pnas.1620993114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Zhang Z, Cenciarini ME, Proietti CJ, Amasino M, Hong T, Yang M, Liao Y, Chiang HC, Kaklamani VG, Jeselsohn R, Vadlamudi RK, Huang TH, Li R, De Angelis C, Fu X, Elizalde PV, Schiff R, Brown M, Xu K. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ERα-GREB1 Transcriptional Axis. Cancer Res. 2018. February 1; doi: 10.1158/0008-5472.CAN-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, Cai Y, Bielski CM, Donoghue MTA, Jonsson P, Penson A, Shen R, Pareja F, Kundra R, Middha S, Cheng ML, Zehir A, Kandoth C, Patel R, Huberman K, Smyth LM, Jhaveri K, Modi S, Traina TA, Dang C, Zhang W, Weigelt B, Li BT, Ladanyi M, Hyman DM, Schultz N, Robson ME, Hudis C, Brogi E, Viale A, Norton L, Dickler MN, Berger MF, lacobuzio-Donahue CA, Chandarlapaty S, Scaltriti M, Reis-Filho JS, Solit DB, Taylor BS, Baselga J. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell. 2018, 34:427–438.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol. 2005. February; doi: 10.1038/modpathol.3800296. [DOI] [PubMed] [Google Scholar]
- 24.Creighton CJ, Massarweh S, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Shou J, Malorni L, Schiff R. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. 2008. September 15; doi: 10.1158/0008-5472.CAN-08-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008. February 1; doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 26.Browne BC, Hochgräfe F, Wu J, Millar EK, Barraclough J, Stone A, McCloy RA, Lee CS, Roberts C, Ali NA, et al. Global characterization of signalling networks associated with tamoxifen resistance in breast cancer. FEBS J. 2013. November; doi: 10.1111/febs.12441. [DOI] [PubMed] [Google Scholar]
- 27.Fu X, Creighton CJ, Biswal NC, Kumar V, Shea M, Herrera S, Contreras A, Gutierrez C, Wang T, et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. 2014. September 11; doi: 10.1186/s13058-014-0430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrison G, Fu X, Shea M, Nanda S, Giuliano M, Wang T, Klinowska T, Osborne CK, Rimawi MF, Schiff R. Therapeutic potential of the dual EGFR/HER2 inhibitor AZD8931 in circumventing endocrine resistance. Breast Cancer Res Treat. 2014. April; doi: 10.1007/s10549-014-2878-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, Zhou XJ. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016. April 20; doi: 10.1093/nar/gkv1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010. May 28; doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerrard DL, Wang Y, Gaddis M, Zhou Y, Wang J, Witt H, Lin S, Farnham PJ, Jin VX, Frietze SE. Three-dimensional analysis reveals altered chromatin interaction by enhancer inhibitors harbors TCF7L2-regulated cancer gene signature. J Cell Biochem. 2019. March, doi: 10.1002/jcb.27449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources.Nat Protoc. 2009; doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008. April; doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hickinson DM, Klinowska T, Speake G, Vincent J, Trigwell C, Anderton J, Beck S, Marshall G, Davenport S, Callis R, Mills E, Grosios K, Smith P, Barlaam B, Wilkinson RW, Ogilvie D. AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: a unique agent for simultaneous ERBB receptor blockade in cancer. Clin Cancer Res. 2010. February 15; doi: 10.1158/1078-0432.CCR-09-2353. [DOI] [PubMed] [Google Scholar]
- 36.Kim MR, Choi HS, Yang JW, Park BC, Kim JA, Kang KW. Enhancement of vascular endothelial growth factor-mediated angiogenesis in tamoxifen-resistant breast cancer cells: role of Pin1 overexpression. Mol Cancer Ther. 2009, 8:2163–2171. [DOI] [PubMed] [Google Scholar]
- 37.Zhou C, Zhong O, Rhodes LV, et al. Proteomic analysis of acquired tamoxifen resistance in MCF-7 cells reveals expression signatures associated with enhanced migration. Breast Cancer Res. 2012, 14:R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osborne CK, Hobbs K, Clark GM. Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res. 1985. February; 45(2):584–90. [PubMed] [Google Scholar]
- 39.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003. March 5; doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 40.Osborne CK, Jarman M, McCague R, Coronado EB, Hilsenbeck SG, Wakeling AE. The importance of tamoxifen metabolism in tamoxifen-stimulated breast tumor growth. Cancer Chemother Pharmacol. 1994; doi: 10.1007/bf00685924. [DOI] [PubMed] [Google Scholar]
- 41.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol l, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014. December 18; doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosagrahara VP, Hansen LK, Beilman GJ, Remmel RP. Evaluation of the effect of culture matrices on induction of CYP3A isoforms in cultured porcine hepatocytes. Chem Biol Interact. 2000. June 15; doi: 10.1016/s0009-2797(00)00163-0. [DOI] [PubMed] [Google Scholar]
- 43.Hagège H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, Forné T Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc. 2007; doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- 44.Dekker J The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat Methods. 2006;3(1):17–21. doi: 10.1038/nmeth823. [DOI] [PubMed] [Google Scholar]
- 45.Wingett S, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, Andrews S. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 2015. November 20;4:1310. doi: 10.12688/f1000research.7334.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, Gong K, Li Q, Alber F, Zhou XJ. Hi-Corrector: a fast, scalable and memory-efficient package for normalizing large-scale Hi-C data. Bioinformatics. 2015. March 15; doi: 10.1093/bioinformatics/btu747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.