

Abstract
Tobacco smoke exposure is associated with multiple diseases including respiratory diseases like asthma and COPD. Tobacco smoke is a potent inflammatory trigger and is immunosuppressive contributing to increased susceptibility to pulmonary infections in smokers, ex-smokers, and vulnerable populations exposed to secondhand smoke (SHS). Tobacco smoke exposure also reduces vaccine efficacy. Therefore, mitigating the immunosuppressive effects of chronic smoke exposure and improving the efficacy of vaccinations in individuals exposed to tobacco smoke, is a critical unmet clinical problem. We hypothesized that specialized pro-resolving mediators (SPMs), a class of immune regulators promoting resolution of inflammation, without being immunosuppressive, and enhancing B cell antibody responses, could reverse the immunosuppressive effects resulting from tobacco smoke exposure. We exposed mice to SHS for 8 weeks, followed by a period of smoke-exposure cessation, and the mice were immunized with the P6 lipoprotein from non-typeable Haemophilus influenzae (NTHI), using 17-HDHA and aspirin-triggered-RvD1 (AT-RvD1) as adjuvants. 17-HDHA and AT-RvD1 used as adjuvants, resulted in elevated serum and bronchoalveolar lavage levels of anti-P6-specific IgG and IgA that were protective, with immunized mice exhibiting more rapid bacterial clearance upon challenge, reduced pulmonary immune cell infiltrates, reduced production of proinflammatory cytokines, and less lung epithelial-cell damage. Further, the treatment of mice with At-RvD1 during a period of smoke-cessation further enhanced the efficacy of SPM-adjuvanted P6 vaccination. Overall, SPMs show promise as novel vaccine adjuvants with the ability to overcome the tobacco smoke-induced immunosuppressive effects.
Introduction
Tobacco smoke in the form of mainstream tobacco smoke (MTS) or secondhand smoke (SHS) is proinflammatory and immunosuppressive and associated with diseases like cancer, cardiovascular disorders and pulmonary disorders like chronic bronchitis and chronic obstructive pulmonary disease (COPD) (1, 2). Around 8 million people die each year due to tobacco smoke exposure (3). Chronic respiratory diseases are the third-most common cause of death in the United States and fifth worldwide, putting a huge socioeconomic burden on society (2-6). Tobacco smoke triggers proinflammatory physiological changes in the airway epithelium, inducing pulmonary damage and immunosuppression that collectively augment susceptibility to respiratory infections and exacerbate infection-induced pulmonary inflammation (1, 7-9). While acute inflammatory responses are self-limiting processes (10), chronic inflammation due to smoking persists long after smoking cessation, due to the production of excess proinflammatory cytokines and reactive oxygen and nitrogen species by the infiltrating activated immune cells themselves (11, 12). It is also established that smokers and ex-smokers, with and without COPD, and children and others involuntarily exposed to SHS in the home or workplace, are at increased risk of respiratory infections (1, 13-15). Respiratory infections among COPD patients account for 75% of acute exacerbation events, which are responsible for the majority of morbidity, mortality and medical costs in this population (16-20). SHS exposure in young children correlates with acute otitis media episodes, while in patients with COPD, increased incidence of infections with NTHI are common and lead to disease exacerbations (1, 13-15). We have previously shown that chronic exposure to MTS or SHS suppresses antigen-specific adaptive immunity to NTHI and leads to increased bacterial burden in the lungs of mice following infection (8, 9). Vaccination is of limited effectiveness, possibly due in part to the immunosuppressive effects of tobacco smoke exposure (1, 8, 9). Thus, effective solutions are urgently needed to treat chronic inflammation and enhance immune responses in these populations.
Acute inflammatory responses, crucial to fight pathogens and critical for host tissue repair and homeostasis, are self-limiting processes (10, 21). A newly identified class of lipid mediators called specialized proresolving mediators (SPMs) actively regulate the resolution phase of inflammation (22-24). SPMs have pro-resolving as well as anti-inflammatory properties without inducing immunosuppression (24-26). SPMs are endogenous mediators derived from the dietary omega-3 polyunsaturated fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), and are found in a variety of tissues and organs including bone marrow, spleen and blood (27-29). SPMs exert multiple functions in promoting resolution of inflammation including decreasing transmigration of neutrophils, enhancing nonphlogistic monocyte recruitment and augmenting macrophage engulfment of apoptotic neutrophils (29-33). SPMs inhibit the production of proinflammatory mediators such as TNF-α, IL-6, CXCL1 and IL-12, while enhancing anti-inflammatory cytokines like IL-10 (26, 34-36).
More recent evidence shows a role for SPMs in antibody responses, by augmenting differentiation of B cells into antibody-secreting plasma cells to enhance human B cell antibody production in vitro (29). 17-hydroxydocosahexaenoic acid (17-HDHA), which has both direct SPM activity and is also a precursor for resolvin D1 (RvD1), can act as a vaccine adjuvant by augmenting antibody responses to recombinant influenza hemagglutinin protein (30). 17-HDHA, RvD1 and protectin D1 were all found in the spleen (29), suggesting that endogenous SPMs play a role in regulating antibody responses, and that treatment with SPMs might be a novel therapeutic option to increase immunity against infections.
Here, we hypothesized that the SPMs 17-HDHA and AT-RvD1 could overcome SHS exposure-induced suppression of pathogen-specific antibody responses following vaccination and infection. We report that SHS-exposed mice immunized with P6 plus 17-HDHA or with P6 plus AT-RvD1 developed increased systemic as well as mucosal antigen-specific antibody titers that translated into rapid bacterial clearance in the lungs of vaccinated mice following acute NTHI infection with reduced epithelial damage. Treatment of SHS-exposed mice with AT-RvD1 prior to immunization, further reduced lung inflammation and enhanced the efficiency of the immunization. Taken together, our results suggest that pro-resolving mediators will be an effective approach to reduce chronic lung inflammation and improve the effectiveness of immunization against lung pathogens, leading to reduced morbidity and mortality among people with lung disease resulting from chronic exposure to MTS or SHS.
Materials and Methods
Mice
Female C57BL/6J mice (8 wks old) were purchased from Jackson Laboratory (Bar Harbor, ME) and used in all experiments. Animals were housed under specific pathogen-free conditions with free access to food and water and a light/dark cycle of 12:12 h. Number of animals used per group in each experiment was 10, unless mentioned otherwise in the figure legends. All animal procedures performed were approved by the Animal Care and Use Committees of both institutions (Roswell Park Comprehensive CancerCenter, Buffalo, NY, USA and the University of Rochester, Rochester, NY, USA), and complied with all state, federal, and National Institutes of Health regulations.
Secondhand smoke (SHS) exposure
Mice housed in the Inhalation Core Facility at the University of Rochester were exposed to SHS 5 h/day, 5 days/wk for 8 wks as described previously (9). Briefly, research cigarettes 3R4F were combusted in an automated smoking machine (TE-10, Teague Enterprises, Woodland California) and then mainstream tobacco smoke was generated in a puff volume of 35 ml in 2 sec duration once per minute (the FTC protocol). SHS was generated by collecting and mixing sidestream smoke with the mainstream tobacco smoke at 89%:11% ratio (37). Total particulate matter (TPM) concentration of 99±3 mg/m3 was achieved for these chronic exposure experiments by adjusting the number of cigarettes loaded. Control group of animals were exposed to filtered air in an identical manner. At the end of the final exposure cycle, animals were immediately transported to Roswell Park Comprehensive Cancer Center for infection and vaccination experiments.
AT-RvD1 treatment Regimen
Aspirin-triggered resolvin D1 (AT-RvD1, 7S,8R,17S-trihydroxy-4Z,9E,11E,13-Z,15E,19Z-docosahexaenoic acid) is an epimer of RvD1 that is produced when cyclooxygenase 2 (Cox-2) is acetylated by aspirin; it has similar in vivo and in vitro effects to RvD1 and is resistant to endogenous degradation (38). AT-RvD1 (Cayman Chemical) was dissolved in ethanol at 200μg/ml, aliquoted, and stored frozen. Immediately prior to use, this was diluted with 7 volumes of normal saline solution, resulting in a solution containing 25μg/ml AT-RvD1 in 12.5% ethanol. A volume of 40μl (1μg AT-RvD1) was administered to SHS-exposed mice by oropharyngeal aspiration (39), twice a week for 8 consecutive weeks. Control animals received 12.5% ethanol in saline. In experiements pertaining to Figure 1E-H, AT-RvD1 treatment on mice was done on wks 5-12 before chronic infection or P6 vaccination, where wks 5-8 represent the second half of smoke exposure period and wks 9-12 represent a period of cessation to smoke exposure.
FIGURE 1. SPM AT-RvD1 treatment rescues chronic secondhand smoke (SHS)-suppressed antigen-specific antibody responses.
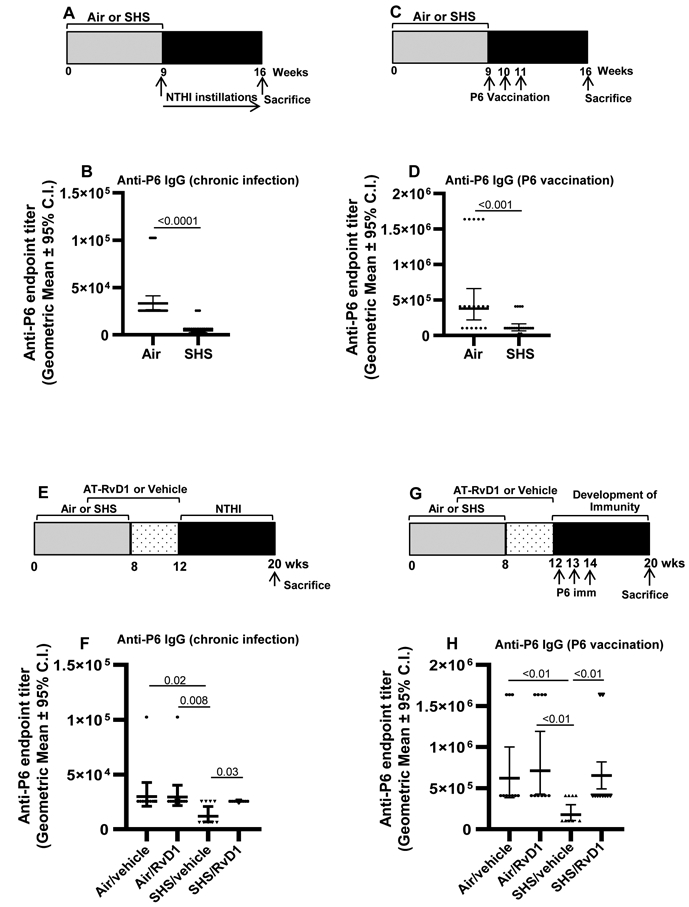
(A) Mice were exposed for 8 wks to SHS or air control, followed by 8 wks of chronic pulmonary NTHI infection. Animals were euthanized 48 h after the final intratracheal NTHI instillation. (B) Total anti-P6 IgG antibodies were measured in endpoint serum collected at wk16 at the time of euthanasia (n=27 for air control and n=29 for SHS-exposed group). (C) Another group of mice exposed to SHS or air for 8 wks were vaccinated with 40 μg P6 lipoprotein using Freund’s adjuvant. All the animals were euthanized 8 wks after the start of vaccination (at wk 16). (D) Total anti-P6 IgG antibodies were measured in endpoint serum collected at the time of euthanasia (n = 18 for air control and n=20 for SHS-exposed groups). (E, F) Mice exposed to air control or SHS were treated with AT-RvD1 for 8 wks before either chronic infection or P6 vaccination as described in methods section. (F,H) Total anti-P6 IgG antibodies were measured in endpoint serum collected at wk20 at the time of euthanasia (n=9-10 animals/group). Data are represented as geometric mean ± 95% C.I. Two-tailed unpaired student’s t test was performed and Mann-Whitney post-test comparison was employed to show the differences between the groups. Differences were taken to be statistically significant at p <0.05.
Chronic and acute pulmonary infection with NTHI
For all the experiments, NTHI bacterial strain 1479 (clinical isolate from a COPD exacerbation) from a single frozen glycerol stock was streaked onto chocolate-agar plates to grow bacterial cultures using a standard SOP to ensure that the NTHI doses were comparable in all chronic and acute pulmonary infection studies as described previously (8, 9). Briefly, for chronic NTHI-mediated pulmonary inflammation, mice received 1 x 106 live bacteria twice per week for 8 consecutive weeks via intratracheal route. For acute bacterial infection, mice were given a single intratracheal challenge with 1 x 106 live bacteria and euthanized 4 and 24 h later. After preparing lung homogenates, multiple serial dilutions were plated onto chocolate agar plates and the resulting colonies are quantitated after overnight incubation.
P6 immunization
Mice were immunized i.m. with 40 μg purified P6 antigen + 1ug SPM (17-HDHA or AT-RvD1) in a final ethanol concentration of 4%. Sham-immunized mice received 4% diluent in PBS. For the experiments in Figure 1, air and SHS-exposed mice were immunized i.p. with 40 μg purified native P6 lipoprotein emulsified in CFA and boosted 1 wk later with P6 in IFA and 2 wks later with Ag alone in PBS (40). Sham-immunized mice received PBS only. To quantify antigen-specific antibodies in serum and BAL, mice were bled on a weekly basis to collect sera and the BAL was harvested at the time of euthanasia.
Bronchoalveolar lavage (BAL)
Upon completion of experimental protocol, mice were euthanized by injecting intra-peritoneally 1 ml of warmed 2.5% avertin solution (2,2,2-tribromethanol). Thoracic cavity was exposed to cannulate trachea and lungs were gently lavaged twice by injecting 750 μl of ice cold 1% BSA solution in PBS as described previously (9). The cell-free BAL fluid was stored at −80°C and used to measure the levels of cytokines, albumin and antigen-specific antibody titers.
Isolation of lung lymphocytes, splenocytes and bone marrow cells
Lung lymphocytes were isolated as described previously (9). Briefly, after euthanasia lungs were excised and minced into small pieces in a petri-dish on ice. The resulting lung slurry was mixed with 1 mg/ml Type IA-S collagenase solution containing 50 U/ml DNase I (Sigma) and incubated for 1 h on a rotator at 37°C. Single-cell suspension was passed through a 40 μm filter to remove debris and undigested tissue, and then centrifuged for 5 minutes and resuspended in complete RPMI-1640 culture media containing 10% FBS. The cell suspension was gently overlaid on top of Ficoll-Paque and centrifuged at 700 x g for 30 minutes with the brake-off. Immune cells at the interface were collected, washed twice with PBS to remove residual Ficoll-Paque and cell count was determined.
Flow cytometry
Immune cell numbers in the lungs of mice was determined by flow cytometry as described previously (9). Briefly, 0.5 million lung lymphocytes from each sample were stained with fluorophore-tagged antibodies in 100 μl of FACS staining buffer (1% BSA in PBS) for 30 minutes at 4°C and then washed in FACS buffer before fixing with Cytofix (BD Biosciences, CA, USA). To stain intracellular markers, cells were first treated with permeabilizing solution (BD Biosciences, CA, USA) and then stained with cell-type specific fluorophore-tagged antibodies. Samples were acquired on LSRII-A flow cytometer and FlowJo software was employed to analyze the data. Gating strategy that was utilized is shown in supplemental Figure 1.
ELISA
ELISA assays were performed as described previously (8, 9) to determine cytokine and albumin levels in the BAL and antigen-specific antibody titers in the serum and BAL. Briefly, the levels of cytokines in the BAL were measured using the eBioscience ELISA kits following the manufacturer’s protocol: IL-17 (cat # 88-7371-77), IL-6 (cat # 88-7064-77), TNF-α (cat # 88-7324-77), IL-1β (cat # 88-7013-77). Mouse albumin levels in BAL fluid (as a surrogate marker of lung-epithelial damage) were quantified using a mouse-specific albumin quantifying ELISA kit from Bethyl Laboratories (Montgomery, TX, USA), with no cross-reactivity to BSA. Antigen-specific antibody titers in the serum and BAL were determined using purified P6 coated ELISA plates as described previously (8, 9). Dilutions of serum and endpoint BAL samples were incubated with P6-coated, BSA-blocked ELISA plates at room temperature, and bound anti-P6 Igs were detected with HRP-conjugated goat anti-mouse Ig(H+L), IgA, IgG1, and IgG2b antibodies (Southern Biotech, Birmingham, AL, USA). Plates were developed with eBioscience 3,3’,5,5’-tetramethylbenzidine (TMB; for HRP) solution, and absorbance was read at 450 nm on an ELISA plate reader (Synergy HTX Multi-Mode Reader, Winooski, VT, USA).
Assessment of NTHI clearance
NTHI bacterial clearance in the lungs of mice following acute infection was performed as described previously (9). Briefly, animals were given an acute intratracheal bacterial challenge with live 1 x 106 CFUs and euthanized 4 and 24 h later. Under aseptic conditions, whole lungs were excised and gently homogenized in 1 ml PBS on ice. Serial dilutions of lung homogenates were plated onto chocolate agar plates and incubated at 35°C at 5% CO2 conditions for 16 h. NTHI colonies were counted the next day and bacterial burden in the lungs was determined and expressed as the total number of bacterial CFUs recovered from the lungs.
Statistical analysis
The statistical objective was to assess the differences in marker measurements across exposure groups and time. Central tendency of the markers was quantified by estimates for the geometric mean, with 2-sided 95% confidence intervals indicating the precision of these estimates. For each marker in experiments with one observation per mouse, an analysis of variance model was specified with fixed effects for exposure group, time, and the interaction terms. When repeated observations per mouse were available (Figures 2B and 3B), Linear Mixed Models were specified with the same fixed effects, and a random subject effect with compound symmetric covariance structure. Throughout, marker measurements were assumed to be lognormally distributed. Compliance with distributional assumptions was confirmed using quantile plots of the model residuals. Each statistical model analyzed one marker within each experiment. For each model, Type 1 error was controlled at the α=0.05 level using Tukey multiple testing methods. All the data are expressed as geometric mean ± 95% confidence intervals. For Figure 1, difference between mean values was tested using a two-tailed unpaired Student t test with a Mann-Whitney post-test comparison using GraphPad Prism 8 software; the difference between two groups was considered significant at P≤0.05.
FIGURE 2. 17-HDHA and AT-RvD1 vaccine adjuvants restore SHS-suppressed systemic and mucosal antigen-specific antibody levels.
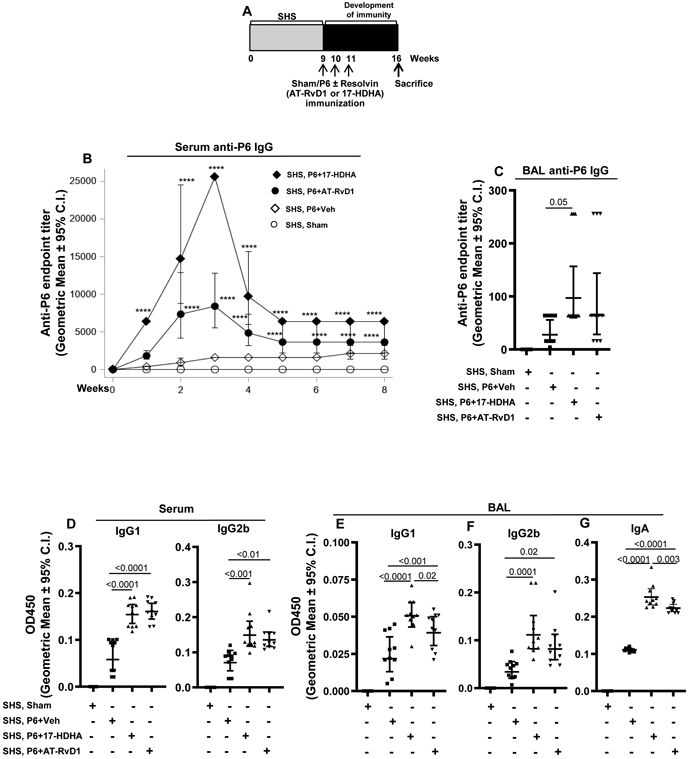
(A) Mice chronically exposed to SHS or air control for 8 wks were vaccinated with P6 antigen either in the presence or absence of SPM adjuvants 17-HDHA or AT-RvD1 as described in the methods section. Sham-immunized mice were vaccinated with PBS alone. All the animals were euthanized 8 wks after the start of vaccination (at wk 16). (B-C) Vaccination efficacy was determined by measuring the total anti-P6 IgG antibody levels in serum and endpoint BAL samples by ELISA (n=10 per group). (D-F) P6-specific IgG1 and IgG2b antibody subclass levels in endpoint serum and BAL, and (G) the levels of mucosal anti-P6 IgA antibody in endpoint BAL were determined by measuring OD values at 450 nm by ELISA. All treatment groups were assayed at the same time, and data represent results generated from a single experiment using n=10 mice per treatment group. Data from individual mice are shown, the line represents geometric mean ± 95% C.I. Statistical significance was determined as described in the methods section. Overall p<0.0001 comparing SHS, P6+vehicle group vs all treatment groups with Tukey’s posttest for multiple comparisons. For Figure 2B, ****p≤0.0001 with Tukey's posttest comparison of SHS, P6+Veh versus SHS, P6+AT-RvD1 or SHS, P6+17-HDHA adjuvant groups (overall p=0.0001 comparing SHS, P6+vehicle group vs all treatment groups).
FIGURE 3. AT-RvD1 treatment of SHS-exposed mice prior to vaccination enhances SPM adjuvant-induced efficacy of P6 immunization.
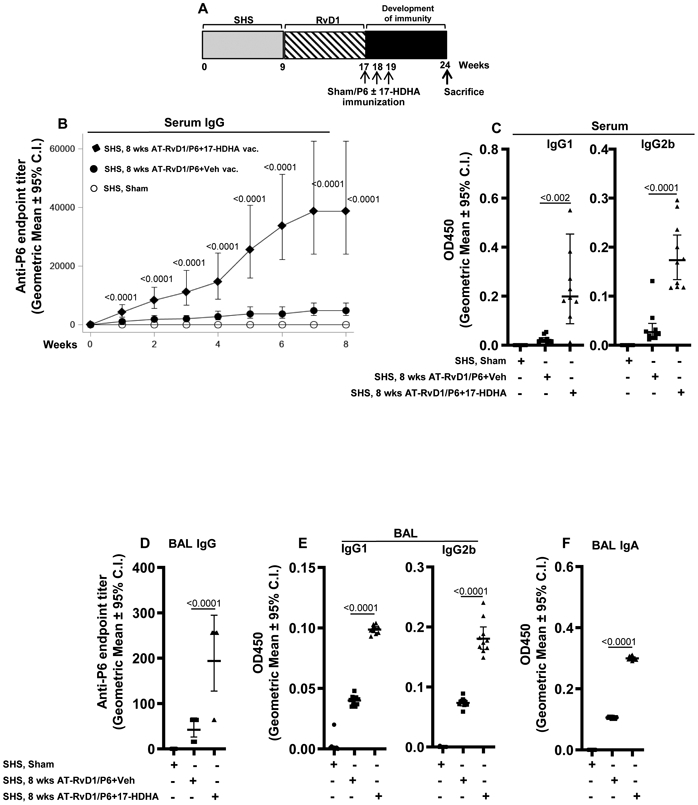
(A) Mice exposed to SHS for 8 wks were first treated with AT-RvD1 and then vaccinated with P6 antigen in the presence or absence of SPM adjuvant 17-HDHA as described in the methods section. Sham-immunized mice were vaccinated with PBS alone. Animals were euthanized at wk 24, 8 wks after the start of vaccination. Efficacy of vaccination was determined by measuring the levels of P6-specific total and subclass IgG antibodies in the serum (B-C) and in endpoint BAL fluid harvested at the time of euthanasia (D-E) (n=10 mice per group). (F) P6-specific IgA antibody levels in endpoint BAL were also determined by measuring OD values at 450 nm by ELISA. All treatment groups were assayed at the same time, and data represent results generated from a single experiment using n=10 mice per treatment group. Data from individual mice are shown, the line represents geometric mean ± 95% C.I. Statistical significance was determined as described in the methods section (overall p=0.0001 comparing SHS, P6+vehicle group vs all treatment groups with Tukey’s posttest for multiple comparisons).
Results
SPMs 17-HDHA and AT-RvD1 overcome immune suppression induced by SHS exposure
First, we confirmed that the model was working as expected. As previously reported (9), chronic exposure of mice to SHS for 8 consecutive weeks markedly suppressed the induction of systemic antigen-specific antibody titers after chronic NTHI infection (Figure 1A-B) and following vaccination with NTHI P6 antigen (Figure 1C-D). We next evaluated if treatment of SHS-exposed mice with AT-RvD1 could rescue the SHS-induced immuno-suppressive effects following infection and vaccination (schema Figure 1E,G). We observed that AT-RvD1 treatment in SHS-exposed mice significantly enhanced the antigen-specific antibody responses following both infection as well as vaccination with NTHI P6 antigen (Figure 1F,H), thus clearly demonstrating in vivo immuno-enhacning potential of SPM AT-RvD1. We next examined the potential of DHA-derived SPMs, 17-HDHA and AT-RvD1 to function as adjuvants to enhance the efficacy of vaccination in SHS-exposed mice. To address this question, mice exposed to SHS for 8 wks were vaccinated with purified P6 antigen plus 17-HDHA or P6 plus AT-RvD1 or vehicle control (Figure 2A) and the efficacy of vaccination was evaluated by measuring the anti-P6 antibody titers in serum and the BAL of mice. Vaccination with P6 in the presence of 17-HDHA or AT-RvD1 accelerated the kinetics of antibody response as well as markedly elevated the absolute serum antibody titers compared to vaccination with P6 plus vehicle control (Figure 2B). The efficacy of vaccination was greater with 17-HDHA than AT-RvD1. Furthermore, endpoint antigen-specific antibody titers in the BAL of SHS-exposed mice were significantly elevated after vaccination with P6 antigen either in the presence of 17-HDHA or AT-RvD1 compared to vaccination with P6 plus vehicle control (Figure 2C). Additionally, the levels of P6-specific IgG1 and IgG2b antibody sub-classes were significantly augmented in the serum and BAL of SHS-exposed mice vaccinated with P6 antigen plus 17-HDHA or P6 antigen plus AT-RvD1 compared to the levels elicited in mice vaccinated with P6 antigen plus vehicle (Figure 2D-F). Importantly we report that vaccinating SHS-exposed mice with P6 antigen in the presence of SPM adjuvants 17-HDHA or AT-RvD1 significantly augmented mucosal P6-specific IgA antibody levels in the BAL (Figure 2G).
AT-RvD1 treatment of SHS-exposed mice prior to vaccination further enhances SPM adjuvant-augmented vaccination efficacy
We next determined if pretreating SHS-exposed mice with AT-RvD1 prior to vaccination would further augment the efficacy of vaccination in the presence of SPM adjuvant 17-HDHA. To examine this, mice chronically exposed to SHS were treated with AT-RvD1 twice a week for 8 consecutive weeks and then immunized with P6 plus 17-HDHA or P6 plus vehicle control (Figure 3A). AT-RvD1 treatment alone resulted in slightly elevated antibody responses when the mice were immunized with P6 plus control vehicle, and this effect was seen in both serum and in lung (BAL) antibody levels. This suggests that AT-RvD1 alone, given after smoke exposure, mitigates the immunosuppressive effects of SHS exposure and primes the mice to make an immune response to the un-adjuvanted P6 antigen. The combination of AT-RvD1 treatment after SHS exposure plus immunization with P6 antigen adjuvanted with 17-HDHA markedly elevated the P6-specific antibody titers in serum (systemic) and BAL (mucosal) following vaccination with P6 plus 17-HDHA compared to vaccination with P6 plus vehicle (Figure 3B,D). Additionally, IgG1 and IgG2b antibody subclasses in the serum and BAL were significantly increased (Figure 3C,E). We also observed markedly augmented levels of antigen-specific IgA antibodies in the BAL of these mice (Figure 3F).
17-HDHA- and AT-RvD1-augmented efficacy of P6 vaccination enhanced bacterial clearance in the lungs of SHS-exposed mice
To demonstrate the functional benefit of higher antibody titers, we measured bacterial clearance of an acute NTHI infection following vaccination. Mice exposed to SHS for 8 wks were either immunized with P6 plus 17-HDHA or P6 plus AT-RvD1 or P6 plus vehicle, and then challenged with acute NTHI infection 8 wks later (Figure 4A). Another group of mice exposed to SHS for 8 wks, were treated with AT-RvD1 for 8 wks and then vaccinated with P6 plus 17-HDHA, and subsequently challenged with an acute NTHI infection at wk 24 (Figure 4B). In both groups of mice, bacterial clearance in the lungs was measured 4 and 24 h following acute challenge with NTHI (Figure 4A,B). We found that, SHS-exposed mice that were vaccinated with P6 plus 17-HDHA or P6 plus AT-RvD1 had significantly improved bacterial clearance at both 4 and 24 h following acute NTHI challenge compared to SHS-exposed mice vaccinated with P6 plus vehicle control (Figure 4C). As pre-treatment with AT-RvD1 augmented antibody responses following immunization (Figure 2), we expected that pre-treatment with AT-RvD1 would also enhance bacterial clearance, and this was indeed what we found. Pre-treatment of the mice with AT-RvD1 markedly improved bacterial clearance, especially at 4 h post-infection, and even in mice immunized with the unadjuvanted P6 antigen (hatched bars vs black bars at 4 h in Figure 4C). However, use of 17-HDHA as an adjuvant significantly improved clearance in AT-RvD1-treated mice at both 4 and 24 h post-infection.
FIGURE 4. 17-HDHA and AT-RvD1 adjuvant-enhanced efficacy of P6 vaccination augments bacterial clearance in the lungs of SHS-exposed mice.
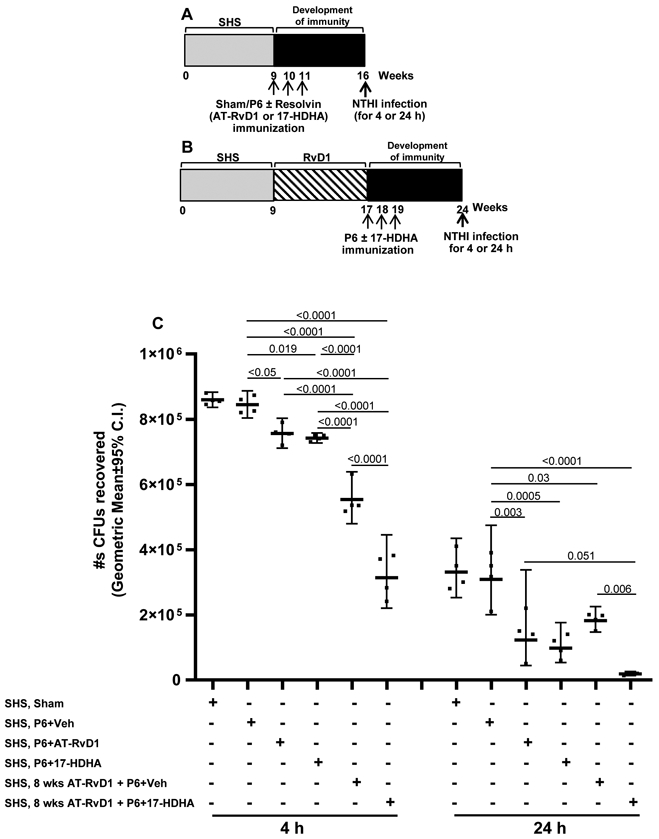
Mice exposed to SHS for 8 wks were either (A) untreated or (B) treated with AT-RvD1 for 8 wks prior to vaccination with P6 ± SPM adjuvant AT-RvD1 or 17-HDHA. All mice received acute intratracheal NTHI challenge 8 wks after the start of vaccination. Pulmonary bacterial burden in SHS-exposed, vaccinated mice was measured at 4 and 24 h following acute infection and data expressed as (C) total number of CFUs recovered. All treatment groups were assayed at the same time, and data represent results generated from a single experiment using a total of n=24 mice at each time point. Data from individual mice are shown, the line represents geometric mean ± 95% C.I. Statistical significance was determined as described in the methods section (overall p<0.0001 comparing SHS, P6+vehicle group vs all treatment groups with Tukey’s posttest for multiple comparisons at 4 and 24 h).
SPM adjuvant-enhanced immunity reduces SHS-exacerbated inflammatory microenvironment following an acute respiratory infection
We next evaluated whether the augmented adaptive immunity induced by the SPMs had an impact on the SHS-exacerbated pulmonary inflammatory microenvironment induced following an acute bacterial infection, with or without AT-RvD1 treatment after SHS exposure (schemas in Figures 5A, B). The total number of pulmonary inflammatory cells was highest in mice exposed to SHS without SPM treatment or immunization. Immunization with P6 plus 17-HDHA or AT-RvD1, but not vehicle, dramatically reduced lung infiltrating cells. Mice pre-treated with AT-RvD1 exhibited reduced numbers of infiltrating leukocytes compared to mice receiving SHS alone, and this was further reduced by immunization with P6 plus 17-HDHA. However, the beneficial effect of immunization with P6 plus 17-HDHA was the same whether or not the mice were pre-treated with AT-RvD1 (Figure 5C). We also observed that the numbers of neutrophils, CD4+IL-17A+ Th17 and CD4+RoRγ+ Th17 T cells in the lungs of SHS-exposed mice vaccinated with P6+ SPM (17-HDHA or AT-RvD1) were markedly reduced after acute bacterial challenge (measured at both 4 and 24 h) compared with the numbers of these inflammatory cells found in the lungs of mice that were either sham or vaccinated with P6 only (Figure 6A-F and Supplemental Table 1). Moreover, the numbers of these cells were further reduced in the lungs of SHS-exposed mice that were treated with AT-RvD1 for 8 wks prior to vaccination with P6 plus 17-HDHA compared to SHS-exposed, vehicle-treated, sham vaccinated mice (Figure 6A-F and SupplementalTable 1).
FIGURE 5. SPM adjuvant-enhanced P6 vaccination efficacy reduces SHS-exacerbated pulmonary immune cell infiltration following acute infection.
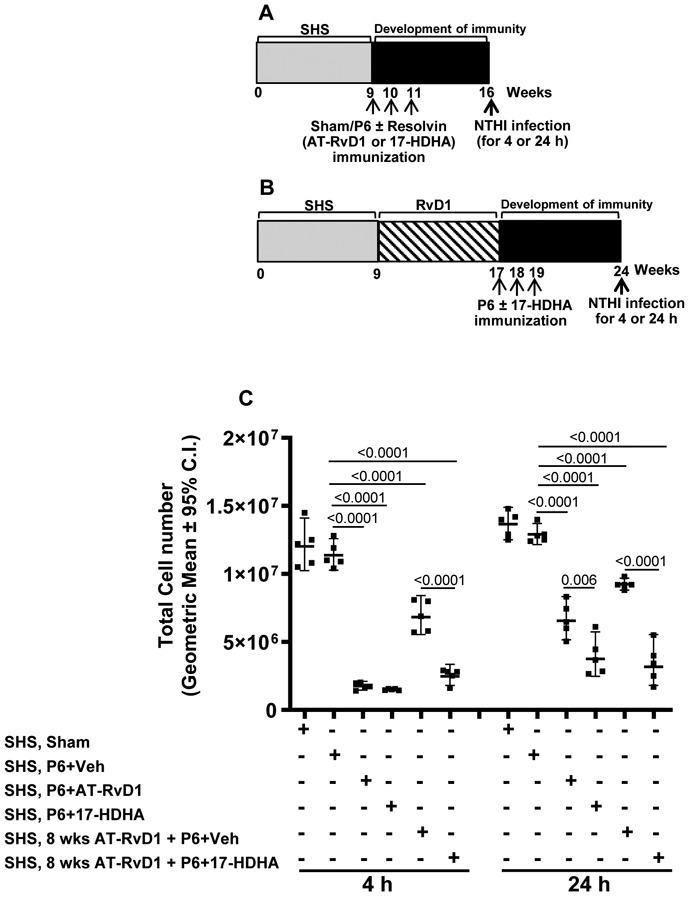
SHS-exposed mice, (A) untreated or (B) treated with AT-RvD1 for 8 wks before vaccination with P6 ± SPM adjuvant 17-HDHA or AT-RvD1, were given acute pulmonary NTHI challenge 8 wks after the start of vaccination. (C) Total pulmonary immune cell infiltration at 4 and 24 h following acute infection was quantified as described in methods. All treatment groups were assayed at the same time, and data represent results generated from a single experiment using a total of n=30 mice at each time point. Data from individual mice are shown and the results are depicted as geometric mean ± 95% C.I. Statistical significance was determined as described in the methods section (overall p<0.0001 comparing SHS, P6+vehicle group vs all treatment groups with Tukey’s posttest for multiple comparisons at 4 and 24 h).
Figure 6: SPM-adjuvanted P6 vaccination decreases SHS-exacerbated pulmonary immune cell infiltration following acute infection.
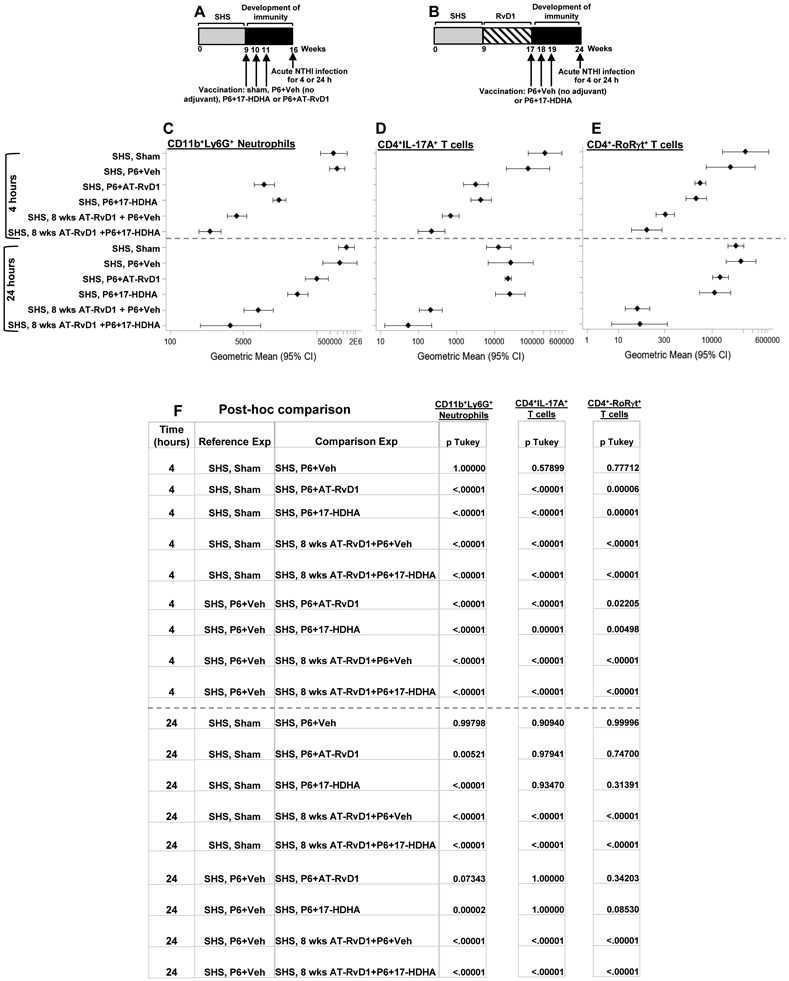
(A-B) SHS-exposed mice, untreated or treated with AT-RvD1 for 8 consecutive wks before vaccination with P6 ± SPM adjuvant (17-HDHA or AT-RvD1), were given acute pulmonary NTHI challenge 8 wks after the start of vaccination. (C) Total numbers of CD11b+Ly6G+ neutrophils, (D) CD4+IL-17A+ T cells and (E) CD4+RoRγt+ T cells accumulating in the lungs at 4 and 24 h following acute infection were determined by flow cytometry using cell-specific markers as described in the methods section. Gating strategy that was utilized is shown in supplemental Figure 1. (F) Multiple comparisons table with Tukey’s post-hoc comparisons and p values are provided. All treatment groups were assayed at the same time, and data represent results generated from a single experiment using a total of 60 mice (n=5 mice for each treatment at each time point. The results are depicted as geometric mean ± 95% C.I. Statistical significance was determined as described in the methods section (overall p<0.0001 [CD11b+Ly6G+ neutrophils, CD4+IL17A+ T cells, CD4+Rorgt+ T cells, 4 and 24 h] comparing SHS, sham or SHS, P6+vehicle group vs all treatment groups with Tukey’s posttest for multiple comparisons.
SPM-enhanced efficacy of P6 vaccination diminished proinflammatory cytokine induction in the BAL and translated into reduced lung-epithelial damage in response to acute infection
We also evaluated the effects of 17-HDHA and AT-RvD1 on the levels of pulmonary pro-inflammatory cytokines. We observed lower levels of proinflammatory cytokines IL-17A, IL-6 and TNF-α measured in the BAL at both 4 and 24 h after acute infection in mice, immunized with P6 plus 17-HDHA or AT-RvD1, compared to sham or P6 plus vehicle-immunized mice (Figure 7A-C). However, IL-1β levels were reduced only 24 h after acute challenge with NTHI (Figure 7D). Pre-treatment with AT-RvD1 alone also reduced levels of inflammatory cytokines to a similar degree, consistent with our previous finding that this SPM accelerated resolution of inflammation after smoke exposure (24). The lowest levels of pro-inflammatory cytokines were seen 24 h after infection in mice that were pre-treated with AT-RvD1 and immunized with P6 plus 17-HDHA.
FIGURE 7. SPM adjuvant-enhanced P6 vaccination efficacy diminishes SHS-exacerbated, infection-induced BAL inflammatory cytokine profile and lung-epithelial damage.
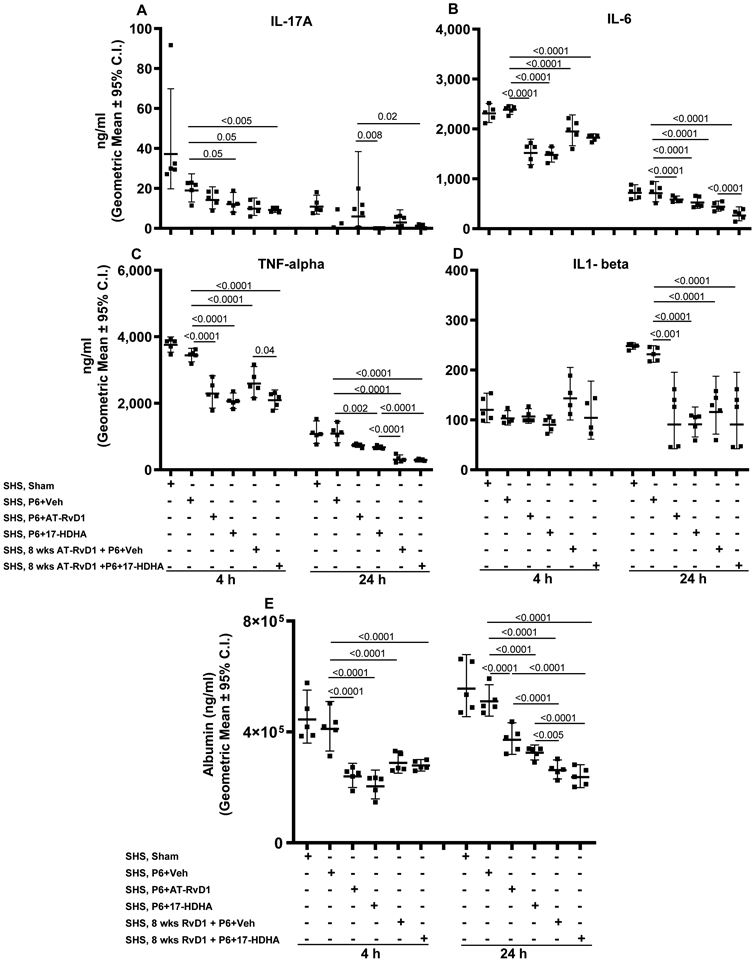
Mice exposed to chronic SHS were either untreated or treated with AT-RvD1 for 8 wks prior to vaccination with P6 ± SPM adjuvant 17-HDHA or AT-RvD1, and then infected in the lungs with acute NTHI 8 wks after the start of vaccination. Concentration of proinflammatory cytokines (A) IL-17A, (B) IL-6, (C) TNF-α and (D) IL1-β in the BAL collected at 4 and 24 h following infection were evaluated by ELISA. (E) The levels of albumin in the endpoint BAL samples as a surrogate marker of lung-epithelial damage were quantified by ELISA. All treatment groups were assayed at the same time, and data represent results generated from a single experiment using a total of 60 mice (n=5 mice for each treatment, at each time point). Data from individual mice are shown and the results are depicted as geometric mean ± 95% C.I. Overall p<0.0001 for IL-17A, IL-6, TNF-alpha, albumin (at 4 and 24 h) and for IL1-beta (at 24 h) comparing SHS, sham or SHS, P6+vehicle group vs all treatment groups with Tukey’s posttest for multiple comparisons.
Finally, we measured the levels of mouse serum albumin in the BAL fluid, a surrogate measure for alveolar epithelial barrier function and integrity. SHS-exposed mice either vaccinated with P6 plus SPM (17-HDHA or AT-RvD1) or pretreated with AT-RvD1 for 8 wks and then vaccinated with P6 plus 17-HDHA, had markedly lower levels of albumin in the BAL than found in SHS-exposed mice that were sham-vaccinated or immunized with P6 + vehicle (Figure 7E).
Discussion
Tobacco smoke, including involuntary secondhand smoke (SHS) exposure, is associated with many human disorders including respiratory diseases like COPD (1, 6, 13-15, 41). In response to toxic exogenous insults like SHS, pulmonary homeostasis is maintained via an array of immune cells including recruited neutrophils and resident macrophages, and secreted cytokines (42). However repeated stimulation with toxic insults results in the persistence of immune mediators that disrupt pulmonary homeostasis and induce chronic lung inflammation (43). These physiological changes induced by tobacco smoke account for the exacerbated pulmonary inflammation observed in smokers with COPD that results in pulmonary damage and impaired lung function (1, 16, 17). Many studies have established that tobacco smoke exacerbates infection-associated pulmonary inflammation, dysregulates immune function and augments susceptibility to respiratory infections, thereby further worsening lung inflammation and function particularly in susceptible individuals like COPD patients (1, 8, 9, 18). Smokers, ex-smokers, children and other vulnerable populations exposed to SHS are shown to be at increased risk for pneumonia and other infections, and this has been replicated in mouse models of smoke exposure and infection (1, 6, 8, 9, 13-15). For instance, SHS exposure in children is known to increase the risk of ear infections, cause acute lower respiratory tract infections like bronchitis and pneumonia in infants and young children, and trigger respiratory symptoms like cough, phlegm, wheezing and breathlessness in school-going children (6, 13-15). Viral and bacterial infections account for up to 75% of acute exacerbations of COPD, and account for much of the morbidity experienced by these patients (19, 20, 44). Various studies have clearly shown that tobacco smoke inhibits innate and adaptive immunity by suppressing functions of immune cells ranging from macrophages, dendritic cells, NK cells and neutrophils to B and T cells (1, 18). Tobacco smoke exposure, both mainstream and secondhand, also reduces vaccine efficacy (8, 9). Therefore, mitigating the effect of chronic tobacco smoke exposure on the immune system, and improving vaccine efficacy in people exposed to tobacco smoke, is a critical unmet clinical problem.
In the present study, we demonstrate that specialized proresolving mediators (SPMs) 17-HDHA and AT-RvD1 as vaccine adjuvants, potently overcome the SHS-induced suppression of protective immune responses to vaccination by augmenting systemic as well as mucosal antigen-specific antibody responses. These SPM adjuvant-augmented antibody responses were functionally relevant as they diminished the bacterial burden and reduced proinflammatory cytokines and alveolar leakage following an acute respiratory infection. Additionally, treatment with AT-RvD1 before vaccination of SHS-exposed mice further enhanced SPM adjuvant-augmented antibody responses that translated into improved protection against acute NTHI pulmonary infection. Our study establishes the utility of SPMs to diminish SHS-exacerbated proinflammatory milieu and enhance antigen-specific systemic and mucosal immunity. As a first step we re-validated our previous published findings that chronic SHS exposure suppresses antigen-specific antibody responses to both chronic NTHI infection as well as NTHI P6 antigen vaccination. Importantly, we found that SPM AT-RvD1 treatment in SHS-exposed mice significantly augmented systemic antigen-specific antibody titers following chronic infection and vaccination, thus highlighting the physiological effects of AT-RvD1 treatment in SHS-exposed, infected or vaccinated mice.
Chronic cigarette smoke exposure-induced immune suppression worsens proinflammatory pulmonary microenvironment to increase bacterial burden in the lungs following infection (8, 9), a phenomenon commonly observed in smokers with COPD during disease exacerbations and associated with increased morbidity and mortality (1, 16-20). SPMs, naturally occurring pro-resolving and anti-inflammatory bioactive agents, are involved in inflammation resolution without causing immunosuppression (22-26). Additionally, SPMs augment adaptive immunity via mechanisms that include upregulation of CD80 and CD86 expression on B cells and promoting differentiation of B cells into antibody-secreting plasma cells to augment antibody responses (29, 30). We thus hypothesized that if SPMs could be utilized to enhance efficacy of vaccination using a candidate vaccine antigen, SHS-suppressed anti-bacterial responses could potentially be restored. Furthermore, we posited that SPM adjuvant-enhanced anti-bacterial responses might have translational relevance in diminishing SHS-exacerbated, infection-induced pulmonary proinflammatory microenvironment, thereby reducing lung-epithelial damage. It has been reported that RvD1 treatment reduces inflammatory cytokines and leads to diminution of bacterial burden (26). We therefore sought to evaluate the beneficial impact of AT-RvD1 treatment in combination with SPM-adjuvant plus antigen vaccination, on antigen-specific immune responses and its impact on SHS-exacerbated, infection-induced pulmonary proinflammatory microenvironment and damage.
It is known that cigarette smoke-suppressed immunity delays resolution of bacterial infection in the lungs of mice (1, 8, 9). By exploiting 17-HDHA and AT-RvD1 as vaccine adjuvants to enhance systemic and mucosal P6 antigen-specific antibody responses, we observed decreased NTHI burden in the lungs of SHS-exposed mice following acute infection. Since cigarette smoke-augmented pulmonary bacterial burden correlates with exacerbated lung inflammation (8, 9, 45), we thus speculated that diminished bacterial burden in the lungs of SHS-exposed mice would lead to diminution in the proinflammatory pulmonary milieu. Indeed, we observed that SPM adjuvant-enhanced immunity by reducing bacterial burden in the lungs of SHS-exposed mice, also helped diminish SHS-exacerbated pulmonary inflammatory microenvironment by decreasing proinflammatory immune cell accumulation which correlated with diminished levels of proinflammatory cytokines following infection.
The translational relevance of our study is depicted by a rapid clearance of bacteria from the lungs of SHS-exposed, SPM-adjuvant plus P6 vaccinated mice with a concomitant reduction in lung-epithelial damage. This beneficial effect of SPM adjuvants is clinically relevant since in susceptible individuals like smokers with COPD, elderly or children exposed to environmental tobacco smoke, increased pulmonary bacterial burden associates with augmented inflammation, enhanced lung damage and impaired lung function (1, 6, 8, 9, 13-20). A novel observation emerging from our study was seen when SHS-exposed mice were treated with AT-RvD1 before P6 + SPM-adjuvant vaccination. AT-RvD1 treatment before P6 + SPM vaccination further enhanced vaccination efficacy that translated into reduced pulmonary bacterial burden, diminished SHS-exacerbated pulmonary inflammatory responses and decreased lung-epithelial damage in SHS-exposed mice following acute bacterial infection. However, we can not rule out the involvelement of other mechanisms associated with resolvin-mediated bacterial clearance including reduction of chronic smoke-induced inflammation (52) and improved macrophage function (26). However our previously published studies on the use of conventional adjuvants with outer membrane NTHI lipoprotein P6 antigen to induce protective immunity to NTHI, amply demonstrates that an appropriate humoral response can have a significant beneficial effect in this model (8, 9). We now demonstrate clearly the beneficial effects of resolvins in the comparison between P6 plus vehicle immunization and P6 immunization with either 17-HDHA or AT-RvD1. It is also of interest that the maximum benefit of immunization with P6 plus 17-HDHA was observed in mice that had also been treated with AT-RvD1 prior to immunization. The results suggest that the beneficial effects of modifying the chronic inflammatory milieu induced by smoke exposure and of improving antibody responses were additive, and resulted in the lowest inflammatory response and most improved bacterial clearance, which is an eventual therapeutic goal in patients with chronic lung inflammation who experience infectious exacerbations. Indeed, utilizing SPMs to treat inflammatory diseases has shown beneficial results in various models including mouse models of COPD, asthma, arthritis, periodontitis, viral infection and cancer (24, 26, 30, 46-52).
The present study confirms that chronic SHS suppresses systemic and mucosal NTHI P6 antigen-specific antibody responses. Long-term immunity is a precondition for durable protection against infections in susceptible individuals. Since alum is the only FDA approved adjuvant currently in use in the United States (53), evaluation of a new class of adjuvants is deemed important to improve the efficacy of human vaccines. Our results establish that SPMs 17-HDHA and AT-RvD1 are potent vaccine adjuvants that can significantly restore SHS-suppressed immunity by augmenting protective antigen-specific systemic and mucosal antibody responses. It is also well understood that some populations, particularly the elderly and immune compromised indivuduals, are at increased risk or morbidity and mortality from respiratory infections, while also being less able to generate protective antibody responses following immunization. Towards this endeavor, SPMs might also be an important new way to augment vaccine responses in the elderly and immune compromised individuals, irrespective of their smoking history (54, 55). Indeed, the current coronavirus pandemic highlights the critical problem of respiratory infections in the elderly, with estimated case fatality rates 10-100-fold higher among the over-60 age group compared to under-60 (56, 57). SPMs have already been theorized to be able to dampen cytokine storm in COVID-19 patients (58); our results suggest that SPMs might also have beneficial effects as adjuvants when a COVID-19 vaccine is developed.
Our present results as well as earlier findings by us and other groups clearly indicate that immuno-enhancing effects of SPMs are not restricted to reducing the immunosuppressive effects of cigarettte smoke on the immune responses to NTHI infection in our murine model of infection and immunity. We have previously reported that 17-HDHA protects against lethal challenge by H1N1 influenza A infection in mice when used as an adjuvant mixed with the HA antigen (30). 17-HDHA adjuvant-mediated immunity provided long-term protection against live influenza viral infection in mice and markedly increased their survival with minimum weight loss compared to non-adjuvanted, immunized mice. Studies in the literature also show that resolvins have protective effects against various infections including Escherichia coli and Staphylococcus aureus via mechanisms like lowering the antibiotic requirements for clearing bacterial infections, thus leading to reduction in local and systemic bacterial loads and enhancing the survival in mice (59, 60). Furtheremore, data from human studies show a benefical association between SPM resolvins and controlling infections including parasites (Trypanosoma cruzi) (61), acute exacerbations of COPD (62), and sepsis (63).
A limitation of our study regarding the adjuvant effect of 17-HDHA and AT-RvD1 with P6 immunization is that we did not include air-only controls in every experiment presented. However, we have previously reported that 17-HDHA and AT-RvD1 are effective adjuvants in mice that were not exposed to smoke when immunized with influenza HA antigen (30). The key goal of the present study was to demonstrate that SPMs can also augment immune responses in a mouse model of immune suppression following 8 weeks of cigarette smoke exposure. Thus, the results from figures 2 and 3 illustrate the beneficial effects of using SPMs as adjuvants on P6 antibody titers. The data from the experiment reported in Figures 4-7 shows that SPMs clearly rescue the immune impairment caused by chronic exposure to cigarette smoke. For example, in Figure 4C, the decreased NTHI load between column 1 and 5 at 4 h time-point are attributable to the rest period with inhaled AT-RvD1 treatment, while the decrease between column 5 and 6 represents the effect of immunization with P6 plus 17-HDHA as an intramuscular injection. The same effect is seen at 24 h. A consistent effect pattern is seen with lung cell counts (Figure 5C), neutrophils (Fig 6C) and Th17 cells (Fig 6D) and cytokines (Fig 7). However, we can not rule out the possibility that some relief from the smoke-induced immune suppression in the experiment reported in Figs 4-7 may be attributable to time lapsed, rather than the effects of inhaled AT-RvD1 and/or adjuvanted immunization. However, our earlier investigations of cigarette smoke-mediated immune suppression in the NHTI model argue that time alone does not restore immune function (9), as does our results in Figure 1E-H. Overall, our current results and previous findings by us and others allow us to conclude that SPMs 17-HDHA and AT-RvD1 show multiple beneficial effects to restore immune function after smoke exposure, and thus indicate that immune enhancing effects of 17-HDHA/AT-RvD1 are physiologically significant.
Collectively, our results establish that SPMs 17-HDHA and AT-RvD1 by acting as pro-resolving mediators and vaccine adjuvants rescue SHS-suppressed immunity to vaccination and infection.
Supplementary Material
Key points:
SPMs as adjuvants augment the efficacy of vaccination with NTHI P6 antigen in mice.
SPM-adjuvanted vaccination efficacy rescues SHS-suppressed anti-bacterial immunity.
SPM-adjuvanted vaccinated mice rapidly clear acute infection with less lung damage.
Acknowledgments
This work was supported by Flight Attendant Medical Research Institute (FAMRI) Clinical Innovators Award to Y.T. and P.J.S. This work was supported in part by National Institutes of Health Grant R01 HL120908 (to P.J.S.). This work was supported by National Cancer Institute Grant P30CA016056 involving the use of Roswell Park Core facilities: Division of Laboratory Animal Resources, Mouse Tumor Model Resource, and Flow Cytometry.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References:
- 1.Stampfli MR, and Anderson GP. 2009. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol 9: 377–384. [DOI] [PubMed] [Google Scholar]
- 2.Oberg M, Jaakkola MS, Woodward A, Peruga A, and Pruss-Ustun A. 2011. Worldwide burden of disease from exposure to second-hand smoke: a retrospective analysis of data from 192 countries. Lancet. 377: 139–146. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2016. Tobacco - fact sheet. Available at: http://www.who.int/mediacentre/factsheets/fs339/en/. Accessed: January 25, 2019. [Google Scholar]
- 4.Eisner MD, Balmes J, Katz PP, Trupin L, Yelin EH, and lanc PD. 2005. Lifetime environmental tobacco smoke exposure and the risk of chronic obstructive pulmonary disease. Environ. Health 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thun MJ, Carter BD, Feskanich D, Freedman ND, Prentice R, Lopez AD, Hartge P, and Gapstur SM. 2013. 50-year trends in smoking-related mortality in the United States. N. Engl. J. Med 368: 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.U.S. Department of Health and Human Services. 2014. The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, Atlanta, GA. [accessed 2020 January 25]. [Google Scholar]
- 7.Bagaitkar J, Demuth DR, Scott DA. 2008. Tobacco use increases susceptibility to bacterial infection. Tob Induc Dis. 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lugade AA, Bogner PN, Thatcher TH, Sime PJ, Phipps RP, and Thanavala Y. 2014. Cigarette smoke exposure exacerbates lung inflammation and compromises immunity to bacterial infection. J. Immunol 192: 5226–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhat TA, Kalathil SG, Bogner PN, Miller A, Lehmann PV, Thatcher TH, Phipps RP, Sime PJ, and Thanavala Y. 2018. Secondhand smoke induces inflammation and impairs immunity to respiratory infections. J. Immunol 200: 2927–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janeway C 2005. Immunobiology: the immune system in health and disease. Garland Science; New York. [Google Scholar]
- 11.Hogg JC 2006. Why does airway inflammation persist after the smoking stops? Thorax. 61(2): 96–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Invernizzi G Persistence of systemic inflammation in COPD in spite of smoking cessation.2011. Multidiscip Respir Med. 6(4): 210–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.U.S. Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2006 [accessed 2020 January 25]. [Google Scholar]
- 14.Csakanyi Z, Czinner A, Spangler J, et al. 2012. Relationship of environmental tobacco smoke to otitis media (OM) in children. Int J Pediatr Otorhinolaryngol. 76:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Centers for Disease Control and Prevention. Health Effects of Secondhand Smoke. February 27, 2020 https://www.cdc.gov/tobacco/data_statistics/fact_sheets/secondhand_smoke/health_effects/index.htm [Google Scholar]
- 16.Sethi S, Evans N, Grant BJ, and Murphy TF. 2002. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N. Engl. J. Med 347: 465–471. [DOI] [PubMed] [Google Scholar]
- 17.Sethi S 2010. Infection as a comorbidity of COPD. Eur Res J. 35: 1209–1215. [DOI] [PubMed] [Google Scholar]
- 18.Bhat TA, Panzica L, Kalathil SG, Thanavala Y. 2015. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 12: S169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sethi S, and Murphy TF. 2001. Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin. Microbiol. Rev 14: 336–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sethi S, and Murphy TF. 2008. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med 359: 2355–2365. [DOI] [PubMed] [Google Scholar]
- 21.Medzhitov R 2008. Origin and physiological roles of inflammation. Nature. 454: 428–435. [DOI] [PubMed] [Google Scholar]
- 22.Serhan CN 2007. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Ann. Rev. Imm 25: 101–137. [DOI] [PubMed] [Google Scholar]
- 23.Serhan CN, Chiang N, and Van Dyke TE. 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. rev. Imm 8: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA, Olsen KC, Pollock SJ, Serhan CN, Phipps RP, and Sime PJ. 2013. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PloS one. 8: e58258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, and Moussignac RL. 2002. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med 196: 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croasdell A, Lacy SH, Thatcher TH, Sime PJ and Phipps RP. 2016. Resolvin D1 dampens pulmonary inflammation and promotes clearance of nontypeable Haemophilus influenzae. J. Immunol. 196: 2742–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dona M, Fredman G, Schwab JM, Chiang N, Arita M, Goodarzi A, Cheng G, von Andrian UH, and Serhan CN. 2008. Resolvin E1, an EPA-derived mediator in whole blood, selectively counter regulates leukocytes and platelets. Blood. 112: 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulsen RC, Gotlinger KH, Serhan CN, Kruger MC. 2008. Identification of inflammatory and proresolving lipid mediators in bone marrow and their lipidomic profiles with ovariectomy and omega-3 intake. Am. J. Hematol 83: 437–445. [DOI] [PubMed] [Google Scholar]
- 29.Ramon S, Gao F, Serhan CN, Phipps RP. 2012. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J Immunol. 189: 1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramon S, Baker SF, Sahler JM, Kim N, Feldsott EA, Serhan CN, Martínez-Sobrido L, Topham DJ, Phipps RP. 2014. The specialized proresolving mediator 17-HDHA enhances the antibody-mediated immune response against influenza virus: a new class of adjuvant? J Immunol. 193: 6031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiurchiù V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, Serhan CN. 2016. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med. 8: 353ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. 2000. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol.164: 1663–1667. [DOI] [PubMed] [Google Scholar]
- 33.Fierro IM, Colgan SP, Bernasconi G, Petasis NA, Clish CB, Arita M, Serhan CN. 2003. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J Immunol. 170: 2688–2694. [DOI] [PubMed] [Google Scholar]
- 34.Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. 2002. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nature immunology. 3: 76–82. [DOI] [PubMed] [Google Scholar]
- 35.Ariel A, Chiang N, Arita M, Petasis NA, Serhan CN. 2003. Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J Immunol. 170: 6266–6272. [DOI] [PubMed] [Google Scholar]
- 36.Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, Pinho V, Nicoli JR, Vieira LQ, Fierro IM, Teixeira MM. 2007. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol. 179: 8533–8543. [DOI] [PubMed] [Google Scholar]
- 37.Hwang JW, Sundar IK, Yao H, Sellix MT, and Rahman I. 2014. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J. 28: 176–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, Colgan SP, Petasis NA, and Serhan CN. 2007. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. 282(13): 9323–9334. [DOI] [PubMed] [Google Scholar]
- 39.Lakatos HF, Burgess HA, Thatcher TH, Redonnet MR, Hernady E, Williams JP, Sime PJ. 2006. Oropharyngeal aspiration of a silica suspension produces a superior model of silicosis in the mouse when compared to intratracheal instillation. Exp Lung Res. 32: 181–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badr WH, Loghmanee D, Karalus RJ, Murphy TF, and Thanavala Y. 1999. Immunization of mice with P6 of nontypeable Haemophilus influenzae: kinetics of the antibody response and IgG subclasses. Vaccine. 18: 29–37. [DOI] [PubMed] [Google Scholar]
- 41.Goldklang MP, Marks SM, D'Armiento JM. 2013. Secondhand smoke and COPD: lessons from animal studies. Front Physiol. 4: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laskin DL 2009. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem. Res. Toxicol 22: 1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorley AJ, Tetley TD. 2007. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis 2: 409–428. [PMC free article] [PubMed] [Google Scholar]
- 44.Wark PA, Tooze M, Powell H, Parsons K. 2013. Viral and bacterial infection in acute asthma and chronic obstructive pulmonary disease increases the risk of readmission. Respirology. 18: 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McEachern EK, Hwang JH, Sladewski KM, Nicatia S, Dewitz C, Mathew DP, Niszet V, and Alexander L. E. Crotty. 2015. Analysis of the effects of cigarette smoke on staphylococcal virulence phenotypes. Infect Immun. 83: 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, Petasis NA, Levy BD, Serhan CN, and Van Dyke TE. 2006. RvE1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB J. 20: 401–403. [DOI] [PubMed] [Google Scholar]
- 47.Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, Israel E, Haley KJ, and Serhan CN. 2007. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J. Immunol 178: 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin Y, Arita M, Zhang Q, Saban DR, Chauhan SK, Chiang N, Serhan CN, and Dana R. 2009. Anti-angiogenesis effect of the novel anti-inflammatory and pro-resolving lipid mediators. Invest. Ophthalmol. Vis. Sci 50: 4743–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y, Hao H, He S, Cai L, Li Y, Hu S, Ye D, Hoidal J, Wu P, and Chen X. 2010. Lipoxin A4 and its analogue suppress the tumor growth of transplanted H22 in mice: the role of anti-angiogenesis. Mol. Cancer Ther 9: 2164–2174. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Zhang X, Wu P, Li H, Jin S, Zhou X, Li Y, Ye D, Chen B, and Wan J. 2008. BML-111, a lipoxin receptor agonist, modulates the immune response and reduces the severity of collagen-induced arthritis. Inflamm. Res 57: 157–162. [DOI] [PubMed] [Google Scholar]
- 51.Zhang B, Jia H, Liu J, Yang Z, Jiang T, Tang K, Li D, Huang C, Ma J, Shen GX, et al. 2010. Depletion of regulatory T cells facilitates growth of established tumors: a mechanism involving the regulation of myeloid-derived suppressor cells by lipoxin A4. J. Immunol 185: 7199–7206. [DOI] [PubMed] [Google Scholar]
- 52.Hsiao HM, Thatcher TH, Colas RA, Serhan CN, Phipps RP, and Sime PJ. 2015. Resolvin D1 Reduces Emphysema and Chronic Inflammation. Am J Pathol. 185: 3189–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.U. S. Food and Drug Administration. 2013. Common Ingredients in U.S. Licensed Vaccines. http://www.fda.gov. [Google Scholar]
- 54.Weinberger B 2018. Vaccines for the elderly: current use and future challenges. Immun Ageing. 15: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Löbermann M, Boršo D, HilgendorfI I, Fritzsche C, Zettl UK, and Reisinger EC. 2012. Immunization in the adult immunocompromised host. Autoimmun Rev. 11: 212–218. [DOI] [PubMed] [Google Scholar]
- 56.Oke J, and Heneghan C 2020. Global Covid-19 Case Fatality Rates. Oxford COVID-19 Evidence Service. Centre for Evidence-Based Medicine. March 17, 2020. (updated 26th May 2020). Accessed from https://www.cebm.net/covid-19/global-covid-19-case-fatality-rates/ [Google Scholar]
- 57.Severe Outcomes Among Patients with Coronavirus Disease 2019 (COVID-19)-United States, February 12-March 16, 2020. MMWR Morb Mortal Wkly Rep 2020;69:343–346. DOI: 10.15585/mmwr.mm6912e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panigrahy D, Gilligan MM, Huang S, Gartung A, Cortés-Puch I, Sime PJ, Phipps RP, Serhan CN, and Hammock BD. 2020. Inflammation resolution: a dual-pronged approach to averting cytokine storms in COVID-19? Cancer Metastasis Rev. 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dalli J, Chiang N, and Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. 2015. Nat Med 21: 1071–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, and Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. 2012. Nature 484: 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.López-Muñoz RA, Molina-Berríos A, Campos-Estrada C, Abarca-Sanhueza P, Urrutia-Llancaqueo L, Peña-Espinoza M, and Maya JD. Inflammatory and pro-resolving lipids in Trypanosomatid Infections: A key to understanding parasite control. 2018. Front Microbiol. 9: 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H, Anthony D, Selemidis S, Vlahos R, and Bozinovski S. Resolving viral-induced secondary bacterial infection in COPD: A Concise Review 2018. Front Immunol. 9: 2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dalli J, Colas RA, Quintana C, Barragan-Bradford D, Hurwitz S, Levy BD, Choi AM, Serhan CN, and Baron RM. Human sepsis eicosanoid and proresolving lipid mediator temporal profiles: Correlations with survival and clinical outcomes. 2017. Crit Care Med. 45: 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.