

Abstract
Severity of cardiovascular disease increases markedly in elderly patients. In addition, many therapeutic strategies that decrease cardiac injury in adult patients are invalid in elderly patients. Thus, it is a challenge to protect the aged heart in the context of underlying chronic or acute cardiac diseases including ischemia-reperfusion injury. The cause(s) of this age-related increased damage remain unknown. Aging impairs the function of the mitochondrial electron transport chain (ETC), leading to decreased energy production and increased oxidative stress due to generation of reactive oxygen species (ROS). Additionally, ROS-induced oxidative stress can increase cardiac injury during ischemia-reperfusion by potentiating mitochondrial permeability transition pore (MPTP) opening. Aging leads to increased endoplasmic reticulum (ER) stress, which contributes to mitochondrial dysfunction, including reduced function of the ETC. The activation of both cytosolic and mitochondrial calcium-activated proteases termed calpains leads to mitochondrial dysfunction and decreased ETC function. Intriguingly, mitochondrial ROS generation also induces ER stress, highlighting the dynamic interaction between mitochondria and ER. Here, we discuss the role of ER stress in sensitizing and potentiating mitochondrial dysfunction in response to ischemia-reperfusion, and the promising potential therapeutic benefit of inhibition of ER stress and / or calpains to attenuate cardiac injury in elderly patients.
Keywords: Aging, ER stress, ischemia-reperfusion, calpain, mitochondria
1. Introduction
Cardiac mitochondria are the primary source to meet the energy demand for a constantly beating heart (Lesnefsky et al., 2016). There are two distinct populations of cardiac mitochondria: subsarcolemmal mitochondria (SSM), which reside under the sarcolemma, and interfibrillar mitochondria (IFM), present between myofibrils (Palmer et al., 1985). Although SSM are more sensitive to ischemia-mediated mitochondrial damage (Lesnefsky et al., 1997), IFM are the population mainly affected in aged hearts (Fannin et al., 1999). Aging-induced mitochondrial defects lead to decreased ATP production and increased ROS generation that potentiate cardiac injury during ischemia-reperfusion (Lesnefsky et al., 2001a). Moreover, most cardiac protective strategies are relatively ineffective in aged hearts (Peart et al., 2014). This review focuses on the potential of novel approaches targeting ER stress and calpain activation to decrease age-related exacerbation of cardiac injury following ischemia-reperfusion.
2. Mitochondrial defects in the Aged Heart
2.1. ETC damage in aged hearts
A detailed description of aging-induced mitochondrial defects can be found in several excellent reviews (Lesnefsky et al., 2016; Lesnefsky et al., 2017; Lesnefsky et al., 2001c). In brief, the rate of oxidative phosphorylation is decreased in IFM isolated from 24 month old Fisher 344 rat hearts. In contrast, oxidative phosphorylation is not markedly altered in SSM from aged rat hearts (Fannin et al., 1999). Direct measurement of enzyme activities shows that aging leads to decreased activities of electron transport chain (ETC) complex III and IV in IFM (Fannin et al., 1999; Lesnefsky et al., 2001b; Moghaddas et al., 2003). In C57BL/6 mice, oxidative phosphorylation is also decreased in aged IFM (Chen et al., 2020), with reduced substrate oxidization by complex I, II, III, and IV, consistent with findings in aged rat IFM. Interestingly in mice, oxidative phosphorylation is also decreased in aged SSM, with reduced substrate oxidation by complex I but not complex II, III, and IV, indicating a potential complex I-specific defect in aged SSM (Chen et al., 2020; Fernandez-Sanz et al., 2014). The complex I activity is decreased in human atrial tissue from aged patients compared to young patients (Emelyanova et al., 2018). Expression of genes encoding for NADH dehydrogenase subunits including NDUFA6, NDUFA9, NDUFB5, NDUFB8, and NDUFS2 are decreased in aged human atrial tissue (Emelyanova et al., 2018). The activity of cytochrome oxidase is also decreased in aged human left ventricular tissue compared to adult patients (Marin-Garcia et al., 1998). However, it is also reported that aging does not alter the activities of mitochondrial respiratory enzymes in cardiac muscle obtained from organ donors (Miró et al., 2000). Thus, issues regarding aging-induced mitochondrial defects in human hearts need to be further clarified.
2.2. Disruption of supercomplexes in aged heart mitochondria
In addition to defects in the individual ETC complexes, a disruption of supercomplexes also contributes to decreased oxidative phosphorylation in aged hearts (Gomez et al., 2009). Supercomplexes are large macromolecular assemblies consisting of complexes I, III and IV in ratio of 1:2:1. In supercomplexes (I1III2IV1), electrons from NADH can quickly transit through the closely connected complex I, coenzyme Q, complex III, cytochrome c, and complex IV and cytochrome c to reduce oxygen (Genova and Lenaz, 2015; Rosca et al., 2011). Supercomplexes increase the efficiency of electron transport and thus decrease ROS generation by reducing electron leakage (Genova and Lenaz, 2014; Rosca et al., 2011). Compared to young rats, supercomplexes are decreased mainly in IFM (Genova and Lenaz, 2015; Gomez et al., 2009). In human skin fibroblasts from centenarians, the complex I-driven ATP synthesis is decreased and H2O2 generation is increased compared to 75 or 27 year old. However, the supercomplexes are not markedly altered in skin fibroblasts from centenarians (Sgarbi et al., 2014). It remains unclear if aging alters supercomplexes in cardiac mitochondria. Age-related reduction in cardiolipin content contributes to decreased supercomplexes in mitochondria (Gomez et al., 2009; Szeto and Liu, 2018). Peroxidation of cardiolipin is also increased in aged hearts that may also affect supercomplex assembly in aged heart mitochondria (Szeto and Liu, 2018). Although cardiolipin deficiency is known to induce mitochondrial dysfunction in Barth syndrome patients (Ghosh et al., 2019), there are few studies regarding cardiolipin alteration in aged human heart tissue. Recent studies show that ER stress contributes to supercomplexes formation. Induction of ER stress through glucose deprivation facilitates supercomplexes assembly by activating the PERK-eIF2a-ATF4 axis activating supercomplexes assembly factor 1 (Balsa et al., 2019; Quintana-Cabrera and Soriano, 2019). This is consistent with the notion that initial ER stress is an adaptive reaction to restore cell function. However, more severe ER stress results in mitochondrial dysfunction (Chen et al., 2017), suggesting prolonged ER stress in the aged heart may dismantle supercomplexes in aged heart mitochondria. Thus, attenuation of ER stress in aged hearts may improve mitochondrial oxidative phosphorylation by promoting supercomplexes assembly (Figure 1).
Figure 1: Schematic of mitochondrial defect in the aged hearts.
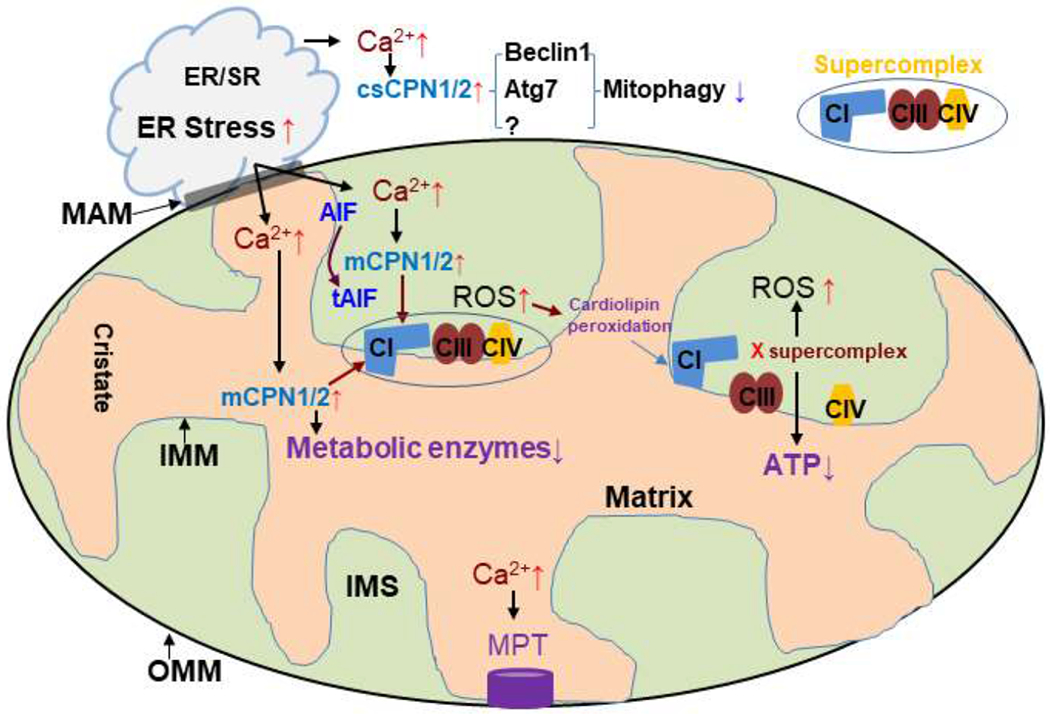
Mitochondria and ER are closely connected through MAM (mitochondria associated membranes). ER stress leads to increased cytosolic calpain1 and 2 (csCPN1/2) activities that cleave beclin1 and Atg7 (autophagy protein 7). The decreased contents of beclin1 and Atg7 impair autophagosome formation that decreases mitophagy. ER stress-induced mitochondrial calcium overload activates mitochondrial calpain 1 and 2 (mCPN1/2) that leads to the degradation of metabolic enzymes and protein subunits of the respiratory complexes. The damaged individual complexes including complex I (CI), III (CIII), and IV (CIV) result in the disruption of supercomplexes. Calcium overload also triggers MPTP opening in aged heat mitochondria. (OMM, mitochondrial outer membrane; IMM, mitochondrial inner membrane; IMS, intermembrane space).
3. Potential mechanisms of the aging-induced mitochondrial defect
3.1. Role of oxidative stress in mitochondrial damage during aging
Oxidative stress plays a key role in aging-induced mitochondrial dysfunction (Moghaddas et al., 2003). Over expression of mitochondria-targeted catalase protects aged hearts, supporting the idea that mitochondrial ROS generation contributes to the mitochondrial dysfunction observed in aged hearts (Bhuiyan and Fukunaga, 2009). The ETC is a key source of ROS generation in cardiac mitochondria (Chen et al., 2003). Decreased complex III activity contributes to increased ROS generation in aged IFM from rat hearts (Barja, 1998; Kwong and Sohal, 1998; Moghaddas et al., 2003; Skulachev, 2001). In addition to complex III, complex I and II are also potential sites of ROS production (Han et al., 2003; Sugioka et al., 1988; Turrens, 2003). There are two ways to generate ROS from complex I, either forward or reverse electron transport-mediated (Brand and Esteves, 2005; Ross et al., 2013). In order to increase ROS generation from complex I through forward electron flow, complex I needs to be kept in the reduced condition with complex I substrate to generate NADH that donates electron into complex I (Kushnareva et al., 2002). Thus, an increase in the forward electron flow-induced ROS generation usually occurs in mitochondria with severe damage in complex I (Chen and Lesnefsky, 2008). Ischemia-mediated complex I damage increases forward flow-mediated ROS generation in isolated mitochondria (Chen and Lesnefsky, 2008). A cytochrome oxidase defect also increases the forward flow-induced ROS generation from complex I (Chen and Lesnefsky, 2006). Additionally, genetic down-regulation of complex I subunit NDUFA11 decreases supercomplexes formation (Jang and Javadov, 2018). The destabilization of supercomplexes increases ROS generation from complex I (Maranzana et al., 2013). Thus, a decreased content of supercomplexes present in aged heart mitochondria may increase ROS generation from complex I. The reverse flow-induced ROS generation from complex I is dependent on inner mitochondrial potential with complex II substrate to generate FADH2 that provides electron flow from complex II to complex I. Depolarization of the inner mitochondrial potential (Brand and Esteves, 2005; Ross et al., 2013) or blockade of electron flow from complex II to I leads to decreased ROS generation from the reverse flow (Chen et al., 2008; Ross et al., 2013). Reverse flow-induced ROS generation contributes to cell injury during ischemia-reperfusion (Chouchani et al., 2014; Pell et al., 2016). However, it is unclear if aging leads to increased ROS generation from the reverse electron flow. In addition to direct complex I damage, destabilization of supercomplexes also increases ROS generation from complex I (Jang and Javadov, 2018; Maranzana et al., 2013). Aging leads to destabilized supercomplexes (Gomez et al., 2009) that may also increase ROS generation from complex I. The potential role of reverse flow-induced ROS generation in aged heart mitochondria needs further studied.
In addition to the ETC, ROS can be generated by other mitochondrial located proteins including p66shc and monoamine oxidase. The p66shc is a redox enzyme located in the mitochondrial intermembrane space. The p66shc generates H2O2 through the oxidation of cytochrome c (Giorgio et al., 2005). Ablation of p66shc increases the resistance to oxidative stress and extends the life span of mice (Migliaccio et al., 1999). Monoamine oxidase is a flavoenzyme located in the outer mitochondrial membrane. Hydrogen peroxide is generated when neurotransmitters including norepinephrine, epinephrine, and dopamine are broken down by monoamine oxidase (Carpi et al., 2009). Inhibition of monoamine oxidase leads to decreased H2O2 generation in the aged heart (Maurel et al., 2003). These results indicate that p66shc and monoamine oxidase may contribute to mitochondrial dysfunction during aging (Lesnefsky et al., 2016).
3.2. ER stress and mitochondrial damage during aging
Mitochondria are closely connected with the ER through mitochondria associated membranes (MAM) (Wieckowski et al., 2009). The ER plays a critical role in protein folding, lipid synthesis, and calcium homeostasis (Sciarretta et al., 2013). Accumulation of misfolded or unfolded proteins within the ER leads to ER stress. Initial ER stress is an adaptive stress to restore the ER function by triggering unfolded protein response (UPR). The UPR activates signal transduction to restore ER homeostasis by upregulating ER chaperone gene expression and heat shock protein synthesis (Jong et al., 2017). However, persistence of the ER stress leads to cell death (Zhang and Ren, 2011). Chronic ER stress contributes to the pathophysiology of various cardiovascular diseases including ischemic heart disease (Jong et al., 2017). Thapsigargin-induced ER stress leads to decreased complex I activity, increased ROS generation, and increased MPTP opening (Chen et al., 2017; Chen et al., 2019a). Attenuation of ER stress using metformin treatment improves mitochondrial function in thapsigargin-treated mice (Chen et al., 2017), indicating that ER stress contributes to mitochondrial dysfunction (Chen et al., 2017; Chen et al., 2019a; Zhang and Ren, 2011). ER stress is progressively increased during aging (Chen et al., 2019b). The increased ER stress occurs earlier than mitochondrial dysfunction in aged mice. In addition, attenuation of ER stress with 4-phenylbutyrate (4-PBA), a chemical chaperone that stabilizes protein conformation in the ER (Basseri et al., 2009; Chen et al., 2020; Jian et al., 2016; Ozcan et al., 2006), improves mitochondrial function in the aged hearts (Chen et al., 2019b). These results indicate that ER stress contributes to mitochondrial dysfunction in aged hearts.
The ER is closely connected with the mitochondria through mitochondria associated membranes (Wieckowski et al., 2009). ER stress results in a disruption of intracellular calcium homeostasis that leads to mitochondrial calcium over load. An increase in mitochondrial calcium favors calcium-dependent protease activation including mitochondrial calpains (Li et al., 2018; Mohsin et al., 2020). The potential role of mitochondrial calpain activation in mitochondrial dysfunction during aging is discussed in Section 3.3. In addition to protease activation, mitochondrial dysfunction during aging can be due to decreased mitophagy, a key process to remove the damaged mitochondria (Gottlieb and Thomas, 2017; Gustafsson and Gottlieb, 2008a). ER stress contributes to decreased mitophagy (Wang et al., 2017) that is shown in aged hearts (Hoshino et al., 2013). Biogenesis is essential to replace the mitochondria removed by mitophagy (Di et al., 2018). Mitochondrial transcription factor A (TFAM) plays a key role in the regulation of mitochondrial biogenesis (Picca and Lezza, 2015). The content of TFAM is decreased in aged hearts (Chen et al., 2020), indicating the potential of decreased biogenesis during aging. Tunicamycin-induced ER stress leads to decreased expression of PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha) and TFAM (Prola et al., 2019), suggesting that ER stress results in decreased biogenesis through inhibition of TFAM. Thus, aging-induced ER stress may contribute to mitochondrial dysfunction by degrading subunits of the ETC, impairing mitophagy, and decreasing mitochondrial biogenesis.
3.3. Mitochondrial proteases and mitochondrial damage during aging
ER plays a key role in maintaining intracellular calcium homeostasis. ER stress leads to intracellular calcium overload that activates calcium-dependent proteases including cytosolic and mitochondrial calpains (Li et al., 2018). Activation of cytosolic calpains increases cardiac injury during ischemia-reperfusion (Cao et al., 2019; Inserte et al., 2008; Li et al., 2018). Activation of mitochondrial calpains contributes to the ETC damage during ischemia-reperfusion (Briston et al., 2019; Chen and Lesnefsky, 2015; Shintani-Ishida and Yoshida, 2015; Thompson et al., 2016). Mitochondrial calpain 1 (mCPN1) exists in the intermembrane space and the matrix of mitochondria (Briston et al., 2019; Thompson et al., 2016). Activation of mCPN1 in the intermembrane space cleaves apoptosis inducing factor (AIF) to truncated AIF which is released from the inner membrane to travel to the cytosol to promote caspase-independent apoptosis (Chen et al., 2011; Ozaki et al., 2007). AIF is a mitochondrial flavoprotein (Vahsen et al., 2004), and though not a component of complex I, reduction of AIF content in Harlequin mice leads to decreased complex I activity (Vahsen et al., 2004). Activation of mCPN1 also leads to the degradation of complex I subunits including NDUFS7 (Briston et al., 2019) and metabolic enzymes including pyruvate dehydrogenase (Thompson et al., 2016), ATP5A1 protein and ATP synthase activity in cardiac mitochondria (Cao et al., 2019), and promotes opening of the MPTP (Shintani-Ishida and Yoshida, 2015). Activation of mitochondrial calpain 2 (mCPN2) cleaves complex I subunit ND6 (Shintani-Ishida and Yoshida, 2015). Taken together, activation of mCPN1/2 leads to decreased complex I activity by cleaving AIF and degrading complex I subunits. Complex I is a key component of supercomplexes (Rosea et al., 2011), and ER stress can disrupt supercomplex formation by inducing complex I damage (Hou et al., 2019; Jang and Javadov, 2018). Thus, ER stress contributes to mitochondrial dysfunction in aged hearts by decreasing individual complex activity and possibly the formation of supercomplexes (Figure 1).[We didn’t show this; you make a deductive reasoning case for this but this has not been proved? Correct?; perhaps you should test in your R01?]
3.4. Mitophagy and mitochondrial dysfunction during aging
Changing mitochondrial dynamics are also involved in the aging process (Biala et al., 2015; Lesnefsky et al., 2016) and have been discussed recently in many excellent reviews (Biala et al., 2015; Costantino et al., 2016; Lesnefsky et al., 2016; Maggiorani et al., 2017; Marin-Garcia et al., 2013). Damaged mitochondria are constantly removed in an autophagic process termed mitophagy (Gottlieb and Carreira, 2010; Gustafsson and Gottlieb, 2008b; Liang et al., 2020). Impaired mitophagy contributes to mitochondrial defects in aged hearts (Hoshino et al., 2013). However, it is unclear if ER stress and / or calpain activation contributes to decreased mitophagy during aging. Beclin1 and autophagy protein 7 (Atg7) are key early components of autophagosome formation (Kim et al., 2013). Interestingly, both Beclin1 and Atg7 are substrates of cytosolic caspases cCPN1/2 (Kim et al., 2008). Activation of cCPN1/2 during ISCHEMIA-REPERFUSION leads to decreased levels of beclin1 and Atg7 (Kim et al., 2008; Kim et al., 2013). Reduced formation of autophagosomes leads to decreased mitophagy and an increased content of damaged mitochondria that likely exacerbate ROS generation. Thus, aging may contribute to cardiac damage via decreased mitophagy through enhanced ER stress-induced activation of cCPN1/2 activity (Figure 1).[good!]
4. Strategic approaches to decrease cardiac injury in aged hearts
4.1. Impairment of endogenous signal transduction in aged hearts
Aged hearts are more susceptible to cardiac injury during ISCHEMIA-REPERFUSION (D’Annunzio et al., 2016; Lesnefsky et al., 1994; Lesnefsky et al., 1996). Classical cardioprotective approaches including ischemic pre/post-conditioning are not effective in aged hearts (D’Annunzio et al., 2016; Wojtovich et al., 2012). Ischemic preconditioning (IPC) activates a number of protective signaling cascades that eventually converge at the mitochondria. The potential mechanisms underlying the loss of IPC protection in the aged heart involve defective IPC signal transduction (Wojtovich et al., 2012), disruption of redox signaling, defects in the ETC (Choksi et al., 2007), and decreased autophagy and mitophagy processes (Huang et al., 2011). Similarly, pharmacologic conditioning strategies, including anesthetic pre/post-conditioning, are also less effective in the aged hearts (Li et al., 2013; Sniecinski and Liu, 2004), though the molecular mechanisms are less well understood.
The loss of endogenous protective mechanisms can be restored in the aged hearts with caloric restriction (Abete et al., 2002) and exercise (Abete et al., 2005). Caloric restriction improves oxidative phosphorylation and complex I activity in aged heart mitochondria (Niemann et al., 2010). In addition, mitochondrial bioenergetics is improved with caloric restriction by reducing oxidative stress and activating sirtuins (Shinmura, 2013). Studies show that sirtuin 1 (Adam et al., 2013) and sirtuin 3 (Quan et al., 2017; Quan et al., 2018) are activated and decrease cardiac injury in aged hearts by caloric restriction, and supplementation with resveratrol, a sirtuin 1 agonist, is also protective. Furthermore, exercise has been shown to restore IPC effectiveness in aged Wistar rat hearts by improving mitochondrial function and the cardiac polyamine pool (Wang et al., 2014). Polyamines, which are decreased with aging, play a key role in promoting cell growth and differentiation and also function as ROS scavengers. Intriguingly, recent studies show that regular exercise decreased the negative consequences of ER stress in aged mice (Belaya et al., 2018), and resveratrol supplementation reduced the ER stress response in the middle aged mice (Pang et al., 2019). Thus, exercise or resveratrol could reverse mitochondrial defects in aged hearts by decreasing ER stress. An impaired autophagic response contributes to decreased protection by IPC (Huang et al., 2011). The decreased mitophagy also contributes to decreased isoflurane-induced anesthetic preconditioning in aged hearts (Huang et al., 2011). Similarly, the protection of isoflurane was restored in aged hearts by improving mitophagy through 4-hydroxy TEMPO treatment (Ma et al., 2017). These results support the idea that age-related, ER stress-induced mitochondrial defects contribute to the loss of endogenous protective mechanisms in aged hearts.
4.2. Direct modulation of mitochondrial function decreases cardiac injury in aged hearts
Mitochondrial dysfunction increases cardiac injury during reperfusion by enhancing ROS generation and sensitizing to MPTP opening (Weiss et al., 2003). Blockade of electron transport at proximal but not distal sites decreased cardiac injury during ischemia-reperfusion (Chen et al., 2010). Reversible inhibition of complex I using amobarbital decreased cardiac injury in isolated adult rat (Chen et al., 2006), guinea pig (Aldakkak et al., 2008) and rabbit (Ambrosio and Flaherty, 1992) hearts. The administration of amobarbital at the onset of reperfusion also attenuates cardiac injury in adult rat (Stewart et al., 2009) and mouse hearts (Xu et al., 2014). Genetic modulation of complex I activity also leads to decreased cardiac injury in adult mouse hearts (Szczepanek et al., 2012). The mechanisms of protection provided by amobarbital involve decreased mitochondrial calcium loading (Aldakkak et al., 2008), less ROS generation (Ambrosio and Flaherty, 1992; Chen et al., 2006), preservation of BCL-2 content in mitochondria (Chen and Lesnefsky, 2011), and inhibition of MPTP opening (Chen et al., 2012a). The blockade of electron transport also decreased cardiac injury in aged hearts either given before ischemia (Tanaka-Esposito et al., 2012) or during early reperfusion (Chen et al., 2012b).
In addition to manipulation of mitochondrial respiration, administration of the agonists of the mitochondrial potassium-ATP channel also decreased cardiac injury in aged hearts (Jahangir et al., 2001; Tani et al., 2001). Cariporide (a sodium-proton exchange inhibitor) treatment also decreased cardiac injury in 24 month old Wistar rat hearts following ischemia-reperfusion by decreasing intracellular calcium content (Besse et al., 2004). Sphingosine 1 phosphate (S1P), a lipid mediator contributing to decreased cardiac injury during ischemia-reperfusion, was decreased in aged rat hearts (Vessey et al., 2009). Supplementation of S1P or administration of sphingosine decreases cardiac injury in aged hearts (Vessey et al., 2009). These studies show that therapeutic interventions bypassing endogenous signal transduction are able to decrease cardiac injury in aged hearts during ischemia-reperfusion.
Reduction of calcium loading decreases cardiac injury during ischemia-reperfusion (Aldakkak et al., 2008; Besse et al., 2004). Ischemia-reperfusion activates calcium-dependent proteases including mitochondrial and cytoplasmic calpains (Chen et al., 2011; Inserte et al., 2008; Thompson et al., 2016). Inhibition of calpains using phamarcologic (Briston et al., 2019; Chen et al., 2011; Shintani-Ishida and Yoshida, 2015; Thompson et al., 2016) or genetic (Cao et al., 2019) approaches decreased cardiac injury by protecting mitochondria during ischemia-reperfusion. Modulation of calpain activivity during ischemia-reperfusion could be a potential approach to decrease cardiac injury in aged hearts.
4.3. Reversal of the mitochondrial defects to decrease cardiac injury
Since the occurrence of myocardial infarction is difficult to predict in patients, the current translational approach is to apply therapeutic interventions at the onset of reperfusion to decrease cardiac injury. However, these approaches have limited beneficial effect in that damage that occurred during ischemia cannot be prevented (Lesnefsky et al., 2004). Mitochondrial dysfunction occurs in aged hearts under basal conditions (Lesnefsky et al., 2001c). The impaired mitochondrial function makes a number of protective approaches ineffective in aged hearts (Downey and Cohen, 2009; Peart et al., 2014). Therefore, the optimal approach to decrease cardiac injury in aged hearts would be to restore mitochondrial function to the “younger” state before a heart attack (Lesnefsky et al., 2006). Reversal of the age-related mitochondrial defects has the potential to not only attenuate mitochondrial dysfunction-induced cardiac injury, but also enhance the protective effect of traditional cardioprotective approaches in aged hearts. Thus, understanding the mechanism of mitochondria dysfunction in aged hearts and developing proper therapeutic strategies are critical to decrease cardiac injury.
Aging impairs the ETC and leads to decreased activities of complex III and IV (Fannin et al., 1999). Treatment of aged rats with acetylcarnitine improved oxidative phosphorylation and the activities of complex III and IV (Lesnefsky et al., 2006; Paradies et al., 1992). Interestingly, administration of acetylcarnitine in the 24 month old Fisher 344 rat in vivo three hours before ischemia significantly decreased cardiac injury in aged hearts following ex vivo ischemia-reperfusion (Lesnefsky et al., 2006) (Figure 2). Since acetylcarnitine was not included in the perfusion buffer, the decreased injury in aged hearts is most likely due to improved mitochondrial function before ischemia (Lesnefsky et al., 2006). Recent study shows that restoration of mitochondrial function with SS-31 treatment improves cardiac function in aged mice (Chiao et al., 2020). These results support the notion that improving mitochondrial function in aged hearts prior to injury is a promising approach to mitigate cardiac injury during ischemia-reperfusion.
Figure 2: Potential approaches to decrease cardiac injury in the aged hearts.
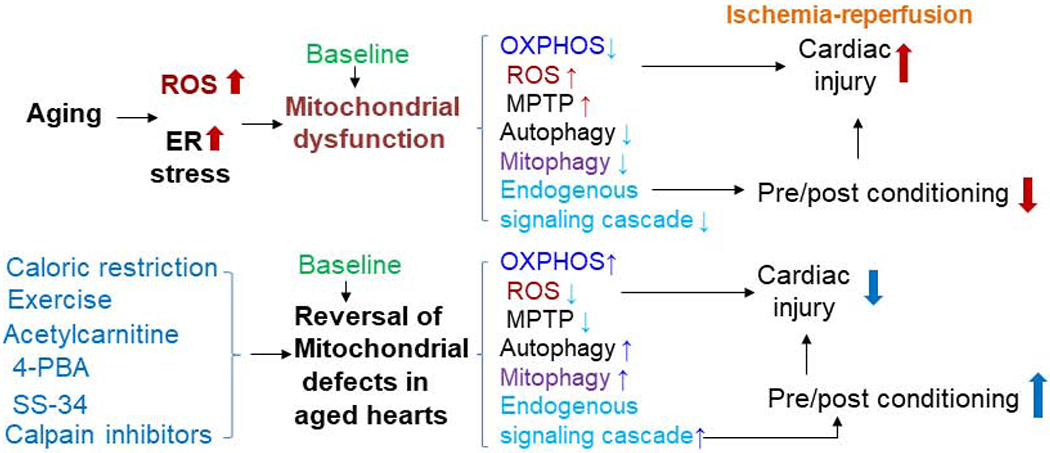
The mitochondrial defect increases cardiac injury in the aged hearts by decreasing oxidative phosphorylation, increasing ROS formation, sensitizing to MPTP opening and decreasing autophagy and mitophagy. The impaired endogenous signal transduction leads to decreased protection from approaches depending on signaling activation. Restoration of the mitochondrial defect in the aged hearts in the baseline condition by using acetylcarnitine or 4-PBA (4-phenylbutyric acid) decreases cardiac injury during ischemia-reperfusion. In addition, restoration of the mitochondrial defect in the aged hearts may decrease cardiac injury by reviving classic therapeutic approaches. Treatment with calpain inhibitors is another potential approach to restore mitochondrial function in the aged heats.
Recent studies show that ER stress also contributes to mitochondrial dysfunction in aged hearts (Chen et al., 2020; Chen et al., 2019b). For example, increased ER stress impaired the mitochondrial respiratory chain in cardiac mitochondria (Chen et al., 2017; Chen et al., 2019a; Zhang and Ren, 2011). Attenuation of ER stress using 4-PBA treatment for two weeks improves mitochondrial function in aged mouse hearts (Chen et al., 2020). Feeding aged mice with metformin for two weeks also leads to decreased ER stress and improved mitochondrial function (Chen et al., 2019b). In addition, metformin feeding also decreased subsequent superimposed cardiac injury in aged mouse hearts (Chen et al., 2019b). These results support that notion that restoration of mitochondrial function in aged hearts decreases cardiac injury. It will be interesting to study if metformin treatment that improves mitochondrial function can restore other cardio-protective interventions, including pre/post-conditioning, in the aged hearts during ischemia-reperfusion. Moreover, the combination of baseline treatments and acute interventions during ischemia-reperfusion may decrease cardiac injury in the high risk aged hearts (Chen et al., 2020; D’Annunzio et al., 2016; Lesnefsky et al., 2016; Lesnefsky et al., 2017; Lesnefsky et al., 1994; Lesnefsky et al., 2006; Lesnefsky et al., 1996) (Figure 2).
Aging leads to impaired autophagy that nullifies a number of protective approaches in the aged hearts (Huang et al., 2011). Activation of calpains led to decreased autophagy during ischemia-reperfusion by calpain-dependent cleavage of beclin1 and ATG7 (Kim et al., 2008; Kim et al., 2013). A decrease in Atg9b content contributes to decreased mitophagy in aged hearts (Liang et al., 2020). However, it is unclear if Atg9b is a potential substrate of calpains. Thus, increased ER stress activates cytosolic and mitochondrial calpains that may impair autophagy. Attenuation of ER stress may be a potential strategy to improve autophagy and mitophagy in the aged heart.
We and others have shown that administration of calpain inhibitors decreased cardiac injury during ischemia-reperfusion by protecting mitochondria (Briston et al., 2019; Chen et al., 2002; Chen et al., 2011; Inserte et al., 2008), especially complex I (Briston et al., 2019). Indeed, an increase in mCPN1 content was demonstrated to lead to dilated heart failure (Cao et al., 2019). Moreover, inhibition of calpain decreased the sensitivity to ischemia-reperfusion injury in diabetic hearts by restoring mitochondrial function and autophagy (Ong et al., 2019). Thus, modulation of calpain activity during aging may be a promising strategy to decrease cardiac injury by restoring mitochondrial function and mitophagy (Ong et al., 2019). Acetylcarnitine treatment in vivo eliminates mitochondrial defects in aged rat heart mitochondria (Lesnefsky et al., 2006). Recent study shows that the attenuation of the ER stress with 4-PBA treatment also improves mitochondrial function in aged mouse hearts (Chen et al., 2020) (Figure 2). Since disruption of supercomplexes contributes to mitochondrial defects during aging, it will be interesting to study the role of acetylcarnitine (Lesnefsky et al., 2006), ER stress reduction (Chen et al., 2020), and / or calpain inhibition (Chen and Lesnefsky, 2015) can restore supercomplexes integrity in aged heart mitochondria.
5. Summary
The presence of dysfunctional mitochondria in aged hearts increases cardiac injury during ischemia-reperfusion. Targeting mitochondrial function in aged hearts by acute intervention at the onset of reperfusion can decrease cardiac injury during ischemia-reperfusion. Obviously, a better approach is to restore mitochondrial function in aged hearts before injury. This approach not only eliminates mitochondrial dysfunction-induced cell injury, but also may enable traditional therapeutic approaches to begin working again in the aged hearts. Current evidence suggests that reduction of ER stress and calpain activation can protect mitochondria during ischemia-reperfusion injury. Thus, the optimal approach to decrease cardiac injury in aged hearts are to both restore basally and acutely damaged mitochondrial function in aged hearts through attenuation of ER stress and concomitant calpain activation during ischemia-reperfusion.
Highlights:
Aging leads to increased ER stress that contributes to mitochondrial dysfunction
ER stress-mediated calpain activation impairs the mitochondrial respiratory chain
The damaged respiratory chain leads to increased injury in aged hearts during ischemia-reperfusion
Intervention to restore mitochondrial function in aged hearts decreases cardiac injury during ischemia-reperfusion
Acknowledgements
The critical review of this manuscript by Dr. Edward Lesnefsky (Pauley Heart Center, Virginia Commonwealth University and Medical Service, McGuire Veterans Affairs Medical Center) is greatly appreciated. This work was supported by R21 AG049461 (Q.C.) from the National Institute on Aging, the Department of Veterans Affairs Office of Research and Development, Medical Research Service Merit Review Award (21O1BX001355-01A2) (QC, EJL), and a Pauley Heart Center Pilot Project from Virginia Commonwealth University (Q.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
The authors state they have no conflict of interest or financial interests to disclose
References:
- Abete P, Testa G, Ferrara N, De Santis D, Capaccio P, Viati L, Calabrese C, Cacciatore F, Longobardi G, Condorelli M, Napoli C and Rengo F, 2002. Cardioprotective effect of ischemic preconditioning is preserved in food-restricted senescent rats. Am J Physiol Heart Circ Physiol. 282, H1978–87. [DOI] [PubMed] [Google Scholar]
- Abete P, Testa G, Galizia G, Mazzella F, Della Morte D, de Santis D, Calabrese C, Cacciatore F, Gargiulo G, Ferrara N, Rengo G, Sica V, Napoli C and Rengo F, 2005. Tandem action of exercise training and food restriction completely preserves ischemic preconditioning in the aging heart. Exp Gerontol. 40, 43–50. DOI: 10.1016/j.exger.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Adam T, Sharp S, Opie LH and Lecour S, 2013. Loss of cardioprotection with ischemic preconditioning in aging hearts: role of sirtuin 1? J Cardiovasc Pharmacol Ther. 18, 46–53. DOI: 10.1177/1074248412458723. [DOI] [PubMed] [Google Scholar]
- Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ and Camara AK, 2008. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc Res. 77, 406–15. [DOI] [PubMed] [Google Scholar]
- Ambrosio G and Flaherty JT, 1992. Effects of the superoxide radical scavenger superoxide dismutase, and of the hydroxyl radical scavenger mannitol, on reperfusion injury in isolated rabbit hearts. Cardiovasc Drugs Ther. 6, 623–32. [DOI] [PubMed] [Google Scholar]
- Balsa E, Soustek MS, Thomas A, Cogliati S, García-Poyatos C, Martín-García E, Jedrychowski M, Gygi SP, Enriquez JA and Puigserver P, 2019. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Mol Cell. 74, 877–890. e6 DOI: 10.1016/j.molcel.2019.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G, 1998. Mitochondrial free radical production and aging in mammals and birds. Ann N Y Acad Sci. 854, 224–38. [DOI] [PubMed] [Google Scholar]
- Basseri S, Lhotak S, Sharma AM and Austin RC, 2009. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J Lipid Res. 50, 2486–501. DOI: 10.1194/jlr.M900216-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaya I, Suwa M, Chen T, Giniatullin R, Kanninen KM, Atalay M and Kumagai S, 2018. Long-Term Exercise Protects against Cellular Stresses in Aged Mice. Oxid Med Cell Longev. 2018, 2894247 DOI: 10.1155/2018/2894247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse S, Tanguy S, Boucher F, Le Page C, Rozenberg S, Riou B, Leiris J and Swynghedauw B, 2004. Cardioprotection with cariporide, a sodium-proton exchanger inhibitor, after prolonged ischemia and reperfusion in senescent rats. Exp Gerontol. 39, 1307–14. DOI: 10.1016/j.exger.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Bhuiyan MS and Fukunaga K, 2009. Stimulation of sigma-1 receptor signaling by dehydroepiandrosterone ameliorates pressure overload-induced hypertrophy and dysfunctions in ovariectomized rats. Expert Opin Ther Targets. 13, 1253–65. DOI: 10.1517/14728220903264064. [DOI] [PubMed] [Google Scholar]
- Biala AK, Dhingra R and Kirshenbaum LA, 2015. Mitochondrial dynamics: Orchestrating the journey to advanced age. J Mol Cell Cardiol. 83, 37–43. DOI: 10.1016/j.yjmcc.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Brand MD and Esteves TC, 2005. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2, 85–93. [DOI] [PubMed] [Google Scholar]
- Briston T, Selwood DL, Szabadkai G and Duchen MR, 2019. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol Sci. 40, 50–70. DOI: 10.1016/j.tips.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Cao T, Fan S, Zheng D, Wang G, Yu Y, Chen R, Song LS, Fan GC, Zhang Z and Peng T, 2019. Increased calpain-1 in mitochondria induces dilated heart failure in mice: role of mitochondrial superoxide anion. Basic Res Cardiol. 114, 17 DOI: 10.1007/s00395-019-0726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpi A, Menabo R, Kaludercic N, Pelicci P, Di Lisa F and Giorgio M, 2009. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta. 1787, 774–80. [DOI] [PubMed] [Google Scholar]
- Chen M, Won DJ, Krajewski S and Gottlieb RA, 2002. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 277, 29181–6. [DOI] [PubMed] [Google Scholar]
- Chen Q and Lesnefsky EJ, 2006. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic Biol Med. 40, 976–82. [DOI] [PubMed] [Google Scholar]
- Chen Q and Lesnefsky EJ, 2008. Ischemic damage to the mitochondrial electron transport chain favors opening of the permeability transition pore. FASEB J. 22, E345 (abstract 750.6). [Google Scholar]
- Chen Q and Lesnefsky EJ, 2011. Blockade of electron transport during ischemia preserves bcl-2 and inhibits opening of the mitochondrial permeability transition pore. FEBS Lett. 585, 921–6. DOI: 10.1016/j.febslet.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q and Lesnefsky EJ, 2015. Heart mitochondria and calpain 1: Location, function, and targets. Biochim Biophys Acta. 1852, 2372–8. DOI: 10.1016/j.bbadis.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL and Lesnefsky EJ, 2006. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 319, 1405–12. [DOI] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL and Lesnefsky EJ, 2008. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 294, C460–6. [DOI] [PubMed] [Google Scholar]
- Chen Q, Paillard M, Gomez L, Li H, Hu Y and Lesnefsky EJ, 2012a. Postconditioning modulates ischemia-damaged mitochondria during reperfusion. J Cardiovasc Pharmacol. 59, 101–8. DOI: 10.1097/FJC.0b013e31823827cc. [DOI] [PubMed] [Google Scholar]
- Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A and Lesnefsky EJ, 2011. Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun. 415, 533–8. DOI: 10.1016/j.bbrc.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Ross T, Hu Y and Lesnefsky EJ, 2012b. Blockade of electron transport at the onset of reperfusion decreases cardiac injury in aged hearts by protecting the inner mitochondrial membrane. J Aging Res. 2012, 753949 DOI: 10.1155/2012/753949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Samidurai A, Thompson J, Hu Y, Das A, Willard B and Lesnefsky EJ, 2020. Endoplasmic reticulum stress-mediated mitochondrial dysfunction in aged hearts. Biochim Biophys Acta Mol Basis Dis. 1866, 165899 DOI: 10.1016/j.bbadis.2020.165899. [DOI] [PubMed] [Google Scholar]
- Chen Q, Thompson J, Hu Y, Das A and Lesnefsky EJ, 2017. Metformin attenuates ER stress-induced mitochondrial dysfunction. Transl Res. 190, 40–50. DOI: 10.1016/j.trsl.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Thompson J, Hu Y, Das A and Lesnefsky EJ, 2019a. Cardiac Specific Knockout of p53 Decreases ER Stress-Induced Mitochondrial Damage. Front Cardiovasc Med. 6, 10 DOI: 10.3389/fcvm.2019.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Thompson J, Hu Y and Lesnefsky EJ, 2019b. Chronic metformin treatment restores mitochondrial function in aged heart. Circulation Abstract (16092) [Google Scholar]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL and Lesnefsky EJ, 2003. Production of reactive oxygen species by mitochondria: Central role of complex III. J Biol Chem. 278, 36027–36031. [DOI] [PubMed] [Google Scholar]
- Chen Q, Yin G, Stewart S, Hu Y and Lesnefsky EJ, 2010. Isolating the segment of the mitochondrial electron transport chain responsible for mitochondrial damage during cardiac ischemia. Biochem Biophys Res Commun. 397, 656–60. DOI: 10.1016/j.bbrc.2010.05.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao YA, Zhang H, Sweetwyne M, Whitson J, Ting YS, Basisty N, Pino LK, Quarles E, Nguyen NH, Campbell MD, Zhang T, Gaffrey MJ, Merrihew G, Wang L, Yue Y, Duan D, Granzier HL, Szeto HH, Qian WJ, Marcinek D, MacCoss MJ and Rabinovitch P, 2020. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife. 9 e55513 DOI: 10.7554/eLife.55513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi KB, Nuss JE, Boylston WH, Rabek JP and Papaconstantinou J, 2007. Age-related increases in oxidatively damaged proteins of mouse kidney mitochondrial electron transport chain complexes. Free Radic Biol Med. 43, 1423–38. DOI: 10.1016/j.freeradbiomed.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T and Murphy MP, 2014. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 515, 431–435. DOI: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino S, Paneni F and Cosentino F, 2016. Ageing, metabolism and cardiovascular disease. J Physiol. 594, 2061–73. DOI: 10.1113/jp270538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Annunzio V, Perez V, Boveris A, Gelpi RJ and Poderoso JJ, 2016. Role of thioredoxin-1 in ischemic preconditioning, postconditioning and aged ischemic hearts. Pharmacol Res. 109, 24–31. DOI: 10.1016/j.phrs.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Di W, Lv J, Jiang S, Lu C, Yang Z, Ma Z, Hu W, Yang Y and Xu B, 2018. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr Issues Mol Biol. 28, 29–46. DOI: 10.21775/cimb.028.029. [DOI] [PubMed] [Google Scholar]
- Downey JM and Cohen MV, 2009. Why do we still not have cardioprotective drugs? Circ J. 73, 1171–7. [DOI] [PubMed] [Google Scholar]
- Emelyanova L, Preston C, Gupta A, Viqar M, Negmadjanov U, Edwards S, Kraft K, Devana K, Holmuhamedov E, O’Hair D, Tajik AJ and Jahangir A, 2018. Effect of Aging on Mitochondrial Energetics in the Human Atria. J Gerontol A Biol Sci Med Sci. 73, 608–616. DOI: 10.1093/gerona/glx160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO and Hoppel CL, 1999. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 372, 399–407. [DOI] [PubMed] [Google Scholar]
- Fernandez-Sanz C, Ruiz-Meana M, Miro-Casas E, Nunez E, Castellano J, Loureiro M, Barba I, Poncelas M, Rodriguez-Sinovas A, Vazquez J and Garcia-Dorado D, 2014. Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 5, e1573 DOI: 10.1038/cddis.2014.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genova ML and Lenaz G, 2014. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta. 1837, 427–43. DOI: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Genova ML and Lenaz G, 2015. The Interplay Between Respiratory Supercomplexes and ROS in Aging. Antioxid Redox Signal. 23, 208–38. DOI: 10.1089/ars.2014.6214. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Iadarola DM, Ball WB and Gohil VM, 2019. Mitochondrial dysfunctions in barth syndrome. IUBMB Life. 71, 791–801. DOI: 10.1002/iub.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F and Pelicci PG, 2005. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 122, 221–33. DOI: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Gomez LA, Monette JS, Chavez JD, Maier CS and Hagen TM, 2009. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch Biochem Biophys. 490, 30–5. DOI: 10.1016/j.abb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA and Carreira RS, 2010. Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol Cell Physiol. 299, C203–10. DOI: 10.1152/ajpcell.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA and Thomas A, 2017. Mitophagy and Mitochondrial Quality Control Mechanisms in the Heart. Curr Pathobiol Rep. 5, 161–169. DOI: 10.1007/s40139-017-0133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson AB and Gottlieb RA, 2008a. Heart mitochondria: gates of life and death. Cardiovasc Res. 77, 334–43. [DOI] [PubMed] [Google Scholar]
- Gustafsson AB and Gottlieb RA, 2008b. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 44, 654–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Antunes F, Canali R, Rettori D and Cadenas E, 2003. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 278, 5557–63. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T and Matoba S, 2013. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 4, 2308 DOI: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- Hou T, Zhang R, Jian C, Ding W, Wang Y, Ling S, Ma Q, Hu X, Cheng H and Wang X, 2019. NDUFAB1 confers cardio-protection by enhancing mitochondrial bioenergetics through coordination of respiratory complex and supercomplex assembly. Cell Res. 29, 754–766. DOI: 10.1038/s41422-019-0208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P and Gottlieb RA, 2011. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 6, e20975 DOI: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inserte J, Barba I, Hernando V, Abellan A, Ruiz-Meana M, Rodriguez-Sinovas A and Garcia-Dorado D, 2008. Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage. Cardiovasc Res. 77, 782–90. [DOI] [PubMed] [Google Scholar]
- Jahangir A, Ozcan C, Holmuhamedov EL and Terzic A, 2001. Increased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener. Mech Ageing Dev. 122, 1073–86. [DOI] [PubMed] [Google Scholar]
- Jang S and Javadov S, 2018. Elucidating the contribution of ETC complexes I and II to the respirasome formation in cardiac mitochondria. Sci Rep. 8, 17732 DOI: 10.1038/s41598-018-36040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian L, Lu Y, Lu S and Lu C, 2016. Chemical Chaperone 4-Phenylbutyric Acid Reduces Cardiac Ischemia/Reperfusion Injury by Alleviating Endoplasmic Reticulum Stress and Oxidative Stress. Med Sci Monit. 22, 5218–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong CJ, Ito T, Prentice H, Wu JY and Schaffer SW, 2017. Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis. Nutrients. 9 DOI: 10.3390/nu9080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Nitta T, Mohuczy D, O’Malley KA, Moldawer LL, Dunn WA Jr. and Behrns KE, 2008. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 47, 1725–36. DOI: 10.1002/hep.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Wang JH, Biel TG, Kim DS, Flores-Toro JA, Vijayvargiya R, Zendejas I and Behrns KE, 2013. Carbamazepine suppresses calpain-mediated autophagy impairment after ischemia/reperfusion in mouse livers. Toxicol Appl Pharmacol. 273, 600–10. DOI: 10.1016/j.taap.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnareva Y, Murphy AN and Andreyev A, 2002. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 368, 545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LK and Sohal RS, 1998. Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch Biochem Biophys. 350, 118–26. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Chen Q and Hoppel CL, 2016. Mitochondrial Metabolism in Aging Heart. Circ Res. 118, 1593–611. DOI: 10.1161/circresaha.116.307505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky EJ, Chen Q, Slabe TJ, Stoll MS, Minkler PE, Hassan MO, Tandler B and Hoppel CL, 2004. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Am J Physiol Heart Circ Physiol. 287, H258–67. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Chen Q, Tandler B and Hoppel CL, 2017. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu Rev Pharmacol Toxicol. 57, 535–565. DOI: 10.1146/annurev-pharmtox-010715-103335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky EJ, Gallo DS, Ye J, Whittingham TS and Lust WD, 1994. Aging increases ischemia-reperfusion injury in the isolated, buffer-perfused heart. J Lab Clin Med. 124, 843–51. [PubMed] [Google Scholar]
- Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ and Hoppel CL, 2001a. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys. 385, 117–28. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda_Saito M, Turkaly PJ and Hoppel CL, 2001b. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J. Mol. Cell. Cardiol. 33, 37–47. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, He D, Moghaddas S and Hoppel CL, 2006. Reversal of mitochondrial defects before ischemia protects the aged heart. Faseb J. 20, 1543–5. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Lundergan CF, Hodgson JM, Nair R, Reiner JS, Greenhouse SW, Califf RM and Ross AM, 1996. Increased left ventricular dysfunction in elderly patients despite successful thrombolysis: the GUSTO-I angiographic experience. J Am Coll Cardiol. 28, 331–7. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J and Hoppel CL, 2001c. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol. 33, 1065–89. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J and Hoppel CL, 1997. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol. 273, H1544–54. [DOI] [PubMed] [Google Scholar]
- Li H, Zhou C, Chen D, Fang N, Yao Y and Li L, 2013. Failure to protect against myocardial ischemia-reperfusion injury with sevoflurane postconditioning in old rats in vivo. Acta Anaesthesiol Scand. 57, 1024–31. DOI: 10.1111/aas.12156. [DOI] [PubMed] [Google Scholar]
- Li S, Ma J, Li JB, Lacefield JC, Jones DL, Peng TQ and Wei M, 2018. Over-expression of calpastatin attenuates myocardial injury following myocardial infarction by inhibiting endoplasmic reticulum stress. J Thorac Dis. 10, 5283–5297. DOI: 10.21037/jtd.2018.08.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Moyzis AG, Lampert MA, Diao RY, Najor RH and Gustafsson Å,B, 2020. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell. e13187 DOI: 10.1111/acel.13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zhu J, Gao Q, Rebecchi MJ, Wang Q and Liu L, 2017. Restoring Pharmacologic Preconditioning in the Aging Heart: Role of Mitophagy/Autophagy. J Gerontol A Biol Sci Med Sci. 72, 489–498. DOI: 10.1093/gerona/glw168. [DOI] [PubMed] [Google Scholar]
- Maggiorani D, Manzella N, Edmondson DE, Mattevi A, Parini A, Binda C and Mialet-Perez J, 2017. Monoamine Oxidases, Oxidative Stress, and Altered Mitochondrial Dynamics in Cardiac Ageing. Oxid Med Cell Longev. 2017, 3017947 DOI: 10.1155/2017/3017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranzana E, Barbero G, Falasca AI, Lenaz G and Genova ML, 2013. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal. 19, 1469–80. DOI: 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Garcia J, Akhmedov AT and Moe GW, 2013. Mitochondria in heart failure: the emerging role of mitochondrial dynamics. Heart Fail Rev. 18, 439–56. DOI: 10.1007/s10741-012-9330-2. [DOI] [PubMed] [Google Scholar]
- Marin-Garcia J, Ananthakrishnan R and Goldenthal MJ, 1998. Human mitochondrial function during cardiac growth and development. Mol Cell Biochem. 179, 21–6. DOI: 10.1023/a:1006839831141. [DOI] [PubMed] [Google Scholar]
- Maurel A, Hernandez C, Kunduzova O, Bompart G, Cambon C, Parini A and Frances B, 2003. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am J Physiol Heart Circ Physiol. 284, H1460–7. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L and Pelicci PG, 1999. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 402, 309–13. [DOI] [PubMed] [Google Scholar]
- Miró O, Casademont J, Casals E, Perea M, Urbano-Márquez A, Rustin P and Cardellach F, 2000. Aging is associated with increased lipid peroxidation in human hearts, but not with mitochondrial respiratory chain enzyme defects. Cardiovasc Res. 47, 624–31. DOI: 10.1016/s0008-6363(00)00122-x. [DOI] [PubMed] [Google Scholar]
- Moghaddas S, Hoppel CL and Lesnefsky EJ, 2003. Aging defect at the Qo site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 414, 59–66. [DOI] [PubMed] [Google Scholar]
- Mohsin AA, Thompson J, Hu Y, Hollander J, Lesnefsky EJ and Chen Q, 2020. Endoplasmic reticulum stress-induced complex I defect: Central role of calcium overload. Arch Biochem Biophys. 683, 108299 DOI: 10.1016/j.abb.2020.108299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann B, Chen Y, Issa H, Silber RE and Rohrbach S, 2010. Caloric restriction delays cardiac ageing in rats: role of mitochondria. Cardiovasc Res. 88, 267–76. [DOI] [PubMed] [Google Scholar]
- Ong SB, Lee WH, Shao NY, Ismail NI, Katwadi K, Lim MM, Kwek XY, Michel NA, Li J, Newson J, Tahmasebi S, Rehman J, Kodo K, Jang HR and Ong SG, 2019. Calpain Inhibition Restores Autophagy and Prevents Mitochondrial Fragmentation in a Human iPSC Model of Diabetic Endotheliopathy. Stem Cell Reports. 12, 597–610. DOI: 10.1016/j.stemcr.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T, Tomita H, Tamai M and Ishiguro S, 2007. Characteristics of mitochondrial calpains. J Biochem. 142, 365–76. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ and Hotamisligil GS, 2006. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 313, 1137–40. DOI: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JW, Tandler B and Hoppel CL, 1985. Biochemical differences between subsarcolemmal and interfibrillar mitochondria from rat cardiac muscle: effects of procedural manipulations. Arch Biochem Biophys. 236, 691–702. [DOI] [PubMed] [Google Scholar]
- Pang J, Xiong H, Ou Y, Yang H, Xu Y, Chen S, Lai L, Ye Y, Su Z, Lin H, Huang Q, Xu X and Zheng Y, 2019. SIRT1 protects cochlear hair cell and delays age-related hearing loss via autophagy. Neurobiol Aging. 80, 127–137. DOI: 10.1016/j.neurobiolaging.2019.04.003. [DOI] [PubMed] [Google Scholar]
- Paradies G, Ruggiero FM, Petrosillo G, Gadaleta MN and Quagliariello E, 1992. The effect of aging and acetyl-L-carnitine on the acitivity of the phophate carrier and on the phospholipid composition in rat heart mitochondria. Biochem Biophys Acta. 406, 136–138. [DOI] [PubMed] [Google Scholar]
- Peart JN, Pepe S, Reichelt ME, Beckett N, See Hoe L, Ozberk V, Niesman IR, Patel HH and Headrick JP, 2014. Dysfunctional survival-signaling and stress-intolerance in aged murine and human myocardium. Exp Gerontol. 50, 72–81. DOI: 10.1016/j.exger.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pell VR, Chouchani ET, Murphy MP, Brookes PS and Krieg T, 2016. Moving Forwards by Blocking Back-Flow: The Yin and Yang of MI Therapy. Circ Res. 118, 898–906. DOI: 10.1161/circresaha.115.306569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picca A and Lezza AM, 2015. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion. 25, 67–75. DOI: 10.1016/j.mito.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Prola A, Nichtova Z, Pires Da Silva J, Piquereau J, Monceaux K, Guilbert A, Gressette M, Ventura-Clapier R, Garnier A, Zahradnik I, Novotova M and Lemaire C, 2019. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc Res. 115, 328–342. DOI: 10.1093/cvr/cvy197. [DOI] [PubMed] [Google Scholar]
- Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X, Cates C, Liu Q, Zheng Y and Li J, 2017. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. Faseb j. 31, 4153–4167. DOI: 10.1096/fj.201700063R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, Wang L, Chen X, Luckett C, Cates C, Rousselle T, Zheng Y and Li J, 2018. Sestrin2 prevents age-related intolerance to post myocardial infarction via AMPK/PGC-1alpha pathway. J Mol Cell Cardiol. 115, 170–178. DOI: 10.1016/j.yjmcc.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Cabrera R and Soriano ME, 2019. ER Stress Priming of Mitochondrial Respiratory suPERKomplex Assembly. Trends Endocrinol Metab. 30, 685–687. DOI: 10.1016/j.tem.2019.08.003. [DOI] [PubMed] [Google Scholar]
- Rosca M, Minkler P and Hoppel CL, 2011. Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim Biophys Acta. 1807, 1373–82. DOI: 10.1016/j.bbabio.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Ross T, Szczepanek K, Bowler E, Hu Y, Larner A, Lesnefsky EJ and Chen Q, 2013. Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: role of complex I inhibition vs. depolarization of inner mitochondrial membrane. Biochim Biophys Acta. 1830, 4537–42. DOI: 10.1016/j.bbagen.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M and Sadoshima J, 2013. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ Res. 113, 1253–64. DOI: 10.1161/circresaha.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgarbi G, Matarrese P, Pinti M, Lanzarini C, Ascione B, Gibellini L, Dika E, Patrizi A, Tommasino C, Capri M, Cossarizza A, Baracca A, Lenaz G, Solaini G, Franceschi C, Malorni W and Salvioli S, 2014. Mitochondria hyperfusion and elevated autophagic activity are key mechanisms for cellular bioenergetic preservation in centenarians. Aging (Albany NY). 6, 296–310. DOI: 10.18632/aging.100654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinmura K, 2013. Effects of caloric restriction on cardiac oxidative stress and mitochondrial bioenergetics: potential role of cardiac sirtuins. Oxid Med Cell Longev. 2013, 528935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani-Ishida K and Yoshida K, 2015. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int J Cardiol. 197, 26–32. DOI: 10.1016/j.ijcard.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Skulachev VP, 2001. The programmed death phenomena, aging, and the Samurai law of biology. Exp Gerontol. 36, 995–1024. [DOI] [PubMed] [Google Scholar]
- Sniecinski R and Liu H, 2004. Reduced efficacy of volatile anesthetic preconditioning with advanced age in isolated rat myocardium. Anesthesiology. 100, 589–97. DOI: 10.1097/00000542-200403000-00019. [DOI] [PubMed] [Google Scholar]
- Stewart S, Lesnefsky EJ and Chen Q, 2009. Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Transl Res. 153, 224–31. [DOI] [PubMed] [Google Scholar]
- Sugioka K, Nakano M, Totsune-Nakano H, Minakami H, Tero-Kubota S and Ikegami Y, 1988. Mechanism of O2- generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochim Biophys Acta. 936, 377–85. [DOI] [PubMed] [Google Scholar]
- Szczepanek K, Chen Q, Larner AC and Lesnefsky EJ, 2012. Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion. 12, 180–9. DOI: 10.1016/j.mito.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH and Liu S, 2018. Cardiolipin-targeted peptides rejuvenate mitochondrial function, remodel mitochondria, and promote tissue regeneration during aging. Arch Biochem Biophys. 660, 137–148. DOI: 10.1016/j.abb.2018.10.013. [DOI] [PubMed] [Google Scholar]
- Tanaka-Esposito C, Chen Q and Lesnefsky EJ, 2012. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Transl Res. 160, 207–16. DOI: 10.1016/j.trsl.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani M, Honma Y, Hasegawa H and Tamaki K, 2001. Direct activation of mitochondrial K(ATP) channels mimics preconditioning but protein kinase C activation is less effective in middle-aged rat hearts. Cardiovasc Res. 49, 56–68. [DOI] [PubMed] [Google Scholar]
- Thompson J, Hu Y, Lesnefsky EJ and Chen Q, 2016. Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release. Am J Physiol Heart Circ Physiol. 310, H376–84. DOI: 10.1152/ajpheart.00748.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF, 2003. Mitochondrial formation of reactive oxygen species. J Physiol. 552, 335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P and Kroemer G, 2004. AIF deficiency compromises oxidative phosphorylation. Embo J. 23, 4679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey DA, Kelley M, Li L and Huang Y, 2009. Sphingosine protects aging hearts from ischemia/reperfusion injury: Superiority to sphingosine 1-phosphate and ischemic pre- and post-conditioning. Oxid Med Cell Longev. 2, 146–51. DOI: 10.4161/oxim.2.3.8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chen X, Nair S, Sun D, Wang X and Ren J, 2017. Deletion of protein tyrosine phosphatase 1B obliterates endoplasmic reticulum stress-induced myocardial dysfunction through regulation of autophagy. Biochim Biophys Acta Mol Basis Dis. 1863, 3060–3074. DOI: 10.1016/j.bbadis.2017.09.015. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhang H, Xue G, Zhang L, Zhang W, Wang L, Lu F, Li H, Bai S, Lin Y, Lou Y, Xu C and Zhao Y, 2014. Exercise training preserves ischemic preconditioning in aged rat hearts by restoring the myocardial polyamine pool. Oxid Med Cell Longev. 2014, 457429 DOI: 10.1155/2014/457429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Korge P, Honda HM and Ping P, 2003. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 93, 292–301. [DOI] [PubMed] [Google Scholar]
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J and Pinton P, 2009. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 4, 1582–90. DOI: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- Wojtovich AP, Nadtochiy SM, Brookes PS and Nehrke K, 2012. Ischemic preconditioning: the role of mitochondria and aging. Exp Gerontol. 47, 1–7. DOI: 10.1016/j.exger.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Szczepanek K, Maceyka MW, Ross T, Bowler E, Hu Y, Kenny B, Mehfoud C, Desai PN, Baumgarten CM, Chen Q and Lesnefsky EJ, 2014. Transient complex I inhibition at the onset of reperfusion by extracellular acidification decreases cardiac injury. Am J Physiol Cell Physiol. 306, C1142–53. DOI: 10.1152/ajpcell.00241.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y and Ren J, 2011. Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: Role of Akt dephosphorylation. Free Radic Biol Med. 51, 2172–84. DOI: 10.1016/j.freeradbiomed.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]