

Abstract
The antimalarial candidate MMV008138 (1a) is of particular interest because its target enzyme (IspD) is absent in human. To achieve higher potency, and to probe for steric demand, a series of analogs of 1a were prepared that featured methyl-substitution of the B- and C-rings, as well as ring-chain transformations. X-ray crystallography, NMR spectroscopy and calculation were used to study the effects of these modifications on the conformation of the C-ring and orientation of the D-ring. Unfortunately, all the B- and C-ring analogs explored lost in vitro antimalarial activity. The possible role of steric effects and conformational changes on target engagement are discussed.
Keywords: Malaria, Plasmodium, MEP pathway, PfIspD
Graphical Abstract
Malaria was estimated to be responsible for 405,000 deaths worldwide in 2018.1 Many prevention methods and drug treatment protocols are available, but emerging resistance to artemisinin and its partner drugs is of great concern. Thus there is a pressing need to develop antimalarials that possess new mechanisms of action.2 Malaria is caused by Plasmodium parasites, of which P. falciparum is the most prevalent, accounting for more than 96% of the malaria cases worldwide.1 Plasmodium sp. contain a relict organelle termed the apicoplast, which is responsible for the biosynthesis of the critical isoprenoid precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP).3 Whereas Plasmodium sp. synthesize these compounds via the methylerythritol phosphate (MEP) pathway, the mevalonate pathway is used to synthesize them in humans.3 This biochemical divergence commends the MEP pathway as a target for antimalarial drug development,4 since inhibitors of MEP target enzymes would not adversely affect IPP biosynthesis in humans.
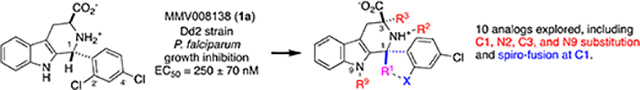
Our initial work in this area5 identified MMV008138 as a MEP pathway inhibitor, by performing a phenotypic screen of the 400-compound Malaria Box6 with the IPP rescue protocol.3 MMV008138 is a tetrahydro-β-carboline, and differentially-functionalized examples of this scaffold are found in a number of other antimalarials,7 and compounds directed towards other indications.8 Subsequent resistance selection studies by Wu et al. demonstrated that 2-C-methyl-D-erythritol-4-phosphate cytidylyltransferase (IspD, E.C.2.7.7.60), the third enzyme in the MEP pathway, is the target of MMV008138.9 Initially, the absolute configuration of MMV008138 was unknown, since it was not disclosed in the Malaria Box; subsequently three independent investigations9–11 demonstrated that the active stereoisomer is (1R, 3S)-configured, as depicted in 1a in Figure 1. Kinetic studies established that 1a competes with cytidine triphosphate (CTP) in its IspD-catalyzed reaction with 2-C-methyl-D-erythritol-4-phosphate.11
Figure 1.

Lead compound 1a and tight D-ring SAR. Note that the X= 2ʹ-Br, 4ʹ-Cl and X= 2ʹ-Cl, 4ʹ-F analogs are also potent in both assays.10
A collection of D-ring analogs of 1a was prepared by the Pictet-Spengler (PS) reaction of l-Trp-OMe·HCl with various benzaldehydes, separation of diastereomers, and hydrolysis.10 Examination of these analogs in both in vitro growth inhibition (SYBR Green) and P. falciparum IspD (PfIspD) inhibition assays show a very close correlation between growth inhibition (EC50) and target engagement (IC50).10b These data also demonstrate a very tight SAR on the D-ring. At least one halogen is required on the 2ʹ- or 4ʹ- position to retain potency in both assays, as shown in Figure 1; substitution at other D-ring positions is not tolerated. It thus appears that the D-ring of 1a binds within a snug, well-defined pocket of PfIspD. In the absence of an X-ray structure for this species of IspD,12 we have speculated10b that halogen-bonding13 contributes to the affinity of 1a for its target.
Since the in vitro potency of 1a was not improved by modulation of the D-ring, we sought to probe the steric requirements for binding around the B- and C-rings, in the hope of identifying more potent analogs. Previously we disclosed analogs 6b and 6d (Scheme 1),10b which feature methyl substitution at C1, but with non-optimal substitution of the D-ring (X = H (6b), X = 4ʹ-Cl (6d)). We attributed the low growth inhibition potency of these compounds to the absence of 2ʹ,4ʹ-dichloro substitution. Synthesis of such C1-Me analogs of 1a requires PS reaction of l-Trp-OMe·HCl 2 with acetophenones 3, which are significantly less electrophilic than benzaldehydes. Thus, the ester precursors to 6b and 6d were prepared by PS reaction with acetophenones 3b and 3d, according to Horiguchi’s protocol:14 ketimine formation in neat Ti(OiPr)4, followed by treatment with TFA and TFAA, all at 70 °C. As we noted in our earlier publication,10b application of the Horiguchi protocol to ortho-substituted acetophenones 3a and 3c did not give the expected products. However, we subsequently found that if ketimine formation was followed by treatment with TFA/TFAA at 0 °C to room temperature, the desired trans-esters 5a and 5c could be isolated in 20% and 17% yield respectively (isopropyl esters result from Ti(OiPr)4-mediated transesterification). As detailed by Horiguchi,14 the trans-relative configuration of 5a and 5c was established by the absence of an NOE correlation between H3 and the C1-methyl; this correlation is visible in the corresponding cis-isomers 4a and 4c (Supplementary Material, Figures S2-S3). Hydrolysis of 5a and 5c afforded the desired amino acids 6a and 6c.
Scheme 1.

Synthesis of C1-Me analogs of 1a.
Unfortunately, the presence of a 2ʹ-Cl substituent in the D-ring of these C1-methyl analogs did not restore antimalarial activity (Table 1, entries 6, 8). Compared to our lead 1a (EC50 = 250 ± 70 nM), C1-methyl analog 6a shows no growth inhibition at 10,000 nM. Similarly, the weakly potent 2ʹ-Cl substituted 1c (EC50 = 3,280 ± 990 nM) loses all growth inhibition potency upon C1-methylation (6c, no growth inhibition at 10,000 nM). Since the mere addition of a methyl group at C1 should not drastically affect permeability or transport of these compounds, we conclude that the loss of growth inhibition potency is due to reduced affinity for PfIspD, the target of 1a (and its potent analogs, cf. Figure 1). In particular, it appears that either there is no room in the PfIspD binding pocket for a methyl group at C1, or that the C1-methyl in 6a induces a conformational change that disfavors binding. We were fortunate to obtain a crystal of 7a (Figure 2), the methyl amide derivative of 6a, and to compare it to 8a, the methyl amide analog of 1a, which we previously crystallized.10a Since 8a is equipotent (Dd2 strain growth EC50 = 190 ± 30 nM) with 1a, comparison of the conformations of 7a and 8a could be informative.
Table 1.
P. falciparum growth inhibition by 1a-d, f, and indicated B- and C-ring analogs.
| Entry | Compound | Dd2 strain P. falciparum Growth EC50 (nM) |
|---|---|---|
| 1 | 1a | 250 ± 70a, c |
| 2 | 1b | >10,000a |
| 3 | 1c | 3,280 ± 990a, d |
| 4 | 1d | 1,170 ± 60a, e |
| 5 | 1f | 700 ± 90a, c |
| 6 | 6a | >10,000 |
| 7 | 6b | >10,000b |
| 8 | 6c | >10,000 |
| 9 | 6d | >10,000b |
| 10 | 8a | 190 ± 30a |
| 11 | 12f | >10,000 |
| 12 | 12i | >10,000 |
| 13 | 16a | 65% inhibition at 10 μM |
| 14 | (±)-20a | >10,000 |
| 15 | 24a | ~8,000 |
| 16 | 25a | >10,000 |
| 17 | 26a | >10,000 |
Reported previously.10a
Reported previously.10b
100% rescued by 200 μM IPP @ 10 μM.
60% rescued by 200 μM IPP @ 10 μM.
50% rescued by 200 μM IPP @ 10 μM.
100% rescued by 200 μM IPP @ 2.5 μM.
Figure 2.

A. Comparison of τ (R-C1-C1ʹ-C2ʹ) dihedral angles in 7a and 8a. B) PyMOL15 overlay of X-ray structures of 7a (cyan: carbon; blue: nitrogen; green: chlorine) and 8a (red). A thermal ellipsoid depiction of 7a is provided in the Supporting Information, Figure S1).
As can be seen in Figure 2, the tetrahydropyridine C-rings of 7a and 8a adopt very similar conformations, featuring a pseudoequatorial C(O)NHMe group and an apparent electrostatic interaction between the amide NH and the tetrahydropyridine nitrogen N2 (Figure 2). There are 4 molecules of 7a in the unit cell, and the average RMSD of the 6 C-ring atoms of them from 8a is 0.041 Å (individual values 0.030, 0.033, 0.046, 0.057 respectively). However, steric strain between the C1-Me and the C2ʹ-Cl in 7a causes the D-ring to adopt a different orientation. We define τ as the dihedral angle between the C1 substituent (CH3 for 7a, H for 8a), C1, C1ʹ, and C2ʹ. For 7a (C1-Me), the average τ value is −63.6° (individual values −59.15°, −62.29°, −64.00°, and −68.91°, respectively), whereas the τ value in 8a (C1-H) is nearly 30° smaller, at −36.5°.
Given the extreme sensitivity of growth and PfIspD inhibition to substitution of the D-ring (see Table 1), it is possible that this dihedral angle change alone, apart from the added steric bulk at C1, could be deleterious to potency. To enforce a smaller τ dihedral angle, we thus proposed to connect C1 and C2ʹ with an ethylene bridge as shown in 12f and 12i (Scheme 2), imparting a cipargamin7b-like spiro-fusion. Compound 12f would thus be a conformationally-constrained mimic of 2ʹ-Me,4ʹ-Cl-substituted 1f, which has significant potency (EC50 = 700 ± 90 nM). The ketone PS reaction between l-Trp-OMe·HCl 2 and indanones 9f/9i was performed according to the original Horiguchi protocol;14 diastereomer separation gave esters 11f and 11i, which mimic the trans-orientation of 5a-d. The trans-configuration of 11f and cis-configuration of 10f was confirmed via 1D NOE experiments (Figure 3).
Scheme 2.

Synthesis of spiro-fused analogs 12f, 12i.
Figure 3.

1D NOE observed in 10f and 11f.
Irradiating C3-H revealed NOE to C7ʹ-H for the trans-isomer 11f, but not for the cis-isomer 10f. When one of the diastereotopic C2ʹ-H is irradiated in 11f, NOE to the N9-H is seen. In contrast, irradiation of one of the diastereotopic C2ʹ-H in the cis-isomer 10f transfers NOE to C3-H (Supplementary Material, Figures S4-S7). Interestingly, 1H NMR analysis of 11f, 11i and their cis-diastereomers (10f, 10i) demonstrated one large and one small coupling constant of H3 to the H4 protons, indicating a near antiperiplanar relationship of H3 and H4β. Thus, regardless of cis- or trans-orientation, the 3-CO2iPr group is pseudoequatorial, and H3 is pseudoaxial. The trans-esters 11f and 11i were then hydrolyzed to give the desired spiro-fused analogs 12f and 12i. Unfortunately, neither compound was potent for growth inhibition (Table 1, entries 11–12). Compound 12f (EC50 > 10,000 nM) differs from 1f (EC50 = 700 ± 90 nM) by substitution of the C2ʹ-CH3 group with an ethylene bridge to C1. Molecular mechanics-based conformational analysis16, 17 of the methyl amide of 11f (11f NHMe), demonstrates that the spiro-ring fusion generates two conformer ensembles defined by narrow ranges in the τ dihedral angle (here C2ʹ-C1-C7aʹ-C3aʹ, Figure 3). One ensemble (13 conformers) features τ = −19 ± 1°, more similar to that of 8a (τ = −36.5 °) than of 7a (τ = −63.8°). The other ensemble (16 conformers) features τ = +17 ± 2°. Since the lowest energy conformers in each ensemble are similar in energy (Supplementary Material, Table S1), both conformers are expected to be populated, and be available to bind PfIspD. Thus, unless the τ dihedral angle exhibited by 8a (and presumably 1a) precisely matches the steric requirement of PfIspD, it seems most likely that the low potency of spiro-fused analog 12f, is due to steric bulk at the C1 position, as we concluded for C1-Me analogs 6a and 6c.
To probe the effect of substitution at other positions, we prepared analogs featuring methylation at N2, C3, and N9 (Scheme 3). Since we have shown that antimalarial potency of 1a and analogs requires trans-(1R,3S)-configuration,10 we took special pains to ensure that we had isolated the correct stereoisomer in each case. The N2-Me analog 16a was prepared from PS reaction of Nα-methyl-l-tryptophan methyl ester 13 and 2,4-dichlorobenzaldehyde 3a. The PS adduct 14a was isolated as an inseparable mixture of diastereomeric methyl esters, hydrolyzed, and separated by reverse phase prep-HPLC into the cis-isomer 15a and the trans-isomer 16a. The relative configuration of these isomers was assigned on the basis of the H3-H4α and H3-H4β coupling constants (3J34α and 3J34β). As Van Linn et. al have shown for 1,2,3-trisubstituted PS adducts, 1,3-trans-configured compounds have 3J34α ≈ 3J34β = 4.4–4.9 Hz.18 In contrast, 1,3-cis-configured-1,2,3-trisubstituted PS adducts have differentiated values for these coupling constants (3J34α =3.8–5.2 Hz and 3J34β = 6.5–8.0 Hz).
Scheme 3.

Synthesis of 16a, (±)-20a, and 24a.
The racemic C3-Me analog (±)-20a was prepared from α-methyl-DL-tryptophan (±)-17. Esterification with SOCl2/MeOH followed by PS reaction with 2,4-dichlorobenzaldehyde 3a gave cis-ester (±)-18a and trans-ester (±)-19a; the relative configuration of these compounds was established by 1D NOE experiments (Figure 4). Irradiating the C3-Me in (±)-18a shows NOE to H1, but no such NOE is seen on irradiation of the C3-Me in (±)-19a. In addition, irradiation of the C3-Me (±)-18a shows NOE signal to H4α, but not H4β. In contrast, irradiation of the C3- Me in (±)-19a shows NOE to both H4α and H4β (Supplementary Material, Figures S8-S9). Hydrolysis of the trans-ester (±)-19a was again accomplished with the catch and release protocol,19 but required elevated temperature, possibly due to steric hindrance caused by the C3-Me.
Figure 4.

Assignment of relative configuration in (±)-18a/(±)-19a and 22a/23a by 1D NOE.
The N9-Me analog 24a was prepared by esterification of 1-Me-l-tryptophan 28, PS reaction with 2,4-dichlorobenzaldehyde 3a, separation of diastereomers 22a and 23a, and hydrolysis of 23a. (Scheme 3). The relative configuration of the diastereomeric esters 22a and 23a was again assigned by 1D NOE spectroscopy. Irradiating H3 of the cis-isomer 22a shows an NOE signal to H1, but this correlation is not seen in trans-isomer 23a (Figure 4). In addition, irradiation of H3 in 23a shows NOE signals to both H4α and H4β, indicating the 3-CO2Me group is pseudoaxial orientation. For 22a a strong correlation of H3 to H4α is observed, as shown in Figure 4 (Supplementary Material, Figures S11-S12).
As can be seen in Table 1, methyl substitution at N2, C3 and N9 (compounds 16a, (±)-20a and 24a, respectively) unfortunately abrogates P. falciparum growth inhibition (EC50 ≥ 8,000 nM, entries 13–15). As we concluded for methylation at C1 (e.g. 6a, entry 6), we consider it unlikely that addition of a methyl group would significantly impact permeability or transport, and we conclude that these modifications reduce affinity for PfIspD. Again, steric hindrance to binding caused by methylation at N2, C3 and N9 may be responsible. However, since studies of the stereoisomers9–10,11 and C3-variants10 of 1a indicate that interaction of the 3-CO2− group with PfIspD is important for affinity, we thought it is important to rule out less direct explanations for the low potency of these compounds. In particular, we sought to determine whether substitution at C1, N2, C3, and N9 could strongly bias a pseudoequatorial- (ψeq-) or pseudoaxial- (ψax-) orientation of the 3-CO2− group in the tetrahydropyridine ring.
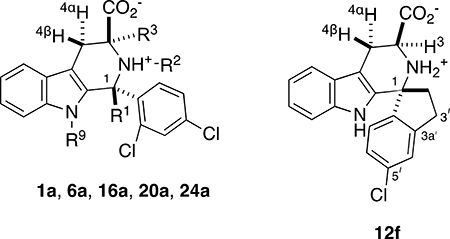
Thus, for 1a, 6a, 12f, 16a and 24a, we examined 1H-1H coupling constants between H3 and H4α, and between H3 and H4β (Table 2). Since (±)-20a lacks a proton at C3, we used NOE to deduce the preferred orientation of the 3-CO2− group, as was done for the methyl ester precursor (±)-19a (Figure 4). Due to reasons of solubility, these experiments were carried out in CD3OD; we do recognize that the conformational thermodynamics could vary somewhat in water. However as described in the supporting information (Supplementary Material, Section F), in large part the conformer preferences seen comport with the principles of maximizing ψeq-substitution, and relief of torsional strain. These effects are summarized in Figure 5.
Table 2.
Predominant orientation of the 3-CO2− group in 1a and analogs, as judged by 1H NMR spectroscopic coupling constants 3J34 and NOE.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Cpd | R1 | R2 | R3 | R9 | 3J34a (Hz) | 3-CO2− |
| 1 | 1a | H | H | H | H | 8.5, 5.5 | ψeq & ψax |
| 2 | 6a | CH3 | H | H | H | 12.0, 5.1 | ψeq |
| 3 | 12f | na | na | na | na | 11.7, 5.0 | ψeq |
| 4 | 24a | H | H | H | CH3 | 10.3, 4.9 | ψeq |
| 5 | 16a | H | CH3 | H | H | 5.0, 5.0 | ψax |
| 6 | (±)-20a | H | H | CH3 | H | NOEb | ψax |
1H NMR spectra measured in CD3OD.
Predominant conformation determined by 1H-1H NOE experiments, since R3 = CH3, see text.
Figure 5.

Effects of C1-, N2, C3- and N9 substitution on the preferred conformation of the tetrahydropyridine C-ring of 1a.
Thus, as anticipated, substitution at C1, N2, C3, and N9 does affect the preferred conformations of these analogs of 1a. However, since neither enforced ψeq-CO2− -orientation (e.g. 6a, 12f, 24a) nor enforced ψax-3-CO2−-orientation (e.g. 16a, (±)-20a) conferred potency, it appears that the low potency of these compounds is indeed steric in origin.
Finally, in addition to installing substituents on the B- and C-rings, we also investigated the shifted C-ring analog 25a and the open C-ring analog 26a (Scheme 4). The reductive amination of l-Trp-OMe·HCl 2 with 3a and sodium cyanoborohydride gives intermediate 27a. Subsequent treatment of 27a with dimethoxymethane and TFA followed by hydrolysis affords the shifted C-ring analog 25a; hydrolysis of 27a gives open C-ring analog 26a. Unfortunately, neither 25a nor 26a was potent for growth inhibition of P. falciparum (Table 1).
Scheme 4.

Synthesis of shifted and open C-ring analogs of 1a.
To conclude, placement of a methyl group on the B-ring (N9, e.g. 24a), C-ring (C1: 6a; N2: 16a; C3: (±)-20a) and installation of a spiro-fusion between the C- and D-rings (e.g. 12f) all drastically reduced in vitro antimalarial potency. In addition to the obvious consequence of increased steric bulk at these positions, these modifications also affected the preferred ψeq- or ψax- orientation of the C3-CO2− group in the tetrahydropyridine C-ring (Table 2), and the orientation of the D-ring (Figure 2 and text). Lastly, shifted and open C-ring analogs of 1a (25a and 26a, respectively) were found to lack in vitro activity. These structural modifications of 1a and 1f are not expected to significantly impact permeation or transport, since they comprise formal addition of a CH2 unit (e.g. 6d, 12f, 12i, 16a, 24a), isomerization (25a), or addition of H2 (26a). We conclude therefore that the loss of P. falciparum growth inhibition potency is due to lack of target engagement. In particular, we propose that the binding pocket of PfIspD features very close contact with the B- and C-rings of 1a, as we had proposed earlier for the D-ring.10b Whether this close contact also extends to all positions of the A-ring of 1a, and whether A-ring modifications could improve potency, work is in progress, and will be reported in due course.
Supplementary Material
Acknowledgments
Funding Sources
Funding from the National Institute of Allergy and Infectious Disease (AI128362 to P.R.C. and M.B.C.; AI108819 to M.B.C.; T32-AI060546 to J.H.B) is gratefully acknowledged. X-ray crystallographic work was supported by the National Science Foundation under CHE-1726077.
Abbreviations
- PS
Pictet-Spengler
- MEP
methylerythritol phosphate
- IPP
isopentenyl diphosphate
- DMAPP
dimethylallyl diphosphate
- Pf
Plasmodium falciparum
- RMSD
root-mean-square deviation
Footnotes
Declaration of Competing Interest
The authors declare no competing interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Material
This information is available free of charge on the Elsevier Publications website, and includes: synthetic procedures and analytical data for all new compounds; NOE spectra; X-ray crystallographic data for 7a; MMFF94 conformer distribution of 11f NHMe; in vitro P. falciparum growth inhibition assay procedures (PDF).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.World Malaria Report 2019. The World Health Organization, available at https://www.who.int/publications-detail/world-malaria-report-2019, last accessed 5/20/20.
- 2.Wells TNC; van Huijsduijnen RH; Van Voorhis WC Nat. Rev. Drug. Discov 2015, 14, 424. [DOI] [PubMed] [Google Scholar]
- 3.Yeh E; DeRisi JL PLoS. Biol. 2011, 9, e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hale I; O’Neill PM; Berry NG; Odom A; Sharma R MedChemComm 2012, 3, 418. [Google Scholar]
- 5.Bowman JD; Merino EF; Brooks CF; Striepen B; Carlier PR; Cassera MB Antimicrob. Agents Chemother 2014, 58, 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spangenberg T; Burrows JN; Kowalczyk P; McDonald S; Wells TNC; Willis P PLOS One 2013, 8, e62906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Gupta L; Srivastava K; Singh S; Puri SK; Chauhan PMS Bioorg. Med. Chem. Lett 2008, 18, 3306; [DOI] [PubMed] [Google Scholar]; (b) Rottmann M; McNamara C; Yeung BKS; Lee MCS; Zou B; Russell B; Seitz P; Plouffe DM; Dharia NV; Tan J; Cohen SB; Spencer KR; González-Páez GE; Lakshminarayana SB; Goh A; Suwanarusk R; Jegla T; Schmitt EK; Beck H-P; Brun R; Nosten F; Renia L; Dartois V; Keller TH; Fidock DA; Winzeler EA; Diagana TT Science 2010, 329, 1175; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Davis RA; Duffy S; Avery VM; Camp D; Hooper JNA; Quinn RJ Tetrahedron Lett. 2010, 51, 583; [Google Scholar]; (d) Beghyn TB; Charton J; Leroux F; Laconde G; Bourin A; Cos P; Maes L; Deprez B J. Med. Chem 2011, 54, 3222; [DOI] [PubMed] [Google Scholar]; (e) Gellis A; Dumètre A; Lanzada G; Hutter S; Ollivier E; Vanelle P; Azas N Biomedicine & Pharmacotherapy 2012, 66, 339; [DOI] [PubMed] [Google Scholar]; (f) Sharma B; Kaur S; Legac J; Rosenthal PJ; Kumar V Bioorg. Med. Chem. Lett. 2020, 30, 126810. [DOI] [PubMed] [Google Scholar]
- 8.(a) Laine AE; Lood C; Koskinen AMP Molecules 2014, 19, 1544; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daugan A; Grondin P; Ruault C; Le Monnier de Gouville A-C; Coste H; Linget JM; Kirilovsky J; Hyafil F; Labaudinière R J. Med. Chem 2003, 46, 4533; [DOI] [PubMed] [Google Scholar]; (c) De Savi C; Bradbury RH; Rabow AA; Norman RA; de Almeida C; Andrews DM; Ballard P; Buttar D; Callis RJ; Currie GS; Curwen JO; Davies CD; Donald CS; Feron LJL; Gingell H; Glossop SC; Hayter BR; Hussain S; Karoutchi G; Lamont SG; MacFaul P; Moss TA; Pearson SE; Tonge M; Walker GE; Weir HM; Wilson Z J. Med. Chem 2015, 58, 8128; [DOI] [PubMed] [Google Scholar]; (d) Singh R; Jaisingh A; Maurya IK; Salunke DB Bioorg. Med. Chem. Lett 2020, 30, 126869. [DOI] [PubMed] [Google Scholar]
- 9.Wu W; Herrera Z; Ebert D; Baska K; Cho SH; DeRisi JL; Yeh E Antimicrob. Agents. Chemother 2015, 59, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Yao Z-K; Krai PM; Merino EF; Simpson ME; Slebodnick C; Cassera MB; Carlier PR Bioorg. Med. Chem. Lett 2015, 25, 1515; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghavami M; Merino EF; Yao Z-K; Elahi R; Simpson ME; Fernández-Murga ML; Butler JH; Casasanta MA; Krai PM; Totrov MM; Slade DJ; Carlier PR; Cassera MB ACS Infect. Dis 2018, 4, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imlay LS; Armstrong CM; Masters MC; Li T; Price KE; Edwards R, L.; Mann KM; Li LX; Stallings CL; Berry NG; O’Neill PM; Odom AR ACS Infect. Dis 2015, 1, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Note that several species of IspD (including Escherichia coli and Arabidopsis thaliana) have yielded high resolution X-ray structures. However, PfIspD features several long and apparently unstructured insertions not present in such crystallized IspDs, and 1a does not potently inhibit any of these enzymes (ref. 9). Therefore, we do not consider it prudent to carry out docking studies of the compounds described in this paper with PfIspD homology models based on these crystallized IspDs.
- 13.Scholfield MR; Zanden CMV; Carter M; Ho PS Protein. Sci 2013, 22, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horiguchi Y; Nakamura M; Saitoh T; Sano T Chem. Pharm. Bull 2003, 51, 1368. [DOI] [PubMed] [Google Scholar]
- 15.The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC. [Google Scholar]
- 16.(a) Shao Y; Molnar LF; Jung Y; Kussmann J; Ochsenfeld C; Brown ST; Gilbert ATB; Slipchenko LV; Levchenko SV; O’Neill DP; DiStasio RA Jr; Lochan RC; Wang T; Beran GJO; Besley NA; Herbert JM; Yeh Lin C; Van Voorhis T; Hung Chien S; Sodt A; Steele RP; Rassolov VA; Maslen PE; Korambath PP; Adamson RD; Austin B; Baker J; Byrd EFC; Dachsel H; Doerksen RJ; Dreuw A; Dunietz BD; Dutoi AD; Furlani TR; Gwaltney SR; Heyden A; Hirata S; Hsu C-P; Kedziora G; Khalliulin RZ; Klunzinger P; Lee AM; Lee MS; Liang W; Lotan I; Nair N; Peters B; Proynov EI; Pieniazek PA; Min Rhee Y; Ritchie J; Rosta E; David Sherrill C; Simmonett AC; Subotnik JE; Lee Woodcock Iii H; Zhang W; Bell AT; Chakraborty AK; Chipman DM; Keil FJ; Warshel A; Hehre WJ; Schaefer Iii HF; Kong J; Krylov AI; Gill PMW; Head-Gordon M Phys. Chem. Chem. Phys 2006, 8, 3172; [DOI] [PubMed] [Google Scholar]; (b) Halgren TA J. Comput. Chem 1996, 17, 490. [Google Scholar]
- 17.Spartan’18; Wavefunction, Inc, Irvine, CA, 2014 [Google Scholar]
- 18.Van Linn ML; Cook JM J. Org. Chem 2010, 75, 3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morwick TM J. Comb. Chem 2006, 8, 649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.