

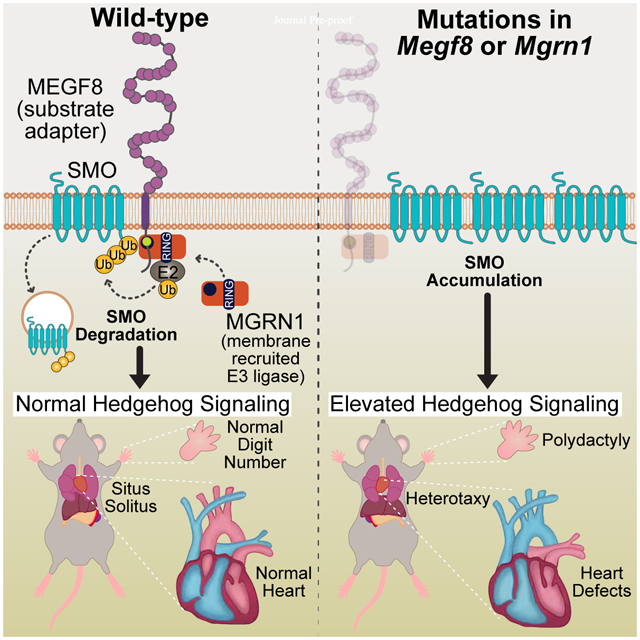
SUMMARY
The etiology of congenital heart defects (CHDs), amongst the most common human birth defects, is poorly understood because of its complex genetic architecture. Here we show that two genes implicated in CHDs, Megf8 and Mgrn1, interact genetically and biochemically to regulate the strength of Hedgehog signaling in target cells. MEGF8, a transmembrane protein, and MGRN1, a RING superfamily E3 ligase, assemble to form a receptor-like ubiquitin ligase complex that catalyzes the ubiquitination and degradation of the Hedgehog pathway transducer Smoothened. Homozygous Megf8 and Mgrn1 mutations increased Smoothened abundance and elevated sensitivity to Hedgehog ligands. While mice heterozygous for loss-of-function Megf8 or Mgrn1 mutations were normal, double heterozygous embryos exhibited an incompletely penetrant syndrome of CHDs with heterotaxy. Thus, genetic interactions can arise from biochemical mechanisms that calibrate morphogen signaling strength, a conclusion broadly relevant for the many human diseases in which oligogenic inheritance is emerging as a mechanism for heritability.
Graphical Abstract
eTOC Blurb
Kong et. al. discovered a membrane-tethered ubiquitination pathway that plays a role in the patterning of multiple tissues during development by dampening Hedgehog signaling strength. Defects in this pathway lead to disrupted left-right patterning (called heterotaxy) of the entire body plan, as well as organ-specific defects in the heart, limb and skeleton.
INTRODUCTION
Morphogens are secreted ligands that influence differentiation, patterning or morphogenesis in a dose-dependent manner. Temporal and spatial gradients of Hedgehog (Hh) ligands (like Sonic Hedgehog, SHH) pattern the spinal cord and limb during development. Varying concentrations or durations of morphogen exposure produce different cellular outcomes by changing the strength or persistence of signaling in target cells (Harfe et al., 2004; Stamataki et al., 2005). The focus in morphogen signaling has largely been on understanding how ligands like SHH are produced and distributed across tissues to form gradients. However, signaling strength in target cells is a function of both ligand exposure and ligand sensitivity. Less is known about the mechanisms in target cells that modulate ligand reception and whether such mechanisms are damaged in developmental disorders.
In CRISPR screens for regulators of Hh signaling, we recently discovered several proteins that attenuate signaling strength in target cells (Pusapati et al., 2018). Because of similarities in their loss-of-function phenotypes, we focus here on three of these proteins: Multiple Epidermal Growth Factor-like Domains 8 (MEGF8), a type I single-pass transmembrane protein, and two paralogous RING superfamily E3 ubiquitin ligases, Mahogunin Ring Finger 1 (MGRN1) and RNF157. Megf8 was identified as a regulator of both left-right patterning and cardiac morphogenesis in mouse genetic screens (Aune et al., 2008; Engelhard et al., 2013; Zhang et al., 2009). Human mutations in MEGF8 result in Carpenter syndrome, an autosomal recessive syndrome similarly characterized by heterotaxy (defects in left-right patterning), severe congenital heart defects (CHDs), preaxial digit duplication, and skeletal defects (Twigg et al., 2012). Unlike many other genes associated with heterotaxy, loss of Megf8 does not result in any detectable defects in either primary or motile cilia (Aune et al., 2008; Pusapati et al., 2018; Zhang et al., 2009). Loss of MGRN1 was also previously shown to cause CHDs and heterotaxy with low penetrance in mice (Cota et al., 2006). How MEGF8 and MGRN1 regulate these critical developmental events has remained unknown for over a decade.
We investigated the biochemical and biological functions of MEGF8, MGRN1 and RNF157 using a combination of mechanistic studies in cultured cells and mouse genetics. MEGF8, MGRN1 and RNF157 anchor a ubiquitination pathway at the cell surface that regulates the sensitivity of target cells to Hh ligands. They assemble into an unusual transmembrane E3 ubiquitin ligase complex that functions as a traffic control system for signaling receptors, including the Hh transducer Smoothened (SMO). Mouse studies revealed striking genetic interactions and gene dosage effects involving Megf8, Mgrn1 and Rnf157 that impact the penetrance of a wide spectrum of birth defects, including CHDs, heterotaxy, skeletal defects, and limb anomalies. Our work shows how genetic interactions between components of a ubiquitin ligase complex that tunes morphogen signaling strength can lead to a birth defect syndrome inherited in an oligogenic pattern.
RESULTS
Megf8 and Mgrn1 are negative regulators of Hedgehog signaling
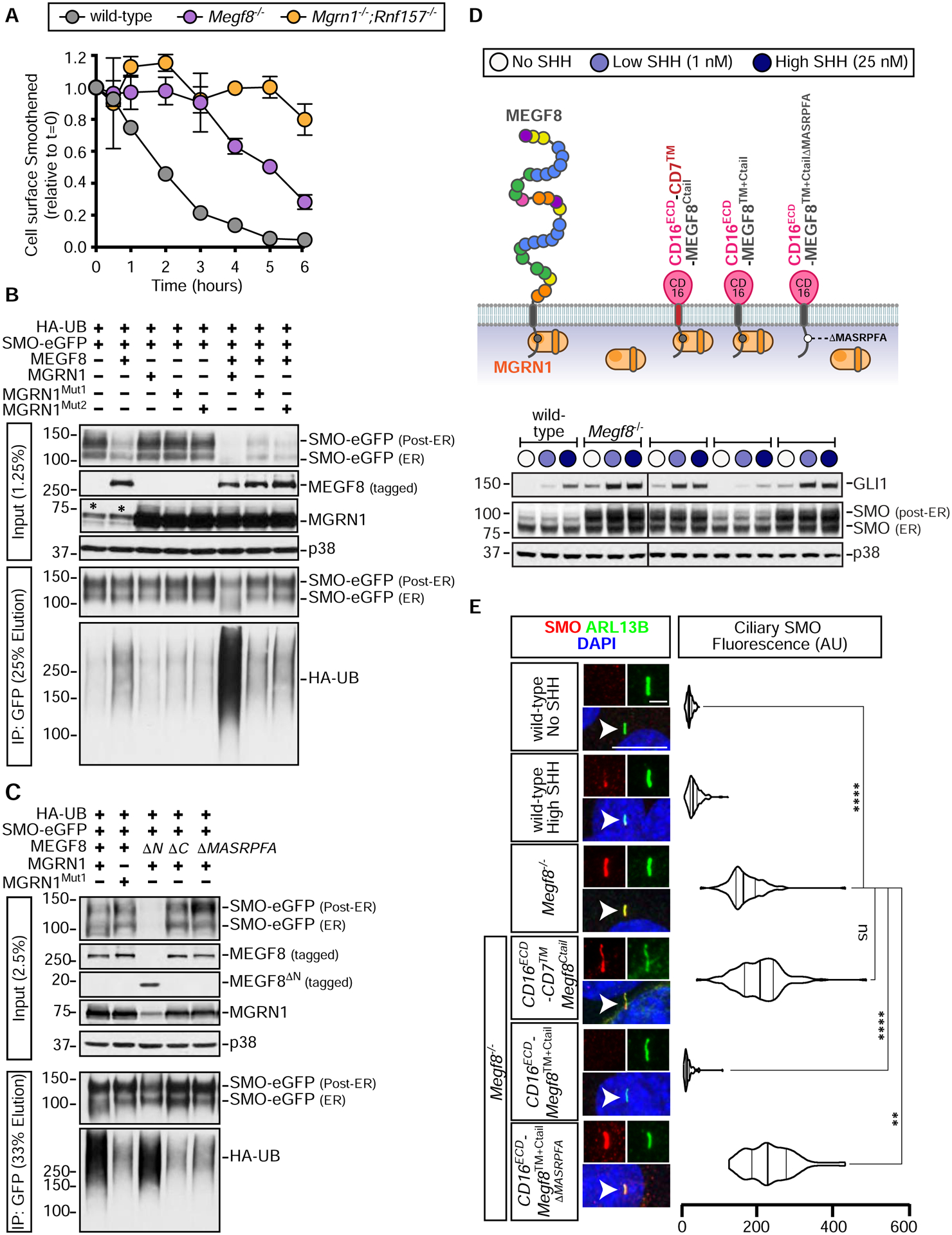
Amongst the top gene hits identified in our genome-wide screen for attenuators of Hh signaling (Pusapati et al., 2018), we pursued a detailed analysis of Megf8 and Mgrn1 (Fig. S1A) because of similarities in their loss of function phenotypes. In both NIH/3T3 fibroblasts and cultured neural progenitor cells (NPCs), loss-of-function mutations in Megf8 and Mgrn1 resulted in an elevated response to Sonic hedgehog (SHH) ligands caused by the accumulation of SMO at the cell surface and primary cilium (Pusapati et al., 2018). To determine if MEGF8 and MGRN1 can attenuate Hh signaling in a more physiological context, we isolated primary mouse embryonic fibroblasts (pMEFs) from embryos homozygous for previously characterized mutant alleles of Megf8 (C193R) or Mgrn1 (md-nc) (Fig. S1A) (He et al., 2003; Phillips, 1963; Zhang et al., 2009). As we observed in NIH/3T3 cells, Megf8C193R/C193R and Mgrn1md-nc/md-nc (hereafter referred to as Megf8m/m and Mgrn1m/m) pMEFs were more sensitive to SHH. When exposed to a sub-saturating concentration of SHH (1 nM), Gli1 (a direct Hh target gene) was only partially induced in wild-type pMEFs, but this same low concentration induced Gli1 to maximum levels in Megf8m/m and Mgrn1m/m pMEFs (Figs. 1A and S1B). Heightened SHH sensitivity was caused by an elevated abundance of SMO in the primary cilia of Megf8m/m and Mgrn1m/m pMEFs, both in the absence and presence of SHH (Figs. 1B and S1C).
Figure 1: Elevated Hh signaling causes birth defect phenotypes in Megf8m/m embryos.
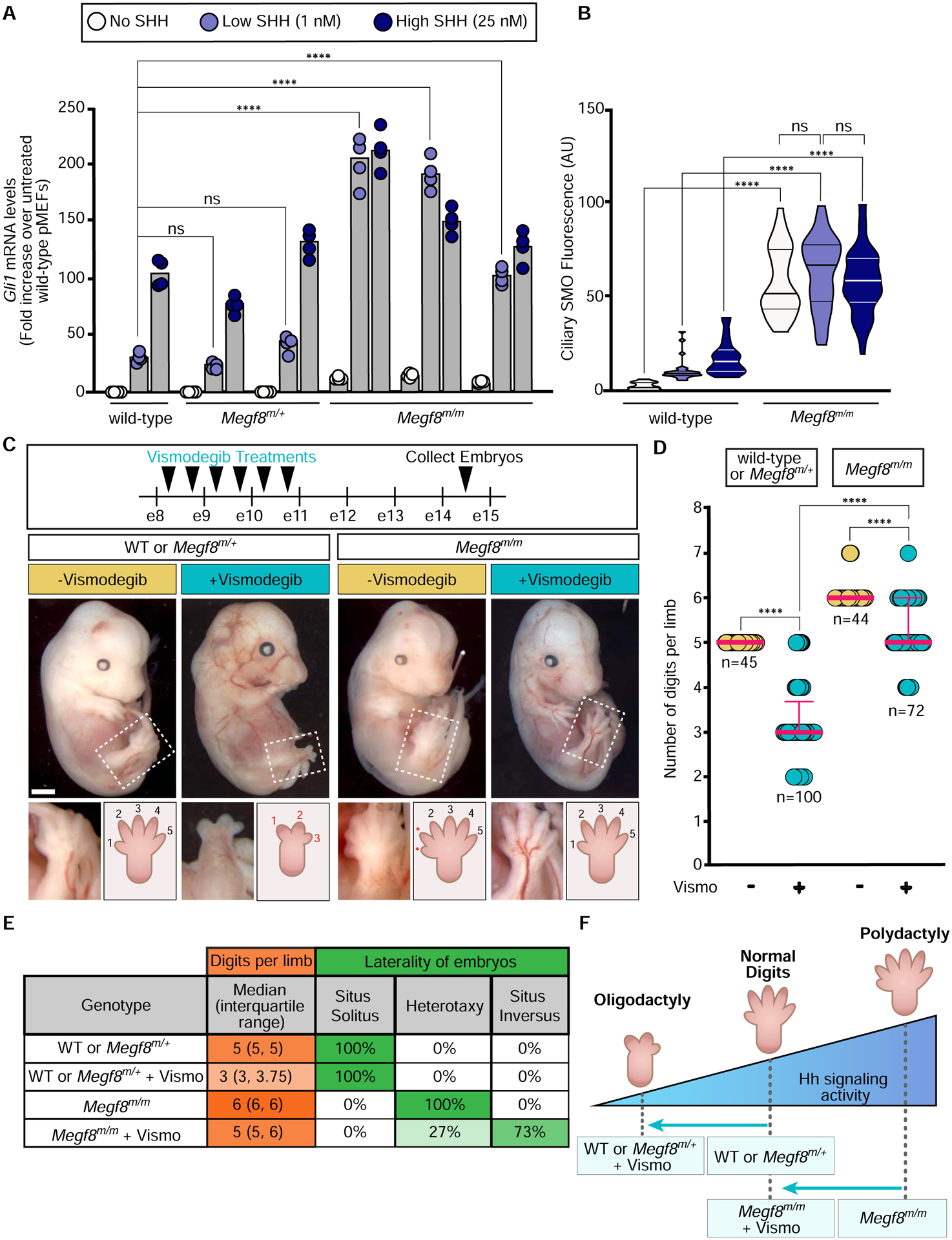
(A and B) Hh signaling strength was assessed using qRT-PCR (A) to measure mRNA for Gli1 (a direct Hh target gene used as a metric for signaling strength) or ciliary SMO abundance (B) in primary mouse embryonic fibroblasts (pMEFs) with the indicated genotypes. Each cell line tested was derived from a different embryo. Bars in (A) denote the median Gli1 mRNA values derived from the four individual measurements shown as circles. Violin plots in (B), with horizontal lines denoting the median and interquartile range, summarize SMO fluorescence at ~15–50 cilia.
(C) Embryos (e14.5) of the indicated genotypes treated with Vismodegib according to the regimen shown at the top. The dotted box marks the hindlimb depicted in zoomed images and cartoons at the bottom show the number of digits. Scale bar, 1mm.
(D) Graph showing the number of digits per limb (forelimb and hindlimb) in embryos of the indicated genotypes treated with Vismodegib. Each circle represents a single limb and the pink lines depict the median with interquartile range.
(E) Table summarizing digit number and left-right patterning phenotypes in embryos of various genotypes, with or without Vismodegib treatment according to the regimen shown in (D).
(F) A model for how the interaction between Vismodegib exposure and genotype influences digit number by altering the strength of Hh signaling
Statistical significance was determined by one-way ANOVA (A) or Kruskal-Wallis (B and D); not-significant (ns) > 0.05 and ****p-value ≤ 0.0001. See also Figures S1 and S2.
The accumulation of ectopic ciliary SMO was also observed in multiple tissues within Megf8m/m and Mgrn1m/m embryos (Fig. S2). In wild-type embryos, Hh signaling activity is restricted to early embryonic development and by e12.5 is turned off in most tissues, resulting in ciliary SMO restricted to cells that were exposed to only the highest concentrations of SHH, like the progenitor cells within the ventral neural tube (Fig. S2) (Corbit et al., 2005; Rohatgi et al., 2007). In contrast, SMO was concentrated in the primary cilia of nearly all Megf8m/m embryonic tissues, regardless of whether it had been exposed to Hh ligands (Fig. S2). Tissues from Mgrn1m/m embryos did not have widespread accumulation of ciliary SMO (Fig. S2). However, ciliary SMO was inappropriately present sporadically in the dorsal neural tube and brain of Mgrn1m/m embryos (Fig. S2), consistent with our observation that Mgrn1−/− NPCs exhibited a moderately elevated response to SHH (Pusapati et al., 2018).
To test if the phenotypes in Megf8m/m embryos were caused by elevated Hh signaling, we employed a pharmacological strategy to attenuate signaling strength in utero using Vismodegib, a specific small molecule SMO inhibitor that is FDA-approved for the treatment of Hh-driven basal cell cancer in humans. Vismodegib is also a teratogen. Its acute administration to pregnant mice can produce “dose-dependent dysmorphology” in embryos, demonstrating that it can be used to tune the strength of Hh signaling during development (not simply to turn it off) (Lipinski et al., 2014). As predicted by the well-known role of Hh signaling in establishing the number of digits in the limb (Litingtung et al., 2002; Riddle et al., 1993; te Welscher et al., 2002), Vismodegib administered between e8.25 and e10.75 induced oligodactyly (reduced digit number) in wild-type and Megf8m/+ embryos (Figs. 1C–F). Remarkably, Vismodegib rescued the preaxial digit duplication universally seen in Megf8m/m embryos, restoring digit numbers to 5 in the majority of treated embryos (Figs. 1C–F). Heterotaxy, defined as randomized organ situs, is another completely penetrant phenotype seen in Megf8m/m embryos (Fig. 1E) (Aune et al., 2008). Megf8m/m embryos exposed to Vismodegib also showed a partial rescue of heterotaxy: ~70% displayed concordant organ situs that was mirror symmetric (situs inversus) (Fig. 1E). Vismodegib treated Megf8m/m embryos still displayed CHDs, likely because heterotaxy was not fully rescued and because Vismodegib was not delivered during some of the critical periods of heart morphogenesis. Future experiments varying the dose and time period of Vismodegib administration should help clarify the role of Hh signaling in all the birth defect phenotypes seen in Megf8m/m embryos.
Mgrn1m/m embryos displayed a very low (<10%) rate of preaxial digit duplication (because of lower levels of SMO in cilia, Fig. S2), making the assessment of Vismodegib rescue challenging. However, compared to wild-type or Mgrn1m/+ embryos, Mgrn1m/m embryos were resistant to Vismodegib-induced oligodactyly (Figs. S1D and S1E), consistent with the fact that they have a higher Hh signaling setpoint (Fig. S1F).
The striking difference in digit number between littermate control and Megf8m/m or Mgrn1m/m embryos exposed to Vismodegib (Fig. 1D and S1D) supports the idea that a just-right “goldilocks” level of Hh signaling is required for proper digit patterning. Vismodegib restores SMO activity to this optimal level in Megf8m/m limbs, but reduces SMO activity below the levels required for normal digit patterning in wild-type embryos (Fig. 1F).
Rnf157 is a genetic modifier of Mgrn1
In both mice and cultured cells, loss of MEGF8 consistently produced stronger phenotypes than the loss of MGRN1 (Figs. 1, S1 and S2), suggesting the involvement of additional genes (Pusapati et al., 2018). The reported penetrance and expressivity of CHDs, heterotaxy, and preaxial digit duplication was much higher in Megf8m/m embryos compared to Mgrn1m/m embryos (Cota et al., 2006; Zhang et al., 2009) (Table S1). Similarly in NIH/3T3 cells, when compared to the loss of MGRN1, the loss of MEGF8 resulted in more Hh signaling activity at baseline and a greater abundance of SMO at the plasma and ciliary membranes (Figs. 2A and 2C). Evolutionary sequence analysis indicated that RNF157, which also encodes a RING superfamily E3 ligase, is a vertebrate-specific paralog of MGRN1 (Fig. 2B). Although MGRN1 is more widely distributed, found amongst almost all major eukaryotic lineages, MGRN1 and RNF157 share a RING domain and a distinctive predicted substrate-binding domain that is unique amongst other members of the RING superfamily (Figs. 2B and S3A).
Figure 2: RNF157 partially compensates for the loss of MGRN1.
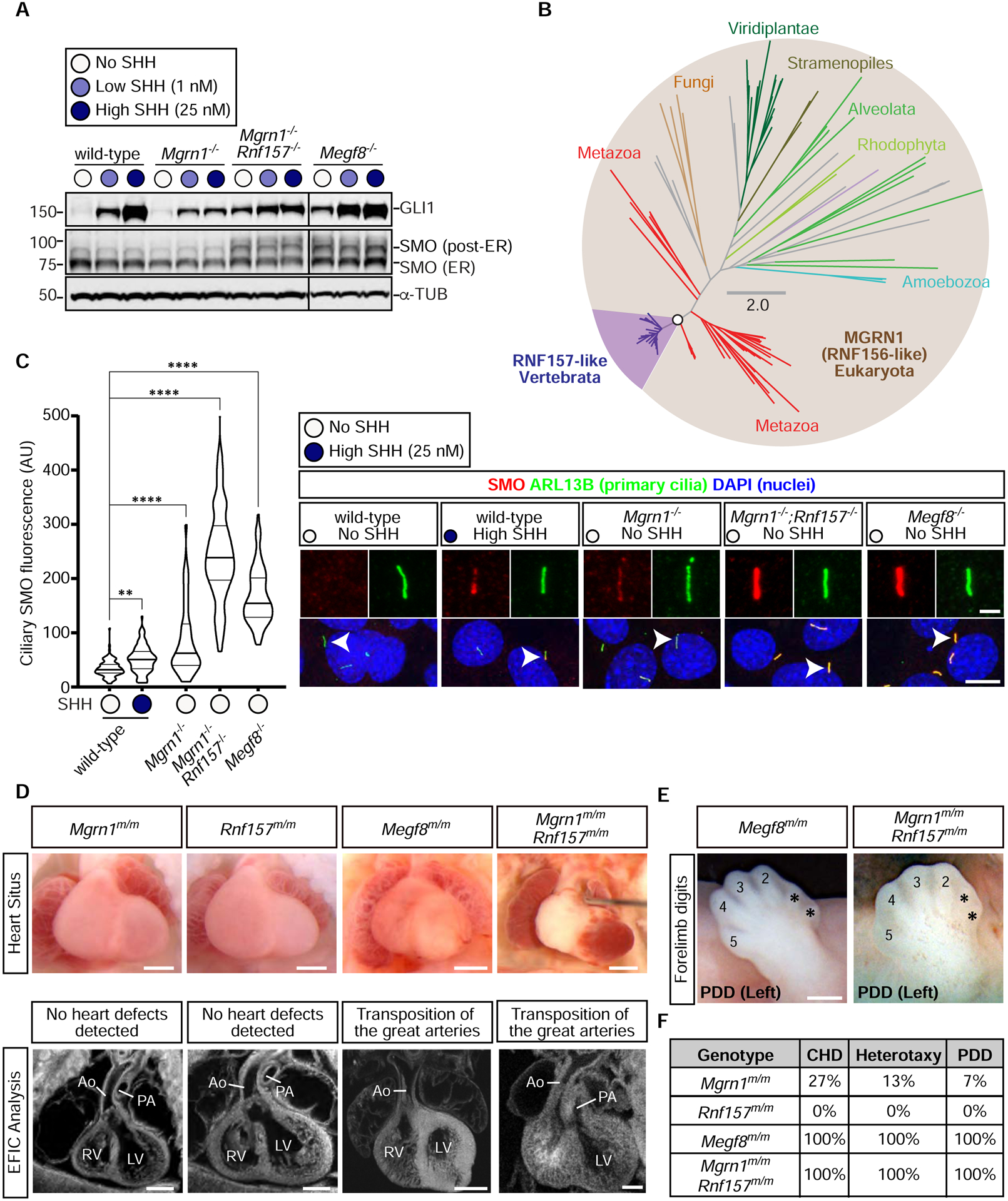
(A) Immunoblots showing GLI1 as a measure of Hh signaling strength and SMO abundance in the indicated NIH/3T3 cell lines treated with various concentrations of SHH. α-Tubulin (α-TUB) is a loading control. Two populations of SMO, localized in the ER or in post-ER compartments, are marked. An analysis of additional clonal cell lines is shown in Fig. S3C.
(B) Unrooted maximum-likelihood tree topology showing the evolutionary relationship between MGRN1 and RNF157, with the vertebrate-specific RNF157 lineage highlighted in purple. The open circle denotes 100% confidence support (1000 replicates) and the scale bar indicates phylogenetic distance. The full Newick tree file is provided in Supplemental File 1.
(C) Violin plots (left) with horizontal lines denoting the median and interquartile range and corresponding representative confocal fluorescence microscopy images (right) of SMO (red) at primary cilia (green, marked by ARL13B) in NIH/3T3 cells with the indicated genotypes (n~70 cilia/condition). Arrowheads identify individual cilia captured in the zoomed images above each panel. Statistical significance was determined by the Kruskal-Wallis test; **p-value ≤ 0.01 and ****p-value ≤ 0.0001. Scale bars, 10 μm in merged panels and 2 μm in zoomed displays. See Fig. S3D for an analysis of additional clonal cell lines.
(D) Necropsy (top row) and episcopic confocal microscopy (ECM, bottom row) images of embryonic hearts from e13.5–14.5 embryos of the indicated genotypes. Scale bars, 200 μm.
(E) Forelimbs of embryos show preaxial digit duplication (PDD). Asterisks (*) mark the duplicated digits. Scale bar, 200 μm.
(F) Table summarizes the frequency of CHDs, heterotaxy, and PDD in Mgrn1m/m (n=15), Rnf157m/m (n=6), Megf8m/m (n=12), and Mgrn1m/m;Rnf157m/m (n=3) embryos. A detailed list of phenotypes observed in each embryo can be found in Table S1. See also Figure S3, Table S1, and File S1.
These analyses raised the possibility that RNF157 may partially compensate for the loss of MGRN1. Depletion of both RNF157 and MGRN1 in NIH/3T3 cells and NPCs using CRISPR methods (Fig. S3B) enhanced Hh signaling activity (Figs. 2A, S3C–E). Mgrn1−/−;Rnf157−/− NIH/3T3 cells constitutively expressed GLI1, even in the absence of SHH (Fig. 2A). In addition, the abundance of SMO carrying mature glycan modifications acquired in the golgi after trafficking from the endoplasmic reticulum (hereafter “post-ER SMO”) and the abundance of SMO in primary cilia was much higher in Mgrn1−/−;Rnf157−/− compared to Mgrn1−/− cells (Figs. 2A and 2C). In all assays, Hh signaling in Mgrn1−/−;Rnf157−/− cells was enhanced compared to Mgrn1−/− cells (and equivalent to Megf8−/− cells) (Figs. 2A, 2C, S3C–E).
To assess the relationship between RNF157 and MGRN1 in vivo, we generated Rnf157−/− mice (hereafter referred to as Rnf157m/m mice) using CRISPR methods (Fig. S3B). Consistent with data collected by the International Mouse Phenotyping Consortium (IMPC) using a different knockout strategy (Dickinson et al., 2016), the Rnf157m/m mice were viable, fertile, and without obvious developmental defects (Figs. 2D and 2F). The penetrance of birth defects in Mgrn1m/m;Rnf157m/m double null embryos was comparable to Megf8m/m embryos (and much higher than single null Mgrn1m/m or Rnf157m/m embryos) (Figs. 2D–F and Table S1).
Based on consequences of the simultaneous disruption of Rnf157 and Mgrn1 in both cultured cells and mice, we conclude that RNF157 can partially compensate for the function of MGRN1 in Mgrn1−/− NIH/3T3 cells, NPCs, and embryos (Figs. 2A, 2C–F, and S3C–E). This compensation is asymmetric, as the loss of RNF157 alone had few developmental consequences (Figs. 2D and 2F), presumably because MGRN1 can fully cover RNF157 functions. In conclusion, Rnf157 is a modifier gene: mutations in Rnf157 are insufficient to cause a phenotype alone, but they increase the penetrance of phenotypes caused by mutations in a different gene (Mgrn1).
MEGF8 binds to MGRN1
Mouse embryos and cells that lack MEGF8 are indistinguishable from those that lack both MGRN1 and RNF157 (Fig. 2 and Table S1), leading us to speculate that MEGF8, MGRN1, and RNF157 may work together to regulate SMO trafficking. We transiently expressed MEGF8 in HEK293T cells and observed that it could be co-immunoprecipitated (co-IP) with either endogenous or over-expressed MGRN1 (Figs. 3A and 3C). Deleting the ~170 amino acid (a.a.) long cytoplasmic tail (hereafter called the “Ctail”) of MEGF8 (MEGF8ΔCtail), but not its large ~2500 a.a. extracellular domain (MEGF8ΔN), abolished the interaction with MGRN1 (Figs. 3A–C). The MEGF8 Ctail contains a peptide motif (with the sequence “MASRPFA”) that is highly conserved across a family of single-pass transmembrane proteins found in Filozoa, animal-like eukaryotes including Filasterea, Choanoflagellatea, and Metazoa (Figs. 3B and S4A) (Gunn et al., 1999; Haqq et al., 2003; Nagle et al., 1999). The deletion of this motif (MEGF8ΔMASRPFA) abrogated the interaction between MEGF8 and MGRN1 (Fig. 3C), establishing an E3 ligase recruitment function for this mysterious sequence element.
Figure 3: The interaction between MGRN1 and MEGF8 is required to attenuate Hedgehog signaling.
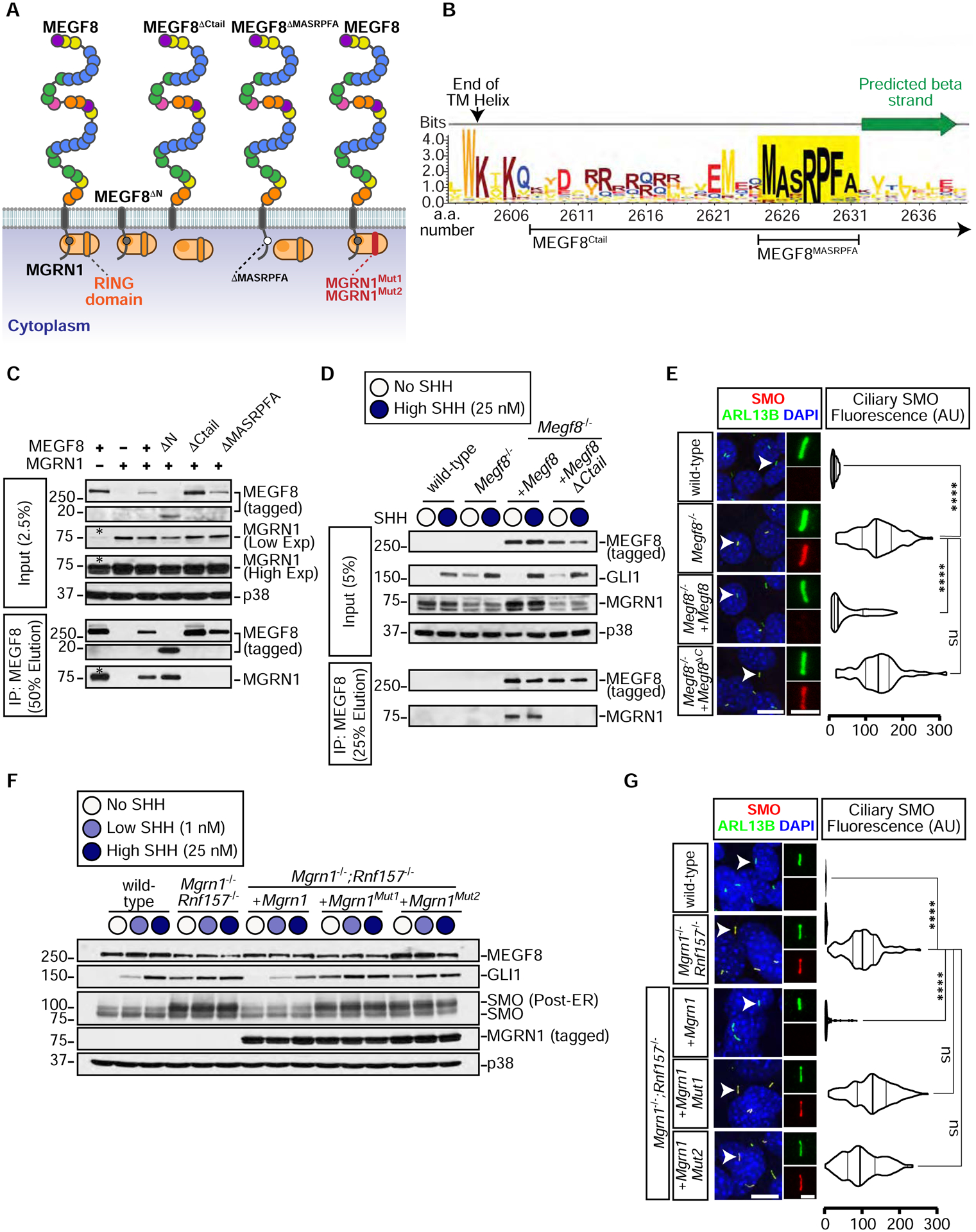
(A) Depictions of full length MEGF8, truncated MEGF8 (MEGF8ΔN, MEGF8ΔCtail, MEGF8ΔMASRPFA), functional MGRN1, and catalytically inactive MGRN1 (MGRN1Mut1 and MGRN1Mut2) proteins. The multiple domains in the extracellular region of MEGF8 are shown as circles and colored as in Fig. S1A.
(B) Sequence logo showing the conservation in sequence entropy bits of the MASRPFA sequence (yellow shading) in the cytoplasmic tail of MEGF8 and related proteins (alignment shown in Fig. S4A). Deletion boundaries for the MEGF8 mutants shown in Fig. 3A are noted below the logo.
(C) The interaction between MEGF8 or MEGF8 mutants (see Fig. 3A, all 1D4 tagged) and MGRN1 (FLAG tagged) was tested by transient co-expression in HEK293T cells, followed by immunoprecipitation (IP) of MEGF8. Asterisk (*) indicates endogenous MGRN1 in HEK293T cells.
(D and E) GLI1 abundance was measured by immunoblotting (D) and SMO ciliary abundance by confocal fluorescence microscopy (E) in Megf8−/− NIH/3T3 cells stably expressing 1D4-tagged MEGF8 or MEGF8ΔCtail (see Fig. 3A). The interaction between MEGF8 and endogenous MGRN1 was tested by co-IP in (D).
(F and G) GLI, SMO and MEGF8 abundances were measured by immunoblotting (F) and SMO ciliary abundance by confocal fluorescence microscopy (G) in Mgrn1−/−;Rnf157−/− NIH/3T3 cells stably expressing wild-type MGRN1 or variants carrying inactivating mutations in the RING domain (MGRN1Mut1 and MGRN1Mut2, see Figs. 3A and S4B).
Violin plots (E, G) summarize the quantification of SMO fluorescence (red) at ~50 individual cilia (green) per cell line from representative images of the type shown immediately to the left.
Statistical significance was determined by the Kruskal-Wallis test; not-significant (ns) > 0.05 and ****p-value ≤ 0.0001. Scale bars, 10 μm in merged panels and 2 μm in zoomed displays. See also Figure S4.
To test if the association between MEGF8 and MGRN1 was relevant for the regulation of Hh signaling, we stably expressed wild-type MEGF8 or the interaction-defective MEGF8ΔCtail mutant in Megf8−/− NIH/3T3 cells (Fig. 3D). Stably expressed MEGF8, but not its truncated MEGF8ΔCtail variant, bound to endogenous MGRN1 (Fig. 3D) and suppressed the elevated basal GLI1 and ciliary SMO seen in Megf8−/− cells (Figs. 3D and 3E). The MEGF8-MGRN1 interaction was unchanged when signaling was activated by the addition of SHH (Fig. 3D). These data establish that MGRN1 in the cytoplasm stably associates with the Ctail of MEGF8 and this interaction is required to suppress ciliary SMO levels and attenuate Hh signaling.
The ubiquitin ligase activity of MGRN1 is required to attenuate Hh signaling
MGRN1 regulates processes ranging from skin pigmentation to spongiform neurodegeneration by directly ubiquitinating multiple substrates (Chakrabarti and Hegde, 2009; Gunn et al., 2013; Jiao et al., 2009). We constructed two variants of MGRN1 (MGRN1Mut1 and MGRN1Mut2) carrying mutations in highly conserved residues of the RING domain (Fig. S4B). These mutations are known to abolish binding between RING domains and their cognate E2 partners, thereby preventing ubiquitin transfer to substrates (Garcia-Barcena et al., 2020; Gunn et al., 2013). We stably expressed wild-type MGRN1, MGRN1Mut1, or MGRN1Mut2 in Mgrn1−/−;Rnf157−/− NIH/3T3 cells and measured the abundance of GLI1, post-ER SMO, and ciliary SMO (Figs. 3F and 3G). In all three assays, wild-type MGRN1 was able to fully attenuate Hh signaling and SMO levels, but the MGRN1Mut1 and MGRN1Mut2 variants were inactive. Importantly, MGRN1Mut1 and MGRN1Mut2 were expressed at equivalent levels as MGRN1 (Fig. 3F) and maintained their stable interaction with MEGF8 (Fig. S4C), demonstrating their integrity. These results support the conclusion that both the stable interaction of MGRN1 with MEGF8 and its E3 ligase function are required to attenuate Hh signaling.
The MEGF8-MGRN1 complex ubiquitinates SMO
At this point our data suggested that MGRN1 functions as a membrane-tethered ubiquitin ligase complex that attenuates Hh signaling by reducing SMO abundance at the cell surface and primary cilium. Trafficking assays revealed that both the steady state abundance and the stability of cell-surface SMO were markedly greater in Megf8−/− and Mgrn1−/−;Rnf157−/− cells compared to wild-type cells (Figs. 4A and S5). Since the endocytosis and degradation of cell-surface receptors is often regulated by their ubiquitination, we sought to test whether SMO is a substrate of the MEGF8-MGRN1 complex.
Figure 4: Smoothened is ubiquitinated by the MEGF8-MGRN1 complex.
(A) Degradation of cell-surface SMO in NIH/3T3 cells of the indicated genotypes. See Fig. S5 for details. Error bars represent the standard error of two independent replicates.
(B and C) SMO ubiquitination was assessed after transient co-expression of the indicated proteins in HEK293T cells (see Fig. 3A). Cells were lysed under denaturing conditions, SMO was purified by IP, and the amount of HA-UB covalently conjugated to SMO assessed using immunoblotting with an anti-HA antibody. An asterisk (*) indicates endogenous MGRN1.
(D and E) Total GLI1 and SMO abundances were measured by immunoblotting (D) and ciliary SMO (n~50 cilia) by fluorescence confocal microscopy (E) in Megf8−/− cells expressing various CD16/CD7/MEGF8 chimeras. The ability of these chimeras to support SMO ubiquitination is shown in Fig. S6D and the abundances of chimeras at the cell surface is shown in Fig. S6E. Statistical significance in (E) was determined by the Kruskal-Wallis test; not-significant (ns) > 0.05, **p-value ≤ 0.01, and ****p-value ≤ 0.0001. Scale bars, 10 μm in merged panels and 2 μm in zoomed displays. See also Figures S5 and S6.
We established an assay to measure SMO ubiquitination by expressing SMO and Hemagglutinin (HA)-tagged ubiquitin (UB) together in HEK293T cells and then measuring the amount of HA-UB conjugated to SMO (Figs. 4B and 4C). SMO was isolated by immunoprecipitation and the attached UB chains detected (as a smear) by immunoblotting with an anti-HA antibody. Co-expression of MGRN1 alone had no effect on SMO ubiquitination, co-expression of MEGF8 alone slightly increased SMO ubiquitination, but the co-expression of both MEGF8 and MGRN1 dramatically increased levels of ubiquitinated SMO and concomitantly reduced SMO abundance (Fig. 4B). A ubiquitin mutant lacking all lysine residues (UBK0) was poorly conjugated to SMO, suggesting that SMO is attached to poly-UB chains, rather than to a single ubiquitin (Fig. S6A). Inactivating mutations in the RING domain of MGRN1 (MGRN1Mut1 and MGRN1Mut2) failed to promote SMO ubiquitination (Fig. 4B). SMO was a selective substrate for MGRN1 and MEGF8 because their co-expression did not change the abundance of a different ciliary GPCR, SSTR3 (Fig. S6B). SMO contains 21 lysine (K) residues exposed to the cytoplasm that could function as acceptors for ubiquitin. Changing all of these lysines to arginines (R) impaired MGRN1-mediated ubiquitination (Fig. S6C), but changing specific clusters of lysines in each of the cytoplasmic loops or the tail of SMO did not reduce ubiquitination (Fig. S6C). Thus, MGRN1 does not seem to favor a particular lysine residue or set of lysine residues on the cytoplasmic surface of SMO, at least in this over-expression based HEK293T assay.
Efficient SMO ubiquitination required both MEGF8 and the E3 ligase function of MGRN1. The small increase in SMO ubiquitination seen in the presence of MEGF8 alone is likely due to presence of endogenous MGRN1 in HEK293T cells (see asterisks in the MGRN1 panel in Fig. 4B). To directly test whether the physical interaction between MEGF8 and MGRN1 was required to mediate SMO ubiquitination, we co-expressed MGRN1 with one of three MEGF8 variants (diagrammed in Fig. 3A): (1) MEGF8ΔCtail, (2) MEGF8ΔMASRPFA (both of which cannot bind to MGRN1, Fig. 3C), or (3) MEGF8ΔN, which lacks the large extracellular domain of MEGF8 but retains its transmembrane (TM) helix and Ctail. MEGF8ΔCtail and MEGF8ΔMASRPFA failed to support SMO ubiquitination (Fig. 4C). In contrast, MEGF8ΔN, which can still bind to MGRN1 (Fig. 3C), efficiently promoted SMO ubiquitination and degradation (Fig. 4C). Interestingly, MEGF8ΔN was much more active than full-length MEGF8 (despite both proteins being expressed at comparable levels), suggesting that the extracellular domain of MEGF8 may negatively regulate the function of the Ctail or interfere with its recognition of SMO. In addition to recruiting MGRN1 to the plasma membrane, the association between MEGF8 and MGRN1 promoted the intrinsic E3 ligase activity of MGRN1, evident through the ability of MEGF8ΔN to reduce the abundance of co-expressed wild-type MGRN1 (Fig. 4C). Most E3 ligases catalyze their own ubiquitination and de-stabilization, a property that reflects their intrinsic catalytic activity.
Unexpectedly, MEGF8ΔN, which includes only the TM helix and Ctail of the protein (232 out of the 2778 amino acids in the full-length protein), was sufficient to promote SMO ubiquitination (Fig. 4C). To further narrow down the region of MEGF8 required for SMO recognition, we constructed a set of chimeric proteins that fused the MEGF8 Ctail, TM helix, or both to heterologous extracellular and transmembrane domains from CD16 and CD7, respectively (diagrammed in Fig. S6D). In the HEK293T assay, both the TM helix and the Ctail of MEGF8 were required to promote SMO ubiquitination; simply tethering the isolated Ctail to the plasma membrane by fusing it to a CD16-CD7 hybrid protein was not sufficient. Abrogating the interaction with MGRN1 by deleting the “MASRPFA” motif abolished the function of these chimeric proteins, demonstrating that they still require MGRN1 to promote SMO ubiquitination (Fig. S6D).
If the biochemical function of MEGF8 in Hh signaling is to ubiquitinate SMO, a key prediction is that the CD16ECD-MEGF8TM+Ctail chimera, a minimal engineered protein that is sufficient to carry out this function, should be able to reverse the enhanced Hh signaling phenotype in Megf8−/− cells. To test this prediction, we stably expressed CD16ECD-CD7™-MEGF8Ctail, CD16ECD-MEGF8TM+Ctail, and CD16ECD-MEGF8TM+CtailΔMASRPFA (a variant carrying the MASRPFA deletion) in Megf8−/− cells (Fig. 4D). All three chimeras were expressed and localized properly to the cell surface as measured by flow cytometry using an antibody against the CD16ECD (Fig. S6E). However, only the CD16ECD-MEGF8TM+Ctail chimera could completely suppress Gli1 expression and both post-ER and ciliary SMO abundance (Figs. 4D and 4E). These experiments again highlighted the importance of the TM helix of MEGF8 for SMO regulation: replacing it with a TM helix from CD7 abolished activity (Figs. 4D and S6D). We conclude that the TM helix and Ctail of MEGF8 function as a minimal membrane-localized substrate adapter to recruit and activate the E3 ligase activity of MGRN1 towards SMO, catalyzing SMO ubiquitination and clearance from both the plasma and ciliary membrane, and consequently dampening sensitivity to Hh ligands.
Genetic interactions between Megf8 and Mgrn1
After identifying the MEGF8-MGRN1 interaction and elucidating the ubiquitination based mechanism through which it regulates the sensitivity of target cells to Hh ligands, we sought to investigate the role of this protein complex in embryonic development using the previously published Megf8m/+ and Mgrn1m/+ mouse lines (He et al., 2003; Phillips, 1963; Zhang et al., 2009). Notably, both Megf8m/m and Mgrn1m/m mutant embryos display CHDs, heterotaxy, and preaxial digit duplication. While these phenotypes are fully penetrant in the Megf8m/m mutants, they show lower penetrance in the Mgrn1m/m mutants (likely due to partial redundancy with Rnf157) (Fig. 2F) (Cota et al., 2006; Zhang et al., 2009). To determine whether the developmental defects exhibited by these two mutants are a product of the same pathway (as predicted by our biochemical studies), we assessed for a genetic interaction by intercrossing the Megf8m/+ and Mgrn1m/+ mice and examining the phenotypes of the resultant double heterozygous Megf8m/+;Mgrn1m/+ embryos.
As reported previously (Cota et al., 2006; Zhang et al., 2009), the single heterozygous Megf8m/+ and Mgrn1m/+ embryos were normal without any developmental defects, consistent with the adult viability of Megf8m/+ and Mgrn1m/+ mice (Figs. 5 and 6, Tables S2 and S3). In contrast, the Megf8m/+;Mgrn1m/+ double heterozygous embryos showed preaxial digit duplication, heterotaxy and CHDs, phenotypes similar to those seen in homozygous Megf8m/m and Mgrn1m/m embryos (Figs. 5 and 6, Table S4). Detailed anatomic phenotyping was conducted on e13.5–14.5 Megf8m/+;Mgrn1m/+ embryos using both necropsies (Figs. 5B and 5C) and episcopic confocal microscopy (ECM) to generate 3D histological reconstructions of intracardiac anatomy (Fig. 6B).
Figure 5: A genetic interaction between Megf8 and Mgrn1 causes heart defects and heterotaxy.
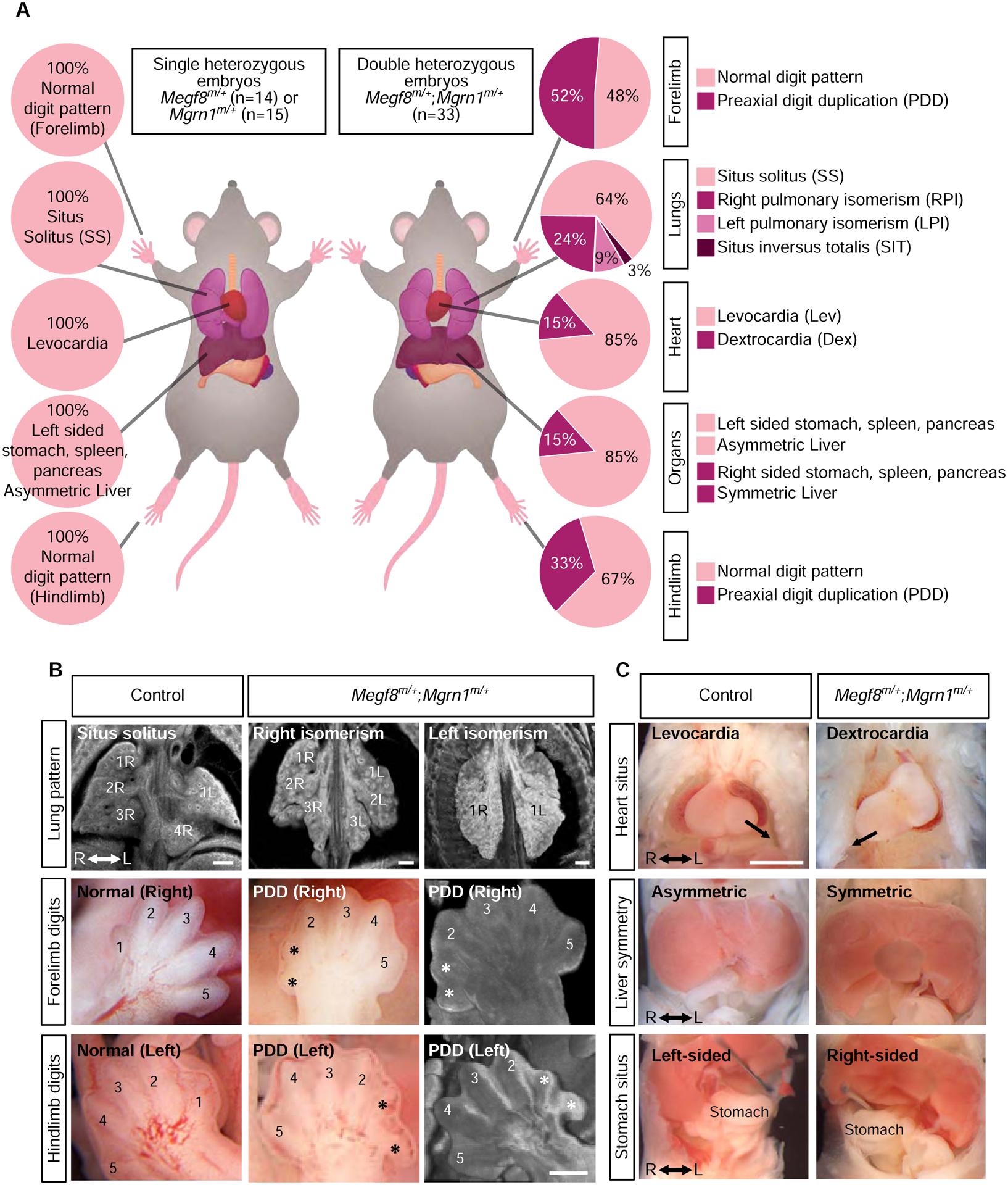
(A) Summary of phenotypes observed in mouse embryos with the indicated genotypes (e13.5–14.5). Dex, dextrocardia; Lev, levocardia; LPI, left pulmonary isomerism; PDD, preaxial digit duplication; RPI, right pulmonary isomerism; SIT, situs inversus; SS, situs solitus. A detailed list of phenotypes observed in each embryo can be found in Tables S2, S3, and S4.
(B) Representative light microscopy and ECM images of the developing lungs and limbs of single (control) and double heterozygous embryos. The normal right lung has 4 lobes (1R, 2R, 3R and 4R) and the left lung has one lobe (1L). Asterisks (*) mark the duplicated preaxial digits.
(C) Representative necropsy images showing the position of the heart, symmetry of the liver, and location of the stomach in single (control) and double heterozygous embryos. Arrow (top row) denotes the direction of the cardiac apex. See also Tables S2, S3, and S4.
Figure 6: Spectrum of heart defects in mice carrying mutant alleles of Megf8 and Mgrn1.
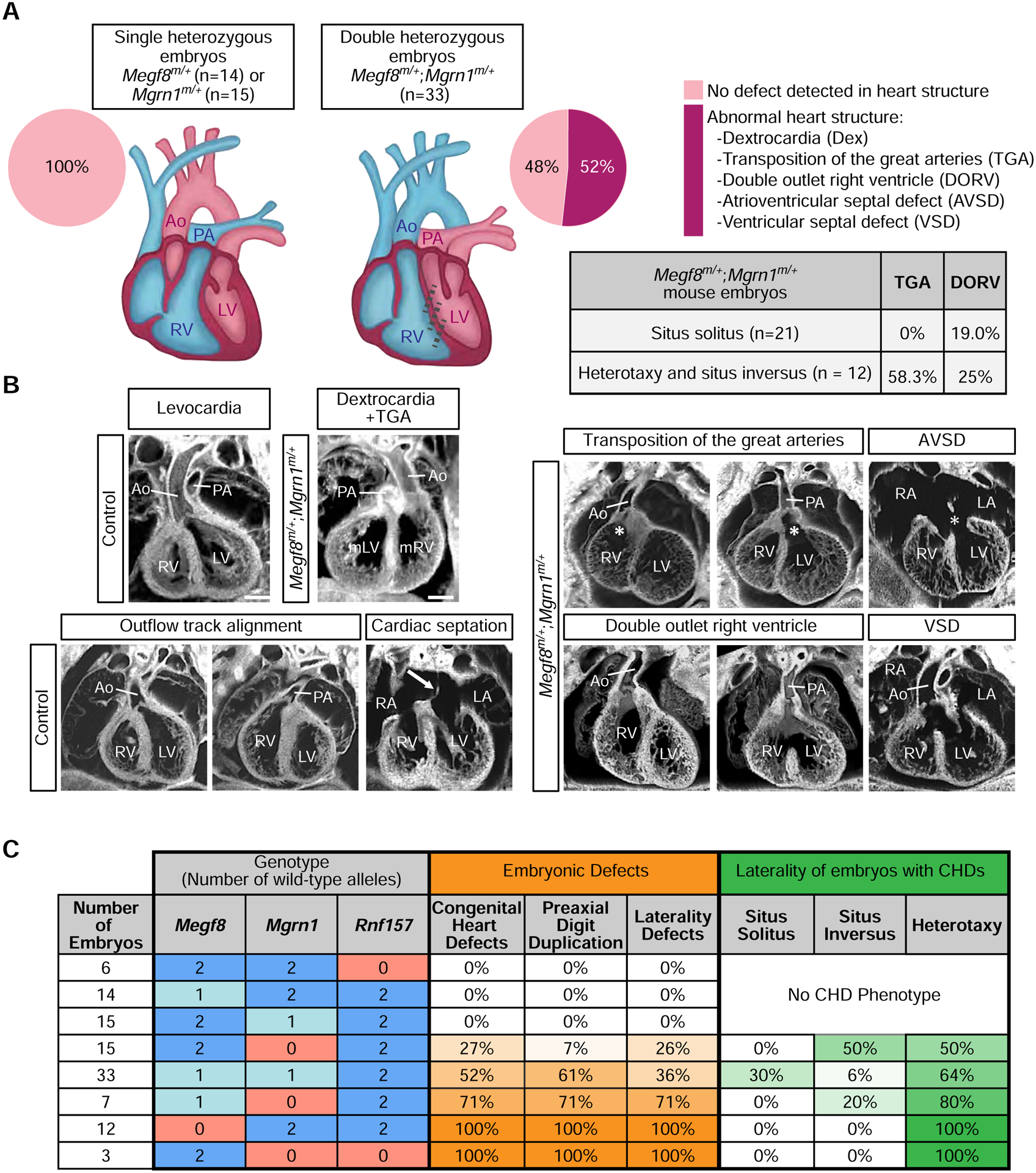
(A and B) Summary of congenital heart defects (CHDs) in mouse embryos of various genotypes (e13.5–14.5) as determined by ECM imaging. (B) Shows representative ECM images of the many defects observed in double heterozygous embryos, along with normal hearts from control (single heterozygous) embryos. Ao, aorta; AVSD, atrioventricular septal defect; Dex, dextrocardia; LA, left atrium; LV, left ventricle; mLV, morphological left ventricle; mRV, morphological right ventricle; PA, pulmonary artery; RA, right atrium; RV, right ventricle; VSD, ventricular septal defect. A detailed phenotypic analysis of each embryo can be found in Tables S2, S3, and S4. Scale bars, 100 μm.
(C) Table shows the frequencies of CHDs, preaxial digit duplication, and laterality defects observed in mouse embryos carrying increasing numbers of mutant alleles of Megf8, Mgrn1, and Rnf157. Darker shades of orange and green indicate a higher penetrance of the indicated birth defect and laterality phenotype, respectively. A detailed phenotypic analysis of every embryo of each genotype can be found in Tables S1–S5 and a full compilation of the penetrance of various phenotypes is provided in Table S6. For a more detailed analysis of the correlation between laterality and CHD phenotypes observed in Megf8m/+;Mgrn1m/+ embryos, refer to Table S7. See also Tables S1–S7.
The limb, heart and left-right patterning defects observed in 100% of Mefg8m/m embryos (Table S1), were incompletely penetrant in Megf8m/+;Mgrn1m/+ double heterozygous embryos (Figs. 5 and 6, Table S4). Preaxial digit duplication, a hallmark of elevated Hh signaling in the limb bud, was observed in only 61% of Megf8m/+;Mgrn1m/+ embryos (Figs. 5A and 6C). Defects in left-right patterning were seen in only 36% of Megf8m/+;Mgrn1m/+ embryos (Figs. 5A and 6C). Heart defects were seen in ~52% of Megf8m/+;Mgrn1m/+ embryos.
In addition to reduced penetrance, the CHDs seen in Megf8m/+;Mgrn1m/+ double heterozygous embryos were also milder compared to Megf8m/m embryos. All Megf8m/m embryos suffered from transposition of the great arteries (TGA), a severe outflow tract (OFT) malalignment defect in which the aorta emerges from the right ventricle and the pulmonary artery from the left ventricle (Figs. 6A and 6B, Table S1). Amongst the 52% of Megf8m/+;Mgrn1m/+ embryos with CHDs, only 41% of these embryos displayed TGA and 47% displayed a milder OFT defect called double outlet right ventricle (DORV) with or without atrioventricular septal defect (AVSD) (Figs. 6A and 6B, Table S4).
Given the known co-occurrence of heterotaxy with severe CHDs in clinical data from human birth registries (Lin et al., 2014; Pradat et al., 2003), we examined the correlation between these two types of birth defects in our mutant mouse embryos. All Megf8m/m embryos had both heterotaxy and TGA (Figs. 2F and 6C, Table S1). In Megf8m/+;Mgrn1m/+ embryos, heterotaxy was associated 100% of the time with CHDs and, conversely, CHDs were associated 64% of the time with heterotaxy (Fig. 6C, Tables S4, S6 and S7). Interestingly, the presence of heterotaxy was also correlated with more severe CHDs: ~60% of these embryos also had TGA (Fig. 6A). In contrast, Megf8m/+;Mgrn1m/+ embryos with normal left-right patterning (situs solitus) did not have TGA and instead had the milder DORV in ~20% of cases (Fig. 6A). These correlations are remarkably similar to data from human birth registries, which report that ~85% of heterotaxy cases are associated with CHDs that include DORV, TGA and AVSD (Lin et al., 2014; Pradat et al., 2003). The tight association between CHD and heterotaxy is also supported by the observation that all seven embryos with only preaxial digit duplication (but no CHD) had normal situs solitus (Table S4). Thus, the double heterozygous Megf8m/+;Mgrn1m/+ embryos recapitulate the known association between severe CHD and heterotaxy seen in human clinical data. The wider spectrum of CHDs seen in these embryos, including DORV, compared to homozygous Megf8m/m embryos resembles the more diverse range of CHDs seen in human patients with heterotaxy (Fig. 6A, Table S7) (Lin et al., 2014; Pradat et al., 2003).
Gene dosage effects involving Mgrn1, Megf8 and Rnf157
Our comparison of double heterozygous Megf8m/+;Mgrn1m/+ embryos to homozygous Megf8m/m embryos suggested that both the penetrance and expressivity of birth defect phenotypes may be determined by precise magnitude of ubiquitin ligase activity, which in turn determines the abundance of SMO and the strength of Hh signaling. This hypothesis predicts that the dosage of Megf8, Mgrn1 and Rnf157 should influence the penetrance of birth defect phenotypes.
We analyzed embryos carrying varying numbers of loss-of-function Megf8m, Mgrn1m, and Rnf157m alleles (Fig. 6C). Megf8m/m and Mgrn1m/m;Rnf157m/m embryos have a 100% penetrance of CHDs, heterotaxy, and preaxial digit duplication, presumably because the functions of both the transmembrane adaptor (MEGF8) and the cytoplasmic E3 ligases (MGRN1 or RNF157) are essential for SMO ubiquitination. Loss of one allele of Megf8 (Megf8m/+ embryos), one allele of Mgrn1 (Mgrn1m/+ embryos) or both alleles of Rnf157 (Rnf157m/m embryos) did not lead to birth defects, likely because the abundance of the MEGF8-MGRN1/RNF157 complex remains above the threshold required for normal development. However, between these two extremes, decreasing the cumulative gene dosage (by increasing the number of mutant alleles) of Mgrn1 and Megf8 led to a progressive increase in the penetrance of CHDs, heterotaxy and preaxial digit duplication (Fig. 6C and Tables S5 and S6). In addition, the incidence of TGA (Table S6), the most severe CHD, and the co-occurance of heterotaxy (Fig. 6C) increased with decreasing gene dosage. These striking gene dosage effects support the model that a progressive decrease in ubiquitin ligase function leads to a progressive increase in the penetrance and expressivity of birth defects, likely by driving a graded increase in Hh signaling strength.
The exquisite sensitivity of heart development to mutations in Megf8, Mgrn1 and Rnf157 seen in mouse embryos prompted us to look for potentially damaging variants in these genes in patients with CHDs. Using whole exome sequencing data from a cohort of 652 CHD patients, we searched for missense variants in all three genes with a Combined Annotation Dependent Depletion (CADD) score >10. We additionally used a stringent mean allele frequency (MAF) filter of < 0.5% for MEGF8 and MGRN1, but a more relaxed MAF filter (< 5%) for RNF157, since the Rnf157m/m mouse has no phenotype. Using these criteria, we identified one patient (7501) with two mutations each in MEGF8 and MGRN1 and one mutation in RNF157 (Figs. S7A and S7B; Table S8). Genotyping the parents of patient 7501 revealed that the two mutations in MEGF8 and MGRN1 were both present in the same allele, with the former transmitted from the mother and the latter from the father (along with the RNF157 variant). Patient 7501 clinically presented with OFT anomalies: pulmonary atresia, a severely hypoplastic right ventricle with an intact interventricular septum, an atrial septal defect, and patent foramen ovale (Figs. 7B and S7C). Primary fibroblasts from patient 7501 displayed increased abundance of ciliary SMO (Fig. 7C) and elevated Gli1 expression (Fig. 7D), both at baseline and in response to SHH, when compared to fibroblasts generated from a subject without CHD. Collectively, our mouse and human data support a model where disruption of the MEGF8-MGRN1/RNF157 ubiquitin ligase complex can lead to elevated SMO, increased Hh signaling strength and, consequently, to the emergence of CHDs.
Figure 7: Damaging variants in MEGF8, MGRN1, and RNF157 are associated with congenital heart defects in humans.
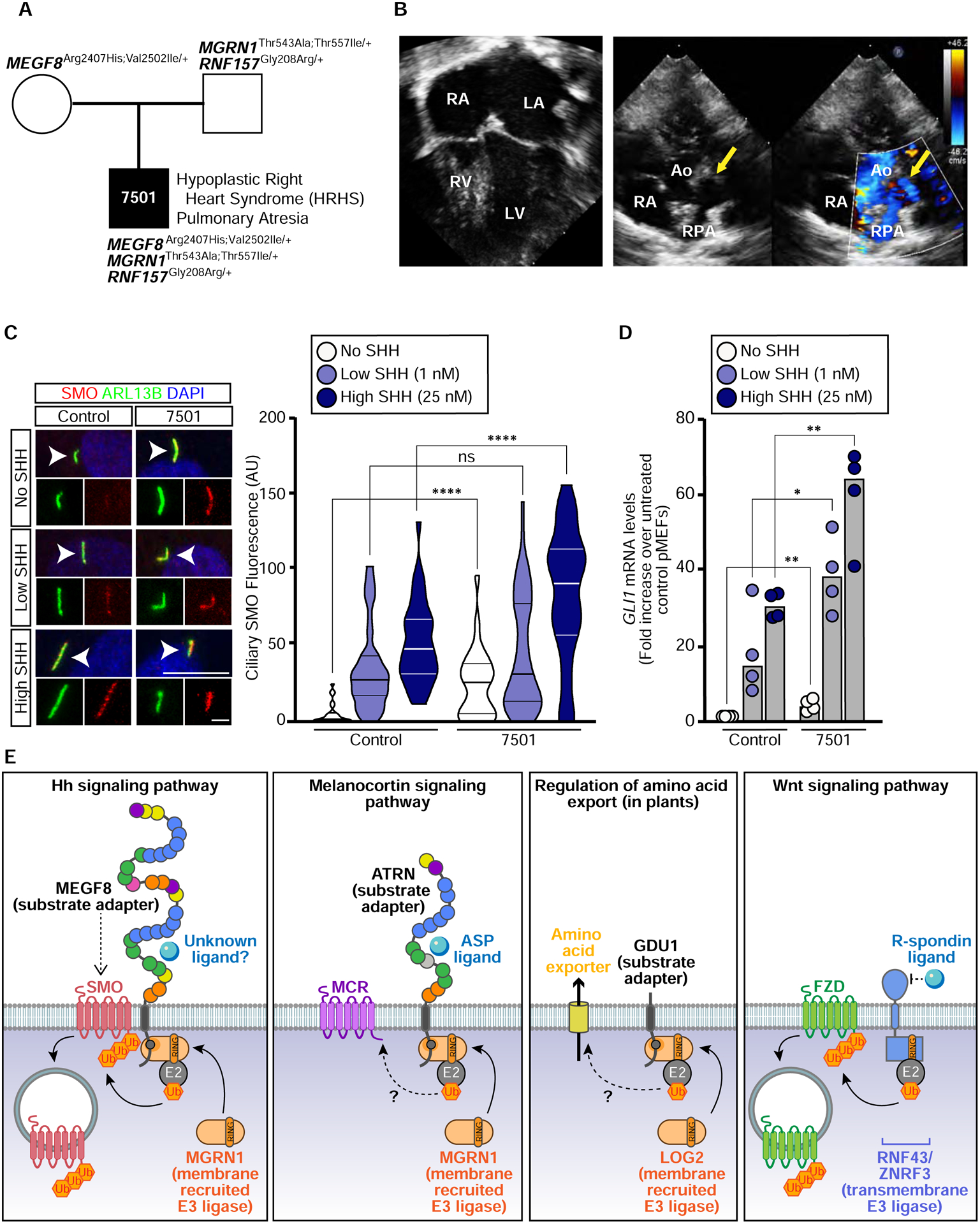
(A) Trio pedigree analysis showing the inheritance of MEGF8, MGRN1, and RNF157 variants from two affected parents to a progeny (patient 7501) with severe CHDs. The position of these variants in MEGF8, MGRN1, and RNF157, their evolutionary conservation, allele frequency and predicted damaging effect on protein function are shown in Figs. S7A and S7B. Whole exome sequencing results can be found in Table S8.
(B) Four-chamber view (left) or short axis view (right) of an echocardiogram from patient 7501 demonstrating a hypoplastic right ventricle (RV) and membranous pulmonary atresia (yellow arrows). RA, right atrium; LA, left atrium; LV, left ventricle; RPA, Right Pulmonary Artery; Ao, Aorta.
(C and D) Ciliary SMO (C) or GLI1 qRT-PCR (D) was used to assess Hh signaling in primary fibroblasts from patient 7501 (A) and from an unaffected control. Scale bars are 10 μm in merged panels and 2 μm in zoomed displays. The violin plot in (C) summarizes the quantification of SMO at ~20–50 cilia for each condition and the bars in (D) denote the median GLI1 mRNA values derived from the four individual measurements shown. Statistical significance was determined by the Mann-Whitney test (C) and unpaired t-test (D); not-significant (ns) > 0.05, *p-value ≤ 0.05, **p-value ≤ 0.01, ****p-value ≤ 0.0001.
(E) Regulation of signaling and transport by receptor-like E3 ubiquitin ligases. A model for the mechanism of SMO regulation by the MEGF8-MGRN1 complex (far left) highlights its conceptual similarity to the regulation of melanocortin receptors (MCRs) by the ATRN-MGRN1 complex (middle left), amino acid export by the GDU1-LOG2 complex in plants (middle right), and Frizzled (FZD) receptors for WNT ligands by the ZNRF3/RNF43 family of transmembrane E3 ligases (far right). MEGF8 functions as a transmembrane substrate adaptor, recruiting MGRN1 (and presumably an unknown E2 enzyme) through its cytoplasmic tail to promote the ubiquitination of SMO. SMO ubiquitination leads to its internalization and degradation, thus attenuating responses to Hh ligands. See also Figure S7 and Table S8.
DISCUSSION
Using a combination of mechanistic studies, mouse genetics, and deep anatomical phenotyping, we uncovered a unique membrane-tethered ubiquitination pathway that regulates developmental patterning in a variety of tissues by controlling the trafficking of signaling receptors. MEGF8 functions as a transmembrane substrate adaptor that recruits a cytoplasmic E3 ligase (MGRN1) to catalyze the ubiquitination of SMO, leading to its endocytosis and degradation (Fig. 7E). This ubiquitination reaction reduces the abundance of SMO at the cell surface and primary cilium and, consequently, dampens Hh signaling in target cells.
While cilia-localized ubiquitination is emerging as a mechanism that regulates ciliary trafficking (Desai et al., 2020; Shinde et al., 2020), the function of the MEGF8-MGRN1 complex is unlikely to be related to cilia or ciliary trafficking. Cell surface biotinylation experiments (see Fig. S5) clearly show that overall cell-surface SMO (not just ciliary SMO) rises dramatically in mutant cells. In addition, we have not been able to detect MGRN1 or MEGF8 in cilia. Hence, the ciliary accumulation of SMO in Megf8m/m and Mgrn1m/m cells and embryos is likely to be a secondary consequence of increased cell-surface SMO.
Receptor-like ubiquitin ligases attenuate signaling strength
The architecture of the MEGF8-MGRN1 complex is notable for the presence of a membrane-spanning component with an extracellular or luminal domain (Fig. 7E). This feature suggests a receptor-like function, conceptually analogous to receptor kinases, to transmit extracellular or luminal signals across the membrane to alter the ubiquitination of substrates in the cytoplasm. Interestingly, Frizzled (FZD) proteins, receptors for WNT ligands that are the closest relatives of SMO in the GPCR superfamily (Bjarnadóttir et al., 2006) are regulated by transmembrane E3 ligases (RNF43 and ZNRF3) in which the RING-containing domain is directly fused to the membrane-spanning component (Fig. 7E). While a ligand for MEGF8 remains unknown, ZNRF3 and RNF43 are regulated by R-Spondin ligands, critical regulators of progenitor cells during development and stem cells in adult tissues (Hao et al., 2012; Koo et al., 2012). The ubiquitination of receptors by membrane-tethered E3 ligases represents an attractive post-transcriptional mechanism to control the sensitivity of tissues to signaling ligands during development or tissue renewal.
Evolutionary sequence analysis supports a widespread role for MGRN1-based transmembrane E3 ligase complexes in ubiquitin signaling. In animals and their immediate sister lineages, MGRN1 and RNF157 likely function as common components of multiple membrane-tethered E3 ligase complexes featuring members of the MEGF8 family of cell-surface proteins, all of which contain an equivalent of the cytoplasmic MASRPFA motif (Fig. S4A) (Gunn et al., 1999; Haqq et al., 2003; Nagle et al., 1999). For example, MGRN1 and a different member of this family, Attractin (ATRN), have been implicated in regulation of melanocortin receptor levels (Fig. 7E) (Cooray et al., 2011; Walker, 2010). A plant ubiquitin ligase, LOG2, which belongs to the MGRN1 family, associates with and ubiquitinates a single TM protein Glutamine dumper-1 (GDU1) which in turn regulates amino acid transport (Fig. 7E) (Guerra et al., 2013). Strikingly, human MGRN1 can functionally replace LOG2 in plants (Guerra et al., 2013). We propose that the MGRN1 family of RING E3 ligases can associate more generally across eukaryotes with single-pass TM proteins, each of which function as a substrate adaptor to target the ubiquitination of specific receptors or transporters (Fig. 7E).
It will be interesting to unravel how these transmembrane E3 ligases are regulated in cells. Key questions include the potential role of phosphorylation in modulating ligase activity or substrate recognition, the type of molecular linkages used in the ubiquitin chains attached to SMO and the identity of the deubiquitinase that opposes the function of MGRN1. Most interesting is the mysterious function of the large MEGF8 extracellular domain, which may allow regulation of Hh signaling by an extracellular ligand, extracellular matrix protein or homotypic interaction (Fig. 7E).
Role of Hh signaling in left-right patterning and heart development
Our Vismodegib rescue experiments (Fig. 1) strongly suggest that preaxial digit duplication phenotypes in mice carrying mutant alleles of Megf8 and Mgrn1 are caused by elevated Hh signaling. However, further work will be required to determine whether the heterotaxy and CHD phenotypes are also caused entirely by increased Hh signaling.
Both decreased and increased Hh signaling have previously been implicated in left-right patterning, a very early event in development that directs the correct asymmetric development of the heart and other visceral organs (Levin et al., 1995; Tsiairis and McMahon, 2009; Zhang et al., 2001). The genetic deletion of SMO, which reduces Hh signaling strength, disrupts left-right patterning and causes a midline heart tube that fails to loop to the right and an embryo that fails to turn (Zhang et al., 2001). Conversely, ablation of the conserved negative regulator SUFU, which causes increased Hh signaling, also leads to left-right patterning defects (Cooper et al., 2005). Sufu−/− embryos fail to undergo embryonic turning, have a ~30–40% frequency of reversed heart looping (L-looping) and show either bilateral or absent expression of Pitx2, an established determinant of organ situs. These seemingly discordant results may be explained by the idea that left-right patterning (like digit patterning, see Fig. 1F) depends on a just-right “goldilocks” level of Hh signal amplitude or duration.
Hh signaling also influences multiple aspects of heart development: development of the secondary heart field and proper septation of the atria and outflow tract (Dyer and Kirby, 2009; Hoffmann et al., 2009; Washington Smoak et al., 2005). Thus, CHDs seen in our mutant mice may be caused by both early defects in left-right patterning and by later defects in Hh-mediated patterning of the cardiac septa and outflow tract. The common link of both processes to precisely-calibrated level of Hh signaling may explain the tight association between heterotaxy and CHDs that has been long-noted in clinical studies and is recapitulated in our mutant mouse embryos (Pradat et al., 2003).
We acknowledge that MEGF8 and MGRN1/RNF157 may regulate signaling receptors other than SMO and some of the birth defect phenotypes we observe may be related to disruption of other signaling pathways. Genetic and pharmacological experiments that attenuate Hh signaling or disrupt the MEGF8-MGRN1 complex at earlier time points in development or in specific tissues (such as the lateral plate mesoderm or developing cardiac outflow tract) will be required to uncover the molecular and cellular mechanisms through which these proteins regulate left-right patterning and heart development.
Oligogenic interactions and gene dosage effects underlie birth defects
While single heterozygous Megf8m/+ and Mgrn1m/+ embryos are normal, double heterozygous Megf8m/+;Mgrn1m/+ embryos display CHDs with heterotaxy. This phenomenon has been called “synthetic haploinsufficiency” and can result in an oligogenic pattern of inheritance, where mutations in one gene affect the phenotypic outcome of mutations in a different gene (Kousi and Katsanis, 2015; Veitia et al., 2013). Synthetic haploinsufficiency is most commonly seen between genes that encode subunits of a protein complex, like MEGF8 and MGRN1 (Veitia, 2010). Pioneering studies of Bardet-Biedl Syndrome (BBS) and other inherited retinopathies have demonstrated the importance of oligogenic interactions for understanding the genetic etiology of human diseases (Badano et al., 2006; Katsanis et al., 2000).
Beyond binary genetic interactions, the penetrance and expressivity of birth defect phenotypes progressively increases as an inverse function of the gene dosage of Megf8, Mgrn1 and Rnf157. We propose that this quantitative effect of mutations in this pathway is explained by the central role of the MEGF8-MGRN1 pathway in calibrating the amplitude of Hh signaling in target cells. The inheritance of increasing numbers of Megf8, Mgrn1 and Rnf157 mutant alleles will lead to a progressive decrease in the abundance (and hence activity) of the MEGF8-MGRN1/RNF157 complex. Decreasing E3 ligase activity will result in progressive increases in cell surface and ciliary SMO and thus increases in target cell sensitivity to Hh ligands. More generally, our results show that developmental patterning events can be tightly regulated by mechanisms in target cells that function to precisely tune sensitivity to extracellular morphogens.
We finish by noting that our genetic analyses highlight how interactions between a small number of genes can produce a complex inheritance pattern (common to many human diseases). Homozygous mutations in Megf8 result in a uniform phenotypic spectrum, with 100% of embryos displaying TGA, heterotaxy, and preaxial digit duplication. However, the co-inheritance of one mutant allele of Megf8 with one mutant allele of Mgrn1 (even in the homogenous genetic background of inbred mice) results in both incomplete penetrance and variable expressivity of phenotypes, manifested by a wider range of CHDs like TGA, DORV, and septal defects. Indeed, whole-exome sequencing studies of human CHD cohorts increasingly support a prominent role for such oligogenic inheritance mechanisms in the genetic etiology of CHDs (Gifford et al., 2019; Jin et al., 2017; Liu et al., 2017, 2020; Priest et al., 2016).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rajat Rohatgi (rrohatgi@stanford.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article contains all datasets generated and analyzed during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
NIH/3T3 and HEK293T cell culture
Flp-In-3T3 (a derivative of NIH/3T3 cells and referred to as “NIH/3T3” cells throughout the text) and HEK293T cell lines were purchased from Thermo Fisher Scientific and ATCC, respectively. Information on the gender of the cell lines is not available. NIH/3T3 and HEK293T cells were cultured in Complete Medium: Dulbecco’s Modified Eagle Medium (DMEM) containing high glucose (Thermo Fisher Scientific, Gibco) and supplemented with 10% fetal bovine serum (FBS) (MilliporeSigma), 2 mM L-Glutamine (Gemini Bio-Products), 1 mM sodium pyruvate (Thermo Fisher Scientific, Gibco), 1x MEM non-essential amino acids solution (Thermo Fisher Scientific, Gibco), and penicillin (40 U/ml) and streptomycin (40 μg/ml) (Gemini Bio-Products). The NIH/3T3 and HEK293T cells were passaged with 0.05% Trypsin/EDTA (Gemini Bio-Products). All cells were housed at 37 °C in a humidified atmosphere containing 5% CO2. Cell lines and derivatives were free of mycoplasma contamination as determined by PCR using the Universal Mycoplasma Detection Kit (ATCC).
Generation of primary mouse embryonic fibroblasts
Primary mouse embryonic fibroblasts (pMEFs) were generated using a modified published protocol (Durkin et al., 2013). Briefly, e12.5–14.5 embryos were harvested and rinsed thoroughly with PBS to remove any excess blood. Using forceps, the head and internal organs (heart and liver) were removed. The embryos were then separated into individual dishes and a sterile razor blade was used to physically mince the tissue in 0.25% Trypsin/EDTA (Thermo Fisher Scientific, Gibco). After pipetting the minced tissue up and down several times to further break up the tissue, the dishes were placed in a 37 °C tissue culture incubator for 10–15 minutes. If there were still large tissue pieces present, the minced tissue was pipetted further and the dish was placed in the incubator for an additional 5–10 minutes. The trypsin was then deactivated using Complete Medium (containing 10% FBS). The cells were then centrifuged, resuspended in fresh Complete Medium, and plated. Each clonal cell line represents pMEFs generated from a single embryo. The gender of the embryos were not determined prior to generating the pMEF cultures. Cells were housed at 37 °C in a humidified atmosphere containing 5% CO2.
Patient recruitment and nasal sampling for patient derived fibroblast cultures
Patients and parents were recruited from the Children’s Hospital of Pittsburgh with informed consent obtained under a human study protocol approved by the University of Pittsburgh Institutional Review Board. Control, CHD patient, and parents recruited had blood drawn for DNA extraction. CHD diagnosis was confirmed with examination of the patient’s medical records. Nasal tissue was obtained from the patient by curettage of the inferior nasal turbinate using a rhino probe. The nasal epithelial tissue was plated in RPMI medium (Thermo Fisher Scientific, Gibco) with 10% FBS (MilliporeSigma) and the fibroblast outgrowths that emerged were expanded and used for Hh signaling assays (below). Both primary fibroblast cell lines (control and patient 7501) were derived from cells collected from female patients.
Hh signaling assays in NIH/3T3 cells and primary fibroblasts
For Hh signaling assays, NIH/3T3 cells, pMEFs, and primary human fibroblasts were first grown to confluence in Complete Medium (containing 10% FBS) and then ciliated by changing the cell medium to Low Serum Medium (Complete Medium containing 0.5% FBS) for 24 hours. Cells were treated with either no SHH, a low concentration of SHH (1 nM), a high concentration of SHH (25 nM), or SAG (100 nM) for at least 4 hours prior to fixation (for NIH/3T3 immunofluorescence assays), 24 hours prior to lysis (for NIH/3T3 Western blot assays or NIH/3T3 RNA extraction for qRT-PCR), or 48 hours prior to experimentation (for pMEF and primary human fibroblast immunofluorescence, western blot, and qRT-PCR assays).
Hh signaling activity was measured using real-time quantitative reverse transcription PCR (qRT-PCR). RNA was extracted from NIH/3T3 cells and mouse pMEFs using TRIzol reagent (Thermo Fisher Scientific, Invitrogen) as previously described (Rio et al., 2010). Equal amounts of RNA were used as template for cDNA synthesis using the iScript Reverse Transcription Supermix (Bio-Rad Laboratories). qRT-PCR for mGli1 and mGapdh was performed on a QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific) with the following custom designed primers: mGli1 (Fwd 5’-CCAAGCCAACTTTATGTCAGGG-3’ and Rev 5’-AGCCCGCTTCTTTGTTAATTTGA-3’) and mGapdh (Fwd 5’-AGTGGCAAAGTGGAGATT-3’ and Rev 5’-GTGGAGTCATACTGGAACA-3’). Similarly, RNA was isolated from primary human fibroblasts using the RNeasy Plus Mini Kit (Qiagen). Equal amounts of RNA were used as template for human cDNA synthesis using the High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific, Applied Biosystems). qRT-PCR for hGLI1 and hGAPDH was performed on a 7900HT Real-Time PCR System (Life Technologies) with the following primers: hGLI1 (Fwd 5’-CAGGGAGGAAAGCAGACTGA-3’ and Rev 5’-ACTGCTGCAGGATGACTGG-3’) and hGAPDH (Fwd 5’-GTCTCCTCTGACTTCAACAGCG-3’ and Rev 5’-ACCACCCTGTTGCTGTAGCCAA-3’). For all qRT-PCR experiments, Gli1 transcript levels were calculated relative to Gapdh and reported as a fold change across conditions using the comparative CT method (ΔΔCT method).
Neural progenitor differentiation assay
Maintenance of HM1 mouse embryonic stem cells (mESCs) harboring the GLI-Venus and OLIG2-mKate dual reporter system and their differentiation into neural progenitor cells (NPCs) was performed as described previously (Pusapati et al., 2018). The parental HM1 mESC line was derived from a male mouse. mESCs were grown and maintained on feeder cells in mESC Medium: Dulbecco’s Modified Eagle Medium (DMEM) containing high glucose (Thermo Fisher Scientific, Gibco) and supplemented with 15% FBS (MilliporeSigma), 2 mM L-Glutamine (Gemini Bio-Products), 1 mM sodium pyruvate (Thermo Fisher Scientific, Gibco), 1x MEM non-essential amino acids solution (Thermo Fisher Scientific, Gibco), 1% penicillin/streptomycin (Gemini Bioproducts), 1% EmbryoMax nucleosides (MilliporeSigma), 55 μM 2-mercaptoethanol (Thermo Fisher Scientific, Gibco), and 1000 U/ml ESGRO LIF (MilliporeSigma). mESCs were differentiated into spinal neural progenitor cells using a previously described protocol (Sagner et al., 2018). mESCs were panned to clear the feeder cells, then plated on 6-well gelatin-coated CellBIND plates (Corning) at a density of 100,000 cells/well. Differentiation was conducted in N2B27 Medium: DMEM/F12 (Thermo Fisher Scientific, Gibco) and Neurobasal medium (Thermo Fisher Scientific, Gibco) (1:1 ratio) supplemented with 1x N-2 supplement (Thermo Fisher Scientific, Gibco), 1x B-27 supplement (Thermo Fisher Scientific, Gibco), 1% penicillin/streptomycin, 2 mM L-Glutamine, 55 μM 2-mercaptoethanol (Thermo Fisher Scientific, Gibco), and 40 μg/ml bovine serum albumin (MilliporeSigma). On Day 0 (the day the cells were plated) and Day 1, the N2B27 medium was supplemented with 10 ng/ml bFGF (R&D Systems). On Day 2, the N2B27 medium was supplemented with 10 ng/ml bFGF (R&D Systems) and 5 μM CHIR 99021 (Axon Medchem). On Day 3, the N2B27 medium was supplemented with 100 nM Retinoic Acid (RA) (MilliporeSigma) and either no SHH, 5 nM (low SHH), or 25 nM (high SHH). The cells were cultured in RA and SHH for a total of 3 days, where the medium was changed every 24 hours. On Day 6, the cells were washed with PBS and trypsinized with 0.25% Trypsin/EDTA (Thermo Fisher Scientific, Gibco) for flow cytometry analysis. GLI-Venus and OLIG2-mKate fluorescence was measured on a FACScan Analyzer at the Stanford Shared FACS Facility. To detect GLI-Venus, a 488 nm (blue) laser was used with a 525/50 filter and B525 detector. To detect OLIG2-mKate, a 561 nm (yellow) laser was used with a 615/25 filter and Y615 detector.
Generation of knockout cell lines
Clonal Mgrn1−/− NIH/3T3 lines were previously generated using a dual single guide (sgRNA) strategy and validated (Pusapati et al., 2018). Clonal double knockout Mgrn1−/−;Rnf157−/− NIH/3T3 lines were generated using the same dual sgRNA strategy to target Rnf157 in Mgrn1−/− NIH/3T3 cells. Briefly, sgRNAs targeting Rnf157 were designed using the Broad Institute Genetic Perturbation Platform sgRNA Designer Tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design): Exon 6, 5’-CCACAGCGTGCACTACCAGA-3’ and Exon 7, 5’-CAAAAGTGCCCAGAAGCACG-3’. The sgRNAs were then were cloned into pSpCas9(BB)-2A-GFP (Addgene) (Ran et al., 2013) and pSpCas9(BB)-2A-mCherry (Pusapati et al., 2018) and transfected into NIH/3T3 cells using X-tremeGENE 9 DNA transfection reagent (Roche Molecular Systems). Five days post transfection, GFP and mCherry double positive single cells were sorted into a 96-well plate using a FACSAria II at the Stanford Shared FACS Facility. To detect the GFP, a 488 nm (blue) laser was used with a 530/30 filter and B530 detector. To detect the mCherry, a 561 nm (yellow) laser was used with a 616/23 filter and G616 detector. Clonal lines were screened by PCR (Fwd 5’-GAGCAGAGAGGAGGTTAGCG-3’ and Rev 5’-CAAGCTAGACCTTCCCGAGG-3’) to detect excision of the genomic DNA (317 bp) between the two sgRNA cut sites (Fig. S3B).
Clonal Mgrn1−/− HM1 mouse embryonic stem cells (mESCs) with both GLI-Venus and OLIG2-mKate reporters were previously created using a dual sgRNA strategy and validated (Pusapati et al., 2018). Similar to what was done in NIH/3T3 cells, clonal double knockout Mgrn1−/−;Rnf157−/− mESC lines were generated using a dual sgRNA strategy to target Rnf157 in Mgrn1−/− mESCs. Briefly, the same sgRNAs used to target Rnf157 in NIH/3T3 cells were used in mESCs, but these sgRNAs were cloned into pSpCas9(BB)-2A-Puro (Addgene) (Ran et al., 2013). Prior to any manipulation, the mESCs were maintained for three passages under feeder free conditions in 2i Medium: DMEM/F12 (Thermo Fisher Scientific, Gibco) and Neurobasal medium (Thermo Fisher Scientific, Gibco) (1:1 ratio) supplemented with 1x N-2 supplement (Thermo Fisher Scientific, Gibco), 1x B-27 supplement (Thermo Fisher Scientific, Gibco), 1% penicillin/streptomycin (Gemini Bioproducts), 2 mM L-Glutamine (Gemini Bioproducts), 55 μM 2-mercaptoethanol (Thermo Fisher Scientific, Gibco), 40 μg/ml bovine serum albumin (MilliporeSigma), 5 μM CHIR 99021 (Axon Medchem), 1 μM PD 98059 (Axon Medchem), and 1000 U/ml ESGRO LIF (MilliporeSigma). Cells were trypsinized in 0.25% Trypsin/EDTA (Thermo Fisher Scientific, Gibco) and rinsed once in PBS. Plasmids were nucleofected into the mESCs using the Lonza Cell nucleofector kit (VAPH-1001) and program A-023 on the Lonza Nucleofector 2b Device (Lonza Bioscience). After the cells were nucleofected, they were plated in 2i Medium onto a 10 cm gelatin-coated CellBIND plate. 24 hours post nucleofection, selection was started and the medium was changed to 2i Medium containing 1.5 μg/ml puromycin (MilliporeSigma) for 48 hours (or until all the cells on the non-nucleofected control plate died). Approximately 1 week after nucleofection, individual mESC colonies were manually picked, expanded, and screened by PCR using the same primers used to screen the Mgrn1−/−;Rnf157−/− NIH/3T3 cells (Fig. S3B).
Generation of stable cell lines expressing transgenes
Clonal Megf8−/− and Mgrn1−/− Flp-In-3T3 cell lines were previously generated and validated (Pusapati et al., 2018). Stable addback cell lines expressing tagged MEGF8 and MEGF8 ΔCtail (featured in Figs. 3D and 3E), were generated using Flp recombinase-mediated DNA recombination (Thermo Fisher Scientific, Invitrogen) as previously described (Pusapati et al., 2014). Briefly, the pOG44 Flp-recombinase expression vector (Thermo Fisher Scientific, Invitrogen) and either pEF5/FRT/V5-DEST-MEGF8-1D4 or pEF5/FRT/V5-DEST-MEGF8 ΔCtail-1D4 were transfected into Megf8−/− NIH/3T3 cells using the X-tremeGENE 9 DNA transfection reagent (Roche Molecular Systems). Approximately 48 hours post transfection the cells were split to 25% confluence and 12–16 hours post split the medium was changed to Complete Medium containing 200 μg/ml Hygromycin B (VWR Life Science). The medium was replenished every 3–4 days and antibiotic selection was conducted for about 2 weeks or until all the cells on the control plate were dead.
Stable addback cell lines expressing tagged MGRN1 (featured in Figs. 3F and 3G) in Mgrn1−/−; Rnf157−/− NIH/3T3 cells or tagged MEGF8 in Megf8−/− NIH/3T3 cells (featured in Figs. 4D and 4E) were generated using the lentiviral expression system. Briefly, to generate lentivirus, four million HEK293T cells were seeded onto a 10 cm plate and 24 hours later these cells were transfected with 1 μg pMD2.G (Addgene), 5 μg psPAX2 (Addgene), and 6 μg of the desired pLenti CMV Puro DEST construct using 36 μl of 1mg/ml polyethylenimine (PEI) (Polysciences). Approximately 48 hours post transfection, the lentivirus was harvested and filtered through a 0.45 μm filter. 2 ml of the filtered lentivirus solution was mixed with 2 ml of Complete Medium containing 16 μg/mL polybrene (MilliporeSigma). The diluted virus was then added to NIH/3T3 cells seeded on 6-well plates. Approximately 48 hours post infection, cells were split and selected with puromycin (2 μg/ml) for 5–7 days or until all the cells on the control plate were dead.
Established mouse lines
All mouse studies were conducted using animal study protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Stanford University, the University of Pittsburgh, and the McLaughlin Research Institute for Biomedical Sciences. Mgrn1md-nc/md-nc null mutant mice (referred to in the paper as Mgrn1m/m) (MGI:3704004) and Megf8C193R/C193R mice (referred to in the paper as Megf8m/m) (MGI:3722325) have been described previously (Gunn et al., 2013; He et al., 2003; Zhang et al., 2009). Mgrn1md-nc/+ animals crossed to Megf8C193R/+ heterozygotes had been outcrossed to FVB/N/Mri and intercrossed for up to 3 generations. Animals were genotyped for the Mgrn1md-nc mutation by allele-specific PCR using the following primers: wild-type (Fwd 5’-GCCTGCATGGATAGATGGAT-3’ and Rev 5’-AGGAAGTTGCCCACAAGAACGCA-3’) and mutant (Fwd 5’-CAAGAACAACCAGGAGACTAAGGA-3’ and Rev 5’-GCCCAAGTCCTAAACCTCT-3’) (Gunn et al., 2019). Amplification was performed using GoTaq Green Master Mix (Promega Corporation), the initial 10 cycles with an annealing temperature of 60 °C, followed by 30 cycles with an annealing temperature of °57 C. Animals were genotyped for the Megf8C193R mutation by either (1) sequencing of a PCR product generated using primers Fwd 5’-ACGACCCATATCTCTGCCTT-3’ and Rev 5’-GCCTCCAGACCCTCCAAG-3’ or (2) using allele-specific PCR with primers Fwd 5’-CTCAGCTCTGCACCCCTAAC-3’ and Rev (wild-type) 5’-TCCCAAGAATCCAGGTTCACA-3’ or Rev (mutant) 5’-CCAAGAATCCAGGTTCACG-3’. Amplification was performed using GoTaq Green Master Mix (Promega Corporation), 30 cycles with an annealing temperature of 62 °C.
Generation and validation of Rnf157−/− mutant mice
Rnf157−/− mutant mice (referred to in the paper as Rnf157m/m mice) were generated by CRISPR/Cas9 mediated genome editing. The website Benchling (www.benchling.com) was used to design sgRNAs that target exon 4 of Rnf157: (5’-CTACTACCAGGCCACTG-3’ and 5’-TGAACTCGACATTGTAG-3’) (Fig. S3B). Synthetic sgRNAs and Cas9 2NLS nuclease were purchased from Synthego and electroporated into one cell mouse embryos following the Easy Electropopration of Zygotes (EEZy) protocol (Tröder et al., 2018). Briefly, fertilized eggs/1-cell embryos were collected from superovulated C57BL/6J females mated to C57BL/6J males into M2 or EmbryoMax Advanced KSOM medium (MilliporeSigma). Cas9/sgRNA ribonucleoproteins (RNPs) were assembled by combining 4 μM Cas9 protein with 4 μM of sgRNAs in 20 μl Opti-MEM reduced serum medium (ThermoFisher Scientific, Gibco) and incubating 10 min at room temperature. For each electroporation, up to 60 embryos were washed through one drop of Opti-MEM and added to the 20 μl of Cas9 RNP mix. The entire solution was immediately transferred to a 1 mm cuvette (Bio-Rad Laboratories) and placed in a Bio-Rad Gene Pulser XCell electroporator. Two square wave pulses were applied (30V, 3 ms pulse duration, 100 ms interval). Embryos were retrieved from the cuvette by flushing twice with 100 μl of pre-warmed KSOM, transferred to a droplet of KSOM under oil and maintained in a 37 °C incubator with 15% CO2 for 1–24 hours. Embryos were subsequently moved through a droplet of M2 medium and transferred to the oviduct of 0.5 dpc (days post coitum) pseudopregnant ICR females. At weaning, a small piece of tail tissue was taken from each pup and the DNA was isolated and genotyped using the following primers to PCR amplify the region around the sgRNA target sequences: Fwd 5’-AACAAAGTCCCGATCCACTG-3’ and Rev1 5’-CAAGCTAGACCTTCCCGAGG-3’ or Rev2 5’-CCTTTCAGCATGGCTTTCTC-3’. Sequence data was analyzed using Synthego’s ICE tool (https://ice.synthego.com/#/) and animals carrying modified alleles predicted to result in a loss of RNF157 function were mated to C57BL/6J animals. Rnf157em1Tmg carries a single nucleotide deletion at each sgRNA target site: a cysteine at position 58 of exon 4 and another cysteine at position 120 (Fig. S3B). Animals carrying this allele were genotyped by sequencing, as described above, or by allele-specific PCR using the following genotyping primers: Fwd (wild-type) 5’-AGGCAAAGCTAAGGTCCACTAC-3’, Fwd (mutant) 5’-AGGCAAAGCTAAGGTCCACTAA-3’, and Rev 5’-CCTGCTATGCCGTCTTACCT-3’).
RT-PCR was used to verify loss of Rnf157 expression in Rnf157em1Tmg mice. Briefly, brains (which express high levels of Rnf157) were collected from wild-type mice and Rnf157em1Tmg heterozygote and homozygote animals (Fig. S3B). DNase-I treated RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Invitrogen) and the Direct-zol RNA miniprep kit (Zymo Research). Equal amounts of RNA were used as template for cDNA synthesis using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, Invitrogen). PCR was performed using GoTaq Green Master Mix (Promega Corporation) the following RT-PCR primers: Fwd 5’-ATCCCGTCCAATTCCGTGTA-3’ and Rev 5’-GTACCAGGTGCGATGTAGGA-3’.
METHOD DETAILS
Constructs
MEGF8 constructs:
Mammalian Gene Collection (MGC) cDNA clone for human MEGF8 (NM_001410.3) was purchased from Transomic Technologies, Inc and used as a template for the generation of all MEGF8 constructs. All MEGF8 constructs were tagged with a C-terminal 1D4 and cloned into pEF5/FRT/V5-DEST (Thermo Fisher Scientific, Invitrogen) or pLenti CMV PURO DEST (Campeau et al., 2009) using Gateway recombination methods (Thermo Fisher Scientific, Invitrogen). MEGF8ΔN (a.a. 2573–2778) was generated using restriction enzymes SrfI and SapI (New England Biolabs) to remove the N-terminal region (a.a. 26–2572 deleted). MEGF8ΔCtail (a.a. 1–2607) and MEGF8ΔMASRPFA (a.a. 2625–2631 deleted) were created using a combination of overlap extension PCR and restriction enzyme cloning methods. MEGF8 chimeras were generated using Gibson assembly methods (New England Biolabs). CD16ECD-CD7™-MEGF8Ctail (a.a. 2604–2778), CD16ECD-MEGF8TM+Ctail (a.a. 2573–2778), and CD16ECD-MEGF8TM+CtailΔMASRPFA (a.a. 2573–2778 with a.a. 2625–2631 deleted) were all cloned using MEGF8ΔN and CD16ECD-CD7™-mCherry-Nck-HA (gift from Bruce Mayer (Rivera et al., 2009)). Lastly, for the bacterial production of MEGF8 Ctail recombinant protein, the C-terminal end of the Ctail (a.a. 2738–2778) was cloned into the pGEX vector using restriction enzyme cloning methods.
Mgrn1 constructs:
Mouse full-length Mgrn1 (NM_001252437.1) with a C-terminal 3xFLAG tag was synthesized as a gBlock (Integrated DNA Technologies) and used as a template for the generation of all Mgrn1 constructs. Overlap extension PCR was used to generate MGRN1Mut1 (C279A;C282A) and MGRN1Mut2 (L307A;R308A). All constructs were cloned into pEF5/FRT/V5-DEST (Thermo Fisher Scientific, Invitrogen) or pLenti CMV PURO DEST (Campeau et al., 2009) using Gateway recombination cloning methods (Thermo Fisher Scientific, Invitrogen).
Smo constructs:
mSmo-EGFP was a gift from Philip Ingham (Zhao et al., 2016). For Fig. S6C, pCS2-mSmo (Byrne et al., 2016) and a mSmo gBlock fragment with all 21 intracellular lysines mutated to arginines (Twist Bioscience) were used as templates to generate the following constructs: untagged full length Smo (WT), intracellular lysine-less Smo (K0), C-tail lysine-less Smo (CtailK0), intracellular loop 2 and 3 lysine-less Smo (ICLK0), intracellular loop 2 lysine-less Smo (ICL2K0), and intracellular loop 3 lysine-less Smo (ICL3K0). Constructs were generated using PCR amplification followed by Gibson assembly methods (New England Biolabs).
Other constructs:
SSTR3-GFP was a gift from Kirk Mykytyn(Berbari et al., 2008) and pRK5-HA-Ubiquitin-WT and pRK5-HA-Ubiquitin-K0 were purchased from Addgene (Lim et al., 2005).
Reagents and antibodies
Recombinant SHH was expressed in bacteria and purified in the lab as previously described (Bishop et al., 2009). Briefly, His-tagged SHH-N (C24II followed by human SHH a.a. 25–193) was expressed in Escherichia coli (BL21 strain; Rosetta2 (DE3)pLysS). Cells were lysed in 10 mM Phosphate Buffer pH 7.5, 500 mM NaCl, 1 mM 2-mercaptoethanol, 1 mM PMSF, and 1xprotease inhibitor cocktail, followed by centrifugation at 20,000xg for 30 min at 4°C. Clarified samples were incubated with Ni-NTA resin (Qiagen) for 1 h at 4°C. The resin was washed with 20 column volumes of wash buffer A (lysis buffer without protease inhibitors), followed by wash buffer B (wash buffer A+10 mM Imidazole) and bound proteins eluted with elution buffer (wash buffer A+250 mM Imidazole). Peak fractions were pooled, concentrated using a 5 kDa cut-off VIVASPIN 15R (Life Technologies), and loaded onto a Superdex 75 gel filtration column (Amersham Biosciences) equilibrated with column buffer (10 mM HEPES pH 7.5, 150 mM NaCl, and 1 mM DTT). The recombinant protein was >98% pure, as assessed from coomassie staining and stored at −80 °C. SAG was purchased from Thermo Fisher Scientific (Enzo Life Sciences). The selection antibiotic puromycin was purchased from MilliporeSigma and hygromycin B from VWR Life Science. The transfection reagent XtremeGENE 9 was purchased from Roche Molecular Systems and polybrene from MilliporeSigma. Bafilomycin A1 was purchased from Cayman Chemical. Vismodegib and Bortezomib were purchased from LC labs. The following primary antibodies were purchased from the following vendors: mouse anti-1D4 (The University of British Columbia, 1:5000); mouse anti-CD16 (clone 3G8, Santa Cruz Biotechnology, 1 μg per 1 million cells in 100ul); mouse anti-CD16 (clone DJ130c, Santa Cruz Biotechnology, 1 μg per 1 million cells in 100ul); mouse anti-FLAG (clone M2, MilliporeSigma, 1:2000); goat anti-GFP (Rockland Immunochemicals, 1:1000); rabbit anti-GFP (Novus Biologicals, 1:5000); mouse anti-GLI1 (clone L42B10, Cell Signaling, 1:1000); mouse anti-HA.11 (clone 16B12, BioLegend, 1:2000); mouse anti-HA (clone 2–2.2.14, Thermo Fisher Scientific, 1:2000); rabbit anti-p38 (Abcam, 1:2000); and rabbit anti-RNF156 (anti-MGRN1, Proteintech, 1:500); mouse anti-ɑ-Tubulin (Clone DM1A, MilliporeSigma, 1:10000); mouse anti-acetylated-Tubulin (MilliporeSigma, 1:10000). The following primary antibodies were generated in the lab or received as a gift: Guinea pig anti-ARL13B (1:1000) (Dorn et al., 2012); rabbit anti-SMO (designed against an intracellular epitope, 1:2000) (Rohatgi et al., 2007); and rabbit anti-SMO-N (designed against an extracellular epitope, 1:2000) (Milenkovic et al., 2009). The anti-MEGF8 rabbit polyclonal antibody was produced against amino acids 2738–2778 of the mouse MEGF8 protein and affinity purified before use (Cocalico Biologicals, Inc., 1:2000). Hoechst 33342 and secondary antibodies conjugated to horseradish peroxidase (HRP) or Alexa Fluor dyes were obtained from Jackson Laboratories and Thermo Fisher Scientific.
Protein Sequence Analysis
Iterative sequence profile searches were performed using the PSI-BLAST program run against the NCBI non-redundant (NR) protein database (Altschul et al., 1997). Multiple sequence alignments were built using the Kalign2 software (Lassmann et al., 2009) and were later manually adjusted based on profile-profile, secondary structure information, and structural alignments. Similarity-based clustering for both classification and discarding of nearly identical sequences was performed using the BLASTClust program (Fig. S4A). Maximum-likelihood (ML) tree topology was derived using an edge-linked partition model as implemented in the IQ-TREE software (Nguyen et al., 2015). ModelFinder (Kalyaanamoorthy et al., 2017) was used to automatically identify the best-fit substitution model and estimated “JTT+F+R9” as the suitable model for the given dataset. Branch supports were obtained using the ultrafast bootstrap (UFBoot) approximation method (1000 replicates) (Hoang et al., 2018). To further assess the branch supports, Shimodaira-Hasegawa(SH-)aLRT branch test was also computed as implemented in the IQ-TREE software (Fig. 2B). The sequence logo was generated using the Logo software (Crooks et al., 2004) (Fig. 3B). An alignment comprising a collection of all unique members of the MEGF8-Attractin family from the RefSeq database was utilized as input. The UniProt align tool was used to compare two protein sequences with the Clustal Omega program (Fig. S3A) (UniProt Consortium, 2019). Sequence analysis of the MGRN1 RING domain was done using ConSurf (Ashkenazy et al., 2016). Briefly, 200 MGRN1 homologs were collected from UniProt using the homolog search algorithm HMMER and a color coded multiple sequence alignment was built using ClustalW (Fig. S4B).
Immunoprecipitation and Western Blotting
Whole cell extracts from HEK293T and NIH/3T3 cells were prepared in Immunoprecipitation (IP) Lysis Buffer: 50 mM Tris at pH 8.0, 150 mM NaCl, 1% NP-40, 1 mM DTT, 1x SIGMAFAST protease inhibitor cocktail (MilliporeSigma), and 1x PhosSTOP phosphatase inhibitor cocktail (Roche). Cells were lysed for 1 hour on a shaker at 4 °C, supernatants were clarified by centrifugation, and 1D4 tagged MEGF8 was captured by a 1D4 antibody (The University of British Columbia) covalently conjugated to Protein A Dynabeads (Thermo Fisher Scientific, Invitrogen). Immunoprecipitates were washed once with IP Wash Buffer A (50 mM Tris at pH 8.0, 150 mM NaCl, 1% NP-40, and 1 mM DTT), once with IP Wash Buffer B (50 mM Tris at pH 8.0, 500 mM NaCl, 0.1% NP-40, and 1 mM DTT), and finally with IP Wash Buffer C (50 mM Tris at pH 8.0, 0.1% NP-40, and 1 mM DTT). Proteins were eluted by resuspending samples in 1xNuPAGE LDS sample buffer (Thermo Fisher Scientific, Invitrogen) supplemented with 100 mM DTT, incubated at 37 °C for 30 min, and subjected to SDS-PAGE (Figs. 3C, 3D, and S4C).
For all other immunoblotting data presented in the manuscript, whole cell extracts were prepared in RIPA lysis buffer: 50 mM Tris at pH 8.0, 150 mM NaCl, 2% NP-40, 0.25% Deoxycholate, 0.1% SDS, 0.5 mM TCEP, 10% glycerol, 1x SIGMAFAST protease inhibitor cocktail (MilliporeSigma), and 1x PhosSTOP phosphatase inhibitor cocktail (Roche). The resolved proteins were transferred onto a nitrocellulose membrane (Bio-Rad Laboratories) using a wet electroblotting system (Bio-Rad Laboratories) followed by immunoblotting.
Flow cytometry of live cells
As described above, a lentiviral expression system was used to stably express CD16/CD7/MEGF8 chimeras in Megf8−/− NIH/3T3 cells (diagramed in Fig. S6D). A modified live cell immunostaining protocol from Santa Cruz Biotechnology and Cell Signaling Technology was used to label and analyze cell surface CD16/CD7/MEGF8 chimeras (Fig. S6E). Briefly, four cell lines were analyzed: Megf8−/−, Megf8−/− with CD16ECD-CD7TM-Megf8Ctail addback, Megf8−/− with CD16ECD-Megf8TM+Ctail addback, and Megf8−/− with CD16ECD-Megf8TM+CtailΔMASRPFA addback. Prior to staining, the cells were serum starved for 24 hours to allow for primary cilia growth. On staining day, the Complete Medium was removed, the cells were rinsed with PBS, and then dissociated in 0.2% EDTA (prepared in PBS) for approximately 5 minutes at 37 °C. Upon seeing the cells lift from the plate, the cells were pipetted up and down five times to create a single cell suspension, and Complete Medium (containing 10% FBS) was added to neutralize the EDTA. A small sample was taken to determine the total number of cells present. The cells were then resuspended in Flow Cytometry (FCM) Blocking Buffer (0.5% bovine serum albumin prepared in PBS) at a concentration of 10 million cells/ml. The cells were blocked for 10 min on ice, 1 million cells (100 ul of the cell suspension) was then transferred to a fresh tube, and 1 ug of an anti-CD16 antibody (Santa Cruz Biotechnology, clones 3G8 and DJ130c) was added directly to the cells. The cells were incubated with primary antibodies for 30 minutes on ice. Primary antibodies were rinsed off with 2 washes in FCM Blocking Buffer. The cells were then incubated for 30 minutes on ice in 1 ug of donkey anti-mouse IgG, Alexa Fluor 488 (Thermo Fisher Scientific, Invitrogen) diluted in FCM Blocking Buffer. The cells were washed 2 times in FCM Blocking Buffer then analyzed on a BD Accuri C6 Flow Cytometer (BD Biosciences).
SMO internalization assay
Cell surface internalization assay for SMO was performed as described previously for Figs. 4A, S5A and S5B (Pusapati et al., 2018). Briefly, wild-type, Megf8−/−, and Mgrn1−/−;Rnf157−/− NIH/3T3 cells were plated on 15 cm plates in Complete Medium (containing 10% FBS). Once the cells were confluent they were switched to Low Serum Medium (Complete Medium containing 0.5% FBS) for 24 hours. On biotinylation day, the cells were removed from the 37 °C incubator and placed on an ice-chilled metal rack in a 4 °C cold room. The medium was removed and cells were quickly washed 3 times with ice-cold DPBS+ buffer (Dulbecco’s PBS supplemented with 0.9 mM CaCl2, 0.49 mM MgCl2.6H2O, 5.6 mM dextrose, and 0.3 mM sodium pyruvate). Biotinylation of cell surface proteins using a non-cell permeable and thiol-cleavable probe was initiated by incubating cells with 0.4 mM EZ-Link Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific) in DPBS+ buffer for 30 min. Unreacted Sulfo-NHS-SS-Biotin was quenched with 50 mM Tris (pH 7.4) for 10 min. Cells were then washed 3 times with a 1x Tris-buffered saline (25 mM Tris at pH 7.4, 137 mM NaCl, and 2.7 mM KCl) and whole cell extracts were prepared in Biotinylation Lysis Buffer A (50 mM Tris at pH 8.0, 150 mM NaCl, 2% NP-40, 0.25% Deoxycholate, 1x SIGMAFAST protease inhibitor cocktail (MilliporeSigma), and 1x PhosSTOP phosphatase inhibitor cocktail (Roche). Biotinylated proteins from clarified supernatants were captured on a streptavidin agarose resin (TriLink Biotechnologies), washed once with Biotinylation Lysis Buffer A, once with Biotinylation Wash Buffer A (Biotinylation Lysis Buffer A + 0.5% SDS), once with Biotinylation Wash Buffer B (Biotinylation Wash Buffer A + 150 mM NaCl), and finally once again with Biotinylation Wash Buffer A. Biotinylated proteins captured on streptavidin agarose resin were eluted in 1x NuPAGE-LDS sample buffer (Thermo Fisher Scientific, Invitrogen) containing 100 mM DTT at 37 °C for 1 hour and assayed by immunoblotting for SMO (Fig. S5B).
Ubiquitination assay
8 million HEK293T cells were plated onto a 15 cm plate. 24 hours after plating, the cells were transfected using PEI. 6 ug of each construct was transfected into the cells (at a DNA:PEI ratio of 1:3). An empty plasmid construct was used as filler DNA to ensure that each plate was transfected with the same amount of DNA. 36 hours post transfection, cells were pre-treated with 10 μM Bortezomib (a proteasome inhibitor) and 100 nM Bafilomycin A1 (a lysosome inhibitor) for 4 hours to enrich for ubiquitinated proteins. Cells were washed twice with chilled 1x PBS and lysed in Ubiquitination Lysis Buffer A comprised of: 50 mM Tris at pH 8.0, 150 mM NaCl, 2% NP-40, 0.25% sodium deoxycholate, 0.1% SDS, 6M urea, 1 mM DTT, 10 μM Bortezomib, 100 nM Bafilomycin A1, 20 mM N-Ethylmaleimide (NEM, MilliporeSigma), and 1x SIGMAFAST protease inhibitor cocktail (MilliporeSigma). Clarified supernatants were diluted ten-fold with Ubiquitination Lysis Buffer B (Ubiquitination Lysis Buffer A prepared without urea) to adjust the urea concentration to 600 mM. For these assays, we assessed ubiquitination on both GFP tagged and untagged SMO. Ubiquitinated GFP tagged SMO (Figs. 4B, 4C, S6A, and S6D) was captured using a GFP binding protein (GBP) covalently conjugated to carboxylic acid decorated Dynabeads (Dynabeads M-270 carboxylic acid, Thermo Fisher Scientific). Untagged SMO (Fig. S6C) was captured using SMO antibody covalently conjugated to Protein A Dynabeads (Thermo Fisher Scientific, Invitrogen). Immunoprecipitates were washed once with Ubiquitination Wash Buffer A (Ubiquitination Lysis Buffer B + 0.5% SDS), once with Ubiquitination Wash Buffer B (Ubiquitination Wash Buffer A + 1 M NaCl), and finally once again with Ubiquitination Wash Buffer A. Proteins bound to dynabeads were eluted in 2x NuPAGE-LDS sample buffer (Thermo Fisher Scientific, Invitrogen) containing 30 mM DTT at 37 °C for 30 minutes and assayed by immunoblotting for GFP or SMO antibodies for GFP tagged SMO and endogenous SMO, respectively.
Immunofluorescence staining of cells and tissue and image quantifications
Mouse embryos (e12.5) were harvested and fixed in 4% (w/v) paraformaldehyde (PFA) in 1x PBS for 2 hours at 4 °C and then rinsed thoroughly in chilled PBS. To cryopreserve the tissue, the embryos were transferred to 30% sucrose in 0.1M PB (pH 7.2) and allowed to equilibrate overnight. To allow for better analysis of the tissue, the embryos were further dissected into five pieces: 2 hands (forelimbs), head, upper body, and lower body. All five pieces were then mounted and frozen into Tissue-Plus OCT (optimal cutting temperature) compound (Thermo Fisher Scientific) and 12–14 μm sections were collected. Prior to staining, the tissue was blocked for 1 hour in immunofluorescence (IF) Blocking Buffer: 1% normal donkey serum (NDS) and 0.1% Triton-X diluted in 1x PBS. In a humidified chamber, the sections were incubated with primary antibodies overnight at 4 °C, rinsed 3 times in PBST (1x PBS + 0.1% Triton-X), incubated with secondary antibodies and Hoescht for 1 hour at room temperature, rinsed 3 times in PBST, and then mounted in Prolong Gold antifade mountant (Thermo Fisher Scientific, Invitrogen).
NIH/3T3 cells, pMEFs, and primary human fibroblasts were fixed in chilled 4% PFA in 1x PBS for 10 minutes and then rinsed with chilled PBS. Cells were incubated in IF Blocking Buffer for 30 minutes, primary antibodies for 1 hour, and secondary antibodies for 30 minutes.
Fluorescent images were acquired on an inverted Leica SP8 confocal microscope equipped with a 63X oil immersion objective (NA 1.4). Z-stacks (~4 μm sections) were acquired with identical acquisition settings (laser power, gain, offset, frame and image format) within a given experiment. An 4–8X optical zoom was used for imaging cilia to depict representative images. For the quantification of SMO at cilia, images were opened in Fiji (Schindelin et al., 2012) with projections of the maximum fluorescent intensities of z-stacks. Ciliary masks were constructed based on ARL13B images and then applied to corresponding SMO images to measure the fluorescence intensity of SMO at cilia.
Vismodegib dosing via oral gavage
Vismodegib treatment was performed as described previously (Heyne et al., 2015). Briefly, Megf8m/+ × Megf8m/+ and Mgrn1m/m × Mgrn1m/+ mouse crosses were set up and monitored daily. E0 was defined as midnight prior to visualization of the copulation plug. Females mice were weighed at ~e0.25 (the morning the plug was visualized) and ~e7.25. Only mice that gained 1.75 grams over 7 days were deemed “likely pregnant” and treated with either vehicle or Vismodegib. For Vismodegib treatment, a 3 mg/ml Vismodegib solution was prepared in 0.5% methyl cellulose (MilliporeSigma) with 0.2% Tween. Vismodegib (40 mg/kg) was administered via oral gavage every 12 hours (~7am and 7pm) for a total of three days (e8.25, e8.75, e9.25, e9.75, e10.25, and e10.75). Embryos were harvested at e14.5, fixed in 4% (w/v) PFA in 1x PBS for 2–3 days, and analyzed for limb and patterning defects.
Mouse embryo phenotyping analysis
Mouse embryos (e13.5–14.5) were fixed in 4% (w/v) PFA in 1x PBS for 2–3 days. Necropsy was performed to determine visceral organ situs (i.e. lung and liver lobation, heart and stomach situs, and spleen and pancreas structure). The samples were embedded in paraffin and processed for episcopic confocal microscopy as previously described (Liu et al., 2013). Briefly, this entailed sectioning of the tissue block using a Leica sledge microtome with serial images of the block face captured with a Leica confocal microscope. The serial two-dimensional (2D) image stacks generated were three-dimensionally (3D) reconstructed using the Osirix software (Rosset et al., 2004) and digitally resliced in different orientations to aid in the analysis of intracardiac anatomy and the diagnosis of congenital heart defects (Liu et al., 2013).
Variant Discovery and Validation
Genomic DNA was extracted from blood using the PAXgene Blood DNA kit (Qiagen). Patient genomic DNA was analyzed using whole exome sequencing performed using the Agilent V5 Exome Capture kit followed by sequencing with the Illumina HiSeq2000 with 150 base paired- end reads with 100X coverage. Reads were aligned to the human reference genome (version hg19) using Burrows-Wheeler Alignment (BWA, version 0.5.9) (Li and Durbin, 2009) with default parameters, and further processed according to the recommendations of the Genome Analysis Toolkit (GATK) Best Practices (Auwera and Others, 2016; DePristo et al., 2011). GATK HaplotypeCaller was used for single-nucleotide polymorphism (SNP) and insertion/deletion mutation (INDEL) discovery and variants that passed the GATK Variant Score Quality Recalibration (VQSR) and standard GATK filters with minor allele frequency <5% based on the Genome Aggregation Database (GnomAD). Only variants with Combined Annotation-Dependent Depletion (CADD) PHRED (Kircher et al., 2014) score of at least 10 were considered, and PolyPhen-2 (Adzhubei et al. 2010) and SIFT (Kumar, Henikoff, and Ng 2009) were used to assess variant pathogenicity. MEGF8/MGRN1/RNF157 variants recovered were validated by Sanger sequencing and heritable transmission was determined by further Sanger sequencing of genomic DNA from the parents.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data analysis and graphs were generated using GraphPad Prism 8. Violin plots were created using the “Violin Plot (truncated)” appearance function. In Prism 8, the frequency distribution curves of the violin plots are calculated using kernal density estimation. By using the “truncated” violin plot function, the frequency distributions shown are confined within the minimum to maximum values of the data set. On each violin plot, the median (central bold line) and quartiles (adjacent thin lines, representing the first and third quartiles) are labeled.
In Prism 8, the statistical significance between two groups was determined using either Mann-Whitney or an unpaired t-test and the significance between three or more groups was determined using either one-way ANOVA or the Kruskal-Wallis test. For each figure, p-values were calculated using Prism 8 and reported in the figure legend. P-values were reported using the following key: not-significant (ns) p-value > 0.05, *p-value ≤ 0.05, **p-value ≤ 0.01, ***p-value ≤ 0.001, and ****p-value ≤ 0.0001 Additional figure details regarding the n-value and statistical test applied were reported in the individual figure legends.
All cell biological and biochemical experiments were performed two to three independent times, with similar results. To validate newly generated Mgrn1−/−;Rnf157−/− NIH/3T3 and neural progenitor cell lines, 3 independent clonal cell lines were analyzed. Analysis of one clonal NIH/3T3 cell line was featured in the main figures (Figs. 2A and 2C) and data from the additional cell lines was presented in the supplementary figures (Figs. S3C, S3D, and S3E). Similarly, 2–3 primary mouse embryonic fibroblast (pMEF) cell lines were analyzed from both Megf8m/m and Mgrn1m/m embryos, where each pMEF cell line was generated from a single embryo (Figs. 1A, 1B, S1B, and S1C).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-1D4 | The University of British Columbia | RRID: AB_325050 |
| Guinea pig polyclonal anti-ARL13B | Dorn et al., 2012 | N/A |
| Mouse monoclonal anti-CD16 (clone 3G8) | Santa Cruz Biotechnology | Cat#sc-19620; RRID: AB_626924 |
| Mouse monoclonal anti-CD16 (clone DJ130c) | Santa Cruz Biotechnology | Cat#sc-20052; RRID: AB_626925 |
| Mouse monoclonal anti-FLAG (clone M2) | MilliporeSigma | Cat#F1804; RRID: AB_26204 |
| Mouse monoclonal anti-GLI1 (clone L42B10) | Cell Signaling | Cat#2643; RRID: AB_2294746 |
| Goat polyclonal anti-GFP | Rockland Immunochemicals | Cat#600-101-215; RRID: AB_218182 |
| Rabbit polyclonal anti-GFP | Novus Biologicals | Cat#NB600–308; RRID: AB_10003058 |
| Mouse monoclonal anti-HA.11 (clone 16B12) | Biolegend | Cat#901513; RRID: AB_256335 |
| Mouse monoclonal anti-HA (clone 2–2.2.14) | Thermo Fisher Scientific (Invitrogen) | Cat#26183; RRID: AB_10978021 |
| Rabbit polyclonal anti-MEGF8 | This paper | N/A |
| Rabbit polyclonal anti-p38 | Abcam | Cat#ab7952; RRID: AB_306166 |
| Rabbit polyclonal anti-RNF156 (anti-MGRN1) | Proteintech | Cat#11285–1-AP; RRID: AB_2143351 |
| Rabbit polyclonal anti-SMO | Rohatgi et al., 2007 | N/A |
| Rabbit polyclonal anti-SMO-N | Milenkovic et al., 2009 | N/A |
| Mouse monoclonal anti-α-Tubulin (clone DM1A) | MilliporeSigma | Cat#T6199; RRID: AB_477583 |
| Mouse monoclonal anti-Tubulin, acetylated | MilliporeSigma | Cat#T6793; RRID: AB_477585 |
| Peroxidase AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Laboratories | Cat#715-035-150; RRID: AB_2340770 |
| Peroxidase AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Laboratories | Cat#111-035-144; RRID: AB_2307391 |
| Peroxidase AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch Laboratories | Cat#705-035-003; RRID: AB_2340390 |
| Donkey anti-Rabbit IgG (H+L) High ly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific (Invitrogen) | Cat#A-21206; RRID: AB_2535792 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific (Invitrogen) | Cat#A-21207; RRID: AB_141637 |
| Donkey anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific (Invitrogen) | Cat#A-21202; RRID: AB_141607 |
| Donkey anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific (Invitrogen) | Cat#A-31571; RRID: AB_162542 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Guinea Pig IgG (H+L) | Jackson ImmunoResearch Laboratories | Cat#706-545-148; RRID: AB_2340472 |
| Alexa Fluor 647 AffiniPure Donkey Anti-Guinea Pig IgG (H+L) | Jackson ImmunoResearch Laboratories | Cat#706-605-148; RRID: AB_2340476 |
| Bacterial and Virus Strains | ||
| Rosetta2(DE3)pLysS competent cells | MilliporeSigma (Novagen) | Cat#71403 |
| Biological Samples | ||
| Patient derived fibroblast cultures | Children’s Hospital of Pittsburgh (University of Pittsburgh) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-Mercaptoethanol | Thermo Fisher Scientific (Gibco) | Cat#31350010 |
| Bafilomycin A1 | Cayman Chemical | Cat#11038 |
| Bortezomib, Free Base | LC Laboratories | Cat#B-1408 |
| Cas9 2NLS Nuclease | Synthego | N/A |
| CHIR 99021 | Axon Medchem | Cat#Axon 1386 |
| Recombinant mouse FGF basic/FGF2/bFGF protein | R&D Systems | Cat#31339-FB |
| Hygromycin B (50 mg/ml in solution, Ultra Pure Grade) | VWR Life Science | Cat#97064–454 |
| Hoechst 33342 solution (20uM) | Thermo Fisher Scientific | Cat#62249 |
| ESGRO recombinant mouse leukemia inhibitory factor (LIF) protein | MilliporeSigma | Cat#ESG1106 |
| EZ-Link Sulfo-NHS-SS-Biotin | Thermo Fisher Scientific | Cat#21331 |
| Gateway LR Clonase II enzyme | Thermo Fisher Scientific (Invitrogen) | Cat#11791020 |
| Gibson Assembly Master Mix | New England Biolabs | Cat#E2611 |
| GoTaq Green Master Mix | Promega Corporation | Cat#M712 |
| Methyl cellulose (viscosity: 4000 cP) | MilliporeSigma | Cat#M0512 |
| N-Ethylmaleimide | MilliporeSigma | Cat#E3876 |
| PD 98059 | Axon Medchem | Cat#Axon 1223 |
| Penicillin:Streptomycin solution | Gemini Bioproducts | Cat#400–109 |
| PhosSTOP | MilliporeSigma | Cat#PHOSS-RO |
| Polyethylenimine (PEI), Linear, MW 25000, Transfection Grade | Polysciences, Inc. | Cat#23966–1 |
| Retinoic Acid | MilliporeSigma | Cat#R2625 |
| Polybrene (Hexadimethrine bromide) | MilliporeSigma | Cat#107689 |
| Puromycin dihydrochloride | MilliporeSigma | Cat#P8833 |
| SAG (Smoothened agonist) | Enzo Life Sciences | Cat#NC9953751 |
| SIGMAFAST protease inhibitor tablets | MilliporeSigma | Cat# S8820 |
| Sonic hedgehog (recombinant) | Bishop et al., 2009 | N/A |
| Vismodegib, Free Base | LC Laboratories | Cat#V-4050 |
| X-tremeGENE 9 DNA transfection reagent | Roche Molecular Systems | Cat#XTG9-RO |
| B-27 Supplement (50x), minus antioxidants | Thermo Fisher Scientific (Gibco) | Cat#10889038 |
| Bovine serum albumin solution (30% in DPBS) | MilliporeSigma | Cat#A9576 |
| Bovine serum albumin | MilliporeSigma | Cat#A3059 |
| Dulbecco’s Modified Eagles Medium (DMEM) high glucose without L-glutamine and sodium pyruvate (Hyclone) | Thermo Fisher Scientific (Gibco) | Cat#SH30081FS |
| Dulbecco’s Modified Eagles Medium (DMEM)/F12, no glutamine | Thermo Fisher Scientific (Gibco) | Cat#21331020 |
| Dynabeads Protein A for immunoprecipitation | Thermo Fisher Scientific (Invitrogen) | Cat#10002D |
| Dynabeads M-270 carboxylic acid | Thermo Fisher Scientific (Invitrogen) | Cat#14305D |
| EmbryoMax advanced KSOM embryo medium | MilliporeSigma | Cat#MR-101-D |
| EmbryoMax nucleosides (100x) | MilliporeSigma | Cat#ES-008-D |
| L-glutamine solution | Gemini Bioproducts | Cat#400–106 |
| MEM Non-essential amino acids solution | Thermo Fisher Scientific (Gibco) | Cat#11140050 |
| N-2 Supplement (100x) | Thermo Fisher Scientific (Gibco) | Cat#17502048 |
| Neurobasal Medium | Thermo Fisher Scientific (Gibco) | Cat#21103049 |
| NuPAGE-LDS sample buffer (4X) | Thermo Fisher Scientific (Invitrogen) | Cat#NP0007 |
| Opti-MEM reduced serum medium | Thermo Fisher Scientific (Gibco) | Cat#3985062 |
| RPMI medium | Thermo Fisher Scientific (Gibco) | Cat#11875135 |
| Sodium pyruvate | Thermo Fisher Scientific (Gibco) | Cat#11360070 |
| Streptavidin agarose ultra performance resin | TriLink Biotechnologies | Cat#N-1000 |
| Tissue-Plus OCT compound | Thermo Fisher Scientific | Cat#23-730-571 |
| TRIzol reagent | Thermo Fisher Scientific (Invitrogen) | Cat#15596026 |
| Trypsin-EDTA solution (0.05%), phenol red | Gemini Bioproducts | Cat#400–150 |
| Trypsin-EDTA solution (0.25%), phenol red | Thermo Fisher Scientific (Gibco) | Cat#25200072 |
| Critical Commercial Assays | ||
| SureSelect XT Human All Exon V4 | Agilent | Cat#5190–4635 |
| Direct-zol RNA miniprep kit | Zymo Research | Cat#R2050 |
| RNeasy Plus Mini Kit | Qiagen | Cat#74134 |
| High-capacity RNA-to-cDNA kit | Thermo Fisher Scientific (Applied Biosystems) | Cat#4387406 |
| iScript reverse transcription supermix for RT-qPCR | Bio-rad Laboratories | Cat#1708840 |
| SuperScript III first-strand synthesis system | Thermo Fisher Scientific (Invitrogen) | Cat#18080051 |
| PAXgene blood DNA kit | Qiagen | Cat#761133 |
| Mouse embryonic stem cell nucleofector kit | Lonza Bioscience | Cat#VAPH-1001 |
| Universal mycoplasma detection kit | ATCC | Cat#30–1012K |
| Experimental Models: Cell Lines | ||
| Flp-In-3T3 | Thermo Fisher Scientific | Cat#R76107 |
| 293T | ATCC | Cat#CRL-3216 |
| HM1 mESC cells with dual reporters (Olig2::T2A-mKate2 and 8xGBS-H2B::Venus) | Sagner et al., 2018 | N/A |
| NIH/3T3-Flp In Megf8−/− | Pusapati et al., 2018 | N/A |
| NIH/3T3-Flp In Megf8−/− + MEGF8-1D4 | This paper | N/A |
| NIH/3T3-Flp In Megf8−/− + MEGF8 ΔN-1D4 | This paper | N/A |
| NIH/3T3-Flp In Megf8−/− + MEGF8 ΔCtail-1D4 | This paper | N/A |
| NIH/3T3-Flp In Megf8−/− + MEGF8 ΔMASRPFA-1D4 | This paper | N/A |
| NIH/3T3-Flp In Megf8−/− + CD16ECD-CD7TM-MEGF8 Ctail-1D4 | This paper | N/A |
| NIH/3T3-Flp In Megf8−/− + CD16ECD- MEGF8 TM+Ctail-1D4 | This paper | N/A |
| NIH/3T3-Flp In Megf8−/− + CD16ECD- MEGF8 TM+CtailΔMASRPFA-1D4 | This paper | N/A |
| NIH/3T3-Flp In Mgrn1−/− | Pusapati et al., 2018 | N/A |
| NIH/3T3-Flp In Mgrn1−/−;Rnf157−/− | This paper | N/A |
| NIH/3T3-Flp In Mgrn1−/−;Rnf157−/− + Mgrn1-3xFLAG | This paper | N/A |
| NIH/3T3-Flp In Mgrn1−/−;Rnf157−/− + Mgrn1Mut1-3xFLAG | This paper | N/A |
| NIH/3T3-Flp In Mgrn1−/−;Rnf157−/− + Mgrn1Mut1-3xFLAG | This paper | N/A |
| HM1 mESC Mgrn1−/−;Rnf157−/− | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Mgrn1m/m: Mgrn1md-nc/md-nc | Phillips, 1963 | MGI#3704004 |
| Mouse: Megf8m/m: Megf8C193R/C193R | Zhang et al., 2009 | MGI#3722325 |
| Mouse: Rnf157m/m: Rnf157em1Tmg em1Tmg | This paper | N/A |
| Oligonucleotides | ||
| mGli1 and mGapdh qRT-PCR Primers, See Table S9 | Pusapati et al., 2018 | N/A |
| hGLI1 and hGAPDH qRT-PCR Primers, See Table S9 | This paper | N/A |
| mRnf157 qRT-PCR Primers, See Table S9 | This paper | N/A |
| mGpi qRT-PCR Primers (mice), See Table S9 | Gunn et al., 2013 | N/A |
| mRnf157 genotyping PCR Primers (3T3 cells, neural progenitor cells, and mice), See Table S9 | This paper | N/A |
| mRnf157 sgRNA target sequence PCR Primer (mice), See Table S9 | This paper | N/A |
| Mgrn1md-nc genotyping PCR primers, See Table S9 | Gunn et al., 2019 | N/A |
| Megf8C193R genotyping PCR Primers, See Table S9 | This paper | N/A |
| Recombinant DNA | ||
| MGC premier cDNA clone for MEGF8 (NM_001410.3) | Transomic Technologies, Inc | Cat#TCHS1003 (BC153880 – glycerol stock) |
| pEF5/FRT/V5-DEST-MEGF8-1D4 | This paper | N/A |
| pEF5/FRT/V5-DEST-MEGF8 ΔCtail-1D4 | This paper | N/A |
| Mgrn1 (NM_001252437.1) gBlock | Integrated DNA Technologies | N/A |
| pEF5/FRT/V5-DEST-Mgrn1-3xFLAG | This paper | N/A |
| SmoK0 gBlock fragment | Twist Bioscience | N/A |
| CD16ECD-CD7™-mCherry-Nck-HA | Rivera et al., 2009 | N/A |
| pCS2-mSmo | Byrne et al., 2016 | N/A |
| pCS2-mSmo-eGFP | Zhao et al., 2016 | N/A |
| pEGFPN3-Sstr3 | Berbari et al., 2008 | Addgene#35623 |
| pEF5/FRT/V5-DEST gateway vector | Thermo Fisher Scientific | Cat#V602020 |
| pLenti CMV Puro DEST | Campeau et al., 2009 | Addgene#17452 |
| pLenti CMV Puro DEST-MEGF8-1D4 | This paper | N/A |
| pLenti CMV Puro DEST-MEGF8 ΔN-1D4 | This paper | N/A |
| pLenti CMV Puro DEST-MEGF8 ΔCtail-1D4 | This paper | N/A |
| pLenti CMV Puro DEST-MEGF8 ΔMASRPFA-1D4 | This paper | N/A |
| pLenti CMV Puro DEST-CD16ECD-CD7TM-MEGF8 Ctail-1D4 | This paper | N/A |
| pLenti CMV Puro DEST-CD16ECD- MEGF8 TM+Ctail-1D4 | This paper | N/A |
| pLenti CMV Puro DEST-CD16ECD- MEGF8 TM+CtailΔMASRPFA-1D4 | This paper | N/A |
| pLenti CMV Puro DEST-Mgrn1-3xFLAG | This paper | N/A |
| pLenti CMV Puro DEST-Mgrn1Mut1-3xFLAG | This paper | N/A |
| pLenti CMV Puro DEST-Mgrn1Mut1-3xFLAG | This paper | N/A |
| pMD2.G | Didier Trono Lab (École Polytechnique fédérale de Lausanne) | Addgene#12259 |
| psPAX2 | Didier Trono Lab (École Polytechnique fédérale de Lausanne) | Addgene#12260 |
| pOG44 Flp-Recombinase expression vector | Thermo Fisher Scientific (Invitrogen) | Cat#V600520 |
| pRK5-HA-Ubiquitin-WT | Lim et al., 2005 | Addgene#17608 |
| pRK5-HA-Ubiquitin-K0 | Lim et al., 2005 | Addgene#17603 |
| pSpCas9(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene#48138 |
| pSpCas9(BB)-2A-mCherry | Pusapati et al., 2018 | N/A |
| pSpCas9(BB)-2A-Puro (PX459) | Ran et al., 2013 | Addgene#48139 |
| pGEX vector (modified with FseI/AscI restriction enzyme cleavage sites) | GE Healthcare | N/A |
| pGEX-MEGF8-Ctail(2738–2778) | This paper | N/A |
| Software and Algorithms | ||
| Adobe Illustrator CS6 | Adobe Systems | https://adobe.com/products/illustrator.html |
| Adobe Photoshop CS6 | Adobe System | https://adobe.com/products/photoshop.html |
| Benchling | Benchling | https://www.benchling.com |
| BlastClust | National Center for Biological Information (NCBI) | ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html |
| Broad Institute Genetic Perturbation Platform (GPP) sgRNA Designer | Broad Institute | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| ConSurf | Ashkenazy et al., 2016 | https://consurf.tau.ac.il |
| Fiji | Schindelin et al., 2012 | https://fiji.sc |
| GraphPad Prism (version 8) | GraphPad Software | https://graphpad.com |
| IQ-TREE | Nguyen et al., 2015 | http://www.iqtree.org |
| Kalign2 | Lassmann et al., 2009 | http://msa.sbc.su.se/ |
| Leica Application Suite X | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Logo software | Crooks et al., 2004 | https://weblogo.berkeley.edu/logo.cgi |
| ModelFinder | Kalyaanamoorthy et al., 2017 | http://www.iqtree.org |
| OsiriX software | Rosset et al., 2004 | https://osirix-viewer.com |
| Position-Specific Iterated BLAST (PSI-BLAST) program | Altschul et al., 1997 | https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins |
| SnapGene (version 4.3) software | GSL Biotech | https://snapgene.com |
| Synthego ICE analysis | Synthego Corporation | https://ice.synthego.com/#/ |
| UFBoot2 program | Hoang et al., 2018 | http://www.iqtree.org |
| UniProt Align software | The UniProt Consortium | https://uniprot.org/align |
Highlights.
A cell-surface ubiquitination pathway negatively regulates Hedgehog signaling strength
This pathway promotes the ubiquitination and downregulation of Smoothened
Defects in this pathway cause limb, heart and left-right patterning defects
Mutations in genes associated with this pathway show oligogenic inheritance
ACKNOWLEDGEMENTS
We thank Robert Lipinski for invaluable advice on the in utero treatment of embryos with Vismodegib, Derek Silvius and Janet Peters for genotyping, the McLaughlin Research Institute Animal Resource staff for animal care, and Connie Chuy for her mouse and heart diagrams. RR was supported by a grant from the National Institutes of Health (GM118082), GP by a postdoctoral fellowship from the American Heart Association (14POST20370057), and JK by a postdoctoral fellowship from the American Heart Association (19POST34380734) and a K99/R00 award from the NIH (GM13251801). CWL, CBY, and SH were supported by grants from the NIH (HL142788 and HL132024) and the DOD (W81XWH-15-1-0649 and W81XWH-16-1-0613).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid s Research 25: 3389––3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aune CN, Chatterjee B, Zhao X-Q, Francis R, Bracero L, Yu Q, Rosenthal J, Leatherbury L, and Lo CW (2008). Mouse model of heterotaxy with single ventricle spectrum of cardiac anomalies. Pediatr. Res 63, 9–14. [DOI] [PubMed] [Google Scholar]
- Auwera G, and Others (2016). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. HHS Public Access, Published online October 15, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, and Katsanis N (2006). The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet 7, 125–148. [DOI] [PubMed] [Google Scholar]
- Berbari NF, Johnson AD, Lewis JS, Askwith CC, and Mykytyn K (2008). Identification of Ciliary Localization Sequences within the Third Intracellular Loop of G Protein-coupled Receptors. Mol. Biol. Cell 19, 1540–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop B, Aricescu AR, Harlos K, O’Callaghan CA, Jones EY, and Siebold C (2009). Structural insights into hedgehog ligand sequestration by the human hedgehog-interacting protein HHIP. Nat. Struct. Mol. Biol 16, 698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnadóttir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, and Schiöth HB (2006). Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 88, 263–273. [DOI] [PubMed] [Google Scholar]
- Byrne EFX, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, et al. (2016). Structural basis of Smoothened regulation by its extracellular domains. Nature Publishing Group 535, 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti O, and Hegde RS (2009). Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell 137, 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, and Bale AE (2005). Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development 132, 4407–4417. [DOI] [PubMed] [Google Scholar]
- Cooray SN, Guasti L, and Clark AJL (2011). The E3 ubiquitin ligase Mahogunin ubiquitinates the melanocortin 2 receptor. Endocrinology 152, 4224–4231. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, and Reiter JF (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- Cota CD, Bagher P, Pelc P, Smith CO, Bodner CR, and Gunn TM (2006). Mice with mutations in Mahogunin ring finger-1 (Mgrn1) exhibit abnormal patterning of the left-right axis. Dev. Dyn 235, 3438–3447. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PB, Stuck MW, Lv B, and Pazour GJ (2020). Ubiquitin links smoothened to intraflagellar transport to regulate Hedgehog signaling. J. Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H, et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn KV, Hughes CE, and Rohatgi R (2012). A Smoothened-Evc2 Complex Transduces the Hedgehog Signal at Primary Cilia. Dev. Cell 23, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin ME, Qian X, Popescu NC, and Lowy DR (2013). Isolation of Mouse Embryo Fibroblasts. Bio Protoc 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer LA, and Kirby ML (2009). Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. Dev. Biol 330, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhard C, Sarsfield S, Merte J, Wang Q, Li P, Beppu H, Kolodkin AL, Sucov HM, and Ginty DD (2013). MEGF8 is a modifier of BMP signaling in trigeminal sensory neurons. Elife 2, e01160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Barcena C, Osinalde N, Ramirez J, and Mayor U (2020). How to Inactivate Human Ubiquitin E3 Ligases by Mutation. Front Cell Dev Biol 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford CA, Ranade SS, Samarakoon R, Salunga HT, de Soysa TY, Huang Y, Zhou P, Elfenbein A, Wyman SK, Bui YK, et al. (2019). Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 364, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra DD, Pratelli R, Kraft E, Callis J, and Pilot G (2013). Functional conservation between mammalian MGRN1 and plant LOG2 ubiquitin ligases. FEBS Lett. 587, 3400–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn TM, Miller KA, He L, Hyman RW, Davis RW, Azarani A, Schlossman SF, Duke-Cohan JS, and Barsh GS (1999). The mouse mahogany locus encodes a transmembrane form of human attractin. Nature 398, 152–156. [DOI] [PubMed] [Google Scholar]
- Gunn TM, Silvius D, Bagher P, Sun K, and Walker KK (2013). MGRN1-dependent pigment-type switching requires its ubiquitination activity but not its interaction with TSG101 or NEDD4. Pigment Cell Melanoma Res. 26, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn TM, Silvius D, Lester A, and Gibbs B (2019). Chronic and age-dependent effects of the spongiform neurodegeneration-associated MGRN1 E3 ubiquitin ligase on mitochondrial homeostasis. Mamm. Genome 30, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, et al. (2012). ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485, 195–200. [DOI] [PubMed] [Google Scholar]
- Haqq AM, Rene P, Kishi T, Khong K, Lee CE, Liu H, Friedman JM, Elmquist JK, and Cone RD (2003). Characterization of a novel binding partner of the melanocortin-4 receptor: attractin-like protein. Biochem. J 376, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, and Tabin CJ (2004). Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517–528. [DOI] [PubMed] [Google Scholar]
- He L, Lu XY, Jolly AF, Eldridge AG, Watson SJ, Jackson PK, Barsh GS, and Gunn TM (2003). Spongiform degeneration in mahoganoid mutant mice. Science 299, 710–712. [DOI] [PubMed] [Google Scholar]
- Heyne GW, Melberg CG, Doroodchi P, Parins KF, Kietzman HW, Everson JL, Ansen-Wilson LJ, and Lipinski RJ (2015). Definition of critical periods for Hedgehog pathway antagonist-induced holoprosencephaly, cleft lip, and cleft palate. PLoS One 10, e0120517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, and Vinh LS (2018). UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol 35, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, and Moskowitz IP (2009). sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 136, 1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Sun K, Walker WP, Bagher P, Cota CD, and Gunn TM (2009). Abnormal regulation of TSG101 in mice with spongiform neurodegeneration. Biochim. Biophys. Acta 1792, 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, and Jermiin LS (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanis N, Beales PL, Woods MO, Lewis RA, Green JS, Parfrey PS, Ansley SJ, Davidson WS, and Lupski JR (2000). Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet 26, 67–70. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B-K, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJR, Maurice MM, et al. (2012). Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488, 665–669. [DOI] [PubMed] [Google Scholar]
- Kousi M, and Katsanis N (2015). Genetic modifiers and oligogenic inheritance. Cold Spring Harb. Perspect. Med 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T, Frings O, and Sonnhammer ELL (2009). Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 37, 858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M, Johnson RL, Stern CD, Kuehn M, and Tabin C (1995). A molecular pathway determining left-right asymmetry in chick embryogenesis. Cell 82, 803–814. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Chew KCM, Tan JMM, Wang C, Chung KKK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, et al. (2005). Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci 25, 2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AE, Krikov S, Riehle-Colarusso T, Frías JL, Belmont J, Anderka M, Geva T, Getz KD, Botto LD, and Study NBDP (2014). Laterality defects in the national birth defects prevention study (1998−−2007): birth prevalence and descriptive epidemiology. Am. J. Med. Genet. A 164, 2581–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski RJ, Holloway HT, O’Leary-Moore SK, Ament JJ, Pecevich SJ, Cofer GP, Budin F, Everson JL, Johnson GA, and Sulik KK (2014). Characterization of subtle brain abnormalities in a mouse model of Hedgehog pathway antagonist-induced cleft lip and palate. PLoS One 9, e102603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Dahn RD, Li Y, Fallon JF, and Chiang C (2002). Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 418, 979–983. [DOI] [PubMed] [Google Scholar]
- Liu X, Tobita K, Francis RJ, and Lo CW (2013). Imaging techniques for visualizing and phenotyping congenital heart defects in murine models. Birth Defects Res. C Embryo Today 99, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yagi H, Saeed S, Bais AS, Gabriel GC, Chen Z, Peterson KA, Li Y, Schwartz MC, Reynolds WT, et al. (2017). The complex genetics of hypoplastic left heart syndrome. Nat. Genet 49, 1152–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chen W, Li W, Priest JR, Fu Y, Pang K, Ma B, Han B, Liu X, Hu S, et al. (2020). Exome-Based Case-Control Analysis Highlights the Pathogenic Role of Ciliary Genes in Transposition of the Great Arteries. Circ. Res 126, 811–821. [DOI] [PubMed] [Google Scholar]
- Milenkovic L, Scott MP, and Rohatgi R (2009). Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol 187, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle DL, McGrail SH, Vitale J, Woolf EA, Dussault BJ Jr, DiRocco L, Holmgren L, Montagno J, Bork P, Huszar D, et al. (1999). The mahogany protein is a receptor involved in suppression of obesity. Nature 398, 148–152. [DOI] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, and Minh BQ (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RJ (1963). New mutant: non-agouti curly. Mouse News Letter 29, 38. [Google Scholar]
- Pradat P, Francannet C, Harris JA, and Robert E (2003). The epidemiology of cardiovascular defects, part I: a study based on data from three large registries of congenital malformations. Pediatr. Cardiol 24, 195–221. [DOI] [PubMed] [Google Scholar]
- Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott D, et al. (2016). De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 12, e1005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusapati GV, Hughes CE, Dorn KV, Zhang D, Sugianto P, Aravind L, and Rohatgi R (2014). EFCAB7 and IQCE regulate hedgehog signaling by tethering the EVC-EVC2 complex to the base of primary cilia. Dev. Cell 28, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusapati GV, Kong JH, Patel BB, Krishnan A, Sagner A, Kinnebrew M, Briscoe J, Aravind L, and Rohatgi R (2018). CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev. Cell 44, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle RD, Johnson RL, Laufer E, and Tabin C (1993). Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401–1416. [DOI] [PubMed] [Google Scholar]
- Rio DC, Ares M Jr, Hannon GJ, and Nilsen TW (2010). Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc 2010, db.prot5439. [DOI] [PubMed] [Google Scholar]
- Rivera GM, Vasilescu D, Papayannopoulos V, Lim WA, and Mayer BJ (2009). A reciprocal interdependence between Nck and PI(4,5)P(2) promotes localized N-WASp-mediated actin polymerization in living cells. Mol. Cell 36, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, and Scott MP (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376. [DOI] [PubMed] [Google Scholar]
- Rosset A, Spadola L, and Ratib O (2004). OsiriX: an open-source software for navigating in multidimensional DICOM images. J. Digit. Imaging 17, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagner A, Gaber ZB, Delile J, Kong JH, Rousso DL, Pearson CA, Weicksel SE, Melchionda M, Mousavy Gharavy SN, Briscoe J, et al. (2018). Olig2 and Hes regulatory dynamics during motor neuron differentiation revealed by single cell transcriptomics. PLoS Biol. 16, e2003127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde SR, Nager AR, and Nachury MV (2020). Lysine63-linked ubiquitin chains earmark GPCRs for BBSome-mediated removal from cilia. [DOI] [PMC free article] [PubMed]
- Stamataki D, Ulloa F, Tsoni SV, Mynett A, and Briscoe J (2005). A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev. 19, 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tröder SE, Ebert LK, Butt L, Assenmacher S, Schermer B, and Zevnik B (2018). An optimized electroporation approach for efficient CRISPR/Cas9 genome editing in murine zygotes. PLoS One 13, e0196891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiairis CD, and McMahon AP (2009). An Hh-dependent pathway in lateral plate mesoderm enables the generation of left/right asymmetry. Curr. Biol 19, 1912–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Lloyd D, Jenkins D, Elcioglu NE, Cooper CD, Al-Sannaa N, Annagur A, Gillessen-Kaesbach G, Huning I, Knight SJ, et al. (2012). Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet 91, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium UniProt (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA (2010). A generalized model of gene dosage and dominant negative effects in macromolecular complexes. FASEB J. 24, 994–1002. [DOI] [PubMed] [Google Scholar]
- Veitia RA, Bottani S, and Birchler JA (2013). Gene dosage effects: nonlinearities, genetic interactions, and dosage compensation. Trends Genet. 29, 385–393. [DOI] [PubMed] [Google Scholar]
- Walker W (2010). Studies of the pigment-type switching genes ATRN and MGRN1: evidence for a defect in endosomal/lysosomal protein trafficking as a cause of hyperpigmentation and spongiform neurodegeneration.
- Washington Smoak I, Byrd NA, Abu-Issa R, Goddeeris MM, Anderson R, Morris J, Yamamura K, Klingensmith J, and Meyers EN (2005). Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol 283, 357–372. [DOI] [PubMed] [Google Scholar]
- te Welscher P, Zuniga A, Kuijper S, Drenth T, Goedemans HJ, Meijlink F, and Zeller R (2002). Progression of vertebrate limb development through SHH-mediated counteraction of GLI3. Science 298, 827–830. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, and McMahon AP (2001). Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell 106, 781–792. [PubMed] [Google Scholar]
- Zhang Z, Alpert D, Francis R, Chatterjee B, Yu Q, Tansey T, Sabol SL, Cui C, Bai Y, Koriabine M, et al. (2009). Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy. Proc. Natl. Acad. Sci. U. S. A 106, 3219–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Lee RTH, Pusapati GV, Iyu A, Rohatgi R, and Ingham PW (2016). An essential role for Grk2 in Hedgehog signalling downstream of Smoothened. EMBO Rep. 17, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article contains all datasets generated and analyzed during this study.