

Abstract
The blossoming field of epitranscriptomics has recently garnered attention across many fields by findings that chemical modifications on RNA have immense biological consequences. Methylation of nucleotides in RNA, including N6-methyladenosine (m6A), 2-O-dimethyladenosine (m6Am), N1-methyladenosine (m1A), and 5-methylcytosine (m5C) and isomerization of uracil to pseudouridine (Ψ), have the potential to alter RNA processing events and contribute to developmental processes and different diseases. Though the abundance and roles of some RNA modifications remain contentious, the epitranscriptome is thought to be especially relevant in stem cell biology and neurobiology. In particular, m6A occurs at the highest levels in the brain and plays major roles in embryonic stem cell differentiation, brain development, and neurodevelopmental disorders. However, studies in these areas have reported conflicting results on epitranscriptomic regulation of stem cell pluripotency and mechanisms in neural development. In this review we provide an overview of the current understanding of several RNA modifications and disentangle the various findings on epitranscriptomic regulation of stem cell biology and neural development.
Keywords: epitranscriptome, m6A, stem cells, brain development, brain disorders
Introduction
The central dogma of biology—that information flows from DNA to RNA to protein—has acquired an increasing number of caveats and fine print (He, 2019). Beyond direct exceptions to the rule, like non-coding RNAs, the details of the process are marred by questions like how much RNA is made from DNA, and how is this RNA processed to make the correct quantities of particular proteins or isomers at the correct time? How might the system change in response to stimuli, and what regulatory systems drive these dynamics? The first step of the process, DNA to RNA, unfolds into many fields, including chromatin regulation, epigenetics, and transcription. For example, reversible chemical modifications are dynamically added to DNA and histones to alter gene expression and RNA levels (Akichika et al., 2019; Guo et al., 2011; Kohli and Zhang, 2013; Strahl and Allis, 2000). The next step, making protein from RNA, has largely been focused on the regulation of translation. However, the recent exploration of post-transcriptional modifications on RNA has shown that RNA is much more than a middleman between DNA and protein (Wang and Yi, 2019). In fact, over 100 “epitranscriptomic” modifications have been identified, though their biological consequences are largely unknown (Cantara et al., 2011; Machnicka et al., 2013). A particular methylation at the N6 position of adenosine, termed m6A, has garnered the most attention for its powerful role in regulating mRNA processing (Roundtree et al., 2017).
While the existence of m6A has been known for almost 50 years (Desrosiers et al., 1974; Lavi and Shatkin, 1975; Rottman et al., 1974; Wei et al., 1975; Wei and Moss, 1977), a combination of events reinvigorated a recent interest in the field. First, Zhong et al. showed in 2008 that m6A is critical for developmental processes in Arabidopsis thaliana, which suggested its importance for multicellular eukaryotes, and pushed for the development of m6A mapping methods (Zhong et al., 2008). Next, the discovery of the m6A demethylases, FTO and ALKBH5, in 2011 and 2013, respectively (Jia et al., 2011; Zheng et al., 2013), suggested that the m6A system may be dynamic, which implies that it can be used as a regulatory system to alter mRNA fate in a context-dependent manner. Concurrently, antibody-based m6A mapping techniques were developed that showed that m6A additions to mRNA are non-random, thus pushing biological interest forward and providing techniques to understand the molecular mechanisms of m6A in the cell (Dominissini et al., 2013; Meyer et al., 2012). In this review, we first introduce several epitranscriptomic marks that have garnered the most attention over recent years, with a focus on m6A. We then discuss the major advancements in our understanding of the epitranscriptome in stem cell biology, especially embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). We also touch on how the epitranscriptome itself is regulated in embryonic stem cells. In addition, we review the role of the epitranscriptome in cortical and cerebellar development, as well as in adult neurogenesis. Finally, we discuss the role of the epitranscriptome in neurological disorders.
RNA Modifications of Particular Interest
N6-methyladenosine: m6A
On a global level, 0.2 to 0.5% of all adenines are m6A modified (Geula et al., 2015). The highest levels occur in the brain, where up to 30% of all transcripts are modified (Chang et al., 2017). Epitranscriptomic detection technologies have been focused on m6A, making it one of the best-studied modifications to date. m6A occurs in various types of RNA, including tRNA, rRNA, noncoding RNA (ncRNA), and mRNA. In 2012, two groups independently reported anti-m6A antibody-based m6A RIP-Seq (MeRIP-Seq) techniques (Dominissini et al., 2012; Meyer et al., 2012). Subsequent mapping of m6A in the transcriptome showed that it is most commonly added at a consensus sequence of DRACH (D = A, U or G; R = G or C; H = A, U, or C). m6A is especially enriched in the 3’UTR and around the STOP codon (Dominissini et al., 2012; Meyer et al., 2012). While m6A does not alter Watson-Crick-Franklin base pairing, it can modify protein binding and affect the mRNA secondary structure (Farre et al., 2003; Roost et al., 2015). Many m6A-binding proteins, or readers, have been identified. Individual readers confer unique downstream fates on m6A-modified mRNA, including altered mRNA stability, translation, localization, and splicing. m6A methylation patterns in the transcriptome appear to be cell/tissue-specific and species-specific (Liu et al., 2019). Current m6A sequencing technologies are not sufficiently sensitive to profile m6A at a single-cell level, which would help quell disagreements in the field on whether m6A is truly different across cell types or dynamic in response to stimuli. For example, Garcia-Campos et al. claimed that m6A is “hard coded” and largely predictable in the yeast genome based on the extended sequence around the modified site (Garcia-Campos et al., 2019). In contrast, studies that have profiled multiple mammalian tissues identified tissue-specific methylation profiles and significant changes over development (Liu et al., 2019; Shi et al., 2018; Weng et al., 2018; Yoon et al., 2017; Zhang et al., 2020). Furthermore, up-regulation of the m6A demethylases FTO and ALKBH5 in response to heat stress and hypoxic stress, respectively, suggests that dynamic changes to the m6A methylome may be a way of modulating cellular responses to stimuli (Zhang et al., 2016a; Zhang et al., 2016b; Zhou et al., 2015). Nonetheless, a lack of reproducibility in m6A sequencing, especially using antibody-based methods, may lead to false conclusions on the variability and dynamic nature of m6A (McIntyre et al., 2020).
A growing list of proteins has been found to form the methyltransferase complex that adds m6A onto mRNA (Figure 1). This complex includes a core heterodimer unit of METTL3 and METTL14 (Liu et al., 2014), with accessory proteins including WTAP (Liu et al., 2014; Ping et al., 2014), HAKAI, KIAA1429 (Schwartz et al., 2014b), and RBM15/B (Patil et al., 2016). This complex has been reviewed elsewhere (Balacco and Sober, 2019; Garcias Morales and Reyes, 2020).
Figure 1. Overview of common epitranscriptomic marks.
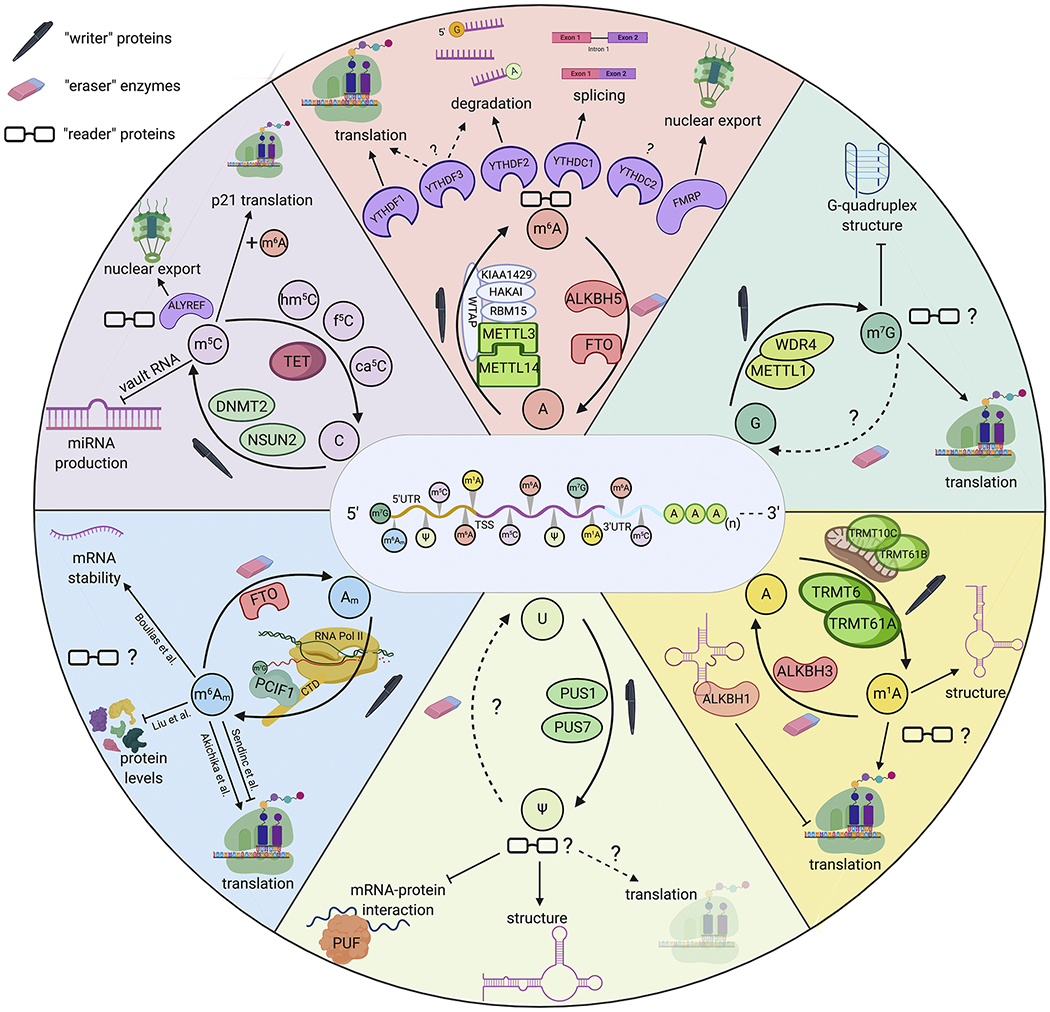
This pinwheel shows the current knowledge for m6A, m7G, m1A, Ψ, m6Am, and m5C (from top, clockwise). Each slice shows the known writer proteins, known eraser enzymes, known reader proteins and downstream functions of the epitranscriptomic mark.
There are two known m6A demethylases, ALKBH5 and FTO (Jia et al., 2011; Zheng et al., 2013). ALKBH5 co-localizes with nuclear speckles, indicating that both methylation and demethylation occur in the nucleus. On the other hand, FTO can act in the nucleus and cytoplasm. However, the in vivo activity of FTO as an m6A demethylase has recently been questioned, with the suggestion that it may instead act on m6Am (Engel et al., 2018; Koh et al., 2019; Linder et al., 2015; Mauer et al., 2017; Schwartz et al., 2014b). On the other hand, one recent study reported that FTO can demethylate m6A, m6Am, and m1A depending on the nuclear or cytoplasmic localization (Wei et al., 2018). Yet another study argued that since loss of FTO has little effect on cytoplasmic mRNA m6A or m6Am levels, FTO likely functions primarily in the nucleus. They further showed that methylation on small nuclear RNAs (snRNAs) is a substrate for FTO demethylation (Mauer et al., 2019). The specificity of FTO remains a major hurdle in the field of m6A; confirming its target is critically important so as not to misattribute a phenotype or biological function to the wrong epitranscriptomic mark. Finally, full knockouts of either ALKBH5 or FTO are not lethal in mice, though they appear to be particularly important in the cellular stress responses (Cao et al., 2019; Engel et al., 2018; Ma et al., 2018).
The highly variable functions of m6A can be attributed to its many distinct reader proteins. The central group of readers is the YTH-domain-containing family of proteins, which bind directly to m6A. These readers have recently been reviewed elsewhere (Patil et al., 2018; Roundtree et al., 2017). Briefly, YTHDC1 is found in the nucleus and regulates splicing, while YTHDF1, YTHDF2, YTHDF3, and YTHDC2 are cytoplasmic and are thought to have distinct functions. YTHDF1 promotes translation, YTHDF2 promotes mRNA degradation, and YTHDF3 seems to promote either translation or degradation in a context-specific manner. Finally, the binding specificity and function of YTHDC2 remain unclear and may only be functional under special cellular conditions (Patil et al., 2018). In contrast to the notion that each YTHDF reader promotes a unique fate of m6A-modified mRNA, two recent studies found that YTHDF-1, 2, and 3 are redundant and jointly promote mRNA degradation in cell lines (Zaccara and Jaffrey, 2020) (BioRxiv: doi: https://doi.org/10.1101/2020.06.03.131441). These divergent conclusions on the function of m6A reader proteins underscore the vital need for additional research into reader protein function and regulation.
N6,2-O-dimethyladenosine: m6Am
Unlike the internal m6A modification, N6,2’-O-dimethyladenosine (m6Am) occurs in the mRNA terminus at the first nucleotide following the N7-methylguanosine (m7G) cap. Approximately 0.0036% to 0.0169% of all adenines are m6Am modified when averaged across multiple human tissue types, corresponding to 526 to 1,028 unique transcripts, depending on the tissue type (Liu et al., 2019). The number of m6Am-modified transcripts was previously thought to be much higher, but improved detection sensitivity has led to the view that m6Am is only moderately abundant (Frye et al., 2016). Three independent studies showed that phosphorylated C-terminal domain (CTD)-interacting factor 1 (PCIF1) is a cap-specific m6Am methyltransferase that targets newly transcribed mRNA by associating with RNA Polymerase II (Akichika et al., 2019; Boulias et al., 2019; Sun et al., 2019). By knocking out PCIF1 in various cell lines, Boulias et al. found that m6Am most strongly correlates with high expression and increased transcript stability (Boulias et al., 2019). However, this was not universally true for all m6Am-modified transcripts, leaving the regulatory capacity of m6Am up for debate. The downstream functions of m6Am are still unclear, with multiple conflicting studies reporting opposite effects on mRNA stability and translation (Akichika et al., 2019; Boulias et al., 2019; Liu et al., 2019; Sendinc et al., 2019). The field would greatly benefit from identification of m6Am reader proteins that could help disentangle its potential downstream functions.
N1-methyladenosine: m1A
The abundance of m1A in cells remains under debate. Some studies found that 0.015% to 0.16% of adenines are m1A modified, corresponding with over 4,000 mRNA transcripts (about 20% of the transcriptome) with high stoichiometries of m1A (Dominissini et al., 2016; Li et al., 2016). Two independent studies claimed that as few as 15 or 53 total m1A sites exist in mRNA and InRNA, and that it mostly occurs at low stoichiometries at these sites (Safira et al., 2017; Schwartz, 2018). In support of this, antibody cross-reactivity with m7G has been shown to produce false positives in m1A identification, and a more recent study found only one high-confidence, high stoichiometry m1A site using a bioinformatic approach, which was validated with an improved m1A antibody (Grozhik et al., 2019). Though our understanding of the modification is limited, progress was made through identification of putative m1A methyltransferases (Figure 1), namely, TRMT6/TRMT61A complex in the cytosol and TRMT10C/TRMT61B complex in the mitochondria (Li et al., 2017c). Currently, ALKBH3 is the only known m1A mRNA demethylase, though it also acts on DNA and m3C in RNA (Aas et al., 2003; E et al., 2016; Ougland et al., 2004). m1A primarily exists in the 5’UTR near the translation initiation site, and its positive charge can induce changes in secondary mRNA structure that may promote translation (Dominissini et al., 2016; Li et al., 2017c). No studies of m1A in the brain have been reported, leaving a major gap in knowledge that will undoubtedly be explored in the coming years.
5-methylcytosine: m5C
m5C is added to tRNA, rRNA, and mRNA by a variety of methyltransferases with specific RNA targets (Motorin et al., 2010). The reported abundance of m5C on mRNA is extremely variable across different studies, with some reporting up to 10,000 m5C sites in a single tissue or cell type (Amort et al., 2017; Squires et al., 2012; Yang et al., 2017), while others using more stringent detection methods showed that zero to only a few hundred sites are modified in mammalian mRNA (Huang et al., 2019b; Legrand et al., 2017). DNMT2 and especially NSUN2 are the most well-characterized m5C methyltransferases that act on both tRNA and mRNA (Khoddami et al., 2019; Li et al., 2017b; Yang et al., 2017) (Figure 1). NSUN2-mediated m5C mRNA methylation promotes mRNA nuclear export through ALYREF, a nuclear m5C reader protein (Yang et al., 2017). Additionally, m5C may cooperate with m6A to enhance translation of particular transcripts like p21 (Li et al., 2017b). Finally, m5C addition to a subset of ncRNAs called vault RNAs (vtRNAs) reduces downstream miRNA production (Hussain et al., 2013). For example, NSUN2 regulates the processing of vault RNA, VTRNA1.1, into small-vault RNAs (svRNAs) that function similarly to miRNAs (Sajini et al., 2019). Though no m5C direct demethylases have been identified, ten-eleven translocation (Tet) enzymes can oxidize m5C to 5-hydroxymethylcytosine (hm5C) and then unmodified cytosine (Ito et al., 2011). The frequency of hm5C is about one hm5C per 5000 m5C (Fu et al., 2014). This is slightly enriched in mRNA, with hm5C occurring on ~7 x 10−6 of the total cytosines (Xu et al., 2016). In Drosophila, hm5C was shown to preferentially mark mRNAs in coding regions and promote their translation (Delatte et al., 2016; Fu et al., 2014). YBX1 is a recently identified m5C reader protein that promotes stabilization of modified mRNAs in early zebrafish embryos (Yang et al., 2019). One study showed that m5C can impair RBP binding, as is the case for SRSF2, which binds to unmethylated vtRNAs with higher affinity than to methylated RNAs (Sajini et al., 2019).
Pseudouridine: Ψ
While pseudouridine (Ψ) is one of the most abundant modifications in ncRNA, its existence in mRNA is a recent finding (Carlile et al., 2014; Schwartz et al., 2014a). PUS1 and PUS7 enzymes isomerize uridine to pseudouridine (Lovejoy et al., 2014) in an mRNA structure-dependent manner (Carlile et al., 2019) (Figure 1). Other PUS-family proteins add Ψ to other types of RNA. On the other hand, no direct readers or removal enzymes have been identified, raising the possibility that Ψ is irreversible. Some downstream effects of Ψ include weakening interactions between mRNA and Pumilio family proteins (PUFs) (Vaidyanathan et al., 2017) and stabilizing RNA structure by improved base stacking and increased hydrogen bonding (Carlile et al., 2014; Davis, 1995; Spenkuch et al., 2014). Ψ has also been hypothesized to promote translation efficiency, though this remains to be proven (Carlile et al., 2014).
While many modifications have been identified, accurately mapping modifications has been difficult. Not only are detection strategies largely limited to antibody-based methods, but different labs mapping modifications in the same cell or tissue type have obtained largely variable results (Table 1). Ongoing efforts to improve reproducibility will be crucial for understanding the biological function of each modification.
Table 1. Compilation of RNA modifications mapped in various cell types.
The number of modified sites (peaks) and total number of unique transcripts that contain at least one modification varies greatly by tissue or cell type, detection method, and lab.
| Mod. | Tissue or Cell Type | Detection Method | # of modified sites (peaks) | # of modified transcripts | Comments | Reference |
|---|---|---|---|---|---|---|
| m6A | Mouse ESCs | m6A RIP-seq | 8,645 | 3,942 annotated | 6,667 and 6,159 peaks in Mettl3 KD and Mettl14 KD cells. | Wang et al., Nat Cell Biology 2015 |
| m6A | Mouse ESCs | m6A RIP-seq | 9,754 | 5,461 mRNAs, 117 ncRNAs | Average 2 peaks per transcript, including on core pluripotency factors | Batista et al., Cell Stem Cell 2015 |
| m6A | Mouse naïve ESCs | m6A RIP-seq | 10,431 | 6,412 | Geula et al., Science 2015 | |
| m6A | Mouse embryonic fibroblasts | m6A RIP-seq | 16,487 | -- | 10,609 peaks are WTAP-dependent (64.3%) | Schwartz et al., Cell Reports 2017 |
| m6A | Total mouse brain | m6A RIP-seq | 13,471 | 4,654 coding 236 non-coding 10,483 mRNAs |
Meyer et al., Cell 2012 | |
| m6A | Mouse cerebellum | m6A RIP-seq with bioinformatic correction for m6Am | 18,594 | 16,576 peaks shared with cortex, and 8,924 mRNAs shared with cortex | Chang et al., Open Biology 2017 | |
| m6A | Mouse cortex | m6A RIP-seq with bioinformatic correction for m6Am | 18,032 | 9,636 mRNAs | 16,989 peaks shared with cerebellum, and 8,924 mRNAs shared with cerebellum | Chang et al., Open Biology 2017 |
| m6A | E13.5 mouse cortex | m6A RIP-seq | 4,055 | 2,179 mRNAs | Yoon et al., Cell 2017 | |
| m6A | PCW11 human cortex | m6A RIP-seq | 10,980 | 5,960 mRNAs | Yoon et al., Cell 2017 | |
| m6A | Mouse P7 cerebellum | m6A RIP-seq | 19,459 | 10,449 total 9,879 mRNA 7,982 |
Increases to 20,722 peaks on 11,215 RNAs upon hypobaric hypoxia exposure | Ma et al., Genome Biology 2018 |
| m6A/m | Mouse cortex | m6A RIP-seq | 14,656 | 25 m6A/m peaks (in 20 genes) were significantly modified in response to 4 h stress | Engel et al., Neuron 2018 | |
| m6A | Human brainstem | m6A RIP-seq | 30,459 | 12,659 | Donor ID 5 | Liu et al., Mol Cell 2019 |
| m6A | Human cerebellum | m6A RIP-seq | 45,975 | 14,048 | Donor ID 5 | Liu et al., Mol Cell 2019 |
| m6A | Human cerebrum | m6A RIP-seq | 28,001 | 11,770 | Donor ID 5 | Liu et al., Mol Cell 2019 |
| m6A | Human hypothalamus | m6A RIP-seq | 26,993 | 11,751 | Donor ID 5 | Liu et al., Mol Cell 2019 |
| m6A | HEK293 | CITS miCLIP, mutation calling | 9,536 | -- | 12,051 residues identified by truncation calling, and 33,157 identified by RIP-seq | Linder et al., Nature Methods 2016 |
| m6A | HEK293T | m6A RIP-seq | 18,756 | 5,768 | 43% conserved transcripts with mouse brain | Meyer et al., Cell 2012 |
| m6A | HeLa cells | m6A-CLIP | 37,557 | -- | ~90% peaks are deposited at pre-mRNA stage, 99% are constant between nucleus and cytoplasm | Ke et al., Genes Dev 2017 |
| m6Am | HEK293 | CITS miCLIP | 487 | -- | Linder et al., Nature Methods 2016 | |
| m6Am | HEK293 | miCLIP | 2,350 | -- | Compared miCLIP in PCIF1 KO cells and WT cells to distinguish m6Am and m6A | Boulias et al, Molecular Cell 2019 |
| m6Am | MEL624 | m6Am-Exo-Seq | -- | 1,521 | Compared PCIF1 WT and KO cells | Sendinc et al., Molecular Cell 2019 |
| m6Am | Human brainstem | m6A RIP-seq peaks at TSS | 511 | 497 annotated | Donor ID 5; one peak per transcript | Liu et al., Mol Cell 2019 |
| m6Am | Human cerebellum | m6A RIP-seq peaks at TSS | 601 | 583 annotated | Donor ID 5; one peak per transcript | Liu et al., Mol Cell 2019 |
| m6Am | Human cerebrum | m6A RIP-seq peaks at TSS | 513 | 494 annotated | Donor ID 5; one peak per transcript | Liu et al., Mol Cell 2019 |
| m6Am | Human hypothalamus | m6A RIP-seq peaks at TSS | 472 | 455 annotated | Donor ID 5; one peak per transcript | Liu et al., Mol Cell 2019 |
| m1A | HeLa | m1A RIP-seq | 7,154 | 4,151 coding 63 non-coding -- |
Average of 1.4 peaks per methylated gene | Dominissini et al., Nature 2016 |
| m1A | HEK293T | m1A RIP-seq | 2,129 | Dominissini et al., Nature 2016 | ||
| m1A | HEK293T | m1A-ID-seq | 901 | 841 mRNAs 46 ncRNAs 226 tRNA 28 mitochondrial 8 rRNA 15 mRNA/other 473 mRNA and lncRNAs 88 tRNA 2 rRNA 1 snoRNA |
1,989 peaks identified in ALKBH3 KO cells | Li et al., Nature Chemical Biology 2016 |
| m1A | HEK293T | m1A RIP-seq | 277 | Safa et al., Nature 2017 | ||
| m1A | HEK293T | m1A-MAP | 740 | Subset of 53 sites is strongly enriched fro GUUCRA motif | Li et al., Molecular Cell 2018 | |
| m1A | HeLa | RBS-Seq | 91 | mRNA peaks identified only if less stringent fdters used | Khoddami et al., PNAS 2019 | |
| m5C | HeLa | RNA-BisSeq | 5,399 | 1,995 mRNAs 336 ncRNAs 3,722 annotated |
45% in CDS | Yang et al., Cell Research 2017 |
| m5C | Mouse small intestine | RNA-BisSeq | 12,201 | Peaks counted that are modified in at least 10% of m1A sites in rep 1 | Yang et al., Cell Research 2017 | |
| m5C | Mouse heart | RNA-BisSeq | 12,397 | 3,915 annotated | ‘’ | Yang et al., Cell Research 2017 |
| m5C | Mouse muscle | RNA-BisSeq | 11,721 | 3,624 annotated | ‘’ | Yang et al., Cell Research 2017 |
| m5C | Mouse brain | RNA-BisSeq | 12,032 | 3,635 annotated | ‘’ | Yang et al., Cell Research 2017 |
| m5C | Mouse kidney | RNA-BisSeq | 12,149 | 3,800 annotated | ‘’ | Yang et al., Cell Research 2017 |
| m5C | Mouse liver | RNA-BisSeq | 11,414 | 3,751 | ‘’ | Yang et al., Cell Research 2017 |
| m5C | Mouse testis 3W | RNA-BisSeq | 11,853 | 2,727 | ‘’ | Yang et al., Cell Research 2017 |
| m5C | HeLa | RBS-Seq | 487 | 340 annotated | High-threshold cutoff | Khoddami et al., PNAS 2019 |
| 5hmC | Drosophila S2 cells | hMeRIP-seq | 3,058 | 1,597 coding | Confirmed via dTet KD | Delatte et al. Science 2016 |
| Ψ | HeLa | RBS-Seq | 754 | 322 mRNA 44 pseudogenes 55 rRNA 246 tRNA 24 snRNA 41 snoRNA 22 ncRNA 88 mRNAs 9ncRNA 238 mRNA 40 ncRNA 168 mRNA 24 ncRNA 46 tRNA 322 mRNA 28 ncRNA |
HeLa cells collected with rRNA depleted total RNA, PolyA selected mRNAs & size selected small RNAs | Khoddami et al., PNAS 2019 |
| Ψ | HeLa | Pseudo-seq | 96 mRNA 12 ncRNA 260 mRNA 74 ncRNA 328 |
Carlile et al., Nature 2014 | ||
| Ψ | S. Cerevisiae (budding | Pseudo-seq | Lower threshold filter shows 466 Ψ on mRNAs. | Carlile et al., Nature 2014 | ||
| Ψ | S. Cerevisiae (midlog) | Ψ-seq | Change under heat shock | Schwartz et al., Cell 2014 | ||
| Ψ | HEK293 and fibroblasts | Ψ-seq | 396 | All human sites were combined into one dataset | Schwartz et al., Cell 2014 | |
| m7G | mESCs | m7G MeRIP | 184 | 184 tRNa | Functionally confirmed in Mettl1 KO mESCs | Lin et al., Mol Cell 2019 |
| m7G | HeLa and HepG2 | m7G meRIP, m7G-seq | 801 | 681 mRNA | present in two replicates in both HeLa and HepG2 cells | Zhang et al., Mol Cell 2019 |
Epitranscriptomics in Stem Cell Biology
Epitranscriptomics appears to be especially important in stem cell biology, as it contributes to self-renewal and differentiation capacity. m6A is by far the most studied RNA modification in stem cells, particularly in ESCs and iPSCs.
m6A in ESCs
Early reports of m6A in ESCs were somewhat conflicting. One study reported that knockdown of Mettl3 and Mettl14 reduces m6A abundance and impairs stem cell self-renewal (Wang et al., 2014b), whereas another study reported that Mettl3 knockout in mESCs improves self-renewal but blocks differentiation (Batista et al., 2014). However, both of these studies examined mESCs in vitro, which muddies our understanding of the exact stage the ESCs are in and what m6A might do to drive embryonic development in vivo. This gap was addressed by Geula et al., who examined m6A in naïve pluripotent mouse ESCs. Naïve mESCs exist in a distinct molecular state compared to more advanced, “primed” epiblast stem cells (EpiSC). By knocking out Mettl3, they identified m6A as a key driver of termination of the naïve state and entry into the primed state, which is necessary for proper lineage differentiation at the post-implantation embryonic stage. The effects of impaired differentiation are so drastic that loss of m6A causes early embryonic lethality (Geula et al., 2015). Importantly, this study further clarified that m6A regulates the genes governing both naïve and primed states, and that loss of m6A causes upregulation of whichever genes are modified in that particular stem cell state. Naïve mESCs show enhanced pluripotency upon Mettl3 knockdown, whereas primed EpiSCs show increased stability of lineage-commitment genes upon loss of m6A (Geula et al., 2015; Zhao and He, 2015). Mechanistically, this study and others determined that m6A primarily functions in development by reducing mRNA stability, which allows for the clearance of key naïve pluripotency-promoting transcripts or pro-differentiation transcripts, depending on the stem cell stage (Figure 2) (Batista et al., 2014; Geula et al., 2015; Wang et al., 2014a; Wang et al., 2014b).
Figure 2. The epitranscriptome in embryonic development and ESCs.
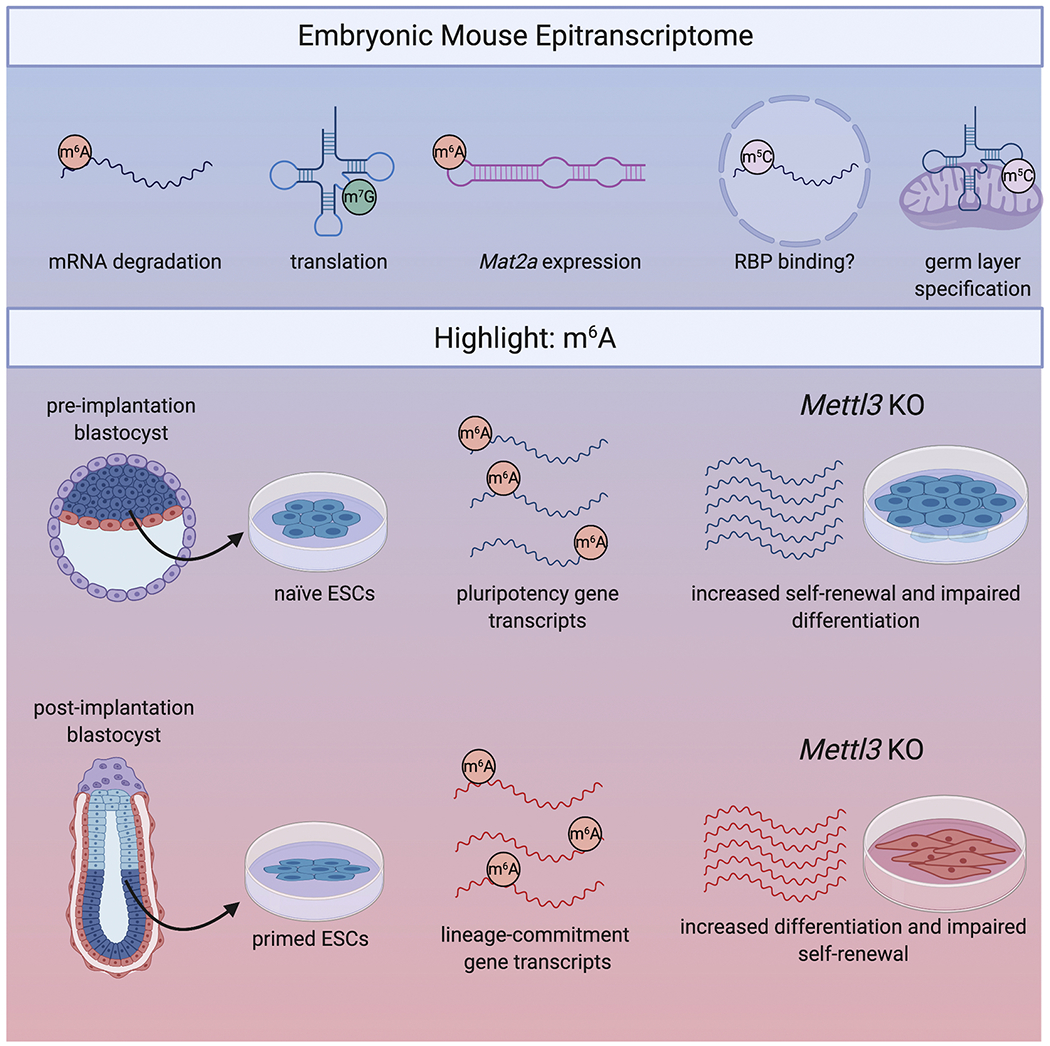
Top: Marks that have been studied in the developing embryo include m6A on mRNA to promote mRNA degradation, m7G on tRNA to promote translation, METTL16-mediated addition of m6A to ncRNA like Mat2a, m5C on mRNA to promote RNA Binding Protein (RBP) mRNA binding, and m5C on mitochondrial tRNA to regulate germ layer specification. Bottom: The most-studied modification in embryonic stem cells (ESCs) is m6A. While several conflicting studies have reported different phenotypes after Mettl3 knockdown, the current consensus is that m6A functions to destabilize whichever gene transcripts are modified at the time. In naïve ESCs purified from preimplantation blastocysts, m6A is primarily added to pluripotency-promoting gen transcripts. Therefore loss of m6A improves self-renewal and impairs differentiation. In contrast, in primed ESCs derived from post-implantation blastocysts, m6A is primarily added to lineage-commitment gene transcripts, so loss of m6A promotes differentiation and impairs self-renewal.
In addition to the traditional METTL3/METTL14-mediated addition of m6A to mRNA, there are several other m6A methyltransferases. In particular, METTL16 was identified in human cells as an m6A methyltransferase that primarily targets small nuclear RNA (snRNA), specifically U6 snRNA, and other non-coding RNAs (Warda et al., 2017). Additionally, METTL16 regulates expression of the SAM synthetase MAT2A (Pendleton et al., 2017), which is highly consequential for all modifications that use SAM as a methyl donor (Figure 2). Accordingly, Mendel et al. found that METTL16-mediated modification of Mat2a mRNA is necessary for proper embryonic development of mouse blastocysts, and homozygous knockout of Mettl16 is embryonic lethal. Analysis of E2.5 Mettl16 KO mouse blastocysts showed that only 20 genes are differentially expressed relative to the wildtype, with Mat2a showing the most significant downregulation. However, by E3.5 the global transcriptome was massively dysregulated (Mendel et al., 2018). While the more common METTL3/METTL14-mediated pathway has garnered the most attention, understanding the complexities of the epitranscriptome and the consequences of mediating highly consequential genes like Mat2a will be necessary to accurately characterize the many roles of m6A.
m5C in mESCs
While the overwhelming focus of research in mESCs has been centered around m6A, m5C has also been examined. In one study, 12,492 m5C sites were identified in nuclear mESC mRNA. Modified mRNAs were enriched for gene ontologies corresponding to cell cycle, RNA processing, chromatin modification, and developmental processes. Though the functionality of m5C in mESCs was not shown, a correlation between m5C sites and RBP sites was identified (Figure 2). Approximately 29% of m5C sites in mESCs overlap with known RBP sites. More specifically, the largest overlaps correspond to UPF1 binding, which regulates nonsense-mediated RNA decay. Additionally, SRSF3 and SRSF3 splicing factors and the PRC2 subunit EZH2 have binding sites that significantly overlap with m5C sites. This led to the hypothesis that m5C may contribute to RBP binding and functionality, though this requires further validation (Amort et al., 2017). Finally, a recent study showed that during the maternal-to-zygotic transition in zebrafish development, the m5C reader, YBX1, binds to m5C-modified mRNA to promote stabilization of maternal mRNAs during the transition (Yang et al., 2019).
As detection of diverse mRNA modifications continues to improve and orphan methyltransferase targets are identified, we expect our understanding of epitranscriptomic regulation of stem cells to grow rapidly. Notably, the low stoichiometry of some modifications relative to m6A should not decrease their perceived importance, as the power of the modification is derived from the strength of its downstream effects, which vary widely among reader proteins.
Induced Pluripotent Stem Cells (iPSCs)
The understanding that m6A contributes to pluripotency and differentiation drove studies of its regulatory capacity in iPSCs. In 2015, Chen et al. showed that high abundance of m6A increases the reprogramming efficiency of mouse embryonic fibroblasts (MEFs) to pluripotent stem cells, in part by altering expression of key pluripotency factors like Oct4, Sox2, and Nanog. This study further found an interplay between microRNA (miRNA) binding to mRNA and enhanced Mettl3 binding to mRNA to promote de novo addition of m6A (Chen et al., 2015). This concept of m6A interplay with noncoding RNAs has been explored with contrasting conclusions, and has been reviewed in-depth elsewhere (Fazi and Fatica, 2019). Furthermore, Chen et al. found thatMettl3 knockdown reduces iPSC colony formation (Chen et al., 2015). However, Geula et al. found thatMettl3 knockdown does not impair reprogramming efficiency, but rather slows proliferation of iPSCs in early reprogramming (Geula et al., 2015). Finally, Wu et al. found thatMettl3 knockdown decreases the proliferation rate of porcine iPSCs (piPSCs) and impairs expression of key pluripotency genes, though they did not test for reprogramming efficiency. This study further identified that m6A promotes YTHDF1-mediated translation of JAK2 in piPSCs, while promoting degradation of SOCS3 via YTHDF2 (Wu et al., 2019). Both of these mechanisms lead to upregulation of the JAK2-STAT3 signaling pathway, which is known to promote stem cell self-renewal by increasing expression of the core pluripotency genes Klf4 and Sox2 (Niwa et al., 1998; Wu et al., 2019). In parallel, YTHDF2 has been shown to be upregulated in iPSCs to destabilize m6A-modified mRNAs related to neural development and thereby promote pluripotency (Heck et al., 2020).
Overall, m6A clearly regulates the pluripotency of iPSCs, but its role in reprogramming likely depends on the cellular context of the starting material or the stage of reprogramming. As was the case in ESCs, m6A may alter expression of the gene transcripts already present. Still, further investigation is needed to identify the fate of m6A-modified transcripts. While m6A-mediated mRNA degradation appears to be a major mechanism, expression of other reader proteins suggests a more complex system. Understanding how m6A reader proteins selectively bind particular mRNA targets will be a major step forward in further elucidating the mechanisms of m6A action in iPSCs.
Regulation of Stem Cell Epitranscriptomes
Upstream regulation of m6A deposition or differential expression of the writers, readers, and erasers contributes to the function of m6A in stem cells. For example, Aguilo et al. showed that zinc finger protein 217 (ZFP217) coordinates epigenetic regulation with m6A deposition. More specifically, ZFP217 is a transcription factor that directly activates transcription of several key pluripotency genes, then blocks m6A modification of these genes by sequestering METTL3 in mESCs and iPSCs. ZFP217 knockdown causes global increases in m6A levels, which correlates with a decreased half-life of Nanog, Sox2, c-Myc, and Klf4 mRNA transcripts. This in turn impairs pluripotency and reprogramming (Aguilo et al., 2015).
Wen et al. found that another zinc-finger protein, Zc3h13, is critical for m6A deposition, and Zc3h13 knockdown significantly impairs self-renewal and maintenance of pluripotency in mESCs (Knuckles et al., 2018; Wen et al., 2018). Zc3h13 can form a complex with WTAP, Virilizer (Kiaa1429), and Hakai, which also contribute to the METTL3-METTL14 m6A methylation complex (Horiuchi et al., 2013; Wan et al., 2015). Wen et al. then showed that Zc3h13 knockdown in mESCs decreases global m6A levels to about 30-40% as in the control, and confirmed m6A dependency on Zc3h13 through MeRIP-seq. More specifically, Zc3h13 promotes m6A deposition by localizing the Zc3h13-WTAP-Virilizer-Hakai complex to nuclear speckles; loss of Zc3h13 causes these complex components, as well as METTL3/METTL14, to significantly shift to localization in the cytoplasm. Functionally, Zc3h13 knockdown impairs mESC self-renewal, decreases expression of pluripotency genes, and increases expression of differentiation markers in correlation with differential m6A modifications of these gene transcripts (Wen et al., 2018). The conclusion that m6A promotes self-renewal is consistent with previous studies (Wang et al., 2014b), and the consequences on pluripotency correspond to studies performed under similar conditions in mESCs (Geula et al., 2015). These two studies on zinc finger proteins are important examples of how m6A may be regulated or targeted to individual transcripts in stem cells. This connection between transcription factors and epitranscriptomic regulation remains an interesting avenue for further research.
Finally, one study has found a direct connection between histone methylation and sites of m6A deposition. Huang et al. showed that histone H3 trimethylation at lysine-36 (H3K36me3) drives m6A methylation by recruiting and promoting interactions between the m6A methyltransferase complex and its target mRNA. More specifically, METTL14 binds to H3K36Me3, chromatin, and RNA, thereby promoting the co-transcriptional addition of m6A to genes with H3K36Me3 epigenetic marks. Knockdown of the H3K36Me3 methyltransferase, SETD2, impairs binding of the m6A methyltransferase complex to sites that lose H3K36Me3, and globally reduces m6A levels. In mESCs, SETD2 knockdown induces higher expression of pluripotency factors (OCT4, SOX2, NANOG) and prevents increased m6A methylation during differentiation. This suggests that H3K36Me3 drives m6A modifications to destabilize pluripotency genes and promote differentiation, and loss of either H3K36Me3 or METTL14 promotes pluripotency over differentiation (Huang et al., 2019a). This corresponds with previous reports that m6A is necessary for proper differentiation of mESCs (Batista et al., 2014; Geula et al., 2015), and provides the first evidence that m6A addition may be directed by epigenetic marks.
While a few studies have identified how the epitranscriptome may be regulated, over 100 putative METTL3 or METTL14 binding proteins have been identified, suggesting that there is much left to be learned about upstream regulation of m6A (Malovannaya et al., 2011). A better understanding of how the methyltransferase complex and demethylases target specific gene transcripts, as well as how writer, reader, and eraser expression is regulated, will drive the field forward.
Epitranscriptomics in Neural Development
Recent work has shown that the epitranscriptome, in particular m6A, is especially important for neural development and brain function (Livneh et al., 2020; Yoon et al., 2018). Lence et al. performed one of the first studies of m6A in the brain, using Drosophila melanogaster as a model organism. This study showed that m6A is enriched in the nervous system and that knockout of the methyltransferase components causes reduced lifespan, severe behavioral defects, and global changes in neural gene expression (Lence et al., 2016). While this work was important for understanding m6A in vivo, it contrasted with mammalian studies in that loss of m6A methyltransferases is not lethal in flies. The next major advances came from studies of conditional knockout of the m6A methyltransferase complex in mice to examine epitranscriptomic regulation during mammalian brain development. Below we provide an in-depth overview of the epitranscriptome in mammalian neural development (Figure 3).
Figure 3. m6A in neural development.
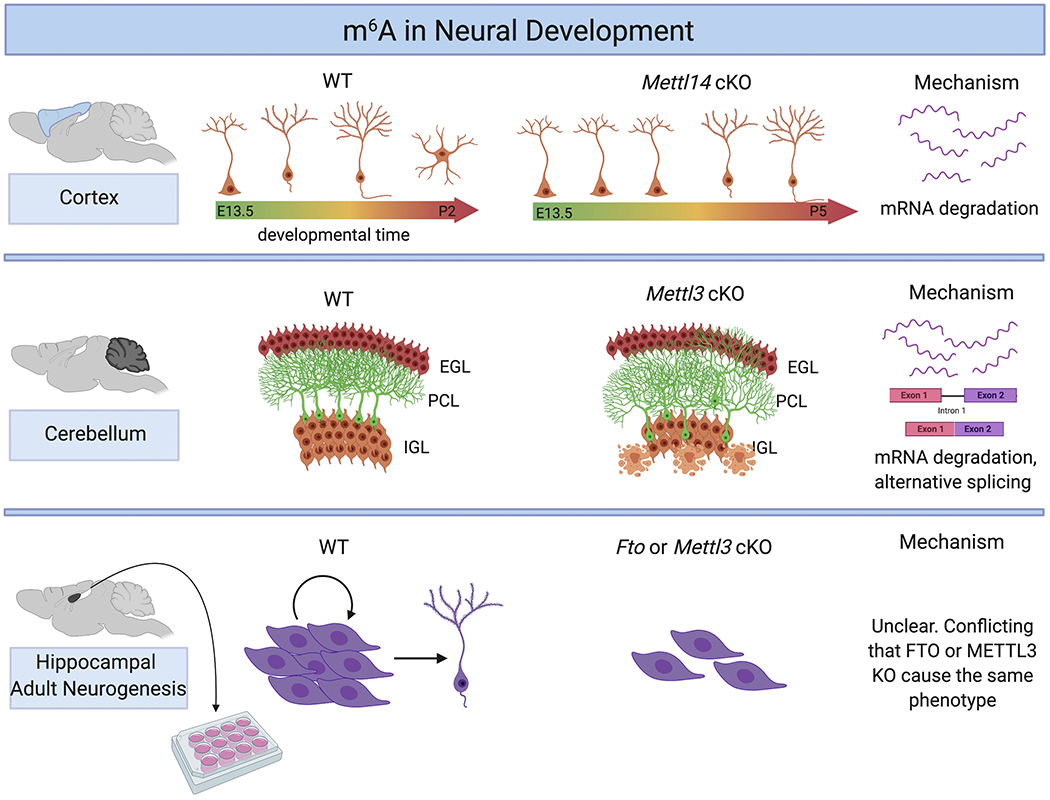
Top: In the cortex, loss of m6A elongates the timeframe of cortical neurogenesis such that the postnatal brain is still generating upper-layer neurons. This is accomplished through altered mRNA degradation rates of key pluripotency and fate-determining gene transcripts. Middle: In the cerebellum, loss of m6A causes disorganization of the Purkinje cell layer (PCL) into the inner granule cell layer (IGL). IGL cells also exhibit a higher rate of apoptosis, resulting in fewer total IGL cells. Mechanistically, m6A has been shown to promote alternative splicing and mRNA degradation in the cerebellum. Bottom: In hippocampal adult neurogenesis, in vitro studies found that loss of m6A impairs self-renewal and neurogenesis, but the mechanism of action remains unclear.
Cortical Development
In 2017, our laboratory showed that conditional knockdown (cKO) of Mettl14 in mice and subsequent loss of m6A in neural progenitor cells (NPCs) drastically impairs brain development in vivo (Yoon et al., 2017). Loss of m6A impairs NPC differentiation, slows cell cycle progression, and elongates the timing of cortical neurogenesis into postnatal stages. Mechanistically, m6A-modified genes are significantly enriched for gene ontologies that correlate with regulation of transcription, neuron differentiation, cell cycle, and stem cell differentiation. These modified transcripts have a shorter half-life than their corresponding unmodified transcripts in Mettl14 cKO mouse NPCs, suggesting that m6A normally destabilizes mRNA in the developing brain. By modifying both multipotency and differentiation-promoting transcripts, the m6A system may contribute to harmonious changes in gene expression that are necessary for the progression of NPCs through the distinct phases of embryonic cortical neurogenesis. To this end, we found that Mettl14 cKO NPCs co-express stem cell and neuronal markers, and that rapid degradation of neuronal markers in wildtype NPCs allow for pre-patterning of differentiation by allowing transcription of pro-neuronal genes but preventing significant protein production (Yoon et al., 2018). Finally, we used iPSC-derived human brain organoids to confirm that m6A also regulates NPC cell cycle progression in humans (Yoon et al., 2017). We then compared m6A-seq analysis among human brain organoids, human post-conception week 11 embryonic brain tissue and E13.5 mouse brains. While many gene transcripts are m6A-modified in both species, the human-specific modifications correlated strongly with disease ontologies for human-specific mental disorders like autism and schizophrenia. This work provided the first in vivo analysis of m6A in mammalian brain development and highlighted the possibility that m6A may contribute to psychiatric or neurodevelopmental disorders in humans.
Shortly thereafter, an independent study by Wang et al. knocked out Mettl14 in the developing forebrain, and also found that loss of m6A slows NPC cell cycle progression. In vitro analysis of Mettl14 cKO NPCs showed that loss of m6A can cause premature differentiation, and in vivo analysis showed that Mettl14 cKO mice had reduced numbers of Pax6+ NPCs and reduced numbers of Satb2+ late-born neurons. This led the authors to suggest that depletion of the NPC pool causes a reduction in neurogenesis (Wang et al., 2018b). This contrasted with our study, which showed an increase in Pax6+ cells in Mettl14 cKO forebrains, but a similar decrease in late-born neurons; we therefore proposed that m6A is necessary for the timely differentiation of NPCs, and loss of m6A causes a build-up of Pax6+ NPCs (Yoon et al., 2017). These differences may stem from different methodologies or antibodies. Nonetheless, the studies agree that m6A regulates mRNA stability to alter gene expression and NPC fate.
Next, Wang et al. identified genome-wide changes in histone modifications upon Mettl14 knockout. Specifically, cKO NPCs show increases in histone H3 acetylation at lysine 27 (H3K27ac), histone H3 trimethylation at lysine 4 (H3K4me3), and histone H3 trimethylation at lysine 27 (H3K27me3). Chemically blocking these epigenetic marks partially rescues cKO NPC proliferation defects. The changes in histone modification were partially attributed to m6A-mediated destabilization of CBP and p300 transcripts, which are stabilized upon loss of m6A. However, this did not apply to transcripts in the PRC2 complex, suggesting there are also other mechanisms at play (Wang et al., 2018b). Overall, the connection between the epitranscriptome and epigenetics in the developing brain is highly intriguing. As single-transcript m6A editing techniques are developed (Wilson et al., 2020), it would be pertinent to edit only CBP and p300 mRNA to quantify the degree to which their methylation contributes to the Mettl14 cKO phenotype, as opposed to the sum of many modified transcripts.
Finally, a third study conditionally knocked out Ythdf2 in the developing forebrain to show that m6A largely functions through YTHDF2-mediated mRNA degradation during cortical development. In this study, Li et al. showed that Ythdf2 KO mice have a very similar phenotype to Mettl14 cKO mice. In particular, loss of Ythdf2 impairs NPC proliferation and differentiation, and causes delays in cortical neurogenesis. They also found that Ythdf2−/− NPCs create fewer primary neurites per neuron and shorter neurites overall when differentiated in vitro, suggesting that m6A also regulates neuron maturation during the differentiation process (Li et al., 2018b). This study was necessary to confirm that m6A regulation of cortical development functions primarily through YTHDF2-mediated mRNA degradation and that m6A promotes NPC proliferation and differentiation.
Beyond m6A, m5C is also crucial for proper neurodevelopment. Flores et al. showed that the m5C methyltransferase NSUN2 is essential for neural stem cell differentiation (Flores et al., 2017). Loss-of-function mutations in Nsun2 caused neurodevelopmental defects such as microcephaly in mouse and human models. In a conditional Nsun2 knockout in the developing mouse brain, there were signs of decreased sizes of the cerebral cortex, hippocampus, and striatum compared to wildtype mouse brains. In addition, Nsun2 cKO mice had lower levels of global protein production and increased cellular stress compared to wildtype brains. These phenotypes are thought to arise due to differentiation delays, lack of neural lineage commitment, and a reduction in upper-layer neurons (Flores et al., 2017). This interesting study broadens the importance of the epitranscriptome in neural development beyond m6A. Indeed, as detection methods for other modifications become more reliable, it will be worthwhile to explore a greater diversity of epitranscriptomic marks in neural development.
Cerebellar Development
The complexity of the brain suggests that epitranscriptomic regulatory systems may have distinct functions in different parts of the brain. Indeed, Chang et al. showed that m6A levels are increased in the adult mouse cerebellum compared to the cerebral cortex, and that there are region-specific methylation patterns (Chang et al., 2017). Even within the cerebellum, methylation patterns change over development. Ma et al. showed that methylation targets change across postnatal day 7 (P7), P14, P21, and P60 mouse cerebella. There are 12,452 m6A peaks that are turned “ON” (emerge at a later stage) over time, and 11,192 that are turned “OFF” (disappear in later stages). The groups of transcripts methylated at each time point correspond with the developmental processes happening at that time. For example, gene transcripts in which m6A is turned OFF from P7 to P14 have gene ontologies enriched for cell cycle. On the other hand, gene transcripts in which m6A is turned ON at P14, P21, or P60 have gene ontologies enriched for signal transduction, cell adhesion, learning, and synaptic plasticity. Overall, m6A modification patterns strongly correlate with the progression from proliferating cells at P7 to mature neuronal activities at P60. This study also examined changes in expression of METTL3, METTL14, WTAP, FTO, and ALKBH5. Though cerebellar expression of all of these genes decreased on average over time, there was a specific reduction in internal granular layers but elevated expression in Purkinje cells. Lentiviral Mettl3 knockdown at P7 lowers the number of Purkinje cells and impairs their organization along the outer surface of the inner granule cell layer. On the other hand, Alkbh5-KO mice had no observable phenotype in the cerebellum under normal conditions, which may be due to redundant actions by FTO. After stressing the developing brain with hypobaric hypoxia, Alkbh5-KO mice had significantly smaller cerebella and fewer mature neurons, yet significantly more proliferating cells. This suggests that ALKBH5 is critical for promoting cerebellar neurogenesis under stress. Finally, this study showed that several important gene transcripts are differentially localized in the cytoplasm over nucleus in Alkbh5-KO cerebella, indicating that m6A promotes nuclear export in this tissue (Ma et al., 2018).
In contrast, Wang et al. used a Mettl3 cKO mouse model to show that m6A promotes mRNA degradation and alternative splicing in the cerebellum. Mettl3 cKO mice have drastically smaller cerebella, significantly fewer cerebellar granule cells (CGCs) in the internal granular layer (IGL), and disordered Purkinje cell organization relative to wildtype controls. Furthermore, loss of m6A causes significantly increased levels of apoptosis of newborn granule cells, which explains the depletion of CGCs. Again, loss of m6A increases mRNA stability; m6A modifications on apoptosis-associated gene transcripts normally restrict their expression. Notably, m6A-mediated regulation of apoptosis appears to be specific to the cerebellum, as these transcripts are not stabilized in the cortex ofMettl3 cKO mice. Finally, Wang et al. identified an additional mechanism of m6A-mediated alternative splicing in the cerebellum. Exon exclusion occurs more frequently upon m6A depletion, especially in transcripts that are normally methylated in the wildtype. These alternatively spliced transcripts are enriched for gene ontologies in synapse-associated pathways and neurotransmitter receptors. Further analysis showed that increases in intracellular calcium concentration in Mettl3 cKO CGCs contributes to their increased apoptosis (Wang et al., 2018a). This work highlights the fact that epitranscriptomic regulation is highly cell-type specific with unique roles in different parts of the brain. How this specificity is regulated will be an interesting avenue of future research.
Adult neurogenesis
The m6A demethylase FTO has been implicated in numerous pathways in the mature brain, from cancer (Cui et al., 2017), to psychiatric and neurodegenerative diseases (Choudhry et al., 2013; Hess et al., 2013; Keller et al., 2011; Li et al., 2018a; Widagdo et al., 2016), to regulation of adult neural stem cells (Gao et al., 2010; Li et al., 2017a). However, understanding the role of FTO remains difficult due to its multiple functions in DNA and RNA demethylation. In fact, the first study on FTO in neurogenesis was published in 2010, before FTO was even identified as an m6A demethylase (Gao et al., 2010; Jia et al., 2011). Gao et al. generated whole-body and neural-specific Fto KO mice and found that the two have very similar phenotypes, indicating that the majority of FTO functions occur in the nervous system (Gao et al., 2010). In 2017, it was shown that FTO is expressed in adult NSCs (aNSCs) and in mature neurons and its expression increases over postnatal time. Fto KO mice show reduced proliferation and differentiation of aNSCs, which functionally impairs learning and memory. Furthermore, loss of FTO results in slightly higher (~15%) levels of m6A, though only 363 genes are both m6A modified and differentially expressed upon loss of FTO (out of 5635 m6A-modified genes and 1862 FTO-dependent genes) (Li et al., 2017a). While FTO does seem to regulate adult neurogenesis, the degree to which this is enacted through m6A remains in question, especially considering that FTO can act on multiple targets in vivo.
Next, Chen et al. found that MellI3 knockdown impairs both proliferation and differentiation of aNSCs cultured in vitro. m6A sequencing showed that the m6A landscape is dynamic between proliferating and differentiating cultured aNSCs; transcripts modified only in proliferating aNSCs correlate with cell cycle, while transcripts modified only in differentiating aNSCs are enriched for protein localization, signaling, and synapse organization (Chen et al., 2019). This study is slightly more direct in studying m6A in adult neurogenesis by knocking down Mettl3, but the use of cultured aNSCs limits the conclusions that can be drawn; aNSCs exist in highly specialized niches in vivo that are difficult to recapitulate in vitro (Ming and Song, 2011; Song et al., 2012).
Finally, a 2019 study found that Fto cKO in aNSCs decreases aNSC proliferation and differentiation into NeuN+ neurons at 4 weeks after FTO knockout. While the fate of m6A-modified transcripts was not tested, individual mRNA transcripts in the Stat3 signaling pathway, Socs5 and Pdgfrα, were shown to play important roles in FTO-mediated regulation of aNSCs. However, Socs5 mRNA and protein decrease in Fto cKO aNSCs, while Pdgfrα mRNA and protein increase (Cao et al., 2019). Therefore, the involvement of m6A and mechanisms of m6A-mediated regulation in aNSC remain unclear. In multiple studies, effects of Fto or Mettl3 KD appear stronger in in vitro cultured cells than in vivo aNSCs. The highly dynamic nature of m6A in response to signaling and stress stimuli suggest that culturing systems need to be incredibly carefully controlled to maintain an accurate representation of the epitranscriptome in in vivo aNSCs.
Epitranscriptomics in Neurodevelopmental Diseases
In accordance with its powerful role in neural development, m6A has been linked to neurodevelopmental defects as well. To date, m6A in Fragile X Syndrome is the best-characterized interaction. Additionally, emerging genome-wide association studies and human genetics studies have linked mutations in epitranscriptomic enzymes with intellectual disability (Figure 4).
Figure 4. The epitranscriptome in neural disorders.
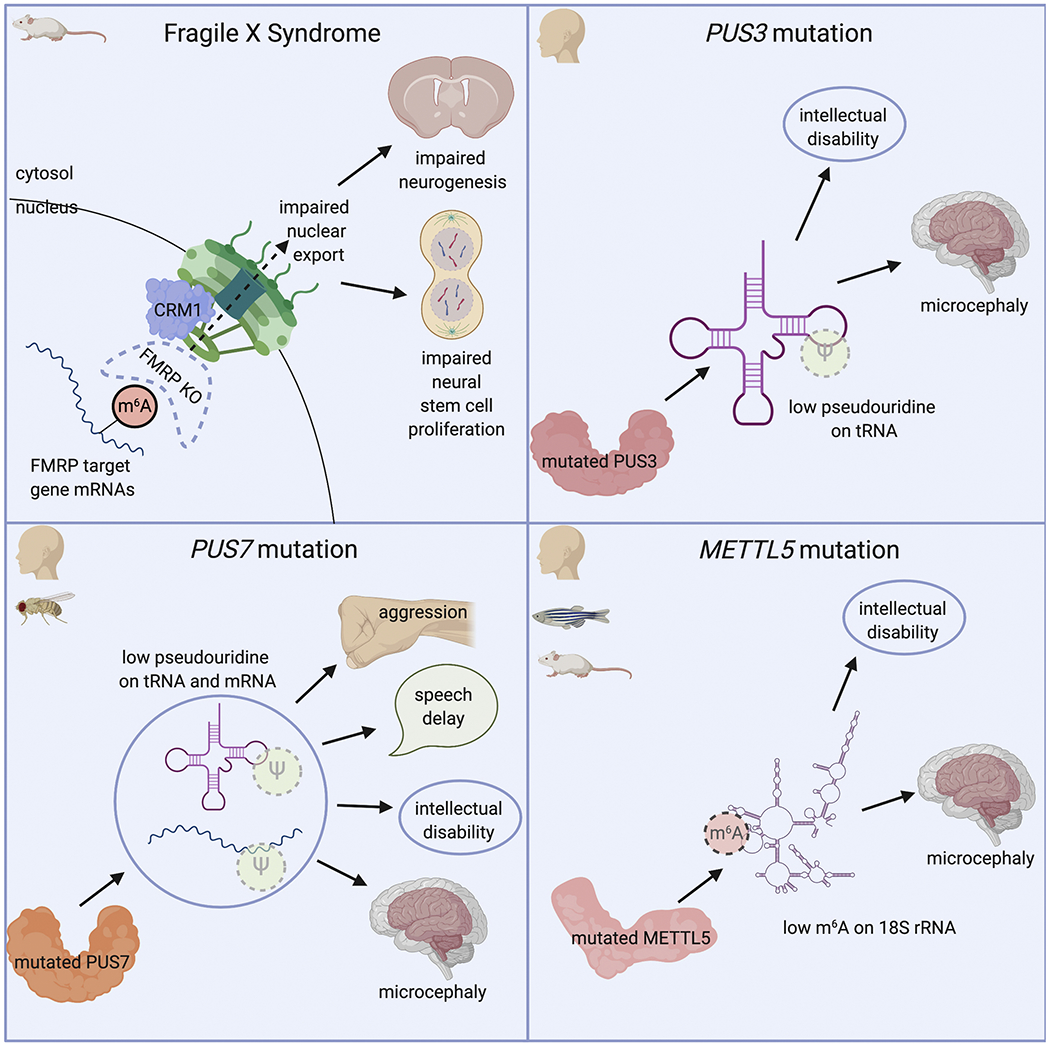
Top left: Fragile X Syndrome is correlated with m6A in that the central protein involved in Fragile X, FMRP, can bind m6A to promote nuclear export of modified mRNAs through interaction with CRM1, a component of the nuclear pore complex. Loss of FMRP in mice impairs this export and causes decreased levels of embryonic neurogenesis and NSC proliferation. Top right: PUS3 mutations in humans significantly correlate with intellectual disability and microcephaly. Though the exact mechanism is unknown, mutations in PUS3 cause significantly lower levels of pseudouridine addition on tRNA relative to wildtype controls. Bottom left: PUS7 mutations identified in humans and validated in drosophila cause increased aggression, speech delay, intellectual disability, and microcephaly through decreased levels of pseudouridine on both tRNA and mRNA. Bottom right: METTL5 mutation in humans and validated in zebrafish and mice cause intellectual disability and microcephaly through decreased levels of m6A on 18S ribosomal RNA.
Fragile X Syndrome
Fragile X mental retardation protein (FMRP), encoded by FMR1, is an RNA-binding protein that is best known for negatively regulating the translation of its target mRNAs (Darnell et al., 2011; Richter et al., 2015) and trafficking mRNA granules (De Diego Otero et al., 2002). Loss-of-function mutations in FMR1 cause Fragile X Syndrome, which is marked by intellectual disability and delayed development. In 2017, Arguello et al. identified FMRP as an m6A binding protein in vitro (Arguello et al., 2017). Zhang et al. then showed that FMRP binds to YTHDF2 and that FRMP target genes are enriched for m6A marks in the mouse cerebral cortex (Zhang et al., 2018). A knockout of Fmrl resulted in the downregulation of some m6A mRNA FMRP target transcripts, suggesting that FMRP stabilizes these m6A modified mRNAs (Zhang et al., 2018). Next, Edens et al. showed that FMRP promotes nuclear export of m6A-modified mRNA by interacting with CRM1, a nuclear export protein. Additionally, Fmr1 KO mice phenocopy Mettl14 cKO mice in terms of delayed embryonic cortical neurogenesis and prolonged NPC cell cycle progression. In both of these mouse knockout models, FMRP target mRNAs are retained in the nucleus (Edens et al., 2019). The binding affinity of FRMP for m6A-modified mRNA and its role in nuclear export was recently confirmed by another study (Hsu et al., 2019).
Intellectual Disability
Recent studies identified correlations between epitranscriptomic modifications and intellectual disability. First, Shaheen et al. found that mutations in human PUS3, a pseudouridinylation enzyme, correlates with intellectual disability and microcephaly in three affected siblings (Shaheen et al., 2016). The affected individuals also have a significant reduction in Ψ-modified tRNA relative to healthy controls in purified lymphoblastoid cells. The PUS3 deficiency phenotype in humans is largely brain-specific, suggesting that PUS3-mediated tRNA Ψ modification is especially important for cognitive function.
Next, both de Brouwer et al. and Shaheen et al. identified mutations in PUS7, a tRNA and mRNA pseudouridinylation enzyme, that cause intellectual disability, microcephaly, speech delay, and aggressive behavior (de Brouwer et al., 2018; Shaheen et al., 2019). Ψ at position 13 in tRNA and PUS7 target mRNAs were significantly reduced in affected individuals compared to healthy controls. Additionally, Pus7 knockout in Drosophila recapitulates the cognitive impairment phenotype and the molecular loss of Ψ at particular target sites (de Brouwer et al., 2018). This provides exciting evidence that Ψ modifications of mRNA and tRNA are not only highly conserved across species, but are critical in neural development. Additional studies using mouse models to investigate the exact mechanism of Ψ in neural development will be an exciting next step.
A 2012 study by Abbasi-Moheb et al. showed that homozygous loss of the m5C methyltransferase, NSUN2, due to nonsense or splicing mutations in the transcript causes memory and learning deficits in humans (Abbasi-Moheb et al., 2012). Further experiments with a knockout of the NSUN2 ortholog, CG6133, in Drosophila showed short-term-memory deficits that could be rescued when the wild type protein was expressed. The fact that humans with NSUN2 mutations show intellectual disabilities and facial dysmorphism, combined with the finding that similar phenotypes are found in Drosophila suggests that m5C may be a fundamental regulator of neural function across species (Abbasi-Moheb et al., 2012). These data strongly suggest that m5C writers are important in neural development and function, but further studies are necessary to understand the mechanistic role of m5C in the brain.
Finally, Richard et al. identified frameshift mutations in METTL5, which adds m6A to 18S rRNA (van Tran et al., 2019), that cause autosomal-recessive intellectual disability and microcephaly. METTL5 is expressed in the human brain from early development and into adulthood, particularly in the cerebellar cortex, hippocampus, and striatum. Analysis in rodents confirmed ubiquitous METTL5 expression in the brain, with increased staining in neural soma and nuclei, as well as in pre- and post-synaptic regions. Finally, Mettl5 knockout in zebrafish recapitulates the microcephaly phenotype and specifically causes a decrease in forebrain and midbrain size (Richard et al., 2019). While mechanistic studies of METTL5 action have only recently begun, this genetic evidence suggests that it is yet another epitranscriptomic modifier that is crucial for proper brain development.
Concluding Remarks and Future Outlook
The field of epitranscriptomics has reached a point where the power of various mRNA modifications has become widely accepted, but the specific mechanisms of their action remain under debate. It is becoming increasingly important to perform extremely careful experiments to detect and validate epitranscriptomics marks to prevent further confusion regarding their downstream functions. Furthermore, expression of multiple reader proteins and multiple published functions of m6A in a single cell type suggest that m6A may differentially regulate various gene transcripts within a single cell. Several important strategies to further elucidate the regulatory capacities of m6A in stem cells and neural development include (1) improved detection techniques for higher sensitivity and accuracy, (2) studies on how reader proteins selectively bind a subset of m6A-modified mRNAs, and (3) careful analyses and conservative interpretations of data to prevent overconfident conclusions that will hinder future studies.
In addition to clarifying studies on m6A, we are particularly excited by the prospects of other epitranscriptomics marks in neural development and disease. Careful mapping of m1A, m5C, m7G, m6Am, and Ψ in the brain alongside generating animal knockouts of their respective modifying enzymes will greatly expand the breadth of knowledge in the field of epitranscriptomics. With an increasing number of scientists working in this field, we expect the next five years to be full of new discoveries with profound implications for basic and translational science.
Highlights.
Overview of current understanding of several RNA modifications
Review of epitranscriptomic regulation of stem cells biology and neural development
Discussion of roles of epitranscriptomics in neurodevelopmental disorders
Acknowledgement:
We thank K. M. Christian for comments. The work in the authors’ laboratories was supported by the National Institutes of Health (R35NS116843 to H.S., and R35NS097370 to G-l.M.), the SAFRI (#575050 to H.S.), and the Sheldon G. Adelson Medical Research Foundation (to G-l.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aas PA, et al. , 2003. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 421, 859–63. [DOI] [PubMed] [Google Scholar]
- Abbasi-Moheb L, et al. , 2012. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am J Hum Genet. 90, 847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilo F, et al. , 2015. Coordination of m(6)A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell Stem Cell. 17, 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akichika S, et al. , 2019. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science. 363. [DOI] [PubMed] [Google Scholar]
- Amort T, et al. , 2017. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 18, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello AE, et al. , 2017. RNA Chemical Proteomics Reveals the N(6)-Methyladenosine (m(6)A)-Regulated Protein-RNA Interactome. J Am Chem Soc. 139, 17249–17252. [DOI] [PubMed] [Google Scholar]
- Balacco DL, Soller M, 2019. The m(6)A Writer: Rise of a Machine for Growing Tasks. Biochemistry. 58, 363–378. [DOI] [PubMed] [Google Scholar]
- Batista PJ, et al. , 2014. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 15, 707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulias K, et al. , 2019. Identification of the m(6)Am Methyltransferase PCIF1 Reveals the Location and Functions of m(6)Am in the Transcriptome. Mol Cell. 75, 631–643 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantara WA, et al. , 2011. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 39, D195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, et al. , 2019. Dynamic effects of Fto in regulating the proliferation and differentiation of adult neural stem cells of mice. Hum Mol Genet. [DOI] [PubMed] [Google Scholar]
- Carlile TM, et al. , 2019. mRNA structure determines modification by pseudouridine synthase 1. Nat Chem Biol. 15, 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile TM, et al. , 2014. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 515, 143–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, et al. , 2017. Region-specific RNA m(6)A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. , 2019. m(6)A Regulates Neurogenesis and Neuronal Development by Modulating Histone Methyltransferase Ezh2. Genomics Proteomics Bioinformatics. 17, 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, et al. , 2015. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 16, 289–301. [DOI] [PubMed] [Google Scholar]
- Choudhry Z, et al. , 2013. Association between obesity-related gene FTO and ADHD. Obesity (Silver Spring). 21, E738–44. [DOI] [PubMed] [Google Scholar]
- Cui Q, et al. , 2017. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 18, 2622–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, et al. , 2011. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 146, 247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DR, 1995. Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res. 23, 5020–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brouwer APM, et al. , 2018. Variants in PUS7 Cause Intellectual Disability with Speech Delay, Microcephaly, Short Stature, and Aggressive Behavior. Am J Hum Genet. 103, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Diego Otero Y, et al. , 2002. Transport of fragile X mental retardation protein via granules in neurites of PC12 cells. Mol Cell Biol. 22, 8332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatte B, et al. , 2016. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science. 351, 282–5. [DOI] [PubMed] [Google Scholar]
- Desrosiers R, et al. , 1974. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 71, 3971–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, et al. , 2013. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc. 8, 176–89. [DOI] [PubMed] [Google Scholar]
- Dominissini D, et al. , 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 485, 201–6. [DOI] [PubMed] [Google Scholar]
- Dominissini D, et al. , 2016. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 530, 441–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E AA, et al. , 2016. ALKBHs-facilitated RNA modifications and de-modifications. DNA Repair (Amst). 44, 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens BM, et al. , 2019. FMRP Modulates Neural Differentiation through m(6)A-Dependent mRNA Nuclear Export. Cell Rep. 28, 845–854 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel M, et al. , 2018. The Role of m(6)A/m-RNA Methylation in Stress Response Regulation. Neuron. 99, 389–403 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre D, et al. , 2003. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazi F, Fatica A, 2019. Interplay Between N (6)-Methyladenosine (m(6)A) and Non-coding RNAs in Cell Development and Cancer. Front Cell Dev Biol. 7, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores JV, et al. , 2017. Cytosine-5 RNA Methylation Regulates Neural Stem Cell Differentiation and Motility. Stem Cell Reports. 8, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, et al. , 2016. RNA modifications: what have we learned and where are we headed? Nat Rev Genet. 17, 365–72. [DOI] [PubMed] [Google Scholar]
- Fu L, et al. , 2014. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J Am Chem Soc. 136, 11582–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, et al. , 2010. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS One. 5, e 14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Campos MA, et al. , 2019. Deciphering the “m(6)A Code” via Antibody-Independent Quantitative Profiling. Cell. 178, 731–747 e16. [DOI] [PubMed] [Google Scholar]
- Garcias Morales D, Reyes JL, 2020. A birds’-eye view of the activity and specificity of the mRNA m(6) A methyltransferase complex. Wiley Interdiscip Rev RNA. e1618. [DOI] [PubMed] [Google Scholar]
- Geula S, et al. , 2015. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 347, 1002–6. [DOI] [PubMed] [Google Scholar]
- Grozhik AV, et al. , 2019. Antibody cross-reactivity accounts for widespread appearance of m(1)A in 5’UTRs. Nat Commun. 10, 5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, et al. , 2011. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 10, 2662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, 2019. Special Issue on Regulating the Central Dogma. Biochemistry. 58, 295–296. [DOI] [PubMed] [Google Scholar]
- Heck AM, et al. , 2020. YTHDF2 destabilizes m(6)A-modified neural-specific RNAs to restrain differentiation in induced pluripotent stem cells. RNA. 26, 739–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ME, et al. , 2013. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 16, 1042–8. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, et al. , 2013. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 288, 33292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PJ, et al. , 2019. The RNA-binding protein FMRP facilitates the nuclear export of N (6)-methyladenosine-containing mRNAs. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, et al. , 2019a. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature. 567, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, et al. , 2019b. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat Struct Mol Biol. 26, 380–388. [DOI] [PubMed] [Google Scholar]
- Hussain S, et al. , 2013. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 4, 255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, et al. , 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333, 1300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, et al. , 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 7, 885–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller L, et al. , 2011. The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer’s disease risk: a prospective cohort study. J Alzheimers Dis. 23, 461–9. [DOI] [PubMed] [Google Scholar]
- Khoddami V, et al. , 2019. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc Natl Acad Sci U S A. 116, 6784–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckles P, et al. , 2018. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 32, 415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CWQ, et al. , 2019. Atlas of quantitative single-base-resolution N(6)-methyl-adenine methylomes. Nat Commun. 10, 5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y, 2013. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 502, 472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi S, Shatkin AJ, 1975. Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc Natl Acad Sci U S A. 72, 2012–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand C, et al. , 2017. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res. 27, 1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lence T, et al. , 2016. m(6)A modulates neuronal functions and sex determination in Drosophila. Nature. 540, 242–247. [DOI] [PubMed] [Google Scholar]
- Li H, et al. , 2018a. FTO is involved in Alzheimer’s disease by targeting TSC1-mTOR-Tau signaling. Biochem Biophys Res Commun. 498, 234–239. [DOI] [PubMed] [Google Scholar]
- Li L, et al. , 2017a. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum Mol Genet. 26, 2398–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, et al. , 2018b. Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol. 19, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, et al. , 2017b. NSUN2-Mediated m5C Methylation and METTL3/METTL14-Mediated m6A Methylation Cooperatively Enhance p21 Translation. J Cell Biochem. 118, 2587–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. , 2016. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. 12, 311–6. [DOI] [PubMed] [Google Scholar]
- Li X, et al. , 2017c. Base-Resolution Mapping Reveals Distinct m(1)A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol Cell. 68, 993–1005 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, et al. , 2015. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 12, 767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, et al. , 2019. Landscape and Regulation of m(6)A and m(6)Am Methylome across Human and Mouse Tissues. Mol Cell. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. , 2014. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 10, 93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh I, et al. , 2020. The m(6)A epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci. 21, 36–51. [DOI] [PubMed] [Google Scholar]
- Lovejoy AF, et al. , 2014. Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One. 9, e110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, et al. , 2018. RNA m(6)A methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biol. 19, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, et al. , 2013. MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res. 41, D262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malovannaya A, et al. , 2011. Analysis of the human endogenous coregulator complexome. Cell. 145, 787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, et al. , 2017. Reversible methylation of m(6)Am in the 5’ cap controls mRNA stability. Nature. 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, et al. , 2019. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol. 15, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre ABR, et al. , 2020. Limits in the detection of m(6)A changes using MeRIP/m(6)A-seq. Sci Rep. 10, 6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel M, et al. , 2018. Methylation of Structured RNA by the m(6)A Writer METTL16 Is Essential for Mouse Embryonic Development. Mol Cell. 71, 986–1000 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, et al. , 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 149, 1635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H, 2011. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 70, 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, et al. , 2010. 5-methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res. 38, 1415–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, et al. , 1998. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ougland R, et al. , 2004. AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol Cell. 16, 107–16. [DOI] [PubMed] [Google Scholar]
- Patil DP, et al. , 2016. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 537, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, et al. , 2018. Reading m(6)A in the Transcriptome: m(6)A-Binding Proteins. Trends Cell Biol. 28, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton KE, et al. , 2017. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 169, 824–835 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, et al. , 2014. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard EM, et al. , 2019. Bi-allelic Variants in METTL5 Cause Autosomal-Recessive Intellectual Disability and Microcephaly. Am J Hum Genet. 105, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, et al. , 2015. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 16, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roost C, et al. , 2015. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc. 137, 2107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottman F, et al. , 1974. Sequences containing methylated nucleotides at the 5’ termini of messenger RNAs: possible implications for processing. Cell. 3, 197–9. [DOI] [PubMed] [Google Scholar]
- Roundtree IA, et al. , 2017. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safra M, et al. , 2017. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 551, 251–255. [DOI] [PubMed] [Google Scholar]
- Sajini AA, et al. , 2019. Loss of 5-methylcytosine alters the biogenesis of vault-derived small RNAs to coordinate epidermal differentiation. Nat Commun. 10, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, 2018. m(1)A within cytoplasmic mRNAs at single nucleotide resolution: a reconciled transcriptome-wide map. RNA. 24, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, et al. , 2014a. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, et al. , 2014b. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 8, 284–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendinc E, et al. , 2019. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol Cell. 75, 620–630 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, et al. , 2016. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum Genet. 135, 707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, et al. , 2019. PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum Genet. 138, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, et al. , 2018. m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature. 563, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, et al. , 2012. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 489, 150–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spenkuch F, et al. , 2014. Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol. 11, 1540–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires JE, et al. , 2012. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40, 5023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD, 2000. The language of covalent histone modifications. Nature. 403, 41–5. [DOI] [PubMed] [Google Scholar]
- Sun H, et al. , 2019. Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res. 29, 80–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan PP, et al. , 2017. Pseudouridine and N(6)-methyladenosine modifications weaken PUF protein/RNA interactions. RNA. 23, 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tran N, et al. , 2019. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 47, 7719–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, et al. , 2015. Panorama of ancient metazoan macromolecular complexes. Nature. 525, 339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CX, et al. , 2018a. METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol. 16, e2004880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yi C, 2019. Coupling Transcription and Translation via the Epitranscriptomic m(6)A Mark. Biochemistry. 58, 297–298. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. , 2014a. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 505, 117–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. , 2014b. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 16, 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. , 2018b. N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci. 21, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warda AS, et al. , 2017. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, et al. , 1975. Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell. 4, 379–86. [DOI] [PubMed] [Google Scholar]
- Wei CM, Moss B, 1977. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 16, 1672–6. [DOI] [PubMed] [Google Scholar]
- Wei J, et al. , 2018. Differential m(6)A, m(6)Am, and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell. 71, 973–985 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, et al. , 2018. Zc3h13 Regulates Nuclear RNA m(6)A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell. 69, 1028–1038 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng YL, et al. , 2018. Epitranscriptomic m(6)A Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron. 97, 313–325 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, et al. , 2016. Experience-Dependent Accumulation of N6-Methyladenosine in the Prefrontal Cortex Is Associated with Memory Processes in Mice. J Neurosci. 36, 6771–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, et al. , 2020. Programmable m(6)A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, et al. , 2019. m(6)A methylation controls pluripotency of porcine induced pluripotent stem cells by targeting SOCS3/JAK2/STAT3 pathway in a YTHDF1/YTHDF2-orchestrated manner. Cell Death Dis. 10, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, et al. , 2016. IDH1/2 Mutants Inhibit TET-Promoted Oxidation of RNA 5mC to 5hmC. PLoS One. 11, e0161261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, et al. , 2017. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 27, 606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. , 2019. RNA 5-Methylcytosine Facilitates the Maternal-to-Zygotic Transition by Preventing Maternal mRNA Decay. Mol Cell. 75, 1188–1202 e11. [DOI] [PubMed] [Google Scholar]
- Yoon KJ, et al. , 2017. Temporal Control of Mammalian Cortical Neurogenesis by m(6)A Methylation. Cell. 171, 877–889 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, et al. , 2018. Epigenetics and epitranscriptomics in temporal patterning of cortical neural progenitor competence. J Cell Biol. 217, 1901–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, Jaffrey SR, 2020. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell. 181, 1582–1595 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. , 2016a. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 113, E2047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. , 2016b. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 7, 64527–64542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, et al. , 2018. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. 27, 3936–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. , 2020. Dynamic landscape and evolution of m6A methylation in human. Nucleic Acids Res. 48, 6251–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, He C, 2015. Fate by RNA methylation: m6A steers stem cell pluripotency. Genome Biol. 16,43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, et al. , 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, et al. , 2008. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 20, 1278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, et al. , 2015. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 526, 591–4. [DOI] [PMC free article] [PubMed] [Google Scholar]