

Abstract
Sepsis and shock states impose mitochondrial stress, and in response, adaptive mechanisms such as fission, fusion and mitophagy are induced to eliminate damaged portions of or entire dysfunctional mitochondria. The mechanisms underlying these events are being elucidated; yet a direct link between loss of mitochondrial membrane potential ΔΨm and the initiation of fission, fusion and mitophagy remains to be well characterized. The direct association between the magnitude of the ΔΨm and the capacity for mitochondria to buffer Ca2+ renders Ca2+ uniquely suited as the signal engaging these mechanisms in circumstances of mitochondrial stress that lower the ΔΨm. Herein, we show that the calcium/calmodulin-dependent protein kinase (CaMK) IV mediates an adaptive slowing in oxidative respiration that minimizes oxidative stress in the kidneys of mice subjected to either cecal ligation and puncture (CLP) sepsis or endotoxemia. CaMKIV shifts the balance towards mitochondrial fission and away from fusion by 1) directly phosphorylating an activating Serine616 on the fission protein DRP1 and 2) reducing the expression of the fusion proteins Mfn1/2 and OPA-1. CaMKIV, through its function as a direct PINK1 kinase and regulator of Parkin expression, also enables mitophagy. These data support that CaMKIV serves as a keystone linking mitochondrial stress with the adaptive mechanisms of mitochondrial fission, fusion and mitophagy that mitigate oxidative stress in the kidneys of mice responding to sepsis.
Keywords: calcium, mitochondria, mitophagy, fission, fusion
Graphical Abstract
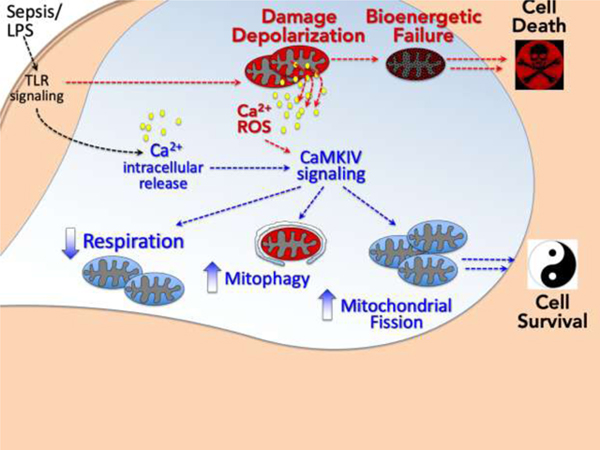
1. Introduction
During sepsis and other shock states, mitochondrial dysfunction occurs that is characterized by a loss in mitochondrial membrane potential ( ΔΨ), the elaboration of toxic reactive oxidant intermediates, and if progressive, opening of the mitochondrial permeability transition pore (MPTP), release of cytochrome C and apoptosis.[1–4] Not surprising, each cell is in possession of adaptive mechanisms that are engaged in such circumstances to prepare for and protect against this stress. These mechanisms include fission, fusion, and mitophagy; processes in which damaged portions of dysfunctional mitochondria are ‘pinched off’ or entire damaged mitochondria are sequestered for elimination.[5–8]
Autophagy is an ancient cytoplasmic process that enables cells to recycle portions of their cytoplasm to support vital functions during periods of stress (i.e., starvation).[9–13] Mitophagy, autophagy directed at the removal of damaged mitochondria, is considered of particular importance in protecting against organ injury.[5–7, 14–17] Prototypically, PINK1 accumulation at the outer mitochondrial membrane selectively recruits Parkin, which in turn promotes the selective degradation of dysfunctional mitochondria by mitophagy.[5, 6, 14, 18–21] Conditions that induce mitochondrial depolarization strongly induce this pathway, which is important for cell survival.[5, 18, 19, 22, 23] In addition, mitochondria undergo fission and fragmentation in response to energetic stress, which isolates dysfunctional from healthy functional mitochondrial components and targets the former for mitophagic elimination.[7, 24–26] This process is regulated, in part, by the coordinated balance of a set of fission (Drp-1, Fis1) and fusion (OPA-1, Mfn1/2) proteins.[7, 24–31] Yet, the mechanisms by which loss of the mitochondrial membrane potential ( ΔΨ) leads to selective targeting of dysfunctional mitochondria for fission and mitophagy remain to be fully characterized.
Mitochondria also influence cellular Ca2+ signaling.[32–36] By taking up and releasing Ca2+, mitochondria determine the spatiotemporal profile of cellular Ca2+ signals and the activity of Ca2+-regulated proteins. Mitochondria provide a high capacity, low affinity Ca2+-buffering system that sequesters Ca2+ predominantly through the negative electrochemical gradient generated by the ΔΨ.[32, 33, 37] Mitochondrial capacity to buffer Ca2+ is dependent upon the ΔΨ, and depolarization reduces buffering capacity, leading to a mitochondrial Ca2+ leak.[32–34, 37] Thus, Ca2+ may be an ideal candidate signal that as a “molecular flare” targets depolarized mitochondria for mitophagy.
The calcium/calmodulin-dependent protein kinases (CaMK),a family of serine/threonine kinases responsive to intracellular Ca2+ concentration [Ca2+], are integral to the immune response, mediating Ca2+-dependent Mφ function and septic inflammation.[38–44] We previously reported that CaMKIV regulates autophagy in the Mφ and kidneys of mice subjected to endotoxemia.[44] In that study loss of CaMKIV was associated with marked destruction of the mitochondrial architecture of renal tubular cells and heightened acute kidney injury. We now propose that this CaMKIV-dependent autophagy is also mitophagy. Specifically, we hypothesize that sepsis perturbs mitochondrial integrity and the ΔΨ, thereby generating a Ca2+ signal that activates CaMKIV to regulate the mechanisms of fission and mitophagy.
2. Results
2.1. CaMKIV regulates mitochondrial dynamics, function and oxidative stress in the kidney during CLP sepsis.
High-resolution respirometry showed that the rates of oxidative phosphorylation within the renal cortex tissue isolated from healthy control wild type (WT) was higher than control CaMKIV−/− mice (Figure 1A). After CLP sepsis, WT kidneys exhibited a significant slowing in oxidative phosphorylation at 6–12 hours, which correlated with significantly reduced tissue ATP content (Figs. 1A, 1B). By contrast, the kidneys of septic CaMKIV−/− mice preserved cellular oxidative metabolism, and tissue ATP content decreased to a lesser extent and trended toward being significantly higher than septic WT mice (Figs 1A, 1B). Mitochondrial complex II-V expression was increased in the kidneys of CaMKIV−/− mice relative to WT mice (Figure 1C), which correlated with a marked increase in oxidatively modified proteins (Figure 1D). There was little difference in superoxide dismutase (SOD) activity between WT and CaMKIV−/− renal tissue (Figure 1E). We measured core temperature (Tc) as a global parameter of animal metabolism and observed that the Tc of CaMKIV−/− mice increased in the first 12 hours after CLP, which contrasted with the slow hypothermia that developed in WT mice (Figure 1F). These data suggest CaMKIV mediates a slowing in oxidative metabolism and attenuation of oxidative stress in the kidney during CLP sepsis.
Figure 1. CaMKIV regulates mitochondrial dynamics, function and oxidative stress in the kidney during sepsis.

C57Bl/6J wild-type and CaMKIV−/− (KO) mice were subjected to cecal ligation and puncture; at the timepoints detailed, the mice were euthanized, and the kidneys harvested. A, total renal tissue isolated from wild-type (WT) and CaMKIV−/− (KO) mice was analyzed for oxidative phosphorylation using a O2k-FluoRespirometer. The following reagents were sequentially added and the specific activity interrogated/analyzed denoted in parentheses: 1mt=cell homogenate (ROX), 1D=ADP (residual oxygen consumption without substrate, ROX), 2M.1=anapleurotic malate (ROX), 3Oct=octanoyl carnitine (oxidative phosphorylation coupled to ATP production, OXPHOS), 3c=cytochrome c (OXPHOS), 4M2= malate (OXPHOS-complex I), 5P=pyruvate (OXPHOS-complex I), 6G=glutamate (OXPHOS-complex I), 7S=succinate (OXPHOS-complexes I and II), 8U=FCCP (Electron transport chain capacity, ET and complex I and II), 9Rot=rotenone (complex II), 10Ama=antimycin A (ROX). (n=5 total mice per group for 2 independent experiments combined) mean±sem; B, total renal tissue ATP concentration was quantified. WT, close circles. KO, open circles. bar=median (n=5–7 total mice per group for 2 independent experiments combined) bar=median; C, D, total renal protein lysate was analyzed by immunoblot for c, mitochondrial complexes I-V and d, oxidative modified proteins. (representative blot of n=2 independent experiments, n=4 mice total/group for 2 experiments combined) E, total renal protein lysate was analyzed for superoxide dismutase (SOD) activity. (n = 2 total mice per group) bar=median; F, Core temperature of WT and KO mice during CLP. line=mean, shaded region=95% confidence intervals (n = 4 total mice per group for 2 independent experiments combined). Data analyzed by Wilcoxon rank sum test.
2.2. CaMKIV regulates PINK1/Parkin-dependent mitophagy during sepsis.
One mechanism regulating a programmed loss in mitochondrial complex mass and a slowing of oxidative metabolism that curtails the generation of reactive oxygen species (ROS) is mitophagy.[6, 17] Mitophagy has been shown to be particularly protective in mitigating cellular injury in models of acute kidney injury (AKI).[6, 17] We observed that the kidneys of WT mice had elevated expression of active p-Ser65-Parkin during CLP, which was lesser in extent in the renal tissues of septic CaMKIV−/− mice (Figure 2A). The canonical pathway for mitophagy involves Parkin recruitment to the mitochondrion.[5, 6, 14, 19, 20, 22, 23] The kidneys of septic WT mice had increased mitochondrial expression of Parkin, which was lesser in extent in CaMKIV−/− mice (Figure 2B). A similar pattern of elevated tissue p-Ser65-Parkin and mitochondrial Parkin was observed in WT mice subjected to endotoxemia; this occurred to a lesser extent in endotoxemic CaMKIV−/− mice (Figs. 2C, 2D).
Figure 2. CaMKIV regulates PINK1/Parkin-dependent mitophagy during sepsis.

A-D, C57Bl/6J wild-type and CaMKIV−/− (KO) mice were subjected to cecal ligation and puncture (CLP) or endotoxemia (5mg/kg); at the timepoints detailed, the mice were euthanized, and the kidneys harvested. A, total kidney protein lysate from 6 and 12 hours of CLP was analyzed by immunoblot. Densitometry of PINK1, p-Parkin and Parkin: solid bars = C57Bl/6J, open bars=CaMKIV-/−. (representative blot of n=3 independent experiments. n= 4 (sham) 6–11 (CLP) mice total/group for both experiments combined). B, total mitochondrial protein lysate from 6 hours of CLP was analyzed by immunoblot. Densitometry of PINK1 and Parkin: solid bars = C57Bl/6J, open bars=CaMKIV-/−. (representative blot of n=3 independent experiments. n=3–5 (sham) 9–12 (CLP) mice total/group for both experiments combined). C, total renal protein lysate from 6, 18 and 48 hours of endotoxemia was analyzed by immunoblot. Densitometry of PINK1, p-Parkin and Parkin: solid bars = C57Bl/6J, open bars=CaMKIV-/−. (representative blot of n=3 independent experiments; n=2–4 (control) 12–14 (endotoxemia) mice total/group for both experiments combined). D, total mitochondrial protein lysate from 3 hours of endotoxemia was analyzed by immunoblot. Densitometry of PINK1 and Parkin: solid bars = C57Bl/6J, open bars=CaMKIV-/−. (representative blot of n=2 independent experiments. n=4 (control) 7–8 (endotoxemia) mice total/group for both experiments combined). Data analyzed by Wilcoxon rank sum test.
The recruitment of Parkin to the mitochondrion is regulated by the upstream mitochondrial kinase PINK1, which is activated by serine phosphorylation.[5, 19–21, 45] Full length PINK1 selectively accumulates on depolarized mitochondria, wherein it recruits Parkin.[5, 21–23] We observed heightened and prolonged expression of full-length PINK1 in the renal tissue of CaMKIV−/− mice subjected to CLP and endotoxemia (Figs. 2A, 2C), relative to the kidneys of WT mice. This elevated PINK1 in the renal tissue of CaMKIV−/− mice correlated with a low expression of PINK1 within the mitochondrial fraction (Figs 2B, 2D). By contrast, the lower levels of tissue PINK1 in WT mice correlated with an elevation in mitochondrial PINK1 that was significantly higher than CaMKIV mice. These data suggest that loss of CaMKIV is associated with a defect in expression of PINK1 at the mitochondrion.
We next over-expressed a full-length FLAG-tagged CaMKIV (CaMKIV-Flag) in human embryonic kidney (HEK293) cells and observed that PINK1 coimmunoprecipitated CaMKIV (Figure 3A). Transfection of HEK293 cells with the constitutively active CaMKIV-dCT, but not the kinase negative mutant dCT-K75E, increased p-Ser-PINK1 (Figure 3B). And finally, using an in vitro kinase assay, we observed that CaMKIV directly phosphorylates PINK1. (Figure 3C). These data suggest that CaMKIV functions as a PINK1 kinase and regulates PINK1/Parkin-dependent mitophagy in the kidney during sepsis.
Figure 3. CaMKIV is a PINK1 kinase.

A, HEK293 cells were transfected with lipofectamine, blank or full length CaMKIV-Flag for 24 hours. Total cell lysate was immunoprecipitated with anti-Flag antibody. Total cell lysate and immunoprecipitated protein were analyzed by immunoblot (representative blot of n=2 independent experiments). B, HEK293 cells were transfected with lipofectamine, a kinase-inactive (dCT-K75E) or a constitutively active (dCT-CaMKIV) CaMKIV mutant for 24 hours. Total cell lysate was isolated immunoprecipitated with anti-PINK1 antibody, and immunoprecipitated protein was analyzed by immunoblot (representative blot of n=2 independent experiments. C, 1μg PINK1 (molecular weight of recombinant protein, 37.9KD) was incubated in the presence or absence of CaMKIV (25 ng, molecular weight of recombinant protein, 70KD) for 10 min at 30°C with the following additions: 10mM MgCl2, 0.2 mM ATP, 1 mM CaCl2 and 1uM CaM. Reactions were terminated by boiling in SDS-2-ME dissociation solution and analyzed by immunoblot. (n=2 independent experiments).
2.3. CaMKIV regulates a transition towards fission and away from fusion.
Mitochondria undergo fission and fragmentation in response to energetic stress; the purported purpose is to isolate dysfunctional from healthy functional mitochondrial components and to target the former for mitophagic elimination.[7, 24–26] Electron microscopy of kidneys isolated from healthy WT and CaMKMIV−/− mice revealed mitochondria of similar morphology at baseline, though the mitochondrial length of CaMIV−/− renal tubular cells (RTC) of the kidney was modestly longer than that of WT renal tubular cells (Figs. 4A, 4B). When mice were subjected to CLP sepsis, WT mitochondria significantly shortened and assumed a spherical morphology relative to baseline: median length 1.24μm vs 0.95μm, p<0.001 (Figs. 4A, 4B). In contrast the mitochondrial architecture of CaMKIV−/− kidneys remained elongated and significantly longer than that of the kidneys of septic WT mice: median length 1.26μm vs. 0.95μm, p<0.001. The proportion of mitochondria exceeding the 90th percentile of 2μm was significantly larger in septic CaMKIV−/− mice vs. WT mice: 12.1% vs. 3.5%, p<0.001. We also noted a paucity of autolysosomes in the kidneys of septic CaMKIV−/− mice relative to WT mice (Figure 4A, right upper and right lower panels), which is in accordance with our prior studies.[44]
Figure 4. CaMKIV regulates a shift towards increased fission and reduced fusion.

C57Bl/6J wild-type and CaMKIV−/− (KO) mice were subjected to cecal ligation and puncture for 8 hours, at which time the mice were euthanized, the kidneys harvested and a) the renal cortex was imaged by electron microscopy, and b) mean mitochondrial length was determined per hpf (bar=2um) using the NIH ImageJ program. solid circles = C57Bl/6J, open circles=CaMKIV-/−. (n=4 renal tubular cells per mouse, n=4 mice per group) bar=median. Data analyzed by Wilcoxon rank sum test.
The balance between mitochondrial fission and fusion is regulated by the coordinated interplay of the fission proteins Drp1 and Fis1 and the fusion proteins OPA1 and Mfn1/2.[7, 24–26] Phosphorylation of Drp1 at Ser616 (p-Ser616-Drp1) promotes fission, whereas Ser637 phosphorylation induces detachment of Drp1 from mitochondria and inhibits fission.[46–52] We observed an elevation in p-Ser616-Drp1 in the kidneys of WT mice exposed to CLP sepsis, whereas CaMKIV−/− mice exhibited low renal p-Ser616-Drp1 (Figure 4A). Furthermore, CaMKIV−/− kidneys possessed elevated OPA-1 expression relative to WT kidneys (Figure 5A). We next evaluated p-Ser616-Drp1 and OPA-1 expression in the mitochondrial fraction of kidneys isolated from endotoxemic mice, 3 hours after LPS administration. We observed that WT kidneys had elevated p-Ser616-Drp1, which was lesser in extent in CaMKIV−/− renal tissues (Figure 5B). CaMKIV−/− mitochondria also exhibited elevated OPA-1 expression.
Figure 5. CaMKIV regulates a shift towards increased fission and reduced fusion during endotoxemia and CLP in vivo.
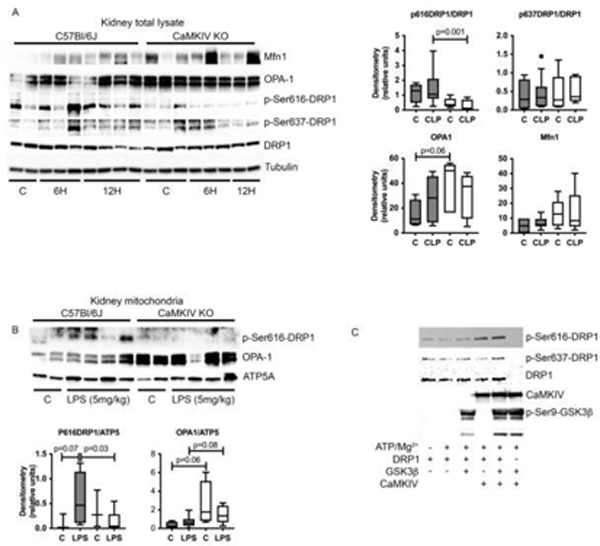
A-D, C57Bl/6J wild-type and CaMKIV−/− (KO) mice were subjected to endotoxemia (5mg/kg) or cecal ligation and puncture; at the timepoints detailed, the mice were euthanized, and the kidneys harvested. A, total kidney protein lysate from 6 or 12 hours of CLP was analyzed by immunoblot. Densitometry of p-Ser616-DRP1, p-Ser637-DRP1, OPA1, and Mfn1: solid bars = C57Bl/6J, open bars=CaMKIV−/−. (representative blot of n=3 independent experiments; n= 4 (sham) 9–10 (CLP) mice total/group for both experiments combined). B, total mitochondrial protein lysate from 3 hours of endotoxemia was analyzed by immunoblot (representative blot of n=2 independent experiments. Densitometry of OPA1 and p-Ser616-DRP1: solid bars = C57Bl/6J, open bars=CaMKIV−/−. (n= 3–4 (control) 7–8 (endotoxemia) mice total/group for both experiments combined). C, 1μg DRP1 (molecular weight of recombinant protein, 104KD) was incubated in the presence or absence of CaMKIV (25 ng, molecular weight of recombinant protein, 70KD) and GSK3β (25 ng, molecular weight of recombinant protein, 38KD) for 10 min at 30°C with the following additions: 10mM MgCl2, 0.2 mM ATP, 1 mM CaCl2 and 1uM CaM. Reactions were terminated by boiling in SDS-2-ME dissociation solution and analyzed by immunoblot. (n=2 independent experiments). Data analyzed by Wilcoxon rank sum test.
The serine phosphorylation of Drp1 at Ser616 promotes Drp1-mediated fission.[46–48] Using an in vitro kinase assay we observed that CaMKIV directly phosphorylates Drp1 on Ser616, suggesting that CaMKIV can function as a DRP1 kinase (Figure 5C). These data suggest that CaMKIV directly mediates p-Ser616-Drp1 and orchestrates a shift towards fission and away from fusion.
3. Discussion
Mitochondrial homeostasis, above and beyond ensuring a source of energy, is integral to many cellular physiologic processes, including the regulation of intracellular Ca2+. Indeed, mitochondrial Ca2+ homeostasis links mitochondrial function to many vital cellular processes including cell bioenergetics, cell death pathways, and the three-dimensionality of Ca2+ signaling itself. It should not be surprising, then, that a host of mechanisms have evolved that function in concert to preserve and restore mitochondrial health; these include mitophagy, fission, and fusion.[5, 6, 8, 10, 12, 14, 17, 24, 53, 54] The relationship between mitochondrial function and mitochondrial Ca2+ homeostasis, render Ca2+ an ideal medium to signal the presence of mitochondrial dysfunction and engage these mechanisms.[32–35] Here we show that the calcium-dependent protein CaMKIV regulates a multitude of these adaptive mechanisms in the response to sepsis. CaMKIV affects a slowing in oxidative metabolism that it may achieve by its capacity as a direct PINK1-kinase to induce Parkin-dependent mitophagy. Concomitantly, CaMKIV induces active p-Ser616-Drp-1 and thereby shifts the balance away from fusion and towards fission, the latter of which reduces oxidative phosphorylation and the resultant elaboration of reactive oxygen species and toxic protein oxidation. Collectively with our prior data, an elegant paradigm emerges in which the CaMKs responding to a mitochondrial Ca2+ ‘flare’, regulate the machinery of mitophagy, mitochondrial fission/fusion, and oxidative metabolism to ensure cell survival.
Mitochondria provide a high capacity, low affinity Ca2+ buffering system. Mitochondrial Ca2+ uptake occurs through the MCU and is driven by the negative electrochemical gradient of the mitochondrial membrane potential (ΔΨ), which is generated by the proton pumping of the electron transport chain.[32–34] The intimate relationship between oxidative phosphorylation, ΔΨ, and mitochondrial Ca2+ handling provides a teleological rationale for the dependence of mitochondrial fission, fusion, and mitophagy on Ca2+ signaling.[33, 34, 37] We recently reported that mitochondrial depolarization and loss in ΔΨ causes an intracellular Ca2+ signal that mediates CaMKI-dependent mitophagy.[55] The collective data now support a paradigm in which the mitochondrion itself participates in maintaining its health, and furthermore, a plausible mechanism by which the machinery of mitophagy, fission, and fusion are selectively targeted to dysfunctional mitochondria and spare those neighboring that are healthy. Of note, members of the CaMKs have also been shown to be redox sensitive, exhibiting increased activity in response to oxidant stress, and functioning at the mitochondrion to mediate mitochondrial ROS generation.[56–60] Further studies are needed to characterize the individual contributions of Ca2+ and ROS in CaMKIV activation and the mechanistic link with fission, fusion and mitophagy.
A ‘sibling’ relationship between CaMKI and CaMKIV has been suggested by experimental data highlighting their ability to work in tandem to coordinate certain cellular processes, such as the nucleocytoplasmic shuttling and subsequent cytoplasmic release of HMGB1.[39, 42] We have shown that both are involve in autophagy[43, 44], and these more recent data suggest that a similar partnership exists in the context of mitophagy. In response to mitochondrial depolarization (i.e., LPS, CCCP), CaMKI functions as a direct PINK1 kinase and localizes to the mitochondrion to recruit Parkin.[55] CaMKIV, though sharing a similar function as a PINK1 kinase, possesses distinct roles in the events of mitophagy. CaMKIV was not observed to translocate to the mitochondrion (data not shown) and our data suggests that it may serve a more integral role for Parkin expression. Many of the pleiotropic functions of Parkin are dependent upon its E3 ubiquitin ligase activity. In fact, it promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin (Mfn), a mechanism we hypothesize underlies the elevated Mfn expression observed in CaMKIV−/− mice.[61–63] The specific mechanisms by which CaMKIV regulates Parkin expression remain to be identified. We previously described that CaMKIV, through an inhibitory serine phosphorylation of GSK-3β and inhibition of FBXW7 recruitment, prevents ubiquitin proteosomal degradation of mammalian target of rapamycin (mTOR) and thereby augments autophagy in the kidney.[44] Alternatively, CaMKIV possesses a nuclear localization sequence, and fundamental roles in gene expression have also been well described.[64, 65]
The ramifications of CaMKIV-dependent mitophagy on organ function and physiology, particularly as it relates to developing novel therapeutics was not a focus of these experiments. The current evidence creates a paradigm in which the early loss of cell function due to mitophagy and dampened mitochondrial function is adaptive and protective.[1, 2, 66] However, if prolonged, irreversible cellular injury and organ failure ensue and contribute to host mortality. Thus, inhibiting CaMKIV, either biochemically or through siRNA, may preserve organ function, however later lead to irreversible cellular and organ injury through a loss of the protective mechanisms of mitophagy and mitochondrial fission. Recovery must occur, however. And thus, persistent CaMKIV activation, a potential consequence of the high prevalence of perturbed Ca2+ regulation during sepsis, may be equally deleterious in suppressing the cellular phenotypes supporting organ function and survival of the organism.
4. Conclusion
Sepsis induces the activation of CaMKIV in the kidney. Active CaMKIV functions as a PINK1 kinase and regulates the PINK1/Parkin pathway, in particular Parkin expression. CaMKIV further functions as a DRP1 kinase to induce DRP1-mediated mitochondrial fission. These mechanisms are operant in vivo during endotoxemia and sepsis. The observation that CaMKIV regulates these mechanisms in non-immune cells underscores the need to further study these mechanisms in the context of tissue-specific injury and dysfunction in response to septic insult. Furthermore, additional studies are needed to translate these cellular events to organ physiology and the host response, in an attempt to identify the clinical utility of CaMKIV modulation during inflammatory states.
5. Methods
Reagents and antibodies.
Ultra Pure LPS (Escherichia coli 0111:B4) was obtained from List Biologicals (Campbell, CA, USA). Antibodies for CaMKIV, Parkin, Mfn1, phospho-serine, ATP5a, and actin were obtained from Abcam (Cambridge, MA, USA). Antibodies for OPA-1, Drp-1, p-Ser616-DRP1, p-Ser637-DRP1, Parkin and tubulin were obtained from Cell Signaling Technology (Danvers, MA, USA). p-Ser65-Parkin was obtained from Biorbyte Ltd (St. Louis MO, USA). Rabbit anti-PINK1 antibody was purchased from Novus Biologicals (Littleton, CO, USA). Anti-Flag antibody was purchased from Sigma Aldrich (St. Louis, MO, USA). Total mitochondrial complex (I-V) expression was analyzed using the total OXPHOS rodent antibody cocktail from Abcam. Total oxidatively modified proteins was analyzed using the OxyBlot Protein Oxidation Detection Kit from Millipore (Burlington, MA, USA).
Animal experimentation
We performed all animal experiments in accordance with the National Institutes of Health guidelines under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh. We purchased 8–12 week old C57BL/6J and B6.129X1-Camk4tm1Tch/J (CaMKIV−/−) male mice from the Jackson Laboratory (Bar Harbor, ME).[39, 44, 67, 68] The CaMKIV−/− strain is derived from a C57BL/6J background; these mice are viable, fertile, normal in size and do not display any gross physical or behavioral abnormalities. We randomly grouped 8–12 week old male C57BL/6J mice and assigned them to a specific experiment. At various time points, mice were euthanized, blood was isolated by cardiac puncture, and the kidneys were harvested. Investigators who treated animals knew the treatment groups and collected samples, which were then analyzed by other investigators blinded to the specific treatment.
Cecal ligation and puncture (CLP).
Mice underwent a 21-gauge double puncture CLP model as previously described.[41, 42, 55] After CLP, all mice received a subcutaneous injection of warmed 0.9% normal saline (30 mL/kg) and were injected intramuscularly with an analgesic (Buprenorphine, 0.10 mg/Kg) and every 6 hrs thereafter. Antibiotics were not administered. For one cohort of mice, an HD-X11 wireless biotelemetry device (Data Sciences International, St. Paul, MN) capable of continuously measuring core temperature was implanted at the time of CLP into the peritoneal cavity as previously published.[69–71] Continuous core temperature was used as a parameter of whole-animal metabolism.
Endotoxemia.
Mice were anesthetized using a mixture of isoflurane and oxygen bled in at 3.5L/min. Ultra Pure LPS (Escherichia coli 0111:B4) from LIST Biologicals (Campbell, CA, USA) was dissolved in DNase/RNase free sterile normal saline and injected intraperitoneally (5 mg/kg).[44, 72]
Cell.
The human embryonic kidney (HEK) 293 cell lines (American Type Culture Collection, Rockville, MD, USA) were grown in DMEM (BioWhittaker, Walkersville, MD, USA) supplemented with 10% fetal calf serum (Sigma, San Diego, CA), 50U/mL penicillin, and 50μg/mL streptomycin (Cellgro Mediatech Inc., Kansas City, MO, USA).
Plasmid construction and transfection.
A plasmid encoding a full-length CaMKIV-FLAG was the generous gift of Dr. Anthony Means. Plasmids encoding a constitutively active CaMKIV (CaMKIV-dCT) or a kinase-inactive CaMKIV-dCTK75E mutant were the generous gifts of Dr. Douglas Black (Yale University, New Haven, CT).[39, 44] For transient transfection, HEK cells were seeded in a 10 cm dish for overnight to reach the 70~80% confluence, and were then transfected with CaMKIV-Flag, CaMKIV-dCT, or CaMKIV-dCTK75E separately using the Lipofectamine 3000 reagent according to the manufacturer instructions (Life Technologies, Carlsbad, CA).
Cellular protein extraction.
Total cellular protein was extracted at 4° C in 1000 μl of lysis buffer as previously described.[42–44, 55] Protein concentration was determined using a bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL, USA).
Mitochondrial Isolation.
Mitochondria were isolated from fresh mouse kidney samples by differential centrifugation, using iced-cold isolation buffer IBI (225mM mannitol, 75mM sucrose, 10mM HEPES, 1 mM EGTA and 0.1% (w/v) fatty acid- free bovine serum albumin (BSA) (pH 7.4) with both phosphatase (Cocktail set II and IV; Calbiochem, USA) and protease (Sigma-Aldrich, USA) inhibitors added. Briefly, the kidneys were cut into small pieces, homogenized, and then centrifuged at 1300 × g for 10 min twice. The supernatant was transferred to a new tube and centrifuged at 10,000 × g for 10min. The crude mitochondrial pellets were washed twice with IBII buffer and then lysed with 2% CHAPS in 1x Cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) with PMSF. The mitochondrial protein concentration was determined using the Pierce BCA Protein Assay kit (Pierce Biotechnology).
Immunoprecipitation.
500μg HEK293 cell lysate was incubated with 30μl Anti-FLAG M2 Magnetic Beads at 4° C overnight. After three times wash with lysis buffer, Flag fused CaMKIV was eluted with 2x SDS-PAGE loading buffer without 2-ME through boiling for 3 minutes. For PINK1, 10 μl of antibody was added to 1 mg of isolated cellular protein within lysis buffer and incubated at 4° C overnight.[43, 55] Thirty μl of 50% slurry of pre-washed Protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were added to each sample and incubated for 2 h at 4° C. The samples were spun at 14,000 rpm and washed 4 times in lysis buffer. Samples were then resuspended in 30 μl of lysis buffer for analysis.
Immunoblot.
Total cellular lysate, mitochondrial lysate, or immunoprecipitated protein was electrophoresed in a 10% SDS-PAGE gel (based on protein of interest) and transferred to a Hybond-enhanced chemiluminescence nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ, USA) as previously performed.[42–44, 55] Densitometry was performed by the NIH Image J64 program (National Institutes of Health, Bethesda, MD, USA).[42, 43, 73]
Electron microscopy.
Transmission electron microscopy (TEM) of kidney samples was performed as previously published.[44] Samples were analyzed using a JEM 1011CX electron microscope 12,000X, bar=2μM (JEOL, Peabody, MA). Images were acquired digitally from a randomly selected pool of 10 to 15 fields for each experimental condition.[74] Sample processing occurred at the Center for Biological Imaging at the University of Pittsburgh. Mitochondrial morphology and length were analyzed using ImageJ (National Institutes of Health, Bethesda, MD, USA). For each condition, at least 6 micrographs were read per sample.[55, 75]
In vitro kinase assay.
An in vitro kinase assay as previous published.[43, 55] Briefly, 1μg PINK1 (ProSpec, East Brunswick, NJ, USA) or DRP-1 protein (Novus Biologicals, Centennial, CO, USA) was incubated in the presence or absence of 25ng CaMKIV (Abcam, Cambridge, MA, USA) for 10 min at 30°C with the following additions: 10mM MgCl2, 0.2 mM ATP, 1mM CaCl2 and 1μM CaM in 50μl reaction system. Reactions were terminated by boiling in SDS-2-ME dissociation solution and analyzed by immunoblot.
Measurement of respiration.
Oxidative phosphorylation of whole kidney tissue homogenate was performed using the O2k-FluoRespirometer (Oroboros Instrument Corp, Innsbruck, Australia). Freshly isolated murine kidney cortex was weighed and homogenized using the PBI Shredder homogenization tool (Presure BioSciences Inc, South Easton, MA, USA) in a mitochondrial respiration buffer (MiR05: 0.5 mM EGTA, 3 mM MgCl2, 60 mM lactobionic acid, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM D-Sucrose, and 1 g/L BSA). Resultant homogenates were adjusted to 2 mg wet weight tissue/mL in MiR05. Following machine equilibration, 2 mL of 2 mg/mL tissue homogenate were instilled into the respiration chambers. The following reagents were sequentially added to analyze a particular aspect or stage of mitochondrial function, which is denoted in parentheses: 1mt=cell homogenate (residual oxygen consumption, ROX), 1D=ADP (ROX); 2M.1=anapleurotic dose malate to ensure Krebs cycle function (ROX); 3Oct=octanoyl carnitine to drive fatty acid oxidation (oxidative phosphorylation coupled to ATP production, OXPHOS-Fatty Acid); 3c=cytochrome c to ensure appropriate cytochrome c levels to facilitate complex II-IV function (OXPHOS-Fatty Acid), 4M2=substrate dose malate (OXPHOS-Fatty Acid + NADH/complex I), 5P=pyruvate (OXPHOS-Fatty Acid + NADH/complex I), 6G=glutamate (OXPHOS-Fatty Acid + NADH/complex I), 7S=succinate (OXPHOS-Fatty Acid + NADH/complex I + succinate/complex II), 8U=FCCP for maximal oxygen consumption (electron transport chain capacity (ET)-not coupled to ATP production, ET-Fatty Acid + NADH/complex I + succinate/complex II + glycerol-3-phosophate dehydrogenase), 9Rot=rotenone to inhibit complex I (ET-succinate/complex II + glycerol-3-phosophate dehydrogenase), 10Ama=antimycin A inhibits complex III (ROX). Steady-state oxygen consumption rates were measured following each substrate according to a pre-established substrate-uncoupler-inhibitor titration (SUIT) protocol.[76]
ATP quantification.
ATP determination kit (Invitrogen, Eugene, OR, USA) was used according to the manufacturer’s instructions. Luminescence was measured using the Soft-MaxPro ATPase Assay program on a Synergy Mx (Biotek) plate reader.[55, 77]
Superoxide dismutase (SOD) activity.
SOD activity was quantified using the Superoxide Dismutase Activity kit from Abcam.
Densitometry.
Densitometry was performed using NIH ImageJ64.
Statistical analysis.
Densitometric data were analyzed using a non-parametric Wilcoxan rank sum test, as the assumption of normality was not met, and sample size was insufficient to invoke the central limit theorem and the use of a parametric analysis. Data are presented as medians with interquartile ranges (IQR). Statistical analyses were performed using Stata 12SE software (College Station, TX, USA). A p<0.05 was considered statistically significant, and a 0.05 ≥ p < 0.10 was considered a statistical trend.
Highlights.
A leading cause of death from sepsis is multiple organ dysfunction (MOD); and, recently, an important role for bioenergetic dysfunction in the development of MODS has been identified.
During sepsis, calcium-calmodulin-dependent protein kinase IV (CaMKIV) links mitochondrial stress with the adaptive mechanisms of mitochondrial fission, fusion and mitophagy.
CaMKIV affects a slowing in oxidative metabolism that is achieved in part through its function as a direct PINK1-kinase and the induction of Parkin-dependent mitophagy.
CaMKIV mediates phosphorylation of an activation Serine616 on the fission protein Drp-1, and thereby shifts the balance away from fusion and towards fission, which correlates with reduced toxic protein oxidation.
Acknowledgements
This work supported by National Institutes of Health grants [R01 GM082852, R01 GM116929]
Disclosure
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome
Abbreviations
- Ca2+
calcium
- CaMK
Calcium/calmodulin-dependent protein kinase
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazine
- CLP
cecal ligation and puncture
- DRP1
Dynamin-related protein 1
- LPS
lipopolysaccharide
- Mφ
macrophage
- MFN
Mitofusin
- MMP (ΔΨ)
mitochondrial membrane potential
- OPA1
Mitochondrial dynamin like GTPase
- PINK1
PTEN-induced kinase 1
Footnotes
Disclosure
The authors do not have any financial or competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Singer M, Brealey D, Mitochondrial dysfunction in sepsis, Biochem Soc Symp, 66 (1999) 149–166. [DOI] [PubMed] [Google Scholar]
- [2].Singer M, Mitochondrial function in sepsis: acute phase versus multiple organ failure, Crit Care Med, 35 (2007) S441–448. [DOI] [PubMed] [Google Scholar]
- [3].Whelan SP, Carchman EH, Kautza B, Nassour I, Mollen K, Escobar D, Gomez H, Rosengart MA, Shiva S, Zuckerbraun BS, Polymicrobial sepsis is associated with decreased hepatic oxidative phosphorylation and an altered metabolic profile, J Surg Res, 186 (2014) 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Crouser ED, Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome, Mitochondrion, 4 (2004) 729–741. [DOI] [PubMed] [Google Scholar]
- [5].Eiyama A, Okamoto K, PINK1/Parkin-mediated mitophagy in mammalian cells, Curr Opin Cell Biol, 33 (2015) 95–101. [DOI] [PubMed] [Google Scholar]
- [6].Ding WX, Yin XM, Mitophagy: mechanisms, pathophysiological roles, and analysis, Biol Chem, 393 (2012) 547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Youle RJ, van der Bliek AM, Mitochondrial fission, fusion, and stress, Science, 337 (2012) 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nunnari J, Suomalainen A, Mitochondria: in sickness and in health, Cell, 148 (2012) 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].He C, Klionsky DJ, Regulation mechanisms and signaling pathways of autophagy, Annu Rev Genet, 43 (2009) 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang J, Klionsky DJ, Autophagy and human disease, Cell Cycle, 6 (2007) 1837–1849. [DOI] [PubMed] [Google Scholar]
- [11].Huang WP, Klionsky DJ, Autophagy in yeast: a review of the molecular machinery, Cell Struct Funct, 27 (2002) 409–420. [DOI] [PubMed] [Google Scholar]
- [12].Klionsky DJ, Autophagy, Curr Biol, 15 (2005) R282–283. [DOI] [PubMed] [Google Scholar]
- [13].Klionsky DJ, Emr SD, Autophagy as a regulated pathway of cellular degradation, Science, 290 (2000) 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Youle RJ, Narendra DP, Mechanisms of mitophagy, Nat Rev Mol Cell Biol, 12 (2011) 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Isaka Y, Kimura T, Takabatake Y, The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells, Autophagy, 7 (2011) 1085–1087. [DOI] [PubMed] [Google Scholar]
- [16].Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y, Autophagy protects the proximal tubule from degeneration and acute ischemic injury, Journal of the American Society of Nephrology : JASN, 22 (2011) 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tang C, He L, Liu J, Dong Z, Mitophagy: Basic Mechanism and Potential Role in Kidney Diseases, Kidney Dis (Basel), 1 (2015) 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK, ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy, Autophagy, 8 (2012) 1462–1476. [DOI] [PubMed] [Google Scholar]
- [19].Springer W, Kahle PJ, Regulation of PINK1-Parkin-mediated mitophagy, Autophagy, 7 (2011) 266–278. [DOI] [PubMed] [Google Scholar]
- [20].Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J, PINK1 controls mitochondrial localization of Parkin through direct phosphorylation, Biochem Biophys Res Commun, 377 (2008) 975–980. [DOI] [PubMed] [Google Scholar]
- [21].Durcan TM, Fon EA, The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications, Genes Dev, 29 (2015) 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Narendra D, Tanaka A, Suen DF, Youle RJ, Parkin is recruited selectively to impaired mitochondria and promotes their autophagy, J Cell Biol, 183 (2008) 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ, PINK1 is selectively stabilized on impaired mitochondria to activate Parkin, PLoS Biol, 8 (2010) e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chan DC, Fusion and fission: interlinked processes critical for mitochondrial health, Annu Rev Genet, 46 (2012) 265–287. [DOI] [PubMed] [Google Scholar]
- [25].van der Bliek AM, Shen Q, Kawajiri S, Mechanisms of mitochondrial fission and fusion, Cold Spring Harb Perspect Biol, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McCoy MK, Cookson MR, Mitochondrial quality control and dynamics in Parkinson’s disease, Antioxid Redox Signal, 16 (2012) 869–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shi SY, Lu SY, Sivasubramaniyam T, Revelo XS, Cai EP, Luk CT, Schroer SA, Patel P, Kim RH, Bombardier E, Quadrilatero J, Tupling AR, Mak TW, Winer DA, Woo M, DJ-1 links muscle ROS production with metabolic reprogramming and systemic energy homeostasis in mice, Nat Commun, 6 (2015) 7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shimizu Y, Lambert JP, Nicholson CK, Kim JJ, Wolfson DW, Cho HC, Husain A, Naqvi N, Chin LS, Li L, Calvert JW, DJ-1 protects the heart against ischemia-reperfusion injury by regulating mitochondrial fission, J Mol Cell Cardiol, 97 (2016) 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Takahashi-Niki K, Ganaha Y, Niki T, Nakagawa S, Kato-Ose I, Iguchi-Ariga SM, Ariga H, DJ-1 activates SIRT1 through its direct binding to SIRT1, Biochem Biophys Res Commun, 474 (2016) 131–136. [DOI] [PubMed] [Google Scholar]
- [30].Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR, DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy, Hum Mol Genet, 20 (2011) 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McCoy MK, Cookson MR, DJ-1 regulation of mitochondrial function and autophagy through oxidative stress, Autophagy, 7 (2011) 531–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Santo-Domingo J, Demaurex N, Calcium uptake mechanisms of mitochondria, Biochimica et biophysica acta, 1797 (2010) 907–912. [DOI] [PubMed] [Google Scholar]
- [33].Rizzuto R, De Stefani D, Raffaello A, Mammucari C, Mitochondria as sensors and regulators of calcium signalling, Nat Rev Mol Cell Biol, 13 (2012) 566–578. [DOI] [PubMed] [Google Scholar]
- [34].Walsh C, Barrow S, Voronina S, Chvanov M, Petersen OH, Tepikin A, Modulation of calcium signalling by mitochondria, Biochim Biophys Acta, 1787 (2009) 1374–1382. [DOI] [PubMed] [Google Scholar]
- [35].Kaftan EJ, Xu T, Abercrombie RF, Hille B, Mitochondria shape hormonally induced cytoplasmic calcium oscillations and modulate exocytosis, The Journal of biological chemistry, 275 (2000) 25465–25470. [DOI] [PubMed] [Google Scholar]
- [36].Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK, Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter, Nature, 476 (2011) 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Luo Y, Bond JD, Ingram VM, Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases, Proc Natl Acad Sci U S A, 94 (1997) 9705–9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rosengart MR, Arbabi S, Garcia I, Maier RV, Interactions of calcium/calmodulin-dependent protein kinases (CaMK) and extracellular-regulated kinase (ERK) in monocyte adherence and TNFalpha production, Shock, 13 (2000) 183–189. [DOI] [PubMed] [Google Scholar]
- [39].Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, Means AR, Billiar TR, Angus DC, Rosengart MR, Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages, Journal of immunology, 181 (2008) 5015–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Means AR, The Year in Basic Science: calmodulin kinase cascades, Mol Endocrinol, 22 (2008) 2759–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Collage RD, Howell GM, Zhang X, Stripay JL, Lee JS, Angus DC, rosengart MR, Calcium supplementation during sepsis exacerbates organ failure and mortality via calcium/calmodulin-dependent protein kinase kinase (CaMKK) signaling, Crit Care Med, IN PRESS (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang X, Guo L, Collage RD, Stripay JL, Tsung A, Lee JS, Rosengart MR, Calcium/calmodulin-dependent protein kinase (CaMK) Ialpha mediates the macrophage inflammatory response to sepsis, J Leukoc Biol, 90 (2011) 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Guo L, Stripay JL, Zhang X, Collage RD, Hulver M, Carchman EH, Howell GM, Zuckerbraun BS, Lee JS, Rosengart MR, CaMKIalpha Regulates AMP Kinase-Dependent, TORC-1-Independent Autophagy during Lipopolysaccharide-Induced Acute Lung Neutrophilic Inflammation, Journal of immunology, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang X, Howell GM, Guo L, Collage RD, Loughran PA, Zuckerbraun BS, Rosengart MR, CaMKIV-dependent preservation of mTOR expression is required for autophagy during lipopolysaccharide-induced inflammation and acute kidney injury, J Immunol, 193 (2014) 2405–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N, PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy, Sci Rep, 2 (2012) 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chang CR, Blackstone C, Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology, J Biol Chem, 282 (2007) 21583–21587. [DOI] [PubMed] [Google Scholar]
- [47].Chang CR, Blackstone C, Drp1 phosphorylation and mitochondrial regulation, EMBO Rep, 8 (2007) 1088–1089; author reply 1089–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chang CR, Blackstone C, Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1, Ann N Y Acad Sci, 1201 (2010) 34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cribbs JT, Strack S, Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death, EMBO Rep, 8 (2007) 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K, Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission, J Biol Chem, 282 (2007) 11521–11529. [DOI] [PubMed] [Google Scholar]
- [51].Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L, Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria, Proc Natl Acad Sci U S A, 105 (2008) 15803–15808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Aguileta MA, Rojas-Rivera D, Goossens V, Estornes Y, Van Isterdael G, Vandenabeele P, Bertrand MJ, A siRNA screen reveals the prosurvival effect of protein kinase A activation in conditions of unresolved endoplasmic reticulum stress, Cell Death Differ, 23 (2016) 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yang Z, Klionsky DJ, Mammalian autophagy: core molecular machinery and signaling regulation, Current opinion in cell biology, 22 (2010) 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Huber TB, Edelstein CL, Hartleben B, Inoki K, Jiang M, Koya D, Kume S, Lieberthal W, Pallet N, Quiroga A, Ravichandran K, Susztak K, Yoshida S, Dong Z, Emerging role of autophagy in kidney function, diseases and aging, Autophagy, 8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang X, Yuan D, Sun Q, Xu L, Lee E, Lewis AJ, Zuckerbraun BS, Rosengart MR, Calcium/calmodulin-dependent protein kinase regulates the PINK1/Parkin and DJ-1 pathways of mitophagy during sepsis, FASEB J, 31 (2017) 4382–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Howe CJ, LaHair MM, Maxwell JA, Lee JT, Robinson PJ, Rodriguez-Mora O, McCubrey JA, Franklin RA, Participation of the calcium/calmodulin-dependent kinases in hydrogen peroxide-induced Ikappa B phosphorylation in human T lymphocytes, J Biol Chem, 277 (2002) 30469–30476. [DOI] [PubMed] [Google Scholar]
- [57].Howe CJ, Lahair MM, McCubrey JA, Franklin RA, Redox regulation of the calcium/calmodulin-dependent protein kinases, J Biol Chem, 279 (2004) 44573–44581. [DOI] [PubMed] [Google Scholar]
- [58].Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME, A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation, Cell, 133 (2008) 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ, Anderson ME, Oxidized CaMKII causes cardiac sinus node dysfunction in mice, J Clin Invest, 121 (2011) 3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sebag SC, Koval OM, Paschke JD, Winters CJ, Jaffer OA, Dworski R, Sutterwala FS, Anderson ME, Grumbach IM, Mitochondrial CaMKII inhibition in airway epithelium protects against allergic asthma, JCI Insight, 2 (2017) e88297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ziviani E, Tao RN, Whitworth AJ, Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin, Proc Natl Acad Sci U S A, 107 (2010) 5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW, Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy, Hum Mol Genet, 19 (2010) 4861–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Glauser L, Sonnay S, Stafa K, Moore DJ, Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1, J Neurochem, 118 (2011) 636–645. [DOI] [PubMed] [Google Scholar]
- [64].Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR, Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation, J Biol Chem, 269 (1994) 15520–15527. [PubMed] [Google Scholar]
- [65].Racioppi L, Means AR, Calcium/calmodulin-dependent kinase IV in immune and inflammatory responses: novel routes for an ancient traveller, Trends Immunol, 29 (2008) 600–607. [DOI] [PubMed] [Google Scholar]
- [66].Singer M, The role of mitochondrial dysfunction in sepsis-induced multi-organ failure, Virulence, 5 (2014) 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Evankovich J, Zhang R, Cardinal JS, Zhang L, Chen J, Huang H, Beer-Stolz D, Billiar TR, Rosengart MR, Tsung A, Calcium/Calmodulin-Dependent Protein Kinase IV limits organ damage in hepatic ischemia/reperfusion injury through induction of autophagy, American journal of physiology. Gastrointestinal and liver physiology, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M, Calcium calmodulin-dependent protein kinase IV is required for fear memory, Nature neuroscience, 5 (2002) 573–579. [DOI] [PubMed] [Google Scholar]
- [69].Lewis A, Zuckerbraun B, Griepentrog J, Zhang X, Rosengart M, Reducing Animal Use with a Biotelemetry-Enhanced Murine Model of Sepsis, Sci Rep, 7 (2017) 6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lewis AJ, Griepentrog JE, Zhang X, Angus DC, Seymour CW, Rosengart MR, Prompt Administration of Antibiotics and Fluids in the Treatment of Sepsis: A Murine Trial, Crit Care Med, 46 (2018) e426–e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lewis AJ, Yuan D, Zhang X, Angus DC, Rosengart MR, Seymour CW, Use of Biotelemetry to Define Physiology-Based Deterioration Thresholds in a Murine Cecal Ligation and Puncture Model of Sepsis, Crit Care Med, 44 (2016) e420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Howell GM, Gomez H, Collage RD, Loughran P, Zhang X, Escobar DA, Billiar TR, Zuckerbraun BS, Rosengart MR, Augmenting autophagy to treat acute kidney injury during endotoxemia in mice, PLoS One, 8 (2013) e69520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang X, Howell GM, Guo L, Collage RD, Loughran PA, Zuckerbraun BS, Rosengart MR, CaMKIV-dependent preservation of mTOR expression is required for autophagy during LPS-induced inflammation and acute kidney injury, Journal of Immunology, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Loughran PA, Stolz DB, Barrick SR, Wheeler DS, Friedman PA, Rachubinski RA, Watkins SC, Billiar TR, PEX7 and EBP50 target iNOS to the peroxisome in hepatocytes, Nitric Oxide, 31 (2013) 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Carchman EH, Whelan S, Loughran P, Mollen K, Stratamirovic S, Shiva S, Rosengart MR, Zuckerbraun BS, Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Doerrier C, Garcia-Souza LF, Krumschnabel G, Wohlfarter Y, Meszaros AT, Gnaiger E, High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria, Methods Mol Biol, 1782 (2018) 31–70. [DOI] [PubMed] [Google Scholar]
- [77].Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS, Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice, Hepatology, 53 (2011) 2053–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]