

Abstract
Targeting epidermal growth factor receptor (EGFR) through an allosteric mechanism provides a potential therapeutic strategy to overcome drug-resistant EGFR mutations that emerge within the ATP binding site. Here, we develop an allosteric EGFR degrader, DDC-01-163, which can selectively inhibit the proliferation of L858R/T790 (L/T) mutant Ba/F3 cells while leaving wildtype EGFR Ba/F3 cells unaffected. DDC-01-163 is also effective against osimertinib-resistant cells with L/T/C797S and L/T/L718Q EGFR mutations. When combined with an ATP-site EGFR inhibitor, osimertinib, the anti-proliferative activity of DDC-01-163 against L858R/T790M EGFR-Ba/F3 cells is enhanced. Collectively, DDC-01-163 is a promising allosteric EGFR degrader with selective activity against various clinically relevant EGFR mutants as a single agent and when combined with an ATP-site inhibitor. Our data suggests that targeted protein degradation is a promising drug development approach for mutant EGFR.
Keywords: EGFR, degrader, allosteric, drug-resistant mutation, combination treatment
Graphical Abstract
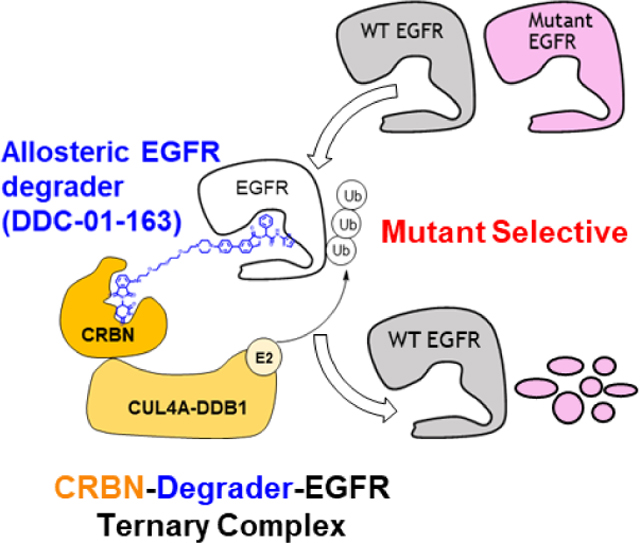
We have developed mutant-selective allosteric EGFR degraders (blue) that mediate the formation of the cereblon (CRBN)-degrader-EGFR ternary complex, resulting in mutant EGFR, but not WT, degradation. The lead degrader, DDC-01-163 is broadly effective against drug-resistant mutants, and exhibits enhanced potency when combined with an inhibitor, osimertinib. The targeted EGFR degradation represents a promising strategy to overcome drug resistance.
Introduction
Epidermal growth factor receptor (EGFR) is a well-known oncogenic driver in many cancers including non-small cell lung cancer (NSCLC).[1] The development of EGFR-targeted small molecule drugs in the past two decades has changed the clinical landscape in which NSCLC patients are now treated.[2] Specifically, targeted therapy directed by genotype-screening has now become the standard practice for treating NSCLC patients harboring oncogenic EGFR mutations. However, earlier generations of tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, and afatinib caused dose-limiting toxicities such as diarrhea and skin rash. Moreover, acquired mutations to EGFR TKI therapy often occur within a year after onset of treatment. One of the most frequent secondary mutations observed in more than half of the patients with acquired drug resistance is the Thr790 to Met790 (T790M) mutation.[3] The T790M mutation increases the affinity of the active site for ATP, thereby allowing the nucleotide to outcompete the binding of reversible EGFR TKIs. This motivated the discovery of the third-generation EGFR inhibitor osimertinib, which circumvents the resistance caused by T790M mutation and also displays improved selectivity for mutant EGFR over wildtype EGFR. As a result, treatment with osimertinib is well tolerated, with marked reduction in adverse drug reactions. Osimertinib was subsequently approved for first-line treatment of metastatic NSCLC harboring the two most common EGFR activating mutations, L858R and exon 19 deletions in 2018.[4]
Despite its efficacy, osimertinib succumbs to the inevitable emergence of acquired drug resistance. Patients treated with osimertinib often develop the Cys797 to Ser797 (C797S) mutation, rendering them resistant to osimertinib and all other irreversible EGFR inhibitors due to the disruption of a potency-conferring covalent bond with Cys797.[5] Various other osimertinib resistant mutations such as G796D/S/R, L792F/Y/H, C797G and L718Q have also been reported in clinic.[6] Given that all these mutations are located within the ATP-binding site where TKIs bind, development of compounds with a different binding mode may represent a viable therapeutic strategy.
Toward this end, we previously reported the rational discovery and characterization of a series of allosteric EGFR inhibitors (EAI001, EAI045 and JBJ-04-125-02)[7] that are active against drug-resistant EGFR mutants. Our mechanistic and structural characterization of EAI001-EGFR interactions showed that the compound could effectively inhibit L858R/T790M/C797S EGFR in vitro, while it required cetuximab, an antibody-based drug that blocks EGFR dimerization[8], for in vivo activity. We established that this was due to EGFR asymmetric dimer formation, resulting in one of the allosteric pockets being harder to occupy. Therefore, EAI001 was only effective when used in combination with a dimer disrupting agent, like cetuximab. Since cetuximab is an anti-EGFR antibody that binds to all forms of EGFR including the wildtype, toxicity-related concerns limit the potential of this therapeutic regimen.
Small molecule mediated targeted protein degradation (TPD) has recently emerged as a promising alternative to inhibition. TPD relies on the use of degrader molecules to bind the protein target of interest and recruit it to an E3 ubiquitin ligase. This proximity with the E3 ligase results in target polyubiquitination and subsequent proteasomal degradation. In this space, small molecules called PROteolysis TArgeting Chimeras (PROTACs) have been of increasing interest. PROTACs are heterobifunctional molecules with two domains, one is used to recognize and bind the target, and the other is used to bind the E3 ligase, connected by a linker. Over the last few years, we and others have used cereblon (CRBN)- or von Hippel Lindau (VHL)-E3 ligase-dependent PROTACs to achieve degradation of a range of protein targets, including several kinases, indicating that this method may be applicable to EGFR.[9] Recently, the first EGFR-targeted PROTACs based on ATP-competitive EGFR inhibitors were developed and reported as effective inducers of EGFR degradation.[9i, 10] However, given that they target the ATP binding site, it remained unclear whether they would be effective against clinically relevant EGFR mutations.
Given the unique binding mode of the mutant selective allosteric EGFR inhibitors, we sought to adapt allosteric compounds to a heterobifunctional degrader to further enhance their ability to block EGFR activation. We postulated that a degrader based on an allosteric EGFR inhibitor would selectively degrade a broad spectrum of drug-resistant EGFR mutants, while leaving wildtype EGFR intact. Here we describe the preparation of a series of mutant-selective allosteric EGFR degraders, and report the complete characterization of our lead compound DDC-01-163.
Results and Discussion
To assess the potency of newly synthesized molecules, we employed a Homogeneous Time Resolved Fluorescence (HTRF) kinase assay (Figure 2A)[7a] and a cell proliferation assay that is broadly used for assessing target-oriented potency of kinase inhibitors. Specifically, Ba/F3 cells transformed by the oncogenic kinase,[11] in our case, the EGFR L858R/T790M mutant, were used as a primary assessment method to examine the cellular potency. Since our allosteric EGFR inhibitor was inactive as a single agent in cell proliferation assays, any observed anti-proliferative activity of these degraders would be mainly due to EGFR degradation and not the potency of the allosteric inhibitor alone. Hence, this allowed us to distinguish the effect of EGFR degradation and anti-proliferative activity simultaneously.
Figure 2.
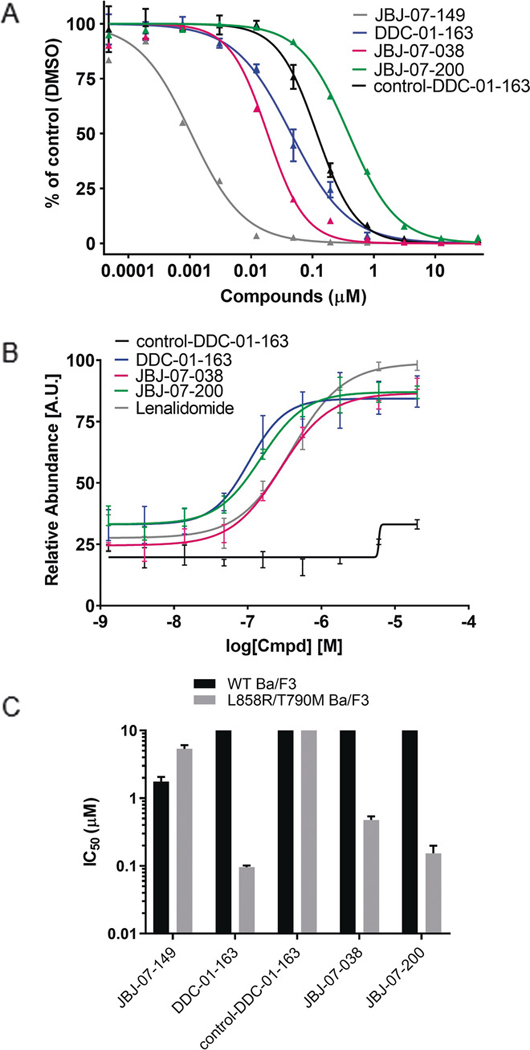
(A) Inhibition of L858R/T790M EGFR by JBJ-07-149, DDC-01-163, control-DC-01-163, JBJ-07-038 and JBJ-07-200. Enzyme activity of purified EGFRL858R/T790M measured using HTRF assay at indicated concentrations of each compounds. (B) Cellular CRBN engagement assay evaluating the ability of DDC-01-163, control-DDC-01-163, JBJ-07-038, JBJ-07-200, and lenalidomide to protect BRD4BD2 from dBET6-mediated degradation measured by three independent experiments. (C) The average IC50 values (μM ± S.D.) of JBJ-07-149, DDC-01-163, control-DDC-01-163, JBJ-07-038 and JBJ-07-200 in wild type EGFR and L858R/T790M EGFR mutant Ba/F3 cells were measured from at least three independent experiments, each performed with six replicates.
We used EAI001 as a starting point for our degrader design.[7a] Since EAI001 is buried deep in the allosteric pocket,[7a] we synthesized JBJ-07-149, which has a 1-(pyridin-2-yl)piperazine at the 6-position of isoindolinone, resulting in the extension of EAI001 to a solvent exposed region (Figure 1B). JBJ-07-149 potently inhibited the catalytic activity of EGFR L858R/T790M in vitro (IC50 = 1.1 nM, Figure 2A), and anti-proliferative activity in the presence of cetuximab (EC50 = 0.148 μM, Figure S1), but remained ineffective as a single agent in proliferation assays (EC50 = 4.9 μM, Figure S1). Using JBJ-07-149 as a starting point, we designed a bifunctional degrader molecule, DDC-01-163, in which JBJ-07-149 was linked to pomalidomide, a CRBN ligand, via a linker conjugated to the piperazine moiety of JBJ-07-149 and positioned it toward the solvent exposed exterior (Figure 1). The structure-activity relationship of allosteric EGFR PROTACs is summarized in Table S1. Among the diverse linkers tested, including 1-, 2-, 3-, or 4- polyethylene glycols (PEGs) and carbon linkers with various lengths, 3-PEG linker displayed the most potent anti-proliferative activity against L858R/T790M-Ba/F3 cells (11g, 11k and DDC-01-163). Changing linker attachment site from ortho- to meta-position on thalidomide (11m and 11n) and replacing the amine linkage with the ether linkage (11o) led to the loss of potency. PROTACs featuring a VHL E3 ligase recruiting ligand, instead of CRBN ligand, were inactive (11p and 11q). Collectively, DDC-01-163 was the most potent analog in the antiproliferative assays.
Figure 1.

(A) A schematic binding mode of allosteric EGFR PROTAC complexed with EGFR and cereblon (CRBN). (B) Chemical structures of an allosteric EGFR inhibitor (JBJ-07-149), allosteric EGFR PROTACs (DDC-01-163, JBJ-07-038, and JBJ-07-200), and a negative control of DDC-01-163 (control-DDC-01-163).
During our PROTACs development process, we identified JBJ-04-125-02 (Figure 1B), an allosteric inhibitor with a 2-hydroxy-5-fluorophenyl group with improved potency, including the ability to inhibit cell proliferation of Ba/F3 cells as a single agent.[7b] Therefore, in addition to DDC-01-163, we synthesized and tested JBJ-04-125-02-based allosteric EGFR degraders. Specifically, we transformed JBJ-04-125-02 to a PROTAC molecule using the same strategy described for DDC-01-163, which resulted in generation of JBJ-07-038 (Figure 1B). In parallel, we substituted the hydroxyl group (Y) on JBJ-04-125-02 with a fluorine and generated JBJ-07-200 (Figure 1B), as a strategy to potentially increase cell membrane permeability.
Our in vitro assay showed that DDC-01-163 exhibited an acceptable biochemical potency with an IC50 value of 45 nM against EGFR L858R/T790M, albeit inferior to its parent allosteric EGFR inhibitor JBJ-07-149 (IC50 = 1.1 nM; Figure 2A). We also observed that JBJ-07-038 (IC50 = 19 nM) was more potent against EGFR L858R/T790M than DDC-01-163 while the fluorine substitution in JBJ-07-200 (IC50 = 390 nM) reduced biochemical activity (9-fold and 21-fold compared to DDC-01-163 and JBJ-07-038, respectively; Figure 2A).
Since PROTACs often exhibit poor cell-penetration due to their high molecular weight and presence of many hydrogen bond acceptors and donors, we sought to evaluate cellular permeability and intracellular CRBN engagement using a previously described cellular assay.[12] Briefly, this is a competition-based assay that measures the ability of a degrader molecule to displace BRD4BD2-directed CRBN ligands, which subsequently leads to the restoration of BRD4BD2-GFP signal. Our results revealed that DDC-01-163 could engage CRBN better than JBJ-07-038 (Figure 2B). As predicted, JBJ-07-200 exhibited more potent CRBN engagement than JBJ-07-038, likely due to better cell permeability (Figure 2B). In addition, further CRBN cellular engagement studies confirmed that JBJ-04-125-02-based degraders had less potent rescue effect due to the presence of free hydroxyl group (Figure S3). These observations were in line with the results of cellular assays where DDC-01-163 and JBJ-07-200 showed superior anti-proliferative activity and effect on EGFR degradation compared to JBJ-07-038 (Figures 2C and 3). This outcome suggests that a highly potent protein ligand is not always ideal for generating an effective PROTAC and that cell membrane permeability is a critical contributing factor to the ultimate potency of the compound.
Figure 3.

Western blot analysis of total EGFR protein levels in wild type EGFR and L858R/T790M BaF3 cells treated with increasing concentrations of DDC-01-163, JBJ-07-038 and JBJ-07-200 for 6, 24, 48 hours. Tubulin was used as a loading control for relative protein expression. Densitometry analysis were performed and shown as a percentage of EGFR protein left compared to DMSO control after data was normalized to tubulin.
As a negative control, we prepared an N-methyl glutarimide analog (control-DDC-01-163) of DDC-01-163 that has diminished affinity for CRBN.[13] Even though control-DDC-01-163 could bind to the EGFR allosteric pocket (Figure 2A), it could not compete for CRBN binding (Figure 2B) and, as expected, displayed no anti-proliferative effects (Figure 2C).
We next examined the ability of these EGFR degraders to selectively inhibit mutant cell proliferation in Ba/F3 cell lines that were either stably transfected with the EGFR L858R/T790M mutant or wildtype EGFR. DDC-01-163 potently inhibited the proliferation of L858R/T790M mutant EGFR Ba/F3 cells (EC50 = 0.096 μM) while sparing the wildtype EGFR Ba/F3 cells (EC50 > 10 μM; Figure 2C and S2). This striking mutant-selectivity likely arises primarily from the parent allosteric inhibitor scaffold.[7a, 7b] Interestingly, DDC-01-163 was 51-fold more potent than its parent allosteric EGFR inhibitor, JBJ-07-149 (0.096 μM vs 4.9 μM) in cellular assays, despite being 45-fold less potent than JBJ-07-149 in biochemical assays. Consistent with our CRBN engagement assays, JBJ-04-125-02-based degrader such as JBJ-07-038 (EC50 = 0.48 μM) displayed an inferior anti-proliferative activity in L858R/T790M EGFR-Ba/F3 cells and JBJ-07-200 (EC50 = 0.15 μM) was more potent than JBJ-07-038.
To assess the effects of our PROTACs on EGFR degradation, we treated wildtype EGFR and EGFR L858R/T790M Ba/F3 cells with increasing concentrations of DDC-01-163, JBJ-07-038 and JBJ-07-200 for 6, 24 and 48 hours and measured EGFR protein abundance at each time point by Western blotting. As illustrated in Figure 3, DDC-01-163, JBJ-07-038 and JBJ-07-200 did not affect protein levels in wildtype Ba/F3 cells. Notably, DDC-01-163 was most effective at reducing protein levels of L858R/T790M EGFR in a dose-dependent manner with a maximum degradation observed at 24 hours (59 – 62%) which was sustained for 48 hours at 0.1 μM. Interestingly, we also observed reduced protein degradation at higher concentrations (1 μM) over time (at 48 hours) for all three EGFR PROTACs, consistent with the hook effect commonly observed in other degraders[9i, 14].
To further confirm the mechanism of action of DDC-01-163, we performed competitive in vitro assays by treating L858R/T790M Ba/F3 cells with the CRBN ligand pomalidomide or the parent allosteric EGFR inhibitor JBJ-07-149 in the presence or absence of 0.1 μM DDC-01-163. Densitometry analyses of Western Blotting showed a marked decrease of EGFR protein to 44% in cells treated with DDC-01-163 alone (Figure 4). However, the presence of pomalidomide and JBJ-07-149 prevented EGFR degradation even in the presence of DDC-01-163, indicating that the engagement to both EGFR and CRBN was essential for EGFR degradation (Figure 4A) and that anti-proliferative activity of DDC-01-163 was highly dependent on EGFR degradation. We also tested whether we could rescue the EGFR degradation effect by treating the cells with an NEDD8 activating enzyme 1 inhibitor, MLN4924, which blocks neddylation of the cullin RING ligase, CUL4, resulting in the disruption of CRBN E3 ligase function. Our Western blot showed that pre-treatment with an increasing concentration of MLN4924 in L858R/T70M EGFR-Ba/F3 cells could indeed rescue EGFR degradation by DDC-01-163 (Figure 4B).
Figure 4.

Western blot analyses of total EGFR protein level in L858R/T790M BaF3 cells after cells were (A) treated in a competitive study with either DMSO, 10 μM pomalidomide or 1 μM JBJ-07-149 for 24 hours; or (B) treated in a rescue study with increasing concentrations of the NEDD8 activating enzyme inhibitor, MLN4924, for 24 hours in the presence or absence of DDC-01-163. Tubulin was used as a loading control for relative protein expression. Densitometry analysis were performed and shown as a percentage of EGFR protein left compared to DMSO control after data was normalized to tubulin.
We next tested whether the same phenomenon was observed in human cancer cells with L858R/T790M EGFR mutation by using H1975 cells. Consistent with our Ba/F3 cells data, treatment with 0.1 μM DDC-01-163 led to increased EGFR degradation over time with maximal degradation occurring between 24 to 48 hours (Figure 5A). Competitive assays using pomalidomide and JBJ-07-149, as well as rescue studies with increasing concentrations of MLN4924 prevented DDC-01-163-induced degradation as observed in Ba/F3 cells (Figure 5B and 5C). This suggests that our allosteric EGFR PROTAC degrader, DDC-01-163, may have potency beyond murine cells and could also be applicable to human cancer cells.
Figure 5.

Western blot analysis of total EGFR protein level in H1975 cells after cells were (A) treated with 0.1 μM of DDC-01-163 for 0, 4, 8, 24, 48, 72 hours; or (B) treated in a competitive study with either DMSO, 10 μM pomalidomide or 1 μM JBJ-07-149 for 24 hours; or (C) treated in a rescue study with increasing concentrations of the NEDD8 activating enzyme inhibitor, MLN4924, for 24 hours in the presence or absence of DDC-01-163. Tubulin was used as a loading control for relative protein expression. Densitometry analysis were performed and shown as a percentage of EGFR protein left compared to DMSO control after data was normalized to tubulin.
Inspired by recent studies showing that combination treatment with allosteric EGFR inhibitor JBJ-04-125-02 and ATP-site inhibitor osimertinib could enhance apoptosis and delay onset of drug resistance,[7b] we examined whether dual modulation of EGFR using an allosteric PROTAC and an ATP-site inhibitor would be more effective than single-agent treatment. To compare the inhibition of cell proliferation in single agent versus combination treatment, we performed an 11-point dose escalation experiment using osimertinib or DDC-01-163 alone or in combination with two constant doses of DDC-01-163 and osimertinib respectively (Figure 6A). Our results showed that the addition of both 0.01 μM and 0.1 μM of DDC-01-163 shifted the dose-response curve of osimertinib to the left, indicating that combination treatment was more effective at inhibiting cell proliferation than single agent treatment. Similarly, the addition of 0.01 μM osimertinib also shifted the dose-response curve of DDC-01-163 to the left; however, the addition of a higher dose (0.1 μM) of osimertinib to dose titration of DDC-01-163 had an antagonistic effect.
Figure 6.

(A) Cell viability assay examining the growth inhibitory effect of L858R/T790M Ba/F3 cells and (B) Western blot analyses assessing the EGFR activity (phospho-EGFR) and protein degradation levels (total EGFR) after treatment with dose escalated osimertinib and DDC-01-163 alone (DR) or in the presence of a constant dose of DDC-01-163 or osimertinib respectively in L88R/T790M Ba/3 cells. Tubulin was used as a loading control for relative protein expression. Densitometry analyses were performed and the percentage of EGFR protein left shown is compared to DMSO control after data was normalized to tubulin. The percentage of EGFR activity shown is compared to the DMSO control after data was normalized to total EGFR.
To further understand how the combination treatment of an allosteric EGFR degrader with osimertinib affects EGFR activity and protein degradation, we performed similar experiments in L858R/T790M EGFR Ba/F3 cells and assessed both EGFR activity and EGFR degradation after treatment for 24 hours by Western blotting with phospho-EGFR and total EGFR antibodies. Consistent with our cell proliferation assays, both inhibition and degradation effects of 0.1 μM DDC-01-163 were apparent in the presence of increasing concentration of osimertinib. Similarly, the addition of 0.01 μM osimertinib to increasing concentration of DDC-01-163 led to an overall enhanced inhibition of EGFR phosphorylation, albeit slightly blunted or less efficient degradation of EGFR (Figure 6B). Collectively, our cell proliferation assays and western blotting studies revealed that the combination of the protein degrader DDC-01-163 and the EGFR TKI, osimertinib, could modulate EGFR together, leading to enhanced anti-proliferative activity compared to either agent alone. However, an antagonistic effect is apparent at higher doses, suggesting that the optimization of both compounds in order to remain within the therapeutic window is important to achieve maximal synergistic effect.
Resistance to current NSCLC drugs was a major motivation for developing EGFR mutant-selective allosteric degrader. Therefore, we evaluated the effect of DDC-01-163 in this context using Ba/F3 cell lines that were stably transfected with L858R/T790M/C797S EGFR (LT/C797S EGFR),[15] and L858R/T790M/L718Q EGFR (LT/L718Q EGFR) (Figure 7).[15b, 16] These tertiary mutations have been found in NSCLC patients whose tumors developed acquired drug resistance after treatment with osimertinib. As anticipated, both cell lines were resistant to clinically approved EGFR inhibitors (gefitinib, afatinib and osimertinib) and the parent allosteric EGFR inhibitor, JBJ-07-149, in our cell proliferation assay (Figure 6A). However, DDC-01-163 was highly effective in inhibiting the cell proliferation of LT/C797S (EC50 = 0.041 μM), and LT/L718Q (EC50 = 0.028 μM) EGFR Ba/F3 cells (Figure 7A), consistent with the result shown in L858R/T790M EGFR-Ba/F3 cells (Figure 2C).
Figure 7.

(A) Cell viability assay examining the growth inhibitory effect of dose escalated gefitinib, afatinib, osimertinib, JBJ-07-149 and DDC-01-163 and; (B) Western blot analyses assessing the EGFR activity (phospho-EGFR) and protein degradation levels (total EGFR) after treatment with either DMSO or 0.1 μM DDC-01-163 or 0.1 μM osimertinib for 24 hours in three different osimertinib resistant Ba/F3 cell lines harboring L858R/T790M/C797S, and L858R/T790M/L718Q EGFR mutations. Tubulin was used as a loading control for relative protein expression. Densitometry analysis were performed and the percentage of EGFR protein left shown is compared to DMSO control after data was normalized to tubulin. The percentage of EGFR activity shown is compared to the DMSO control after data was normalized to total EGFR.
We next examined the effect of DDC-01-163 on EGFR degradation in osimertinib-resistant cell lines with either DMSO, 0.1 μM DDC-01-163 or 0.1 μM osimertinib for 24 hours followed by Western blot analysis. As expected, osimertinib treatment did not induce EGFR degradation or alter the phosphorylation of EGFR compared to DMSO, indicating that these cell lines were osimertinib resistant (Figure 7B). Conversely, the degradation of EGFR was apparent when cells were treated with 0.1 μM of DDC-01-163. Consequently, 74% and 71% degradation were observed in LT/C797S EGFR and LT/L718Q EGFR cells respectively after DDC-01-163 treatment, which led to the subsequent inhibition of EGFR activity (Figure 7B).
Conclusion
DDC-01-163 is a mutant-selective allosteric EGFR degrader that inhibits proliferation of L858R/T790M EGFR mutant Ba/F3 cells while sparing wildtype EGFR-Ba/F3 cells. Moreover, the allosteric degrader activity was cetuximab-independent, therefore representing a significant advance over the parental allosteric inhibitor. We found that DDC-01-163 induced selective degradation and inhibited proliferation of mutant EGFR cells in a dose- and time-dependent manner and notably, without affecting wildtype EGFR. These results were consistent with our studies in the human cancer cell line, H1975. Similar to other published studies, we also observed the hook effect, a decrease in degradation at higher concentrations,, which reinforces prior conclusions that higher concentrations of PROTAC degraders are not necessarily more efficacious. Moreover, our studies with DDC-01-163 and osimertinib revealed that combination treatment with an allosteric degrader and a ATP-site tyrosine kinase inhibitor was possible and more effective than either agent alone, hence, suggesting that dual modulation of target proteins by the enzymatic inhibitor and protein degrader could enhance their efficiency to inhibit overall cell growth. However, it is important to note, as seen in our studies that synergy can be concentration-dependent; therefore, it is imperative to identify the optimal combined doses of both compounds when characterizing PROTACs with other drugs.
One of the most challenging issues in targeted therapy is that all patients eventually acquire mutations that render them resistant to therapy. Here we show that mutant-selective allosteric EGFR degrader DDC-01-163 was efficacious in not only L858R/T790M mutant EGFR cells but also in cells that have acquired resistance to osimertinib. Furthermore, because DDC-01-163 is a degrader targeted at an allosteric site, we anticipate that the onset of resistance could be delayed through combination and/or sequential treatment regimen as the mutations that emerge would presumably be different from that of TKIs. Therefore, it is evident that further development of allosteric EGFR PROTACs could present a valuable approach that could benefit a broader range of patients harboring EGFR mutations and warrants further characterization and optimization.
Supplementary Material
Acknowledgements
This work was supported by NIH P01 CA154303 (Gray, Jänne, Eck), NIH R01 CA218278 (Fischer, Gray), NCI grant R50 CA221830 (Park), and the Linde Program for Chemical Biology.
Conflict of interest
Nathanael Gray is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, B2S, Soltego and a SAB member of Aduro. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield and Sanofi. Pasi Jänne has received consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Merrimack Pharmaceuticals, Chugai Pharmaceuticals, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Daiichi-Sankyo, Takeda Oncology, Novartis, Voronoi, SFJ Pharmaceuticals, Biocartis and LOXO Oncology; receives post-marketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp and has stock ownership in Gatekeeper Pharmaceuticals and LOXO Oncology. The Jänne lab receives or has received research funding from AstraZeneca, Daiichi-Sankyo, Boehringer Ingelheim, PUMA, Eli Lilly and Company, Astellas Pharmaceuticals, Revolution Medicines and Takeda Oncology. The Eck lab receives or has received research funding from Novartis, Takeda, and Her2llc. Eric Fischer is a founder, science advisory board member (SAB) and equity holder in Civetta therapeutics, and an equity holder and SAB member in C4 Therapeutics. The Fischer lab receives or has received research funding from Novartis, Astellas and Deerfield. Gray, Janne, Eck, Jang are inventors of a patent covering the compounds in this publication that has been licensed to C4.
Contributor Information
Jaebong Jang, Department of Cancer Biology, Dana-Farber Cancer Institute.; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
Ciric To, Lowe Center for Thoracic Oncology, Dana-Farber Cancer Institute.; Department of Medical Oncology, Dana-Farber Cancer Institute. Department of Medicine, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston Massachusetts 02215 USA.
Dries J. H. De Clercq, Department of Cancer Biology, Dana-Farber Cancer Institute. Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
Eunyoung Park, Department of Cancer Biology, Dana-Farber Cancer Institute.; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
Charles M. Ponthier, Department of Cancer Biology, Dana-Farber Cancer Institute. Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
Bo Hee Shin, Lowe Center for Thoracic Oncology, Dana-Farber Cancer Institute.; Department of Medical Oncology, Dana-Farber Cancer Institute. Department of Medicine, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston Massachusetts 02215 USA.
Mierzhati Mushajiang, Lowe Center for Thoracic Oncology, Dana-Farber Cancer Institute.; Department of Medical Oncology, Dana-Farber Cancer Institute. Department of Medicine, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston Massachusetts 02215 USA.
Radosław P. Nowak, Department of Cancer Biology, Dana-Farber Cancer Institute. Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
Eric S. Fischer, Department of Cancer Biology, Dana-Farber Cancer Institute. Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
Michael J. Eck, Department of Cancer Biology, Dana-Farber Cancer Institute. Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
Pasi A. Jänne, Lowe Center for Thoracic Oncology, Dana-Farber Cancer Institute. Department of Medical Oncology, Dana-Farber Cancer Institute. Department of Medicine, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston Massachusetts 02215 USA. Belfer Center for Applied Cancer Science. Longwood Center, 360 Longwood Avenue, Boston Massachusetts 02215 USA.
Nathanael S. Gray, Department of Cancer Biology, Dana-Farber Cancer Institute. Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School. Longwood Center, 360 Longwood Avenue, Boston, Massachusetts 02215 USA.
References
- [1].a) Tanaka T, Matsuoka M, Sutani A, Gemma A, Maemondo M, Inoue A, Okinaga S, Nagashima M, Oizumi S, Uematsu K, Nagai Y, Moriyama G, Miyazawa H, Ikebuchi K, Morita S, Kobayashi K, Hagiwara K, Int. J. Cancer 2010, 126, 651–655; [DOI] [PubMed] [Google Scholar]; b) Choi YL, Sun JM, Cho J, Rampal S, Han J, Parasuraman B, Guallar E, Lee G, Lee J, Shim YM, PLoS One 2013, 8, e56011; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Smits AJ, Kummer JA, Hinrichs JW, Herder GJ, Scheidel-Jacobse KC, Jiwa NM, Ruijter TE, Nooijen PT, Looijen-Salamon MG, Ligtenberg MJ, Thunnissen FB, Heideman DA, de Weger RA, Vink A, Cell Oncol. 2012, 35, 189–196; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Juchum M, Gunther M, Laufer SA, Drug Resist. Updat. 2015, 20, 12–28. [DOI] [PubMed] [Google Scholar]
- [2].a) Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA, N. Engl. J. Med. 2004, 350, 2129–2139; [DOI] [PubMed] [Google Scholar]; b) Khozin S, Blumenthal GM, Jiang X, He K, Boyd K, Murgo A, Justice R, Keegan P, Pazdur R, Oncologist 2014, 19, 774–779; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR, J. Pharmacol. Exp. Ther 2012, 343, 342–350; [DOI] [PubMed] [Google Scholar]; d) Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M, N. Engl. J. Med 2015, 372, 1689–1699; [DOI] [PubMed] [Google Scholar]; e) Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W, Cancer Discov. 2014, 4, 1046–1061; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, Bradbury RH, Brown SJ, Butterworth S, Campbell A, Chorley C, Colclough N, Cross DA, Currie GS, Grist M, Hassall L, Hill GB, James D, James M, Kemmitt P, Klinowska T, Lamont G, Lamont SG, Martin N, McFarland HL, Mellor MJ, Orme JP, Perkins D, Perkins P, Richmond G, Smith P, Ward RA, Waring MJ, Whittaker D, Wells S, Wrigley GL, J. Med. Chem 2014, 57, 8249–8267. [DOI] [PubMed] [Google Scholar]
- [3].a) Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, Ladanyi M, Miller VA, Pao W, Clinical Cancer Ressearch 2006, 12, 6494–6501; [DOI] [PubMed] [Google Scholar]; b) Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T, Clin. Cancer Res. 2006, 12, 5764–5769; [DOI] [PubMed] [Google Scholar]; c) Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ, Clin. Cancer Res. 2013, 19, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, Okamoto I, Zhou C, Cho BC, Cheng Y, Cho EK, Voon PJ, Planchard D, Su WC, Gray JE, Lee SM, Hodge R, Marotti M, Rukazenkov Y, Ramalingam SS, Investigators F, N. Engl. J. Med 2018, 378, 113–125. [DOI] [PubMed] [Google Scholar]
- [5].a) Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Janne PA, Oxnard GR, Nat. Med. 2015, 21, 560–562; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yu HA, Tian SK, Drilon AE, Borsu L, Riely GJ, Arcila ME, Ladanyi M, JAMA Oncol. 2015, 1, 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tang ZH, Lu JJ, Cancer Lett. 2018, 420, 242–246. [DOI] [PubMed] [Google Scholar]
- [7].a) Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys PY, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong KK, Janne PA, Eck MJ, Nature 2016, 534, 129–132; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, Xu M, Wang S, Cameron MD, Heppner DE, Shin BH, Gero TW, Yang A, Dahlberg SE, Wong KK, Eck MJ, Gray NS, Janne PA, Cancer Discov. 2019, 9, 926–943; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) De Clercq DJH, Heppner DE, To C, Jang J, Park E, Yun CH, Mushajiang M, Shin BH, Gero TW, Scott DA, Janne PA, Eck MJ, Gray NS, ACS Med Chem Lett 2019, 10, 1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM, Cancer Cell 2005, 7, 301–311. [DOI] [PubMed] [Google Scholar]
- [9].a) Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE, Science 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green JL, Haas W, Nomanbhoy T, Fischer ES, Young RA, Bradner JE, Winter GE, Gray NS, Nat. Chem. Biol 2018, 14, 163–170; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ, Winter GE, Erb MA, Scott TG, Xu M, Seo HS, Dhe-Paganon, Kwiatkowski NP, Perry JA, Qi J, Gray NS, Bradner JE, Nat. Chem. Biol 2018, 14, 405–412; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) de Wispelaere M, Du G, Donovan KA, Zhang T, Eleuteri NA, Yuan JC, Kalabathula J, Nowak RP, Fischer ES, Gray NS, Yang PL, Nat. Commun. 2019, 10, 3468; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, Dastjerdi S, Wuhr M, Dhe-Paganon S, Armstrong SA, Bradner JE, Angew. Chem. Int. Ed 2017, 56, 5738–5743; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, Gray NS, Angew. Chem. Int. Ed 2019, 58, 6321–6326; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Powell CE, Gao Y, Tan L, Donovan KA, Nowak RP, Loehr A, Bahcall M, Fischer ES, Janne PA, George RE, Gray NS, J. Med. Chem 2018, 61, 4249–4255; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Hatcher JM, Wang ES, Johannessen L, Kwiatkowski N, Sim T, Gray NS, ACS Med. Chem. Lett 2018, 9, 540–545; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, Crew AP, Hines J, Crews CM, Cell Chem. Biol. 2018, 25, 67–77 e63; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong H, Siu K, Winkler JD, Crew AP, Crews CM, Coleman KG, Proc. Natl. Acad. Sci. U S A 2016, 113, 7124–7129; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Faelth-Savitski M, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, den Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I, Crews CM, Nat. Chem. Biol. 2015, 11, 611–617; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AMK, Wang J, Chen X, Dong H, Siu K, Winkler JD, Crew AP, Crews CM, Coleman KG, Proc. Natl. Acad. Sci. U S A 2016, 113, 7124–7129; [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, Chen J, Yang CY, Liu Z, Wang M, Liu L, Jiang H, Wen B, Kumar P, Meagher JL, Sun D, Stuckey JA, Wang S, Cancer Cell 2019, 36, 498–511 e417; [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, Liu X, Thummuri D, Yuan Y, Wiegand JS, Pei J, Zhang W, Sharma A, McCurdy CR, Kuruvilla VM, Baran N, Ferrando AA, Kim YM, Rogojina A, Houghton PJ, Huang G, Hromas R, Konopleva M, Zheng G, Zhou D, Nat. Med. 2019, 25, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Cheng M, Yu X, Lu K, Xie L, Wang L, Meng F, Han X, Chen X, Liu J, Xiong Y, Jin J, J. Med. Chem 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang H, Zhao HY, Xi XX, Liu YJ, Xin M, Mao S, Zhang JJ, Lu AX, Zhang SQ, Eur. J. Med. Chem 2020, 189, 112061. [DOI] [PubMed] [Google Scholar]
- [11].Warmuth M, Kim S, Gu XJ, Xia G, Adrian F, Curr. Opin. Oncol 2007, 19, 55–60. [DOI] [PubMed] [Google Scholar]
- [12].Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, Nabet B, Gero TW, Feru F, Li L, Gondi S, Ombelets LJ, Quan C, Janne PA, Kostic M, Scott DA, Westover KD, Fischer ES, Gray NS, Cell Chem. Biol 2020, 27, 19–31 e16. [DOI] [PubMed] [Google Scholar]
- [13].Huang HT, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho JH, Ko E, Jang J, Shi K, Choi HG, Griffin JD, Li Y, Treon SP, Fischer ES, Bradner JE, Tan L, Gray NS, Cell Chem. Biol 2018, 25, 88–99 e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE, Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Janne PA, Oxnard GR, Nat. Med. 2015, 21, 560–562; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, Janne PA, Clin. Cancer Res. 2015, 21, 3913–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Callegari D, Ranaghan KE, Woods CJ, Minari R, Tiseo M, Mor M, Mulholland AJ, Lodola A, Chem. Sci. 2018, 9, 2740–2749; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bersanelli M, Minari R, Bordi P, Gnetti L, Bozzetti C, Squadrilli A, Lagrasta CA, Bottarelli L, Osipova G, Capelletto E, Mor M, Tiseo M, J. Thorac. Oncol 2016, 11, e121–123 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.