

Abstract
We previously demonstrated that AKR vs. DBA/2 mouse bone marrow derived macrophages have higher levels of free cholesterol and lower levels of esterified cholesterol after cholesterol loading, and that AKR, but not DBA/2, macrophages induced C/EBP homologous protein (CHOP) expression after cholesterol loading. We earlier determined that the free and esterified cholesterol level effect is due to a truncation in the sterol O-acyltransferase 1 (Soat1) gene, encoding acetyl-coenzyme A acetyltransferase 1 (ACAT1). Here we examined the mechanism for the differential induction of CHOP by cholesterol loading. CHOP was induced in both strains after incubation with tunicamycin, indicating both strains have competent endoplasmic reticulum stress pathways. CHOP was induced when DBA/2 macrophages were cholesterol loaded in the presence of an ACAT inhibitor, indicating that the difference in free cholesterol levels were responsible for this strain effect. This finding was confirmed in macrophages derived from DBA/2 embryonic stem cells. Cholesterol loading of Soat1 gene edited cells, mimicking the AKR allele, led to increased free cholesterol levels and restored CHOP induction. The upstream pathway of free cholesterol induced endoplasmic reticulum stress was investigated; and, RNA-dependent protein kinase-like endoplasmic reticulum kinase (PERK) and inositol-requiring enzyme 1 α protein kinase (IRE1α) pathways were required for maximal CHOP expression.
Keywords: Cholesterol metabolism, endoplasmic reticulum stress, macrophage
1. Introduction
Atherosclerosis is an important contributor to cardiovascular diseases. The hallmark of atherosclerosis is the formation and accumulation of lipid-laden macrophages in the subendothelial intimal region. These lipid-loaded macrophages promote inflammatory responses in the arterial wall and multiple pathological consequences may ensue[1]. ApoE-deficient mice bred onto different inbred mouse strains have varying aortic root atherosclerotic lesion areas, demonstrating genetic effects that modify the atherosclerosis phenotype[2]. We previously found that the lesion areas in 16-week old chow diet-fed apoE-deficient mice on the DBA/2 genetic background were ~10-fold larger than apoE-deficient mice on the AKR background[3]. As macrophages play a key role in early atherosclerosis lesion development, we performed transcriptomics on bone marrow derived macrophages (BMDM) from both strains in the presence or absence of cholesterol loading using the non-physiological scavenger receptor ligand acetylated LDL (AcLDL)[4]. We found that CHOP expression, a downstream modulator of endoplasmic reticulum (ER) stress encoded by the Ddit3 (DNA-damage inducible transcript 3) gene, was differentially induced in the two strains, with robust CHOP mRNA induction in the AKR BMDM, but a small reduction in the DBA/2 BMDM, leading to a highly significant mouse strain-cholesterol loading interaction effect[4]. Tabas and colleagues, in an elegant series of papers, have shown that free cholesterol loading of C57BL/6 mouse macrophages, by incubation with AcLDL and an inhibitor of acyl-coenzyme A:cholesterol acyltransferase 1 (ACAT1), induces ER stress via activation of the unfolded protein response (UPR), leading to CHOP induction, and if severe to apoptotic cell death [5–7].
We previously demonstrated that after AcLDL loading, AKR cells accumulate more free cholesterol (FC), while DBA/2 cells accumulate more cholesteryl esters (CE)[8]. Using mouse genetics and Crispr-Cas9 gene editing, we determined that the major genetic effect leading to this FC/CE phenotype was due to an N-terminal truncation of the Soat1 gene on chromosome 1, encoding ACAT1, the enzyme that converts FC to CE[9]. Here we performed studies to probe the mechanism that accounts for the differential induction of CHOP by cholesterol loading in AKR and DBA/2 BMDM.
ER stress signaling is triggered by three upstream proteins: inositol requiring 1 (IRE1), activating transcription factor 6 (ATF6) and, RNA-dependent protein kinase-like endoplasmic reticulum kinase (PERK)[10]. We also interrogated which of these pathways is responsible for the cholesterol loading induction of CHOP expression in embryonic stem cell derived macrophages.
2. Materials and Methods
2.1. Bone marrow-derived macrophages
AKR (stock #648) and DBA/2 (stock #671) mouse strains were obtained from JAX. Mouse studies were performed under a protocol approved by the Cleveland Clinic Institutional Animal Care and Use Committee and were in accordance with the National Institutes of Health guide for the care and use of laboratory animals. Bone marrow cells were flushed from femur bones of apoE-deficient mice bred onto the AKR and DBA/2 genetic background[8], resuspended, and plated in macrophage growth medium (DMEM, 10% FBS, 20% L-cell conditioned media as a source of MSCF) and maintained in a 37°C incubator with 5% CO2. The media was renewed three times per week. Cells were used for experiments 10 to 14 days after plating, when the bone marrow cells were confluent and fully differentiated into BMDMs, as indicated by avid uptake of fluorescently labeled AcLDL by >95% of the cells[9, 11].
2.2. DBA/2 embryonic stem cell gene editing and derived macrophages
CRISPR/Cas9 editing of the Soat1 gene in DBA/2J embryonic stem cells (line AC173/GrsrJ from Jackson Laboratory) was previously described[9]. Briefly, a Cas9 expression plasmid encoding puromycin resistance was transfected into these cells and puromycin resistant colonies were selected after plating onto puromycin resistant mouse embryonic fibroblast feeder cells. After expansion, Cas9 expression was confirmed by western blot and immunohistochemistry. Two sgRNAs were designed at positions proximal and distal to the AKR strain deletion endpoints to generate an ~ 6.6kb deletion of Soat1, making the DBA/2 Soat1 exon2 deleted allele (Soat1Δ) that was almost identical to the AKR Soat1 allele. Cas9 stably transfected DBA/2 ES cells were transfected by nucleofection with 0.75 μg of each sgRNA and individual colonies were expanded and screened for homozygous deletion by PCR and agarose gel electrophoresis using primers that flanked the deletion endpoints. Embryonic stem cell derived macrophages (ESDM) were generated using an embryoid body intermediate and characterized as previously described[9].
2.3. Cholesterol loading and CHOP expression
AcLDL was prepared from de-identified human LDL isolated from outdated blood bank plasma treated with acetic anhydride as previously described[12]. Cholesterol loading was performed by incubating BMDM or ESDM with of 50 μg/ml AcLDL for 16–24 hours in the presence or absence of 10 to 30 μg/ml ACAT inhibitor (Sandoz 58–035, S9318, Sigma-Aldrich). Unloaded cells were used as controls. All studies were performed in triplicate wells. When indicated, tunicamycin (1 μg/ml, Sigma Aldrich, T7765) was incubated with BMDM for 16 hrs to induce ER stress. In some experiments the ER stress pathway was disrupted using the PERK inhibitor GSK2606414 (20 μM, SelleckChem, S7307), the IRE1α kinase inhibitor Kira6 (20 μM, SelleckChem, S8658), or the ATF6 processing inhibitor AEBSF (300 μM, Thermo Fisher, 78341). The cells were incubated with these inhibitors for 4–6 hrs before the addition of AcLDL plus ACAT inhibitor. Cells were then washed 2× in PBS and lysed on ice in Pierce RIPA buffer (Thermo Fisher, #UG286842) supplemented with HALT Protease Cocktail Inhibitor (Thermo Fisher, #SC246590) and HALT Phosphatase Inhibitor Cocktail (Thermo Fisher, #Q1221826). After pelleting debris, the soluble protein concentration was determined using Pierce BCA Protein Assay (Thermo Fisher, # TJ272657). For western blot analysis, 5 μg of protein samples were separated by SDS-PAGE and then transferred to polyvinylidene difluoride membranes, followed by blocking for 1 h at room temperature with Casein Blocker in TBS (Thermo Fisher, #37532). Primary antibody incubation was performed at 4°C overnight with slight agitation using a 1:500 dilution (in Casein Blocker) of mouse monoclonal anti-CHOP antibody (Thermo Fisher, #MA1–250), or a 1:20,000 dilution of an HRP-conjugated mouse anti β-actin antibody (Santa Cruz Biotechnology, #SC-47778), used as a loading control. Secondary antibody incubation for CHOP was performed using 1:5,000 dilution (in Casein blocker) of an HRP conjugated goat anti-mouse IgG antibody (Thermo Fisher # A16066). Protein bands were visualized by Amersham ECL reagent (GE Healthcare, #RPN2232). For CHOP mRNA expression, total RNA was isolated from cell pellets using the RNeasy Mini Kit (Qiagen), following the manufacturer’s protocol. On-column digestion with RNase-free DNase (Qiagen) was performed to remove genomic DNA. DNase was removed in the subsequent washing steps. The expression levels of Ddit3 mRNA, encoding CHOP, were quantified relative to the endogenous control gene β-actin using pre-designed TaqMan gene expression assays (Applied Biosystems). Mean fold changes for each sample were calculated using the 2−ΔΔcycle threshold method as previously described[11].
2.4. Cholesterol quantification
BMDM or ESDM in 6-well dishes were cholesterol loaded as described above, and washed with PBS. Total and free cholesterol levels were assayed using a sensitive fluorescent method, and normalized to total cell protein, as previously described[11]. CE levels were calculated as total cholesterol - FC. Three to four wells were used for each condition.
3. Results
3.1. CHOP induced after cholesterol loading in AKR but not DBA/2 BMDM
BMDM derived from AKR and DBA/2 mice were incubated overnight with AcLDL in order to load them with cholesterol. CHOP protein induction was determined as an indicator of ER stress, which was observed by western blot only in the AKR macrophages after AcLDL loading (Figure 1). To determine if the DBA/2 macrophages were competent to induce CHOP using an alternative ER stress inducer, the BMDMs were incubated with tunicamycin, which specifically leads to the accumulation of unfolded proteins in the ER. CHOP was induced robustly by tunicamycin in BMDM from both strains (Figure 1), indicating that the defect in DBA/2 cells may be related more to the difference in cholesterol metabolism than to a difference in the ER stress pathways.
Figure 1. Induction of CHOP in AKR and DBA/2 BMDM.

Cells were incubated with 50 μg/ml AcLDL or with 1 μg/ml tunicamycin for 16 hrs. CHOP and the loading control β-actin were observed by western blot. Con, control; AcLDL, acetylated low-density lipoprotein; tun, tunicamycin; CHOP, C/EBP homologous protein.
We previously determined that AKR vs. DBA/2 BMDM have more FC and less CE after cholesterol loading due to a genomic deletion in the Soat1 gene in the AKR strain leading to an N-terminal truncation of the ACAT1 protein [9]. Thus, both strains of BMDM were incubated overnight with AcLDL in the absence or presence of an ACAT inhibitor. The ACAT inhibitor led to increased FC and decreased CE in the DBA/2 cells, while not significantly altering these levels in the AKR cells (Figure 2A). RNA was prepared after treating the BMDM in a similar fashion. Ddit3 mRNA, encoding CHOP, was determined by qPCR, and we found that Ddit3 expression was significantly increased by AcLDL loading only in AKR BMDM; however, in the presence of ACAT inhibitor Ddit3 mRNA was significantly induced in both AKR and DBA/2 BMDMs (Figure 2B). A similar finding was obtained for CHOP protein observed by western blot (Figure 2C). This result suggests that the accumulation of free cholesterol was required to induce ER stress, thus explaining the strain difference in CHOP induction by AcLDL loading.
Figure 2. Effects of ACAT inhibition on cholesterol loading and CHOP induction.
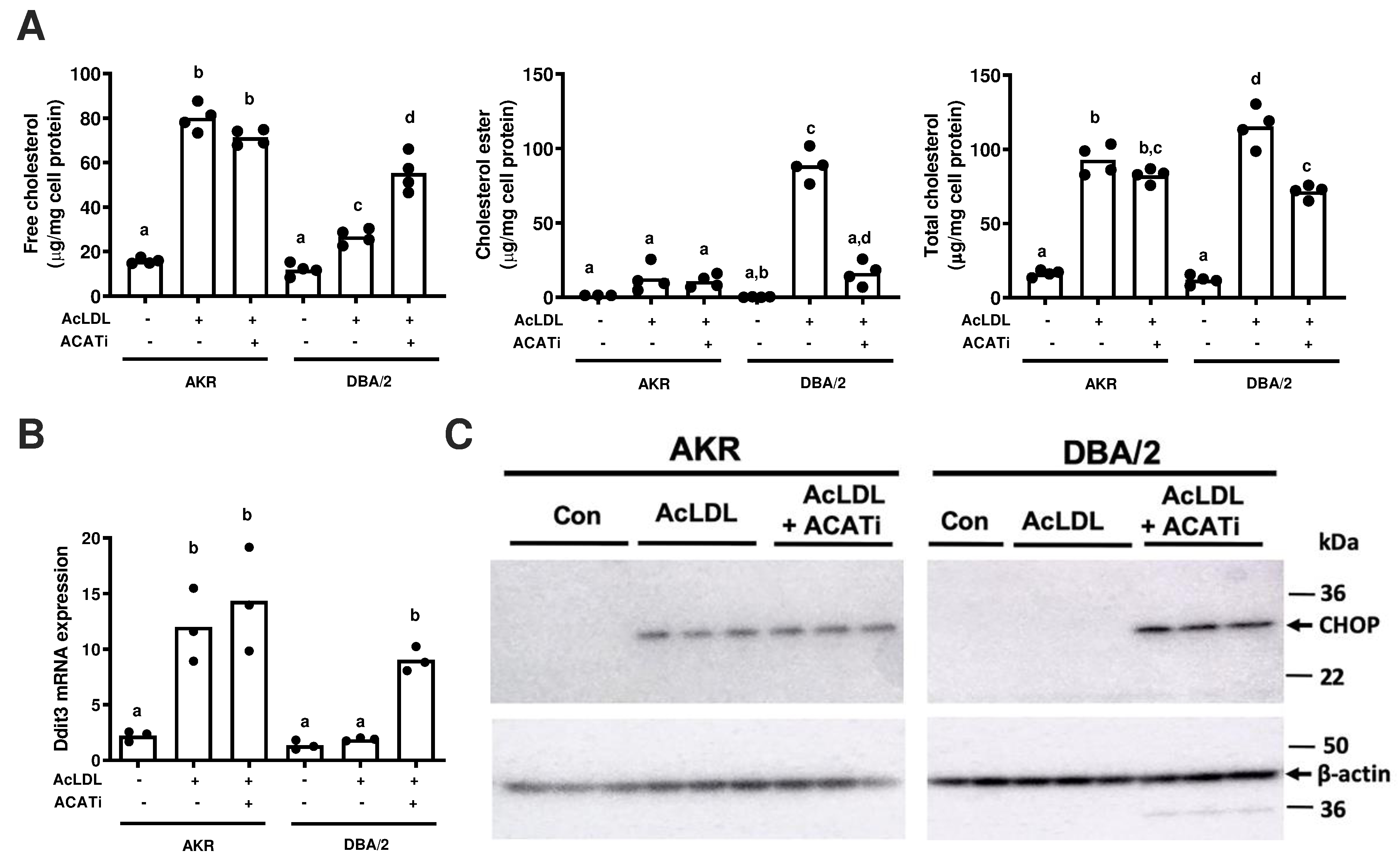
AKR and DBA/2 BMDM were incubated with 50 μg/ml AcLDL without or with the addition of 10 μg/ml Sandoz 58–035, an ACAT inhibitor, for 16–24 hrs. A. Free, esterified, and total cholesterol levels, normalized to cell protein. B. Ddit3 mRNA levels measured by qPCR. C. CHOP protein expression was observed by western blot, with β-actin as an internal control. AcLDL, acetylated low-density lipoprotein; ACATi, ACAT inhibitor. Statistical difference of <0.05 was determined by ANOVA with Tukey’s multiple comparison test and is shown by different letters above the bars.
3.2. Induction of CHOP in DBA/2 embryonic stem cell derived macrophages by AcLDL after Soat1 gene editing
CRISPR/Cas9 gene editing was performed in DBA/2 embryonic stem cells to replicate the AKR strain Soat1 exon 2 deletion[9]. Wild type and homozygous Soat1 edited (Δ) embryonic stem were differentiated into ESDM. After incubation with AcLDL, more FC accumulated in the Soat1 Δ vs. wild type ESDM (Figure 3A). This increase in FC loading was sufficient to induce Ddit3 mRNA and CHOP protein in DBA/2 Soat1 Δ ESDM after incubation with AcLDL (Figure 3B, C). Ddit3 mRNA and CHOP protein were induced in both WT and Δ ESDM after incubation with AcLDL and ACAT inhibitor (Figure 3B, C).
Figure 3. Induction of CHOP expression by AcLDL after Soat1 editing in DBA/2 embryonic stem cell derived macrophages.
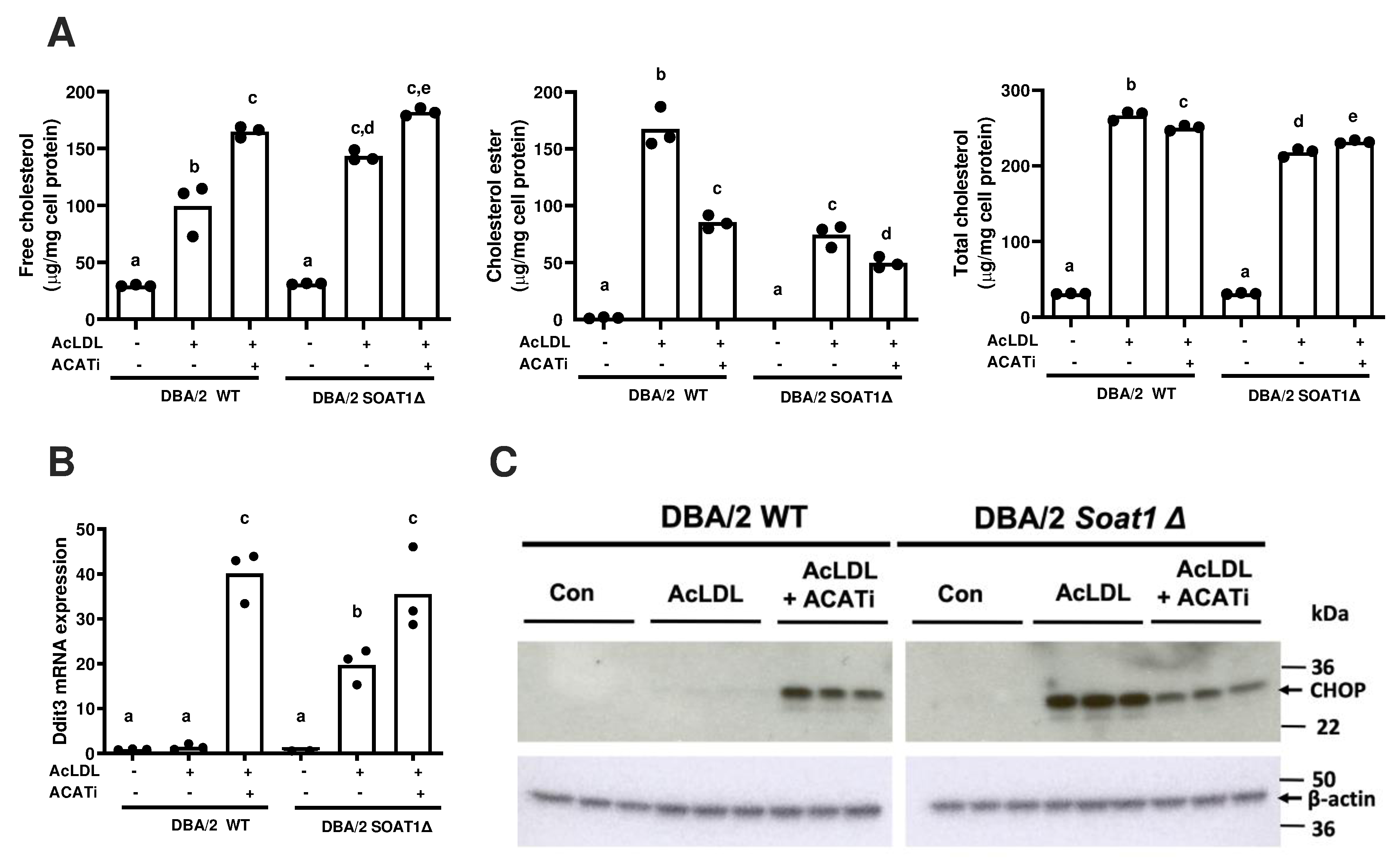
DBA/2 wildtype or Soat1 Δ DBA/2 ESDM were incubated with 50 μg/ml AcLDL without or with the addition 30 μg/ml ACAT inhibitor for 16–24 hrs. A. Free, esterified, and total cholesterol levels, normalized to total cell protein. B. Ddit3 mRNA levels measured by qPCR. C. CHOP protein expression was observed by western blot, with β-actin as an internal control. AcLDL, acetylated low-density lipoprotein; ACATi, ACAT inhibitor. Statistical difference of <0.05 was determined by ANOVA with Tukey’s multiple comparison test and is shown by different letters above the bars.
3.3. PERK and IRE1 pathways mediate AcLDL induction of CHOP
To find out the upstream molecules of CHOP induction, we applied inhibitors of PERK (GSK2606414), IRE1α (Kira 6), or ATF6 (AEBSF) to the DBA/2 Soat1 Δ ESDM treated with either AcLDL+ACAT inhibitor or tunicamycin. CHOP protein was induced by AcLDL+ACAT inhibitor; and, this induction most potently suppressed by the PERK inhibitor, moderately suppressed by the IRE1α inhibitor, and not suppressed at all by the ATF6 inhibitor (Figure 4). Tunicamycin induction of CHOP was suppressed most potently by the IRE1α inhibitor, and moderately suppressed by the PERK inhibitor (Figure 4).
Figure 4. Pathways involved with the induction of ER stress.

DBA/2 Soat1 Δ ESDM were incubated with 50 μg/ml AcLDL and 30 μg/ml ACAT inhibitor or with tunicamycin (1 μg/ml) for 16 hrs. CHOP and β-actin as a loading control were observed by western blot. Con, control; AcLDL, acetylated low-density lipoprotein; GSK, GSK2606414 PERK inhibitor; Kira 6, IRE1 inhibitor; AEBSF, ATF6 inhibitor.
4. Discussion
In the study, we investigated the mechanism by which two mouse strains respond differently to cholesterol loading in the induction of ER stress, as monitored by the expression of CHOP. The Ddit3 gene encodes CHOP, a transcription factor whose expression is upregulated by ER stress response[10]. We previously noted that the Ddit3 mRNA was highly upregulated by cholesterol loading with AcLDL in AKR BMDM, but not DBA/2 BMDM[4]. Here, we confirmed this result on the mRNA level, and extended it to show that CHOP protein was induced by AcLDL in AKR, but not DBA/2, BMDM. To exclude the possibility that DBA/2 BMDM were defective in mounting an ER stress response, we treated both strains with the ER stressor tunicamycin, and we determined that CHOP expression was induced in both AKR and DBA/2 cells.
When macrophages take up modified LDL, it is degraded in the lysosome, the LDL associated CE is hydrolyzed to FC, and the FC is transferred, in an NPC1 stimulated pathway to the endoplasmic reticulum, where it can be re-esterifed to CE by ACAT1 for storage in lipid droplets[7]. The CE can be re-hydrolyzed back to FC, completing the cholesterol ester cycle[13]. The regenerated FC can be used for cell membrane biogenesis or for cholesterol efflux in the reverse cholesterol transport pathway[14]. We previously showed that AKR vs. DBA/2 BMDM accumulate more FC and less CE after AcLDL treatment[8], and that this is due to an N-terminal truncation of ACAT1 protein caused by a genomic deletion of the exon 2 in the Soat1 gene[9]. Thus, we tested whether DBA/2 BMDM could induce CHOP upon AcLDL treatment in the presence of an ACAT inhibitor. Indeed, the ACAT inhibitor led the DBA/2 BMDM to accumulate more FC, similar to what was observed in the AKR cells in the absence of the ACAT inhibitor; and, this was accompanied by CHOP induction in the DBA/2 cells. Therefore, the strain difference in AcLDL mediated induction of CHOP was due to the amount of FC accumulation. In order to verify this, we utilized CRISPR/Cas9 gene editing to replicate the AKR strain Soat1 exon 2 deletion in DBA/2 embryonic stem cells. After differentiation into macrophages and treating with AcLDL, the Soat1 exon 2 Δ cells accumulated more FC and less CE compared to wild type DBA/2 macrophages, which resulted in a marked induction of CHOP expression. The novelty of our findings is that natural variation in different mouse strains in macrophage ACAT1 activity can lead to differential CHOP induction by AcLDL treatment, while prior studies on mouse macrophage induction of CHOP relied on pharmacological inhibition of ACAT1[5–7].
ACAT inhibition has long been recognized as an attractive therapeutic target for cardiovascular disease[15, 16]. However, global deficiency of Soat1 does not prevent the development of atherosclerosis in a mouse model, but led to side effects including xanthomas[17]. Myeloid specific Soat1 deficiency has yielded inconsistent outcomes with reports of both smaller and larger lesions [18, 19]. In addition, human clinical trials with ACAT inhibitors did not reduce lesion progression[20, 21]. Based on the current study, the potential therapeutic effect of ACAT inhibitors may be contra-indicated by the increase of intracellular FC and accompanying ER stress. If ER stress is not resolved it can lead to macrophage cell death, which in the contexts of an advanced atherosclerotic lesion is considered detrimental, as dead macrophages cannot emigrate out of the lesion or participate in cholesterol efflux/reverse cholesterol transport[6].
We also verified the upstream pathway of CHOP induction. Our results showed that PERK and IRE1 were essential for the AcLDL induction of CHOP, as inhibitors of these proteins blocked CHOP induction. Activated PERK phosphorylates eukaryotic translation initiation factor 2 alpha, which induces the activation of transcription factor-4(ATF4) [22, 23]. ATF4 has been shown to directly bind to the Ddit3 gene promoter by chromatin immunoprecipitation-next generation sequencing in mouse embryonic fibroblasts treated with tunicamycin [24]. Active IRE1 phosphorylates c-Jun N-terminal protein kinase-1(JNK1) and upregulates CHOP expression [23]. JNK1 inhibition has been shown to block CHOP induction by an ER-stress inducing drug in a gastric cancer cell line[25]. Although Yao et al. previously demonstrated that ATF6 was the upstream regulator of CHOP induction in macrophages that were cholesterol loaded by treatment with oxidized LDL [26], in our study using AcLDL loading, an ATF6 inhibitor did not block CHOP induction. Since both PERK and IRE1 pathways are required for CHOP induction by AcLDL, this implies that neither pathway alone is sufficient, but both must cooperate for this response.
The role of CHOP in atherosclerosis has been well studied. CHOP activation is a key step in cholesterol-induced ER stress as well as macrophage apoptosis[7]. When apoE-deficient mice were crossed to CHOP-deficient mice, there was ~30% reduction in aortic root lesion area with a reduction in necrotic lesion area, even after matching for total lesion area[27]. A similar finding was observed using LDL receptor deficient mice[27]. A separate group confirmed the effect of CHOP-deficiency to decrease brachiocephalic artery atherosclerosis modestly in apoE-deficient mice; and, they also observed that CHOP-deficiency decreased plaque rupture in a carotid artery ligation and cuff model[28]. Bone marrow transplantation was used to determine if the CHOP-deficiency effect was mediated through the donor myeloid lineage cells or through the host. Although the donor cells had a small, but not significant, effect on en face aortic lesion area, the host effect was significant, leading to the conclusion that endothelial or smooth muscle cell ER stress and CHOP induction have a stronger atherosclerosis promoting effect than ER stress in macrophages[29]. To unravel this, vascular smooth muscle cell specific CHOP-deficient mice were bred onto the LDL receptor-deficient background, and both total lesion and necrotic area were decreased by vascular smooth muscle CHOP-deficiency, associated with increased Klf4 expression and less smooth muscle cell proliferation [30]. It is of interest that upon crossing onto the apoE-deficient background, the AKR mouse strain, with less ACAT1 activity and increased ER stress and CHOP expression after cholesterol loading, is one of the several inbred mouse strains that have small lesions, especially compared to the C57Bl/6 and DBA/2 strains[2, 3]. Thus, genetic factors other than ACAT1 and its downstream effect on ER stress and CHOP induction play a role in suppressing atherosclerosis in the AKR strain, as its high level of macrophage CHOP after cholesterol loading would be expected to promote atherosclerosis.
5. Conclusion
The genetic alteration in the Soat1 gene in AKR macrophages is sufficient to induce the ER stress response after cholesterol loading with AcLDL, confirming the role of FC in the ER stress induction of CHOP expression in macrophages. This explains our prior differential response to AcLDL in mediating gene expression comparing macrophages derived from the AKR and DBA/2 mouse strains.
Highlights.
AKR and DBA/2 mouse strains differ in many phenotypes including atherosclerosis
CHOP induced by cholesterol loading in AKR but not DBA/2 macrophages
Difference is due to Soat1 gene truncation in AKR strain, proven by gene editing
Soat1 gene truncation leads to more free vs. esterified cholesterol accumulation
Natural variation in the Soat1 gene among mouse strains is sufficient to lead to CHOP induction without pharmacological inhibition of ACAT1
Funding
This research was supported by National Institutes of Health [grant P01HL029582] to J.D.S., by American Heart Association Scientist Development Grant [#15SDG25310009] to P.R., and by China Scholarship Council to M.L.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data statement
All original data and full western blot scans are available upon request to the corresponding author.
References
- 1.Lu H and Daugherty A, Atherosclerosis. Arterioscler Thromb Vasc Biol, 2015. 35(3): p. 485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith JD, et al. , In silico quantitative trait locus map for atherosclerosis susceptibility in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol, 2003. 23(1): p. 117–22. [DOI] [PubMed] [Google Scholar]
- 3.Smith JD, et al. , Atherosclerosis susceptibility loci identified from a strain intercross of apolipoprotein E-deficient mice via a high-density genome scan. Arterioscler Thromb Vasc Biol, 2006. 26(3): p. 597–603. [DOI] [PubMed] [Google Scholar]
- 4.Berisha SZ, et al. , Transcriptome analysis of genes regulated by cholesterol loading in two strains of mouse macrophages associates lysosome pathway and ER stress response with atherosclerosis susceptibility. PLoS One, 2013. 8(5): p. e65003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devries-Seimon T, et al. , Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J Cell Biol, 2005. 171(1): p. 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabas I, Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol, 2005. 25(11): p. 2255–64. [DOI] [PubMed] [Google Scholar]
- 7.Feng B, et al. , The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol, 2003. 5(9): p. 781–92. [DOI] [PubMed] [Google Scholar]
- 8.Robinet P, Ritchey B, and Smith JD, Physiological difference in autophagic flux in macrophages from 2 mouse strains regulates cholesterol ester metabolism. Arterioscler Thromb Vasc Biol, 2013. 33(5): p. 903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hai Q, et al. , Quantitative Trait Locus Mapping of Macrophage Cholesterol Metabolism and CRISPR/Cas9 Editing Implicate an ACAT1 Truncation as a Causal Modifier Variant. Arterioscler Thromb Vasc Biol, 2018. 38(1): p. 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ron D and Walter P, Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol, 2007. 8(7): p. 519–29. [DOI] [PubMed] [Google Scholar]
- 11.Robinet P, et al. , A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J Lipid Res, 2010. 51(11): p. 3364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basu SK, et al. , Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc Natl Acad Sci U S A, 1976. 73(9): p. 3178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown MS, Ho YK, and Goldstein JL, The cholesteryl ester cycle in macrophage foam cells. Continual hydrolysis and re-esterification of cytoplasmic cholesteryl esters. J Biol Chem, 1980. 255(19): p. 9344–52. [PubMed] [Google Scholar]
- 14.Wang MD, et al. , Different cellular traffic of LDL-cholesterol and acetylated LDL-cholesterol leads to distinct reverse cholesterol transport pathways. J Lipid Res, 2007. 48(3): p. 633–45. [DOI] [PubMed] [Google Scholar]
- 15.Kusunoki J, et al. , Acyl-CoA:cholesterol acyltransferase inhibition reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation, 2001. 103(21): p. 2604–9. [DOI] [PubMed] [Google Scholar]
- 16.Rong JX, et al. , ACAT inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arterioscler Thromb Vasc Biol, 2013. 33(1): p. 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Accad M, et al. , Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest, 2000. 105(6): p. 711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang LH, et al. , Myeloid Acyl-CoA:Cholesterol Acyltransferase 1 Deficiency Reduces Lesion Macrophage Content and Suppresses Atherosclerosis Progression. J Biol Chem, 2016. 291(12): p. 6232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakabayashi T, et al. , Inflammasome Activation Aggravates Cutaneous Xanthomatosis and Atherosclerosis in ACAT1 (Acyl-CoA Cholesterol Acyltransferase 1) Deficiency in Bone Marrow. Arterioscler Thromb Vasc Biol, 2018. 38(11): p. 2576–2589. [DOI] [PubMed] [Google Scholar]
- 20.Nissen SE, et al. , Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med, 2006. 354(12): p. 1253–63. [DOI] [PubMed] [Google Scholar]
- 21.Meuwese MC, et al. , ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: the CAPTIVATE randomized trial. JAMA, 2009. 301(11): p. 1131–9. [DOI] [PubMed] [Google Scholar]
- 22.Tabas I, The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res, 2010. 107(7): p. 839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cubillos-Ruiz JR, Bettigole SE, and Glimcher LH, Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell, 2017. 168(4): p. 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J, et al. , ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol, 2013. 15(5): p. 481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim TW, et al. , Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis, 2018. 9(9): p. 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao S, et al. , Activating transcription factor 6 mediates oxidized LDL-induced cholesterol accumulation and apoptosis in macrophages by up-regulating CHOP expression. J Atheroscler Thromb, 2013. 20(1): p. 94–107. [DOI] [PubMed] [Google Scholar]
- 27.Thorp E, et al. , Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab, 2009. 9(5): p. 474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsukano H, et al. , The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol, 2010. 30(10): p. 1925–32. [DOI] [PubMed] [Google Scholar]
- 29.Gao J, et al. , Involvement of endoplasmic stress protein C/EBP homologous protein in arteriosclerosis acceleration with augmented biological stress responses. Circulation, 2011. 124(7): p. 830–9. [DOI] [PubMed] [Google Scholar]
- 30.Zhou AX, et al. , C/EBP-Homologous Protein (CHOP) in Vascular Smooth Muscle Cells Regulates Their Proliferation in Aortic Explants and Atherosclerotic Lesions. Circ Res, 2015. 116(11): p. 1736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]