

Abstract
Persistent neutrophilic inflammation is a hallmark of cystic fibrosis (CF). However, the mechanisms underlying this outstanding pathology remain incompletely understood. Here we report that CFTR in myeloid immune cells plays a pivotal role in control of neutrophilic inflammation. Myeloid CFTR-Knockout (Mye-Cftr−/−) mice and congenic wild-type (WT) mice were challenged peritoneally with zymosan particles at different doses, creating aseptic peritonitis with varied severity. A high-dose challenge resulted in significantly higher mortality in Mye-Cftr−/− mice, indicating an intrinsic defect in host control of inflammation in mice whose myeloid cells lack CFTR. The low-dose challenge demonstrated an impaired resolution of inflammation in Mye-Cftr−/− mice, reflected by a significant over-production of pro-inflammatory cytokines, including neutrophil chemokines MIP-2 and KC, and sustained accumulation of neutrophils. Tracing neutrophil mobilization in vivo demonstrated that myeloid CF mice recruited significantly more neutrophils than did WT mice. Pulmonary challenge with zymosan elicited exuberant inflammation in the lung and recapitulated the findings from peritoneal challenge. To determine the major type of cell that was primarily responsible for the over-recruitment of neutrophils, we purified and cultured ex vivo zymosan-elicited peritoneal neutrophils and macrophages. The CF neutrophils produced significantly more MIP-2 than did the WT counterparts, and peripheral blood neutrophils isolated from myeloid CF mice also produced significantly more MIP-2 after zymosan stimulation in vitro. These data altogether suggest that CFTR dysfunction in myeloid immune cells, especially neutrophils, leads to hyper-inflammation and excessive neutrophil mobilization in the absence of infection. Thus, dysregulated inflammation secondary to abnormal or absent CFTR in myeloid cells may underlie the clinically observed neutrophilic inflammation in CF.
GRAPHIC ABSTRACT
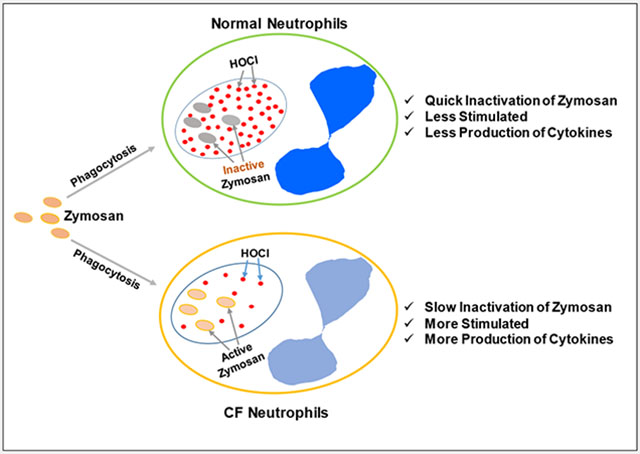
Cystic fibrosis (CF) neutrophils are deficient in hypochlorous acid (HOCl) production, leading to slow inactivation of inflammatory agonists, prolonged stimulation, more production of cytokines, and greater inflammation.
Introduction
Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator gene (cftr) that encodes CFTR, a cAMP-activated chloride channel [1]. The most severe and life-threatening pathology occurs in the lung where chronic bacterial infection, persistent neutrophilic inflammation and purulent small airway obstruction lead to bronchiectasis and pulmonary failure [2]. The lungs of CF newborns are free from inflammation [3, 4], suggesting that CF lung inflammatory disease is acquired after birth. As infection and inflammation are highly associated, it is difficult to study CF lung inflammation without the interference of infection by using the commonly used lung infection model. Thus, outstanding questions remain in the field. First, is the CF lung hyper-inflammation caused by host hyper-responsiveness to stimulation or by unrestrained pathogen infection secondary to defective local host defense? Second, what is the primary mechanism underlying the CF-characteristic neutrophilic inflammation? Third, which type of cell in the lung is mainly responsible for the neutrophilic inflammation? Answering these questions is critical to a complete understanding of CF pathogenesis and to rational design of effective therapies for the disease.
In CF lungs, the predominant inflammatory cells are neutrophils. Excessive neutrophil presence indicates that patients are capable of mounting an aggressive inflammatory response [5]. Neutrophils are dedicated microbial killer cells that phagocytose invading organisms and employ both pre-made and de-novo produced toxic agents to attack ingested microbes [6]. The most efficient mechanism employed by neutrophils to kill organisms within phagosomes relies on hypochlorous acid (HOCl), generated by the action of the azurophil granule protein myeloperoxidase (MPO) [7] to oxidize chloride anion in the presence of hydrogen peroxide (H2O2) [8, 9]. Optimal HOCl generation requires a ready source of H2O2, MPO, and chloride anion, and CFTR serves as a key transporter of chloride from cytoplasm to phagosomes [10, 11]. Thus, CFTR dysfunction limits chloride supply to this organelle, compromising HOCl production and microbial killing by neutrophils [12, 13]. Such a host defense defect may be further deteriorated in the CF-diseased airway microenvironment [14]. In addition to their central role in antimicrobial host defense, neutrophil-derived oxidants are essential to the regulation of inflammatory responses [15]. NADPH oxidase-deficient mice encounter greater neutrophil infiltration and inflammation than do WT mice when aseptic inflammation is created by intraperitoneal injection with zymosan [16, 17]. Interestingly, mice with MPO deficiency, when exposed to zymosan intranasally, also demonstrate greater neutrophil infiltration and inflammation relative to WT mice [18], suggesting the involvement of MPO and its derived HOCl oxidant in suppression of inflammation. Research from our laboratory found that experimental Pseudomonas infection induces sustained neutrophilic inflammation in the lungs of Mye-Cftr−/− mice [19], which lack CFTR only in cells of myeloid origin. However, CF mice have defective microbicidal activity and clearance, thus resulting in sustained stimulation of host defense by the surviving bacteria. Consequently, we could not determine in that experimental setting if the observed neutrophilic inflammation reflected sustained microbial stimulation or failure to initiate resolution of the inflammatory response.
In the current study, we induced aseptic inflammation by administration of sterile zymosan particles into the peritoneal cavities or the lungs of Mye-Cftr−/− and WT mice. Under identical levels of stimulation, degrees of inflammation in these mice were compared to determine if CFTR dysfunction in myeloid immune cells per se predisposes the host to neutrophilic inflammation even in the absence of infection.
Materials and Methods
Chemicals and Reagents:
Zymosan A from Saccharomyces cerevisiae was purchased from Sigma-Aldrich (St. Louis, MO). Anti-mouse Ly-6G-APC antibody and its isotypic control were from BioLegend (San Diego, CA), and anti-mouse F4/80-PE-Cy7 antibody and its isotypic control were from eBioscience (ThermoFisher Scientific, Waltham, MA). Mouse cytokine ELISA kits were obtained from R&D Systems (Minneapolis, MN).
Animals:
All procedures involved with animals were reviewed and approved by the Louisiana State University Health Sciences Center Institutional Animal Care and Use Committee. The breeders with Cftr-Exon-10 floxed, referred to as Cftrfl10, were orignally created by Hodges et al. [20]. Cftrfl10 homozygous mice carrying the LysM-Cre transgene were generated to produce myeloid CFTR-inactivated (Mye-Cftr−/−) mice, as we published previously [19]. The sibling Cftrfl10 homozygotes with no LysM-Cre gene were used as the wild-type (WT) control animals.
Zymosan preparation and induction of peritonitis and pneumonia:
Zymosan dry particles were resuspended in water, boiled for 30 min, sonicated and thoroughly washed for use. For induction of peritonitis, a high dose (1 g/kg) or a low dose (0.7 g/kg) of the prepared zymosan was used to inject intraperitoneally. The high dose produced a lethality of ~50% in myeloid CF mice in a pilot experiment. To compare inflammogenicity of non-treated and HOCl-treated zymosan, zymosan preparations were first incubated with PBS or 5 mM HOCl for 10 minutes at room temperature. After washing twice, the conditioned zymosan particles (1 g/kg) were injected into Mye-Cftr−/− mice for survival test. For induction of pneumonia, a high dose (50 mg/kg) or a low dose (25 mg/kg) of the prepared zymosan was introduced intratracheally.
Peritoneal lavage and bronchoalveolar lavage:
For peritoneal lavage, assigned animals were sacrificed by CO2 asphyxiation, and 2 ml of flushing solution (PBS containing proteinase inhibitors) was injected intraperitoneally into each mouse. After repeated abdominal massage, peritoneal lavage fluid (1.5 ml) was recovered, and spun at 400x g for 10 minutes. The cell pellet was resuspended for cell differential counting. The supernatant was stored at −80°C for cytokine measurements. For bronchoalveolar lavage, the trachea of each sacrificed animal was cannulated, and intubated with 1 ml of the flushing solution. After holding for 1 minute for equilibration, the lavage fluid was recovered and spun to separate the cell and liquid components for further tests as for the peritoneal lavage fluid.
Cell differential counting:
Each cell pellet from lavage was resuspended in PBS, diluted appropriately, and cytospun onto a microscopic slide, followed by Wright-Giemsa staining. Of each slide, multiple random fields were examined microscopically and greater than 100 leukocytes were surveyed to determine cell differential.
Isolation and ex vivo culture of peritoneal neutrophils and macrophages:
WT and Mye-Cftr−/− mice were intraperitoneally challenged with 0.7 g/kg zymosan for different time points as indicated. Total peritoneal lavage cells were obtained, and immunostained with Anti-mouse Ly-6G-APC and Anti-mouse F4/80-PE-Cy7 antibodies. Neutrophils and macrophages were then sorted by FACS, and cultured ex vivo in RPMI supplemented with 10% fetal calf serum. The culture media were sampled at different time points for ELISA determination of MIP-2 and KC levels.
Isolation and in vitro activation of peripheral blood neutrophils and monocytes
Peripheral blood neutrophils and monocytes from WT and Mye-Cftr−/− mice were isolated by Percoll-gradient centrifugation according to our previously published procedure [19]. The purified cells were, respectively, cultured in RPMI supplemented with 10% fetal calf serum, and incubated with 50 µg/ml zymosan particles which had been pre-opsonized by 10% mouse normal serum. At different time points, as indicated, culture media were sampled for MIP-2 and KC measurements by ELISA measurement.
Proinflammatory cytokine measurements by ELISA:
ELISA measurements of TNF-α, IL-6, IL-1β, KC and MIP-2 were carried out using the commercial kits from R&D Systems according to the manufacturer’s instructions (R & D Systems). Pilot tests were performed first to determine the optimal dilution of each original sample.
Comparisons of neutrophil phagocytosis rate in vitro and in vivo.
For in vitro phagocytosis assay, 1×106 neutrophils isolated from peripheral blood were mixed with 25 µg pre-opsonized pHrodo green zymosan A (Life technologies) in Ringer’s buffer in the presence or absence of myeloperoxidase inhibitor 4-amino-benzoic acid hydrazide (ABHA) or sodium azide (NaN3). After phagocytosis for 1 hour, the cells were fixed in 4% paraformaldehyde and immunostained with the anti-Ly6G antibody. Fluorescence intensity of internalized zymosan in the Ly6G-positive cells was determined by flow cytometry. For in vivo phagocytosis assay, pHrodo green zymosan A (0.1 g/kg) was intraperitoneally injected into WT and Mye-Cftr−/− mice, respectively. Twenty-four hours post-injection, peritoneal lavage cells were harvested and immunostained with the Ly6G antibody. Phagocytosis rates were determined similarly by flow cytometry.
Bone marrow cell labeling and neutrophil mobilization assay:
Mye-Cftr−/− mice were sacrificed, and their bone marrow cells were flushed out. After red blood cell lysis, the nucleated cells were cultured in RPMI supplemented with 10% fetal calf serum. CellTracker™ Green CMFDA (5-chloromethylfluorescein diacetate; 4 µM), a live cell-labeling dye, was added to the cells for 30 minutes. The fluorescently labeled cells (1×107) were infused via tail veins into WT and Mye-Cftr−/− mice that had been pre-challenged with intraperitoneal administration of zymosan (0.7 g/kg) for 24 hours. Six hours post cell injection, peritoneal cells were lavaged out and immunostained with the Ly6G antibody. The CMFDA-labeled Ly6G-positive cells were determined by flow cytometry. Neutrophil recruitment was compared between the zymosan-challenged WT and Mye-Cftr−/− mice.
Statistics:
For mortality comparison, Log-rank (Mantel-Cox) test was used. For other statistical comparisons, Student’s t-test was employed. P-value smaller than 0.05 was considered significantly different statistically.
Results
Myeloid CF mice are impaired in protection against zymosan-induced peritonitis.
Myeloid CF mice (Mye-Cftr−/−) were produced by breeding Cftr Exon-10-floxed mice [20] with LysMCre transgenic mice that had Lysozyme 2 gene (Lyz2) promoter-driving Cre expression [21]. These myeloid CF mice have CFTR deletion specifically and selectively in myeloid lineages (monocytes, mature macrophages and granulocytes), as previously confirmed [19, 22]. CFTR expression is normal in non-myeloid cells in the myeloid CF mice. In order to study CF inflammation in the absence of infection, we selected the zymosan-induced aseptic peritonitis model. Myeloid CF mice and their congenic WT mice were challenged intraperitoneally with sterile zymosan particles (1g/kg). Approximately 65% of Mye-Cftr−/− mice and 100% of WT mice survived the peritoneal challenge (Fig. 1A). The survived animals were continuously observed for 2 weeks post-challenge, and no additional deaths were observed. These data indicate that myeloid CFTR dysfunction led to intrinsic defect in host protection against severe inflammation.
Figure 1 – Kaplan-Meier survival curves.
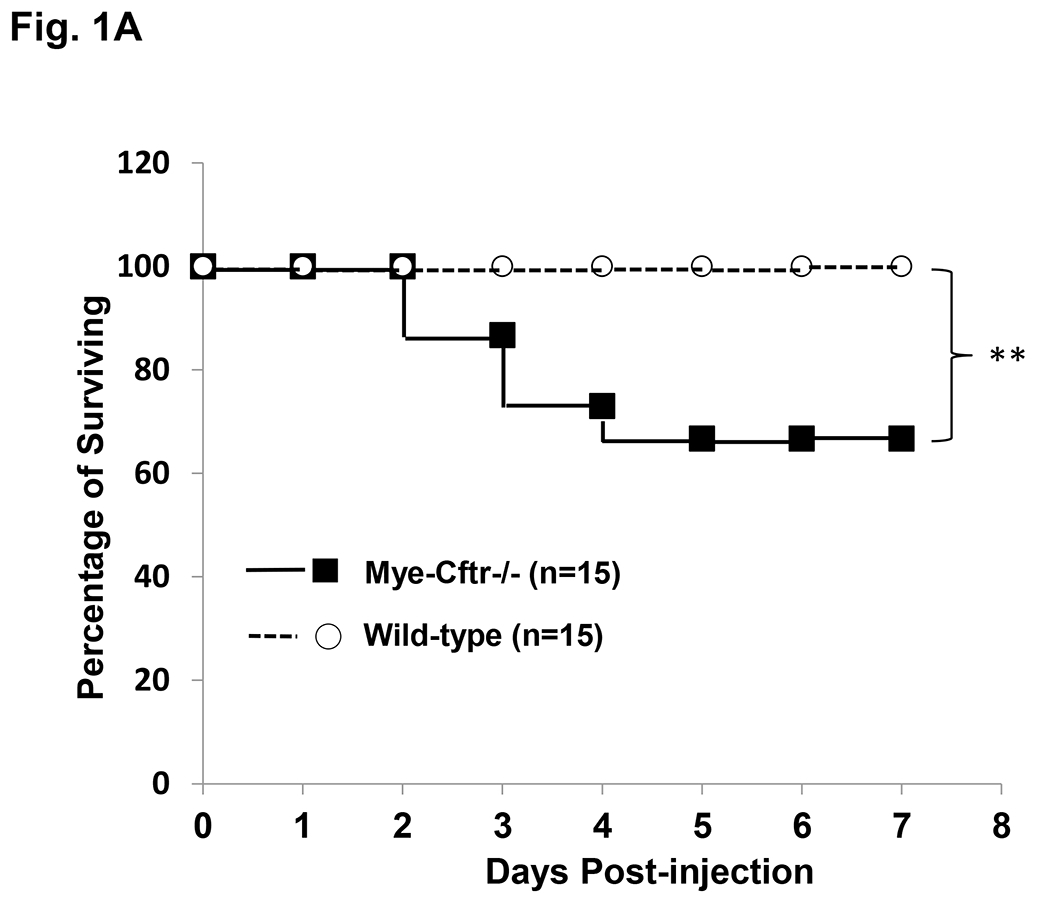
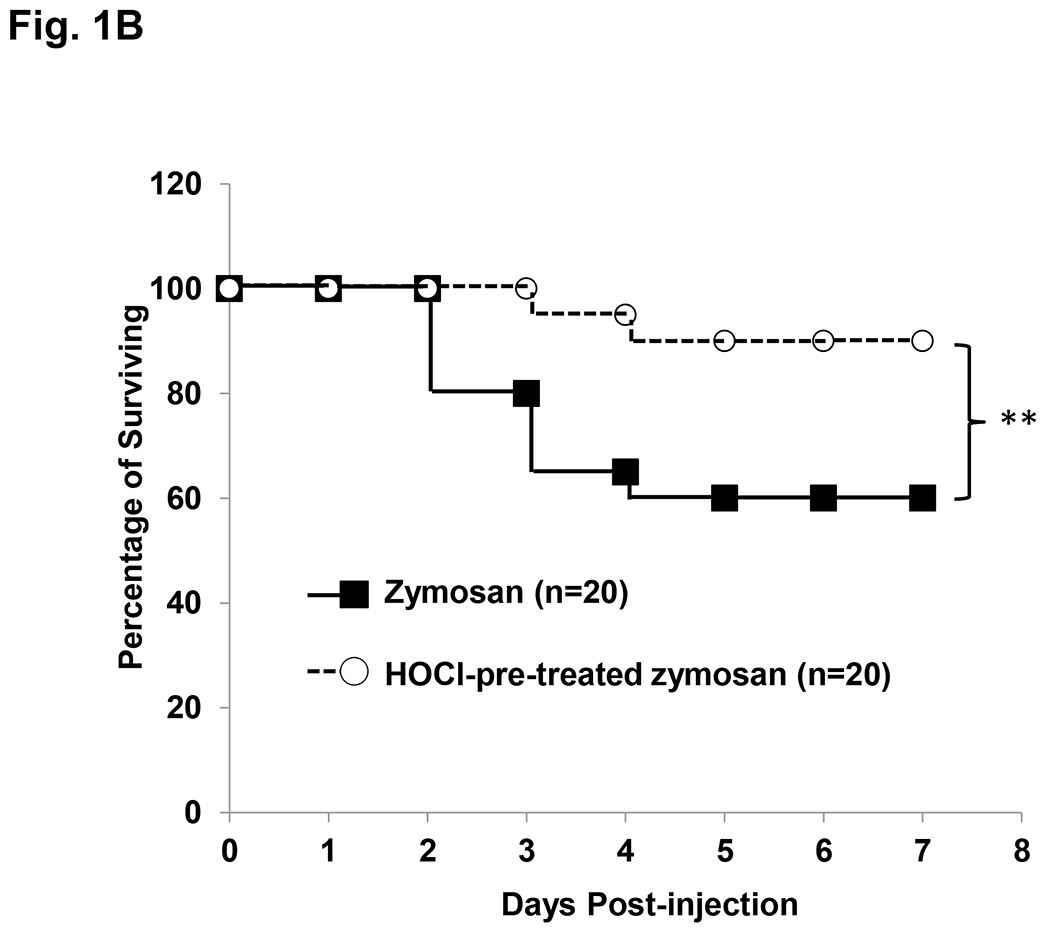
A) Survival comparison between WT and Mye-Cftr−/− mice. WT mice and Mye-Cftr−/− mice were intraperitoneally challenged with sterile zymosan particles at a dose of 1 g/kg. Casualties were recorded and survival curves were traced. The log-rank test was performed to compare the survival rates. Double asterisks indicate a significant difference (P<0.01, n=15). B) Survival comparison of Mye-Cftr−/− mice receiving non-treated or HOCl-treated zymosan challenge. The log-rank test was conducted. Double asterisks indicate a significant difference (P<0.01, n=20).
Because CFTR chloride channel dysfunction renders CF neutrophils unable to generate HOCl [12, 19], we reasoned that Mye-Cftr−/− mice would be unable to use HOCl to disable zymosan inflammogenicity and thus suffer more severe peritonitis in our experimental model. If these two predictions were correct, pre-exposure of zymosan to HOCl should eliminate the exaggerated inflammation seen in CF mice. We thus pre-treated zymosan particles with 5 mM HOCl for 10 minutes. After repeated washes, the non-treated control zymosan or the HOCl-treated zymosan particles (1 g/kg) were used to challenge Mye-Cftr−/− mice intraperitoneally. Survival curves for both conditions show that HOCl-pre-exposed zymosan lost its inflammogenicity and its lethal effect in the myeloid CF mice (Fig. 1B), while the non-treated zymosan resulted in a similar mortality rate as in Fig. 1A. These data clearly suggest that HOCl is a key reactive oxygen species (ROS) that ablates the inflammatory properties of zymosan, thereby preventing protracted stimulation and prolonged response.
Myeloid CFTR dysfunction leads to persistent neutrophilic inflammation.
To confirm that myeloid CFTR loss-of-function affects host resolution of inflammation, we intraperitoneally challenged Mye-Cftr−/− mice and WT mice with a low dose of zymosan (0.7 g/kg, i.p.). At Days 0, 1, 3 and 5, the designated animals were sacrificed, and peritoneal inflammatory cells were collected and examined. Cell differential counting (Fig. 2) demonstrated that at Day 0 before zymosan administration, over 80% of the peritoneal lavage cells were macrophages in both Mye-Cftr−/− and WT mice. However, after zymosan challenge, the population of cells recruited to the peritonea became highly neutrophilic. Importantly, the inflammatory profile in Mye-Cftr−/− mice was persistently neutrophil-predominant throughout the 5 days, while WT mice had transitioned to macrophage-dominant inflammation by Days 3 and 5, consistent with normal resolution. These data demonstrate that myeloid CFTR dysfunction predisposed the host to persistent neutrophilic inflammation even in the absence of infection.
Figure 2 – Zymosan-induced peritoneal inflammation in WT and Mye-Cftr−/− mice.
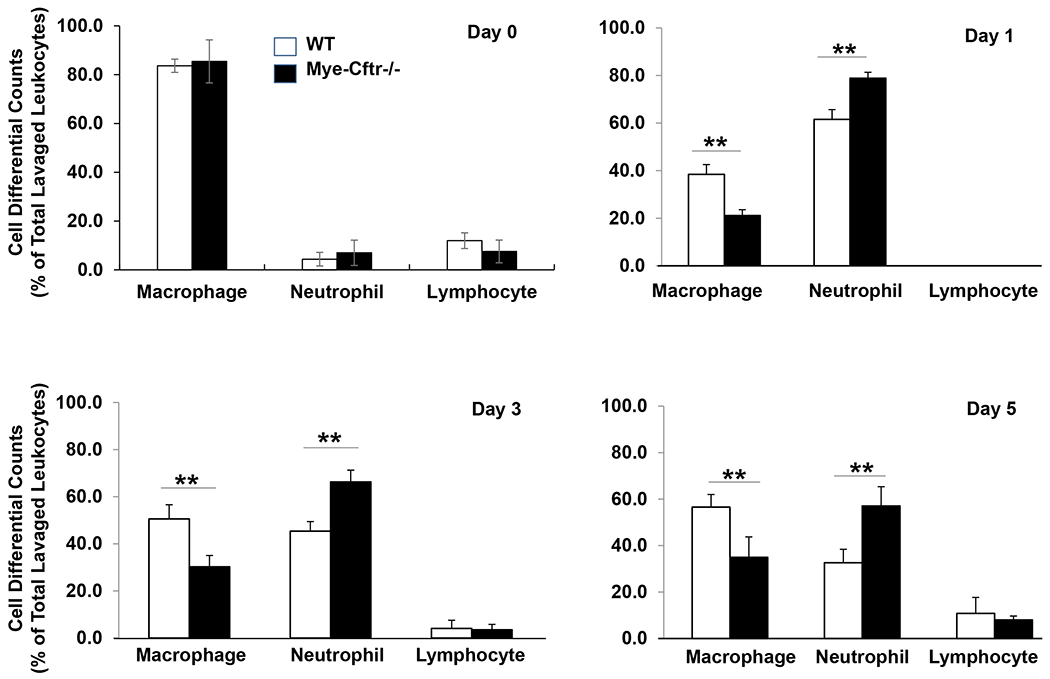
WT mice and Mye-Cftr−/− mice were challenged with a sub-lethal dose of zymosan (0.7 g/kg, i.p.). At different time points after challenge (Days 1, 3 and 5), inflammatory cell differentials were determined. Percentage of macrophages, neutrophils or lymphocytes in peritoneal inflammatory cells at each time point. Double asterisks denote significant difference by Student’s t-test (P<0.01, n=4).
Myeloid CF mice produce significantly more inflammatory cytokines than WT mice in response to a comparable level of stimulation.
To investigate if myeloid CFTR loss-of-function renders the host hyper-responsive to stimulation, which may result in excessive neutrophil recruitment, we challenged Mye-Cftr−/− and WT mice intraperitoneally with the sub-lethal zymosan dose (0.7 g/kg). At Days 0, 1, 3 and 5, peritoneal lavage fluids were assessed for the presence of major pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, KC and MIP-2) by ELISA. At Day 0 before zymosan challenge, all mice had negligibly low cytokine levels. TNF-α, IL-6 and KC were below their detection thresholds. MIP-2 was ~23 pg/ml and IL-1β ~45 pg/ml for both strains of mice. However, after challenge, significantly high levels of these cytokines were produced by both types of mice. Fig. 3A–E show that all the tested cytokines from the myeloid CF mice were significantly higher than those from WT mice at Day 1, MIP-2, KC and TNF-α higher at Day 3, and MIP-2 at Day 5. These data suggest that myeloid CF mice were intrinsically hyper-responsive to stimulation. Noteworthy is that MIP-2, a key neutrophil-recruiting chemokine, was consistently higher in Mye-Cftr−/− mice at all time-points. To determine if over-production of neutrophil chemokines in myeloid CF mice results in an excessive neutrophil recruitment, we performed an in-vivo neutrophil mobilization assay. Bone marrow nucleated cells from Mye-Cftr−/− mice were isolated, labelled with CellTracker™ Green CMFDA, and infused intravenously into Mye-Cftr−/− or WT mice that had been pre-challenged with sub-lethal zymosan (0.7 g/kg, i.p.) for 24 hours. This experimental approach examines how the stimulated peritoneal environment in WT or Mye-Cftr−/− mice mobilized CF neutrophils. Six hours after cell transfusion, peritoneal inflammatory cells were harvested, immunostained with the neutrophil-specific Ly6G antibody, and analyzed by flow cytometry. The number of mobilized neutrophils with CMFDA and Ly6G double-positivity was measured as a fraction of the total number of infused cells. The result (Fig. 3F) shows that myeloid CF mice had significantly more neutrophil recruitment as compared to that in the WT mice.
Figure 3 – Cytokine levels in peritoneal lavage fluid from WT and Mye-Cftr−/− Mice.
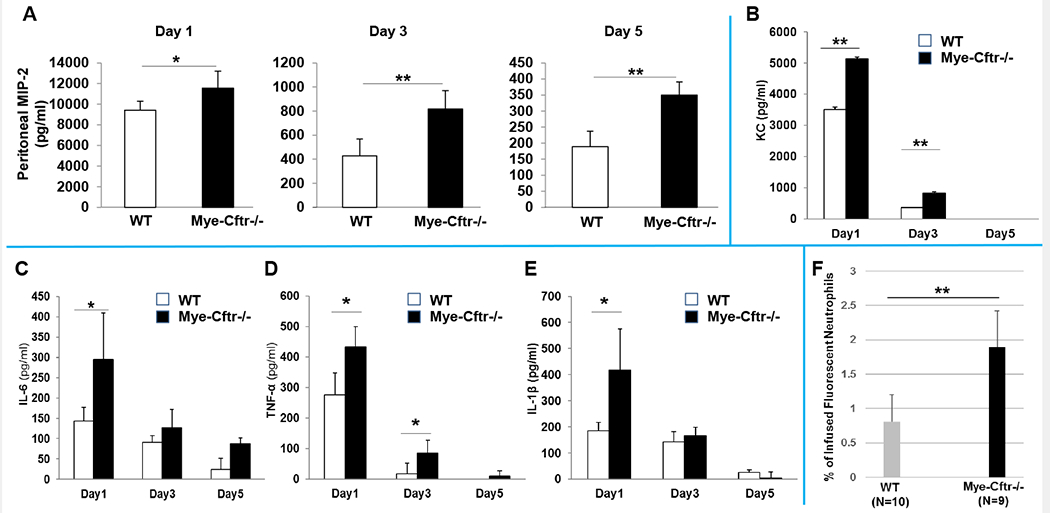
WT and Mye-Cftr−/− mice were challenged with zymosan (0.7g/kg, i.p.). Peritoneal lavage fluid was collected from each assigned animal for Days 1, 3 & 5 post-challenge using 2 ml of PBS. Multiple pro-inflammatory cytokines (A: MIP-2; B: KC; C: IL-6; D: TNF-α; E: IL-1β) were measured using ELISA. Mye-Cftr−/− mouse lungs were more inflamed and produced significantly higher levels of pro-inflammatory cytokines at different time points after zymosan challenge. F) Neutrophil recruitment assay. Bone marrow nucleated cells (1×107) from Mye-Cftr−/− mice were fluorescently labeled and infused via tail veins into WT or Mye-Cftr−/− mice that had been pre-challenged with intraperitoneal administration of zymosan (0.7 g/kg) for 24 hours. Six hours post cell injection, peritoneal cells were lavaged out and immunostained with the Ly6G antibodyand transfused into either WT or Mye-Cftr−/− mice that had been conditioned by peritoneal zymosan challenge. Fluorescent neutrophils recruited to the peritoneal cavity of each tested animal were determined by flow cytometry. Student’s t-test was used to judge any difference in neutrophil recruitment between the two mice (P<0.05, n=4).
Myeloid CF mice exhibit more severe inflammation than WT mice after zymosan pulmonary challenge.
In addition to peritoneal challenge, we further asked if a pulmonary challenge with zymosan recapitulates the peritoneal findings. Mye-Cftr−/− and WT mice were instilled intratracheally with different doses of zymosan. The lung was found more sensitive to zymosan than the peritoneum. A dose of 50 mg/kg zymosan challenge resulted in significantly higher mortality in Mye-Cftr−/− mice than in WT mice, and the difference in mortality paralleled that of the peritoneal challenge (data not shown). Moreover, a low dose (25 mg/kg) gave rise to persistent neutrophilic lung inflammation (Fig. 4A) and significantly more neutrophil-recruiting chemokines MIP-2 and KC (Fig. 4B–C).
Figure 4 -. Zymosan-induced pulmonary inflammation in WT and Mye-Cftr−/− mice.
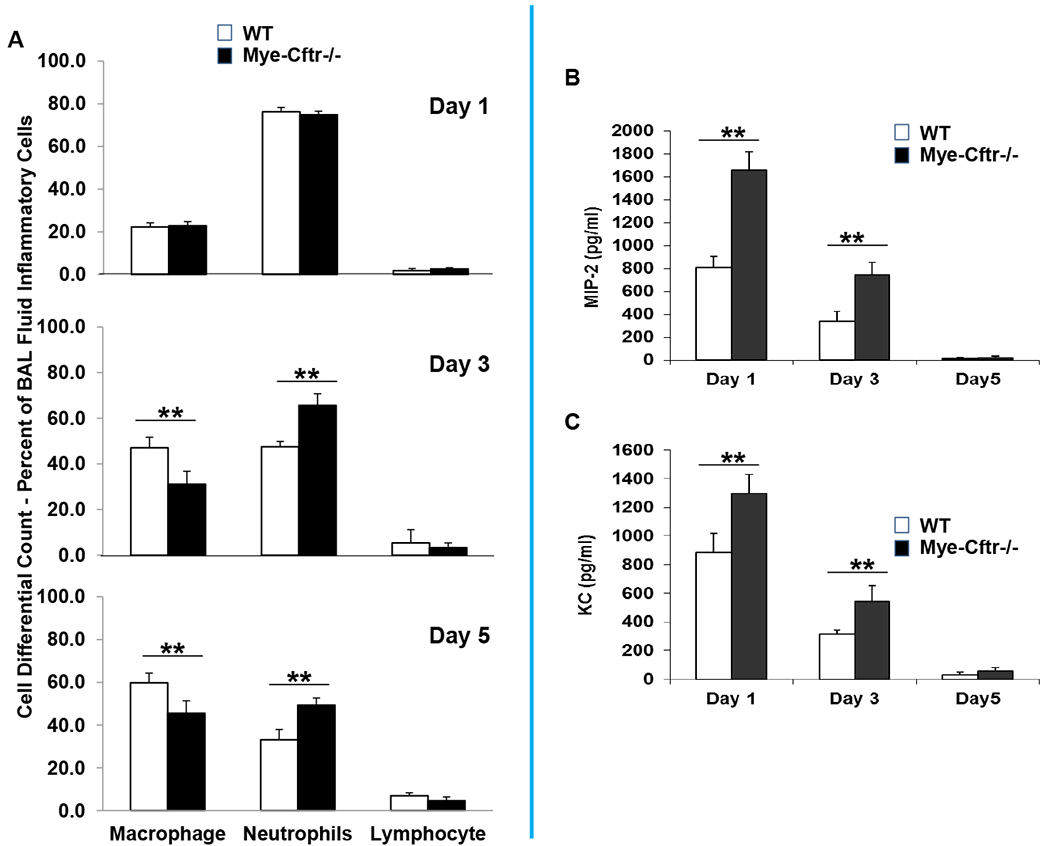
WT and Mye-Cftr−/− mice were challenged with a sub-lethal dose of zymosan (25 mg/kg) intratracheally. At the different time points after challenge (Days 1, 3 and 5), inflammatory cell differential counting of macrophages, neutrophils or lymphocytes (A), and neutrophil-recruiting chemokines MIP-2 (B) and KC (C) were determined. Double asterisks denote significant difference by Student’s t-test (P<0.01, n=4).
CF neutrophils produce excessive neutrophil chemokine MIP-2.
The genetic difference between Mye-Cftr−/− mice and WT mice is the absence or presence of CFTR function in myeloid immune cells, respectively. Thus, we reasoned that the observed exuberant production of neutrophil chemokines by myeloid CF mice must be imparted by abnormal CFTR in myeloid immune cells. To test this prediction, we harvested peritoneal inflammatory cells from Mye-Cftr−/− and WT mice, both of which had been challenged with 0.7 g/kg zyomsan for different times (1, 3 or 5 days). Neutrophils and macrophages were immunostained with Anti-Ly6G antibody and Anti-F4/80 antibody, and sorted by FACS. The sorted cells were cultured, and the culture media were sampled at 2, 4 or 8 hour. Production of MIP-2 and KC by each sample at each time point was measured by ELISA. While all the cells produced abundant MIP-2 (Fig. 5A & B), KC was below the detection threshold of our test. Strikingly, all the CF neutrophils secreted significantly more MIP-2 than did the WT counterparts at any examined time points (Fig. 5B), while all CF macrophages produced comparable or even less MIP-2 than did the WT control cells, except the Day-1 CF macrophages at the 8-hour culture time point. To confirm this unexpected result, we isolated peripheral blood neutrophils and monocytes from Mye-Cftr−/− and WT mice via Percoll-gradient centrifugation. These cells were non-primed and not pre-stimulated. After culture and stimulation with zymosan in vitro for 4 or 8 hours, MIP-2 and KC levels in the culture media were similarly measured. Fig. 5C shows that the CF neutrophils produced significantly more MIP-2 than did the WT neutrophils. In contrast, the CF monocytes produced comparable levels of MIP-2 as did the WT monocytes. Again, KC was not detectable, suggesting that our short-term culture condition supported limited KC production. To rule out the possibility that the observed difference in MIP-2 production by neutrophils was due to possible differences in phagocytosis, we compared neutrophil phagocytosis in vivo and in vitro between Mye-Cftr−/− mice and WT mice. For in vivo comparison, fluorescently labeled zymosan particles were intraperitoneally injected into Mye-Cftr−/− and WT mice. Twelve hours later, peritoneal lavage cells were immunostained with the Ly6G antibody followed by flow cytometric analyses. Fig. 5D shows that neutrophils from the two types of mice had no difference in phagocytosis of the fluorescent zymosan in vivo. For in vitro comparison, neutrophils isolated from the peripheral blood of WT and Mye-Cftr−/− mice were mixed with pre-opsonized fluorescently labeled zymosan in the presence or absence of myeloperoxidase inhibitor 4-amino-benzoic acid hydrazide (ABAH) or sodium azide (NaN3). Inclusion of these inhibitors was to exclude the possibility that HOCl could bleach zymosan fluorescence, which might confound the intepreation of observed differences in phagocytosis. After phagocytosis for 1 hour, the cells were immunostained with the Ly6G antibody for flow cytometry. Neutrophils from WT and Mye-Cftr−/− mice had a comparable phagocytosis rate, regardless of the presence of any MPO inhibitors (Fig. 5E). From these data, we conclude that CFTR loss-of-function in neutrophils lead to significantly more MIP-2 production, which contributed to excessive neutrophil recruitment and neutrophilic inflammation in CF.
Figure 5 – In vitro production of MIP-2 by neutrophils and macrophages.
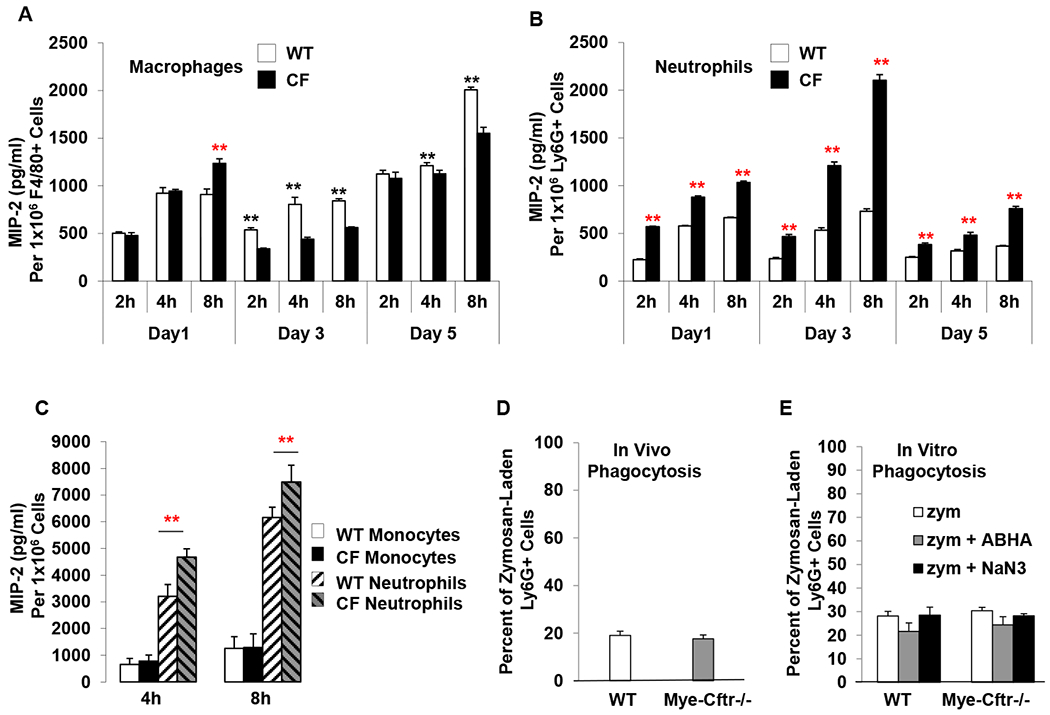
A & B) MIP-2 production by zymosan-pre-activated neutrophils and macrophages from WT and Mye-Cftr−/− mice that had been intraperitoneally challenged with sub-lethal dose zymosan (0.7 g/kg) for different times (1, 3 or 5 days). Peritoneal inflammatory cells were harvested and immunostained with the Ly6G and Anti-F4/80 antibodies, followed by FACS-sorting of neutrophils and macrophages. After culturing for 2, 4 and 8 hours, the media of each type of cells were sampled for MIP-2 measurements. Statistical analyses by Student’s t-test were performed to determine differences in MIP-2 production between the WT and Mye-Cftr−/− cells. Black asterisks indicate that the level of MIP-2 produced by WT cells is greater than that of Mye-Cftr−/− cells (P<0.01, n=4), while red asterisks indicate the opposite (P<0.01, n=4). C) MIP-2 production by neutrophils and monocytes isolated from peripheral blood and activated in vitro with zymosan. Asterisks indicate significant differences in MIP-2 production by Student’s t-test (P<0.01, n=4). D & E) Neutrophil phagocytosis comparison in vivo and in vitro. Fluorescently labeled zymosan particles were administered intraperitoneally into WT and Mye-Cftr−/− mice, respectively. Neutrophil phagocytosis was determined by flow cytometry (D). Peripheral blood neutrophils were isolated from WT and Mye-Cftr−/− mice. Opsonized fluorescent zymosan particles were allowed to phagocytose in the presence or absence of MPO inhibitor 4-amino-benzoic acid hydrazide (ABHA) or sodium azide (NaN3). Phagocytosis rates were compared by flow cytometry (E). No significant difference was detected by Student’s t-test between the two types of neutrophils.
Discussion
The lung is an organ open to the environment and continuously challenged by inhaled microbes. In response, the host has developed a delicate host defense system that involves multiple types of cells, including permanent lung-resident cells, such as pulmonary epithelial cells and tissue macrophages, and transient lung-recruited immune cells, notably neutrophils and monocytes/macrophages. As CF lung disease claims the majority of morbidity and mortality in CF patients, the prevailing theory posits that the defective airway epithelia are the primary cause of neutrophilic inflammation in CF [23]. However, recent research using genetically matched airway epithelial cell lines or gene-complemented primary human airway epithelial culture provides contradicting data [24], suggesting that CF airway epithelial cells are not necessarily inherently hyper-inflammatory. Thus, the identity of the cell that plays the major role in the development of CF neutrophilic inflammation remains uncertain.
Inflammation is a normal host pathophysiological response to an insult, with the integrated aims of removal of the challenge and restoration of tissue homeostasis. Whereas infection represents a very common inducer of inflammatory responses, a wide variety of sterile events, including autoimmune disorders, coronary artery disease, and trauma, elicit inflammation. Inflammation is a well-orchestrated and programmed process, beginning with predominant neutrophil infiltration, the hallmark of acute inflammation. If the challenge is successfully met and the danger eliminated, acute inflammation transitions to resolution, characterized by macrophage and later lymphocyte infiltration and culminating in restoration of tissue homeostasis. However, if the challenge remains, chronic inflammation develops. CF lungs exhibit an early, sustained and intense inflammatory process that no other known lung diseases can match. Additionally, the lung is predisposed to infection with a select number of bacterial species that adversely affect CF patients in several ways. First, untamed bacterial infection in CF lungs underscores the inherent impairment in the phagocytic microbicidal function of CF neutrophils, as a normal host defense against extracellular bacterial infection heavily relies on phagocytic innate immunity. Second, CF lung inflammation stalls at the acute phase with a sustained and excessive neutrophil accumulation. Third, purulent small airway obstruction reflects dysregulated neutrophil cell death and subsequent clearance. In a sense, all CF lung complications reflect abnormal phagocytic innate immunity.
Over-accumulation of neutrophils at inflammatory loci defines clinical neutrophilic inflammation [25], which lies in the imbalance between neutrophil recruitment and neutrophil clearance. CF lungs are highly neutrophilic, theoretically reflectingincreased recruitment, decreased clearance, or a combination of both. Our data clearly demonstrate that myeloid CFTR dysfunction leads to significantly more neutrophil recruitment, which undoubtedly facilitates CF neutrophilic inflammation. Previous research has demonstrated that neutrophil programmed cell death (apoptosis) and macrophage efferocytosis of neutrophils are impaired in CF [26–28]. A slower apoptotic rate favors neutrophil accumulation [29, 30]. CF neutrophils undergo non-programmed necrosis, which leads to release of danger signals, additional inflammation, and suppuration. The necrotic neutrophils release large amounts of intracellular contents, including granular enzymes and toxins that damage airway structure, cytoskeletal actin, and long-stranded DNAs, which contribute to the viscosity of CF sputum, and small airway obstruction.
MIP-2 (CXCL2) is a major inflammatory mediator for neutrophil recruitment in mice. There are several human homologs of rodent MIP-2, including human growth-related oncogenes (GRO-β and -γ), MIP-2α, and MIP-2β [31, 32]. MIP-2 is produced by a variety of cell types, including macrophages, monocytes and epithelial cells, in response to infection or injury. Our data demonstrate that activated neutrophils generated abundant MIP-2, even more than did monocytes and macrophages, suggesting the involvement of neutrophils in regulating neutrophilic inflammation. KC (CXCL1), with human homologs including GRO-α and melanoma growth stimulatory activity (MGSA), is another important neutrophil chemoattractant cytokine in mice [31, 32]. Mouse resident macrophages are reported to be a major source for KC. Although MIP-2 alone is sufficient to recruit neutrophils, the presence of both KC and MIP-2 maximizes the process [33]. The synthesis of MIP-2 and KC is rapidly regulated at the transcriptional level by signaling through TLR2, TLR3, and TLR4 that each recognizes distinct pathogen patterns. Our data demonstrate that significantly more neutrophil chemokines (MIP-2 and KC) were produced by Mye-Cftr−/− mice upon stimulation, suggesting that myeloid CFTR loss-of-function leads to an intrinsic defect in regulation of inflammation.
It is noteworthy that CFTR loss-of-function in Mye-Cftr−/− mice gave rise to only ~40% mortality in the peritonitis experiments (Fig. 1A & B), which might reflect the activities of non-CFTR chloride transporters. In addition to CFTR, mammalian cells express voltage-gated Cl− channels or H+/ Cl− exchangers such as ClC family members [34], calcium-activated Cl− channels (CaCCs) such as TMEM16A and TMEM16B [35–40], ligand-gated Cl− channels (GABA- and glycine-gated) [41, 42], and volume-regulated Cl− channels [43], some of which might transport chloride to neutrophil phagosomes [10, 11]. For example, ClC3 was identified in phagocytes including neutrophils [44, 45], but the contribution of ClC3 to phagocyte HOCl production has not been explored.
In summary, prominent in neutrophil-mediated inflammation and central to differences between normal and CFTR-deficiency is HOCl production, which is deficient in CF due to defective chloride transport into neutrophil phagosomes. We demonstrate that normal CFTR function in myeloid immune cells, especially neutrophils, was required to control neutrophilic inflammation. CFTR loss-of-function in neutrophils results in over-production of neutrophil-recruiting chemokines and excessive recruitment of neutrophils to inflammatory sites, thus serving as a mechanism for persistent neutrophilic inflammation in CF. This study provides the first evidence suggesting that CF neutrophil defect is directly responsible for CF neutrophilic inflammation, a finding that reveals novel targets for future therapeutic development for the treatment of CF.
Acknowledgements
This work was supported by Grant AI140088 (to GW) from the U.S. National Institutes of Health.
References
- 1.Collins FS (1992) Cystic fibrosis: molecular biology and therapeutic implications. Science 256, 774–9. [DOI] [PubMed] [Google Scholar]
- 2.Davis PB, Drumm M, Konstan MW (1996) Cystic fibrosis. Am J Respir Crit Care Med 154, 1229–56. [DOI] [PubMed] [Google Scholar]
- 3.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson G. A. t., Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB Jr., Zabner J, Welsh MJ (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2, 29ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong DS, Hook SM, Jamsen KM, Nixon GM, Carzino R, Carlin JB, Robertson CF, Grimwood K (2005) Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol 40, 500–10. [DOI] [PubMed] [Google Scholar]
- 5.Marteyn BS, Burgel PR, Meijer L, Witko-Sarsat V (2017) Harnessing Neutrophil Survival Mechanisms during Chronic Infection by Pseudomonas aeruginosa: Novel Therapeutic Targets to Dampen Inflammation in Cystic Fibrosis. Front Cell Infect Microbiol 7, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winterbourn CC, Kettle AJ, Hampton MB (2016) Reactive Oxygen Species and Neutrophil Function. Annu Rev Biochem 85, 765–92. [DOI] [PubMed] [Google Scholar]
- 7.Nauseef WM (2018) Biosynthesis of human myeloperoxidase. Arch Biochem Biophys 642, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM (2013) Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol 93, 185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babior BM (2004) NADPH oxidase. Curr Opin Immunol 16, 42–7. [DOI] [PubMed] [Google Scholar]
- 10.Wang G and Nauseef WM (2015) Salt, chloride, bleach, and innate host defense. J Leukoc Biol 98, 163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G (2016) Chloride flux in phagocytes. Immunol Rev 273, 219–31. [DOI] [PubMed] [Google Scholar]
- 12.Painter RG, Valentine VG, Lanson NA Jr., Leidal K, Zhang Q, Lombard G, Thompson C, Viswanathan A, Nauseef WM, Wang G (2006) CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 45, 10260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, Wang G (2008) The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol 83, 1345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laval J, Ralhan A, Hartl D (2016) Neutrophils in cystic fibrosis. Biol Chem 397, 485–96. [DOI] [PubMed] [Google Scholar]
- 15.Pittman K and Kubes P (2013) Damage-associated molecular patterns control neutrophil recruitment. J Innate Immun 5, 315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitmore LC, Hilkin BM, Goss KL, Wahle EM, Colaizy TT, Boggiatto PM, Varga SM, Miller FJ, Moreland JG (2013) NOX2 protects against prolonged inflammation, lung injury, and mortality following systemic insults. J Innate Immun 5, 565–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitmore LC, Goss KL, Newell EA, Hilkin BM, Hook JS, Moreland JG (2014) NOX2 protects against progressive lung injury and multiple organ dysfunction syndrome. Am J Physiol Lung Cell Mol Physiol 307, L71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeuchi K, Umeki Y, Matsumoto N, Yamamoto K, Yoshida M, Suzuki K, Aratani Y (2012) Severe neutrophil-mediated lung inflammation in myeloperoxidase-deficient mice exposed to zymosan. Inflamm Res 61, 197–205. [DOI] [PubMed] [Google Scholar]
- 19.Ng HP, Zhou Y, Song K, Hodges CA, Drumm ML, Wang G (2014) Neutrophil-mediated phagocytic host defense defect in myeloid cftr-inactivated mice. PLoS One 9, e106813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodges CA, Cotton CU, Palmert MR, Drumm ML (2008) Generation of a conditional null allele for Cftr in mice. Genesis 46, 546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF (2005) Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 25, 1647–53. [DOI] [PubMed] [Google Scholar]
- 22.Bonfield TL, Hodges CA, Cotton CU, Drumm ML (2012) Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J Leukoc Biol 92, 1111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen TS and Prince A (2012) Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med 18, 509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hampton TH, Ballok AE, Bomberger JM, Rutkowski MR, Barnaby R, Coutermarsh B, Conejo-Garcia JR, O’Toole GA, Stanton BA (2012) Does the F508-CFTR mutation induce a proinflammatory response in human airway epithelial cells? Am J Physiol Lung Cell Mol Physiol 303, L509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serhan CN and Savill J (2005) Resolution of inflammation: the beginning programs the end. Nat Immunol 6, 1191–7. [DOI] [PubMed] [Google Scholar]
- 26.Nichols DP and Chmiel JF (2015) Inflammation and its genesis in cystic fibrosis. Pediatr Pulmonol 50 Suppl 40, S39–56. [DOI] [PubMed] [Google Scholar]
- 27.Vandivier RW, Richens TR, Horstmann SA, deCathelineau AM, Ghosh M, Reynolds SD, Xiao YQ, Riches DW, Plumb J, Vachon E, Downey GP, Henson PM (2009) Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am J Physiol Lung Cell Mol Physiol 297, L677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandivier RW, Fadok VA, Ogden CA, Hoffmann PR, Brain JD, Accurso FJ, Fisher JH, Greene KE, Henson PM (2002) Impaired clearance of apoptotic cells from cystic fibrosis airways. Chest 121, 89S. [DOI] [PubMed] [Google Scholar]
- 29.McKeon DJ, Condliffe AM, Cowburn AS, Cadwallader KC, Farahi N, Bilton D, Chilvers ER (2008) Prolonged survival of neutrophils from patients with Delta F508 CFTR mutations. Thorax 63, 660–1. [DOI] [PubMed] [Google Scholar]
- 30.Moriceau S, Lenoir G, Witko-Sarsat V (2010) In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J Innate Immun 2, 260–6. [DOI] [PubMed] [Google Scholar]
- 31.Konrad FM and Reutershan J (2012) CXCR2 in acute lung injury. Mediators of inflammation 2012, 740987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi Y (2008) The role of chemokines in neutrophil biology. Front Biosci 13, 2400–7. [DOI] [PubMed] [Google Scholar]
- 33.De Filippo K, Henderson RB, Laschinger M, Hogg N (2008) Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J Immunol 180, 4308–15. [DOI] [PubMed] [Google Scholar]
- 34.Zifarelli G and Pusch M (2007) CLC chloride channels and transporters: a biophysical and physiological perspective. Rev Physiol Biochem Pharmacol 158, 23–76. [DOI] [PubMed] [Google Scholar]
- 35.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–4. [DOI] [PubMed] [Google Scholar]
- 36.Hartzell C, Putzier I, Arreola J (2005) Calcium-activated chloride channels. Annu Rev Physiol 67, 719–58. [DOI] [PubMed] [Google Scholar]
- 37.Schroeder BC, Cheng T, Jan YN, Jan LY (2008) Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–5. [DOI] [PubMed] [Google Scholar]
- 39.Pifferi S, Dibattista M, Menini A (2009) TMEM16B induces chloride currents activated by calcium in mammalian cells. Pflugers Arch 458, 1023–38. [DOI] [PubMed] [Google Scholar]
- 40.Stohr H, Heisig JB, Benz PM, Schoberl S, Milenkovic VM, Strauss O, Aartsen WM, Wijnholds J, Weber BH, Schulz HL (2009) TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J Neurosci 29, 6809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sigel E and Steinmann ME (2012) Structure, function, and modulation of GABA(A) receptors. J Biol Chem 287, 40224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avila A, Nguyen L, Rigo JM (2013) Glycine receptors and brain development. Front Cell Neurosci 7, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffmann EK, Lambert IH, Pedersen SF (2009) Physiology of cell volume regulation in vertebrates. Physiol Rev 89, 193–277. [DOI] [PubMed] [Google Scholar]
- 44.Moreland JG, Davis AP, Bailey G, Nauseef WM, Lamb FS (2006) Anion channels, including ClC-3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. J Biol Chem 281, 12277–88. [DOI] [PubMed] [Google Scholar]
- 45.Alex P, Ye M, Zachos NC, Sipes J, Nguyen T, Suhodrev M, Gonzales L, Arora Z, Zhang T, Centola M, Guggino SE, Li X (2010) Clcn5 knockout mice exhibit novel immunomodulatory effects and are more susceptible to dextran sulfate sodium-induced colitis. J Immunol 184, 3988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]