

Abstract
Immunomodulation of macrophages against cancer has emerged as an encouraging therapeutic strategy. However, there exist two major challenges in effectively activating macrophages for antitumor immunotherapy. First, ligation of signal regulatory protein alpha (SIRPα) on macrophages to CD47, a ‘don’t eat me’ signal on cancer cells, prevents macrophage phagocytosis of cancer cells. Second, colony stimulating factors, secreted by cancer cells, polarize tumor-associated macrophages (TAMs) to a tumorigenic M2 phenotype. Here, we report that genetically engineered cell membrane-coated magnetic nanoparticles (gCM-MNs) can disable both mechanisms. The gCM shell genetically overexpressing SIRPα variants with remarkable affinity efficiently blocks CD47-SIRPα pathway while the MN core promotes the M2 TAM repolarization, synergistically triggering potent macrophage immune responses. Moreover, the gCM shell protects the MNs from immune clearance; and in turn, the MN core delivers the gCMs into tumor tissues under magnetic navigation, effectively promoting their systemic circulation and tumor accumulation. Furthermore, in melanoma and breast cancer models, we show that gCM-MNs significantly prolong overall mouse survival by controlling both local tumor growth and distant tumor metastasis. The combination of cell membrane coating nanotechnology and genetic editing technique offers a safe and robust strategy in activating body’s immune responses for cancer immunotherapy.
Keywords: cancer immunotherapy, gene engineering, cell membrane coating, macrophage immune response, CD47-SIRPα, tumor-associated macrophages
Graphical Abstract
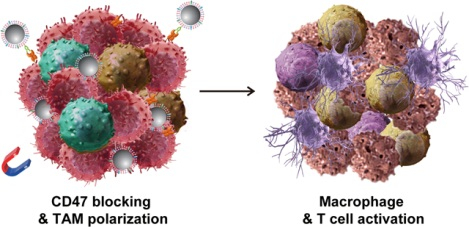
A novel genetically edited nanoparticle was developed to trigger macrophage-mediated antitumor immunity through a powerful two-step strategy: blocking CD47-SIRPα pathway in the first step followed by repolarizating tumor-associated macrophages in the second. The work presented here offers a simple, safe and effective strategy in activating body’s immune responses for cancer immunotherapy.
Cancer immunotherapy for the purpose of activating the body’s immune system against cancer has recently garnished significant attention and stood ready as an adjuvant treatment to join traditional treatment modalities in the clinic.[1, 2] Macrophages are a significant constituent of innate immune system and have an indispensable impact in activating body’s first-line defense against infection and cancer.[3] Effectively activating macrophage-mediated immunity holds great potential in cancer immunotherapy.[4] However, cancer cells are masters of immunomodulation and express ‘don’t eat me’ signal CD47 on the cellular surface, protecting them from the phagocytosis via binding to signal regulatory protein alpha (SIRPα) receptor on macrophages.[5, 6] Blockade of CD47-SIRPα signaling pathway has been widely studied and dozens of CD47 antagonists are being actively tested in clinical trials.[7, 8] CD47 checkpoint inhibitors have been demonstrated to not only promote macrophages to directly ‘eat’ cancer cells but also trigger potent T-cell immune responses.[9] Although promising, systemic infusion of these CD47 inhibitors can cause sginificant side effects, such as thrombocytopenia and anemia.[2, 10] Meanwhile, similar to other checkpoint inhibitors, the clinical benefit rate and objective response rate of these antagonists need to be further improved.[10] Thus, addressing these concerns are extremely important in paving the way for clinical application of CD47-SIRPα immunotherapy.
Recent advances in cancer immunology have revealed that the compromised efficacy of CD47-SIRPα blockades might be caused by immunosuppressive tumor microenvironment (TME).[11, 12] In particular, cancer cells secret colony stimulating factors, which polarize tumor-associated macrophages (TAMs) towards M2 phenotype.[11, 12] M2 TAMs can recruit regulatory T cells (Tregs) to hinder CD47-blockade-induced T-cell immune activation.[2, 13, 14] Therefore, repolarizing TAMs from protumorigenic M2 phenotype to antitumor M1 one may regain the antitumor immunity of CD47 blockades. Moreover, the limited efficacy and side effects may be owing to nonspecific accumulation of systemically infused CD47 antagonists in normal tissues.[10, 12] Thus, it would be ideal for CD47-SIRPα-mediated antitumor immunotherapy to concentrate on the TME whilst avoiding off-target immune activation in other sites.
Rapid progress in nanotechnology and materials science, especially in functionalized cellular membrane nanovesicles (CM-NVs), offers many promising opportunities for cancer immunotherapy.[15–18] Additionally, biomimetic synthetic strategies involving directional expression of specific proteins on cell surface enable efficient development of CM-NVs displaying membrane proteins with native orientation, structure, and activity.[15, 19–22] Although CM-NVs have shown desirable feasibility and safety in both of preclinical experiments and clinical trials, the beneficial effects of these nanovesicles on cancer treatment were not very significant.[18, 20, 21] We and others have recently developed various cell membrane-coated nanoparticles,[23–28] of which the cell membrane shell and nanoparticle core can both be customized, providing a high level of freedom in nanoparticle functionalization.[29–32] Thus, it is conceivable to utilize the functionalized nanoparticle core to synergistically amplify CM-NVs-induced antitumor immunity for enhanced cancer immunotherapy.
Herein, we combined biomimetic synthetic strategy with cell membrane coating nanotechnology and demonstrated that genetically edited cell membrane-coated magnetic nanoparticles (gCM-MNs) can elicit potent macrophage immune responses for cancer immunotherapy. In this system, the gCM shell genetically overexpressing SIRPα variants with 50,000-fold enhanced affinity to CD47 efficiently blocks CD47-SIRPα signaling pathway while the MN core promotes the M2-to-M1 repolarization within the TME, synergistically facilitating macrophage phagocytosis of cancer cells as well as triggering antitumor T-cell immunity (Figure 1). In addition, the biomimetic gCM shell protects the MN core from immune clearance; and in a feedback manner, the MN core delivers the gCM shell into the TME under magnetic navigation, improving the tumor accumulation of gCM-MNs and reducing the off-target immune over-activation.
Figure 1.
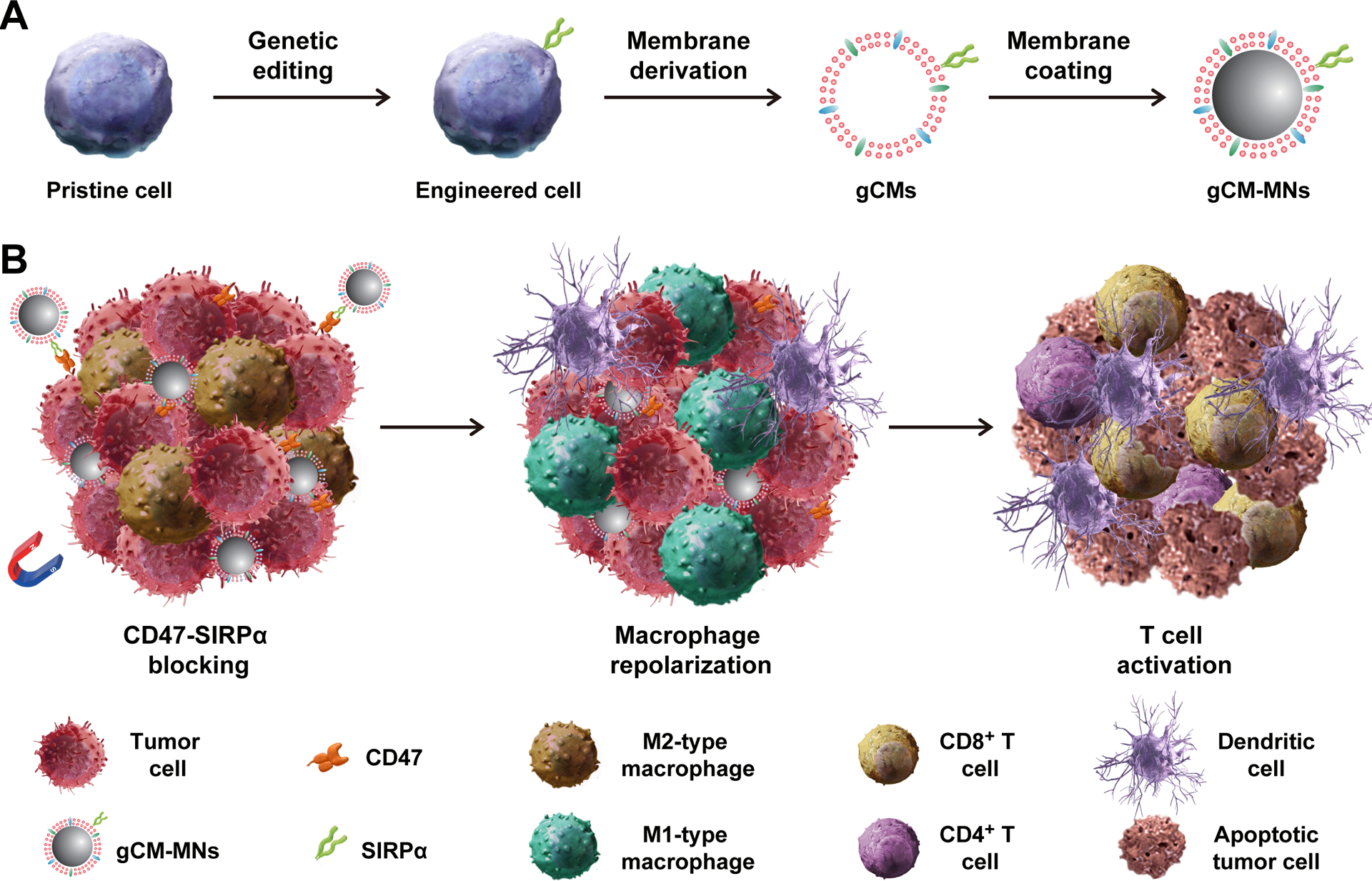
Scheme of genetically edited cell membrane-coated magnetic nanoparticles (gCM-MNs) elicit potent macrophage immune responses for cancer immunotherapy. A) Cell membranes were isolated from the genetically engineered cells that overexpressing SIRPα variants and then coated onto magnetic nanoparticles (MNs). B) Under external magnetic field, gCM-MNs efficiently accumulate in the tumor microenvironment, block the CD47-SIRPα ‘don’t eat me’ pathway, and repolarize tumor-associated macrophages (TAMs) towards M1 phenotype, promoting macrophage phagocytosis of cancer cells as well as boosting antitumor T-cell immunity.
The preparation of gCM-MNs briefly contains two steps: preparing genetically engineered cell membrane vesicles gCMs in the first step and followed by coating gCMs onto the MNs in the second.[27] SIRPα variant, CV1,[33] with enhanced affinity to CD47 was transduced onto murine melanoma B16F10 cells and murine mammary carcinoma 4T1 cells by lentivirus. We demonstrated the CV1 expression on cells by immune fluorescence and flow cytometry (Figure 2A,B). To obtain the cellular membrane components, the intracellular contents were eliminated by serial treatments of hypotonic lysis, physical extraction, and gradient centrifugation.[27] Subsequently, gCMs were prepared by successive sonication and extrusion of the resulting cellular membrane components on a mini extruder equipped with a nanopore membrane (Figure S1, Supporting Information). After then, MNs with a diameter of ~80 nm (Figure S2) were wrapped with a layer of gCMs by extruding the mixture of gCMs and MNs through 200 nm and 100 nm pores orderly.[29]
Figure 2.
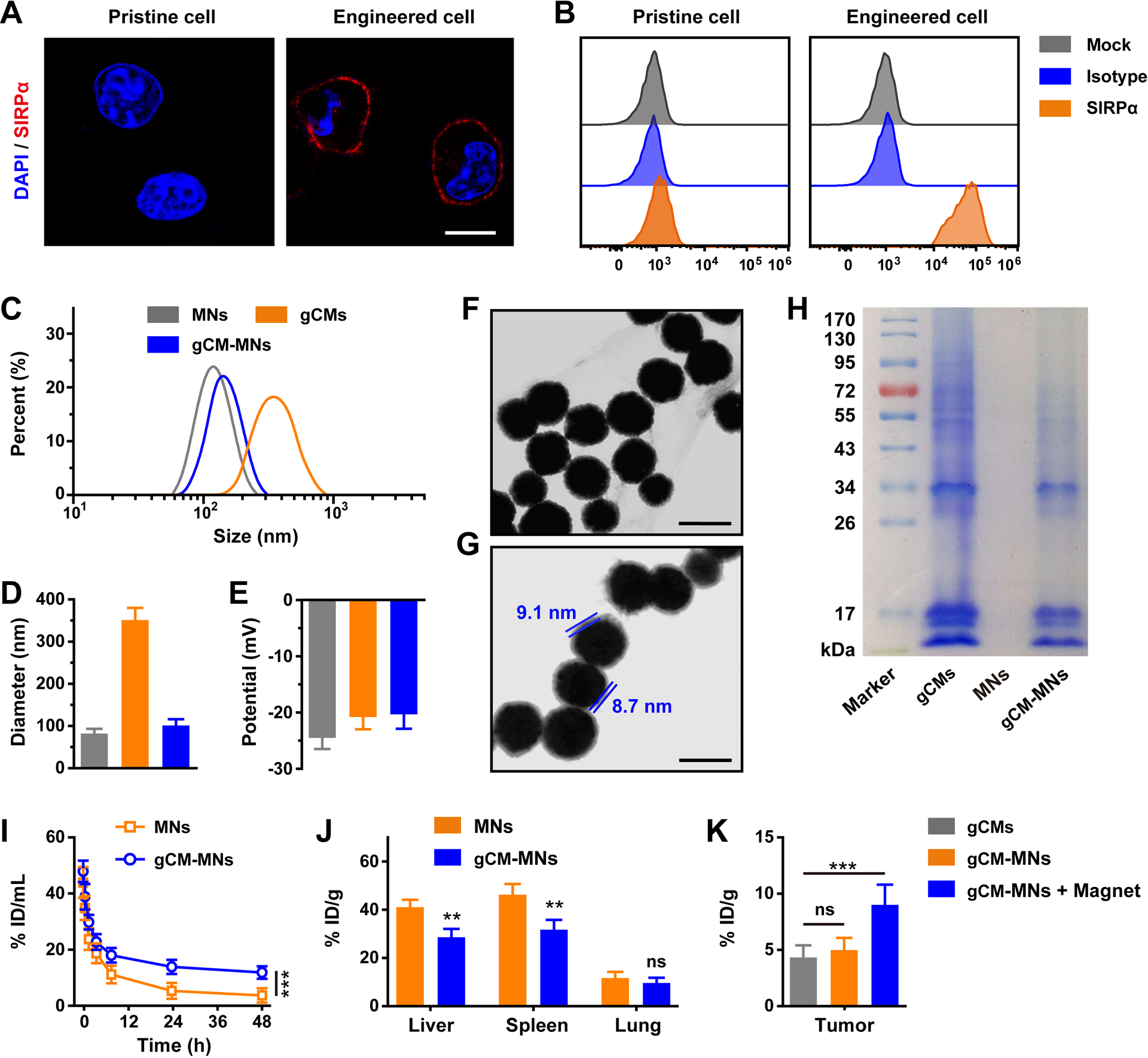
Preparation and characterization of gCM-MNs. A) Immunofluorescence imaging and B) flow cytometry quantification of SIRPα variants on pristine and engineered cells. Scale bar, 10 μm. C) Size curves, D) mean diameter, and E) zeta potential of MNs, gCMs and gCM-MNs. TEM images of F) MNs and G) gCM-MNs. Scale bars, 100 nm. H) SDS-PAGE analysis of gCMs, MNs, and gCM-MNs. I) In vivo pharmacokinetic curves of MNs and gCM-MNs. J) In vivo distribution of MNs and gCM-MNs in liver, spleen and lung. K) Tumor accumulations of gCMs and gCM-MNs without or with an external magnet. All data are presented as mean ± S.D. (D,E, n = 3; I-K, n = 4). Statistical significance was calculated via 2way ANOVA with a Tukey’s test (I) or unpaired t test (J) or ordinary one-way ANOVA with a Tukey’s test (K). *P < 0.05; ***P < 0.001.
Dynamic light scattering (DLS) characterizations suggested that the diameter of gCM-MNs were ~100 nm and the zeta potential improved roughly to the level of gCMs (Figure 2C–E). Moreover, transmission electron microscopy (TEM) visualization demonstrated a ~80 nm MNs core and a layer of ~10 nm outer lipid shell (Figure 2F,G),[34] suggesting successful cellular membrane coating. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was further employed to examine protein contents in gCM-MNs and the gel demonstrated successful migration of cellular membrane proteins from gCM to MNs (Figure 2H). The obtained gCM-MNs remained stable for at least 15 days (Figure S3), ensuring the technical feasibility of downstream experiments. We also showed that the cellular membrane coating does not compromise the powerful magnetism (Figure S4). Furthermore, we tested the in vivo pharmacokinetics and biodistribution of gCM-MNs. As compared with MNs, the gCM-MNs exhibited improved blood retention (Figure 2I) and reduced enrichment in liver, spleen, and lung (Figure 2J), suggesting that the gCM shell provides cloaking ability for the MNs allowing them to reduce non-specific clearance. In a feedback manner, the inner MN core stabilizes the gCM shell enhancing survival in the circulatory system and promotes the tumor accumulation of gCMs under magnetic navigation (Figure 2K).
Blocking the CD47-SIRPα signaling pathway has been explored to be an encouraging antitumor immunotherapy.[35] Many kinds of SIRPα or CD47 blockades, including SIRPα-Fc fusion proteins and anti-CD47 monoclonal antibodies (anti-CD47 mAbs), have shown promising results in both preclinical experiments and clinical trials.[2] Given the weak binding between native SIRPα and CD47, SIRPα variants have been developed to markedly promote the affinity to CD47.[36] To determine whether SIRPα variant-engineered gCMs could promote the phagocytosis by macrophages, B16F10 cells were incubated with gCMs and then co-cultured with bone marrow-derived macrophages (BMDMs). Confocal imaging revealed that CD47 blocking by genetically edited gCMs significantly stimulated the cytophagy of B16F10 cells by BMDMs (Figure 3A–C). CD47 is broadly expressed on cell surfaces, especially red blood cells (RBCs) and platelets (PLTs). We found that the tight binding between gCM-MNs and RBCs and/or PLTs effectively protects gCM-MNs from the phagocytosis by BMDMs (Figure S5), indicating a potential new mechanism for enhanced circulation of these genetically edited nanoparticles. The in vivo phagocytosis effects induced by gCMs were further examined based on a B16F10 mouse model. An observable increase in dendritic cells (CD11c+) within the TME was noticed and these cells exhibited higher expression of CD103, CD80, and CD86 (Figure 3D,E), suggesting their mature status. After the treatment with gCMs, a reduction in Tregs (CD4+Foxp3+) was observed as well (Figure 3F), indicating a down-regulation of tumor immunosuppression. By using the B16F10 mouse model, we also compared the in vivo effects of different concentrations of gCMs and anti-CD47 mAbs on tumor inhibition (Figure S6).
Figure 3.
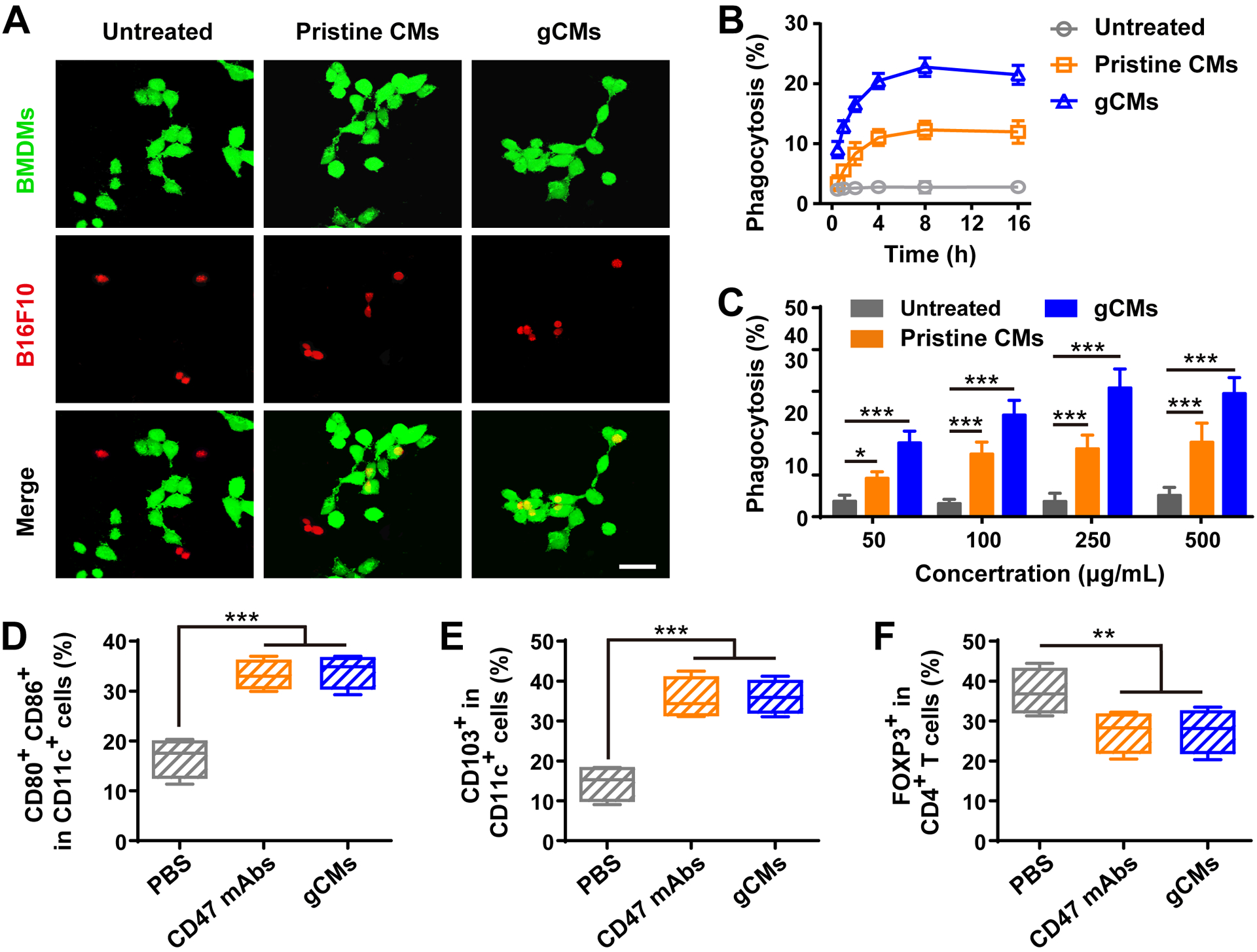
gCMs increase macrophage phagocytosis in vitro and improve antigen presentation in vivo. A) Fluorescence images of the phagocytosis of B16F10 cells by BMDMs. Scale bar, 50 μm. Quantification analysis of the phagocytosis of B16F10 cells by BMDMs with B) different incubation time or C) different concentrations. Flow cytometric analysis of D) CD80+CD86+ dendritic cells and E) CD103+ dendritic cells in the tumor tissues gating on CD45+CD11c+ cells. F) Flow cytometric analysis of CD4+Foxp3+ T cells in tumor tissues gating on CD3+CD4+ cells. All data are presented as mean ± S.D. (n = 4). Statistical significance was calculated via ordinary one-way ANOVA with a Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001.
TAMs have been considered as promising therapeutic targets due to their significant roles in tumor progression.[13, 37] Among therapies targeting TAMs, reprogramming TAMs toward antitumor M1-like phenotype is a prominent strategy.[38, 39] Recently, Zanganeh et al. have showed that an U.S. Food and Drug Administration (FDA) approved dextran-coated magnetic nanoparticles (named as Ferumoxytol) could inhibit breast cancer progression and lung and liver metastasis by inducing the repolarization of TAMs towards antitumor M1-type ones,[40] which was mediated by reactive oxygen species generated via Ferumoxytol-mediated Fenton reactions.[41, 42] To test whether our MNs could repolarize M2 TAMs to M1 ones, we measured the mRNA levels in M2 macrophages after the treatment with MNs. Higher levels of M1 markers (i.e., iNOS and IL-6) and lower levels of M2 markers (i.e., IL-4 and IL-10) were noticed in the MNs group as compared with the untreated one (Figure 4A,B). Immunofluorescence imaging and cytokine measurement further demonstrated that MNs could effectively repolarize TAMs from M2 phenotype towards M1 ones (Figure 4C,D and Figure S7). We subsequently test the in vivo effects of MNs on TAM repolarization by using the B16F10 mouse model. Flow cytometry results demonstrated that, M1 TAMs (CD80+CD11b+F4/80+) increased while the M2 ones (CD206+CD11b+F4/80+) decreased within the tumor (Figure 4E). This repolarization was also confirmed by the increased level of IL-12 (M1 marker) and decreased level of IL-10 (M2 marker) within the TME (Figure 4F).
Figure 4.
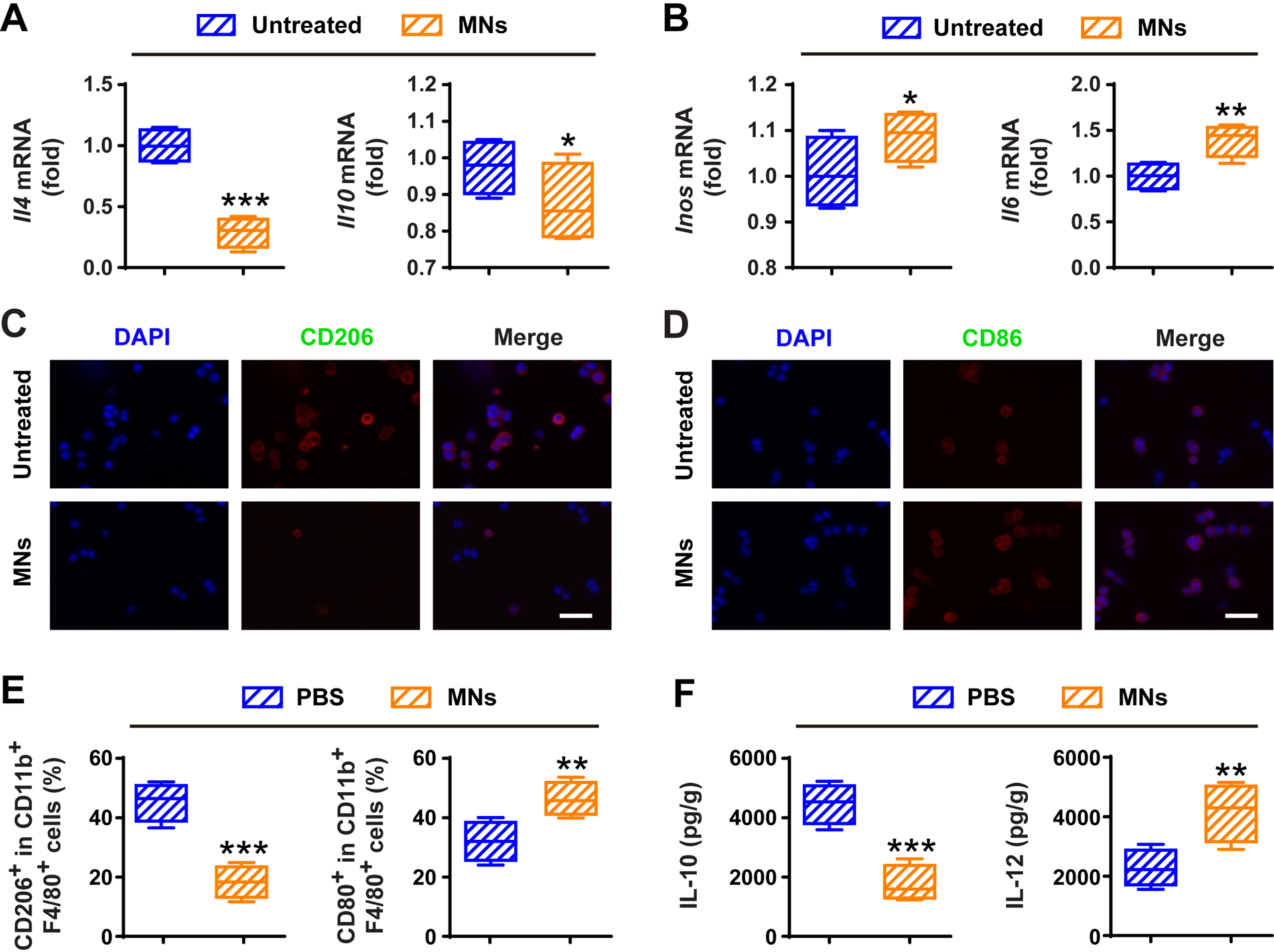
MNs alter macrophage polarization and relieve immunosuppressive tumor microenvironment in vivo. Relative mRNA expressions of A) M2 markers (i.e., IL-10 and IL-4) and B) M1 markers (i.e., IL-6 and iNOS) in M2 macrophages treated without or with MNs for 24 h. Immunofluorescence staining of C) M2 marker (i.e., CD206) and D) M1 marker (i.e., CD86) in M2 macrophages treated without or with MNs for 24 h. Scale bars, 100 μm. E) Flow cytometric analysis of CD206+ M2 TAMs and CD80+ M1 ones in tumor tissues gating on F4/80+CD11b+CD45+ cells. F) Secretion of IL-12 and IL-10 in PBS and MNs groups. All data are presented as mean ± S.D. (n = 4). Statistical significance was calculated via unpaired t test. *P < 0.05; **P < 0.01; ***P < 0.001.
We then investigated the in vivo antitumor effects of gCM-MNs by using the B16F10 tumor-bearing mice, which were intravenously (i.v.) injected with three doses of phosphate buffered saline (PBS), MNs, gCMs, gCMs + MNs or gCMs-MNs every other day. We demonstrated that the MNs and gCMs treatments showed limited antitumor effects; in contrast, the gCMs-MNs treatment significantly inhibited the tumor growth, which was even better than co-administration of gCMs and MNs in a cocktail manner (Figure 5A,B). It should be pointed out that the improved antitumor effects can be partly attributed to the improved tumor accumulation of gCMs-MNs (Figure S8). Benefiting from effective tumor suppression, the survival rate of the mice treated with gCMs-MNs was improved to about 57% after 50 days (Figure 5C). We enriched and analyzed tumor tissues on day 5 after the injection. An increased polarization of TAMs towards an M1 phenotype was noticed by flow cytometry (Figure 5D,E). Although a decreased level of M2 TAMs was observed in the gCMs group in vivo, the gCMs alone could not trigger the TAM repolarization in vitro (Figure S9). Markedly increased CD8+ T cells was also observed within the TME after the treatment with gCMs-MNs (Figure 5F,G). In addition, promoted secretion of certain cytokines (i.e., IFN-γ and TNF-α) further indicated potent innate and adaptive antitumor immunity induced by gCMs-MNs (Figure 5H,I). By combining the repolarization of TAMs and the blockade of CD47-SIRPα pathway, the gCMs-MNs significantly improved the phagocytosis of cancer cells by macrophages as well as boosted antitumor T-cell immunity within the TME.
Figure 5.
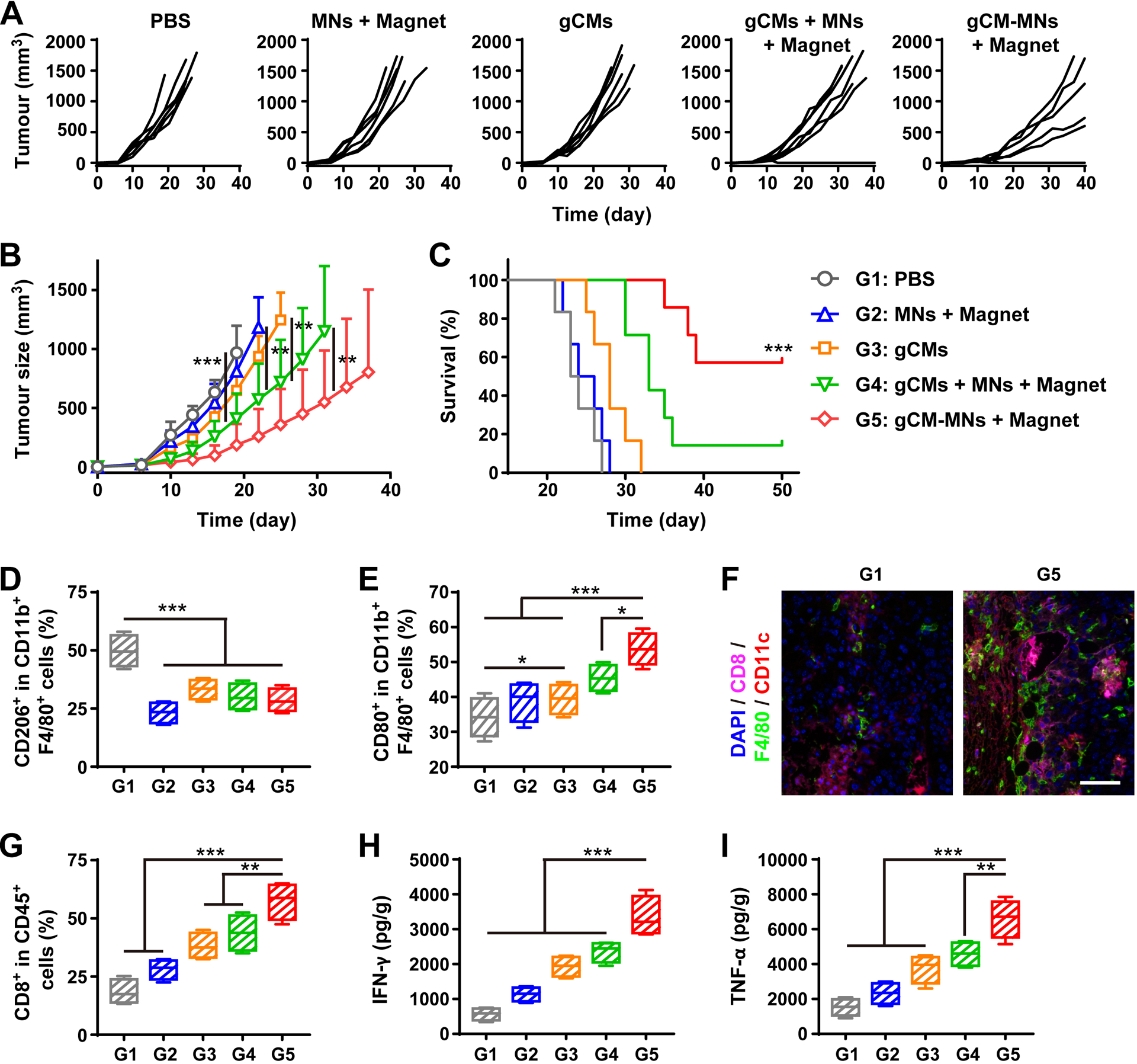
gCM-MNs inhibit B16F10 tumor growth and trigger robust antitumor immunity. A) Individual and B) average tumor growth kinetics after indicated treatments. C) Survival curves for different treatment groups. Flow cytometric analysis of D) CD206+ M2 macrophages and E) CD80+ M1 macrophages in tumor gating on F4/80+CD11b+CD45+ cells. F) Multiplex IHC images of F4/80+ macrophage and CD8+ T cell infiltration in tumor tissues. Scale bar, 100 μm. G) Flow cytometric analysis of CD8+ T cells in tumor gating on CD45+ cells. H) IFN-γ and I) TNF-α levels in tumor tissues collected from mice on day 5 after different treatments. All data are presented as mean ± S.D. (B,C, n = 6 for the groups treated with PBS, MNs + Magnet, and gCMs, and n = 7 for the other groups; D-I, n = 4). Statistical significance was calculated via 2way ANOVA with a Tukey’s test (B) or the log-rank (Mantel–Cox) test (C) or ordinary one-way ANOVA with a Tukey’s test (D-I). *P < 0.05; **P < 0.01; ***P < 0.001.
In terms of the mechanism, we demonstrated that once the nanoparticles are bound to the cancer cell surface, most of gCM-MNs remain on the surface of cells rather than being internalized inside the cells via endocytosis (Figure S10). After the binding by gCM-MNs, there are two main strategies to trigger the TAM repolarization within the TME: 1) although parts of gCM-MNs are bound to the cancer cell surface, there’s still parts of gCM-MNs that can be directly taken up by macrophages to result in the M2-M1 repolarization; 2) after the binding of gCM-MNs to cancer cell surface, cancer cells along with gCM-MNs can be taken up by macrophages, resulting in the M2-M1 repolarization. Furthermore, we demonstrated that the gCM-MNs lowered the levels of M2 markers (i.e., IL-4 and IL-10) and promoted the levels of M1 markers (i.e., iNOS and IL-6) in a dose-dependent manner (Figure S11), suggesting successful repolarization induced by gCM-MNs.
Subsequently, we further evaluated the effectiveness of gCMs-MNs in inhibiting cancer metastasis in a triple negative breast cancer 4T1 tumor model. The mice were i.v. injected with three doses of PBS, MNs, gCMs, gCMs + MNs or gCMs-MNs every other day. Compared with gCMs, MNs or gCMs + MNs, the gCMs-MNs showed more effective inhibition of tumor growth (Figure 6A,B). The mice receiving gCMs-MNs exhibited substantially prolonged survival, with more than 60% of them still alive on day 60 post the tumor inoculation (Figure 6C). After the treatment with gCMs-MNs, only very few metastatic foci were noticed in the lungs (Figure 6D,E) and Ki-67 staining further suggested effective control of tumor cell proliferation (Figure 6F). Notably, the lung metastasis rate decreased from 83.33% to 37.5% after the gCMs-MNs treatment (Figure 6G), indicating the great potential of gCMs-MNs in the treatment of cancer metastasis.
Figure 6.
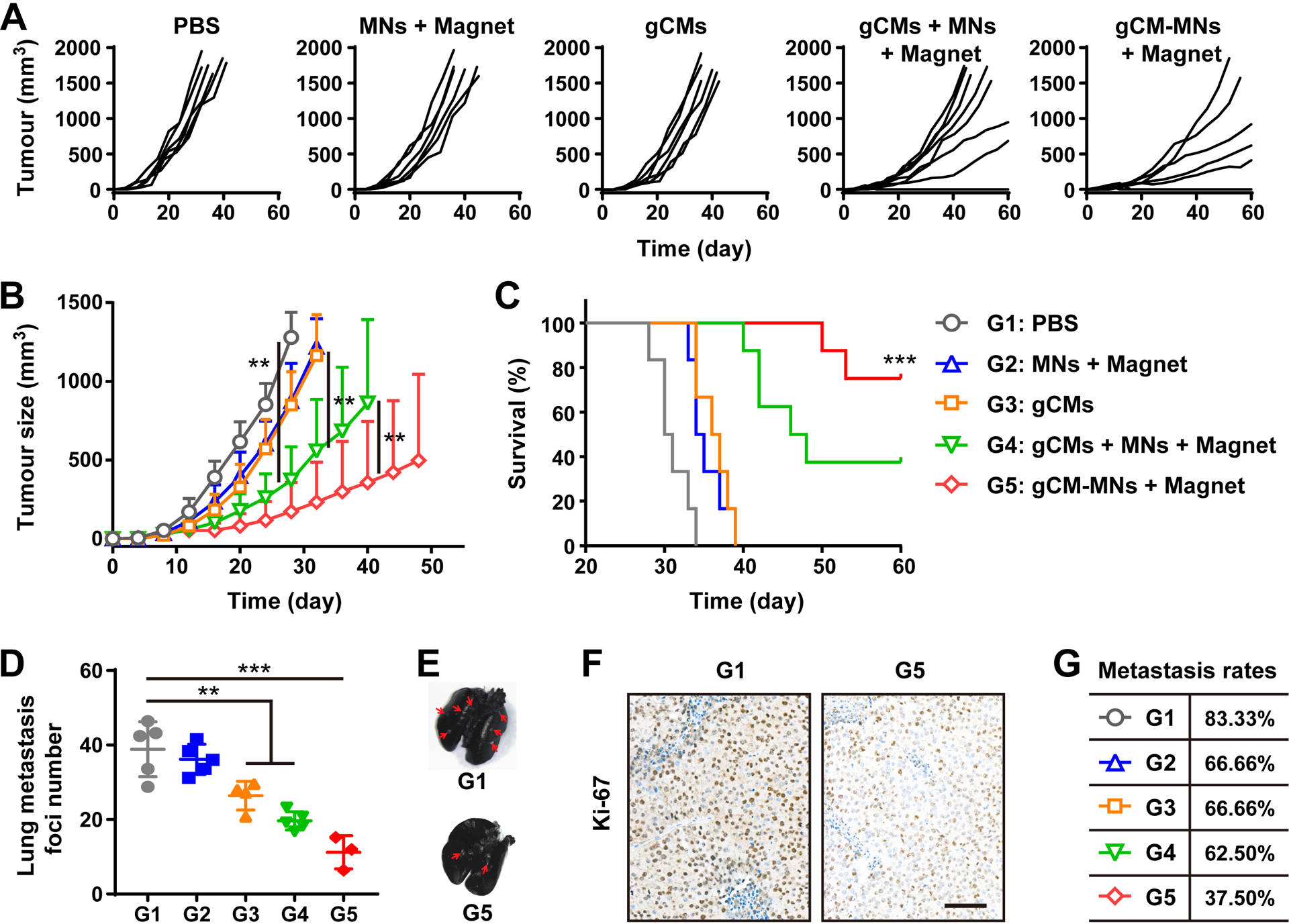
gCM-MNs suppress 4T1 tumor growth and lung metastasis. A) Individual and B) average tumor growth kinetics after indicated treatments. C) Survival curves for different treatment groups. D) Lung metastatic foci after indicated treatments. E) Ink-stained lung photographs collected from mice after indicated treatments. Red arrowheads indicate tumor foci in the lung. F) Ki-67-stained lung slices for different treatment groups. Scale bar, 100 μm. G) Metastasis rates after indicated treatments. All data are presented as mean ± S.D. (B,C, n = 8 for the groups treated with gCMs + MNs + Magnet and gCM-MNs + Magnet, and n = 6 for the other groups; D, n = 4 for the gCMs group, n = 3 for the gCM-MNs + Magnet group, and n = 5 for the other groups). Statistical significance was calculated via 2way ANOVA with a Tukey’s test (B) or the log-rank (Mantel–Cox) test (C) or ordinary one-way ANOVA with a Tukey’s test (D). **P < 0.01; ***P < 0.001.
In vivo toxicity is always a critical concern for nanomaterials used in biomedical applications.[43] Despite that the employment of cell membrane coating could mitigate immune rejection caused by synthetic foreign materials, immunogenicity remains to be a significant concern.[44] We first demonstrated that gCMs-MNs had little influence on cell viability (Figure S12) and then further investigated the in vivo toxicity by i.v. infusing PBS or PBS containing gCMs-MNs into the mice. Neither death nor evident weight difference was monitored over 30 days, suggesting that no significant side effects were caused by gCMs-MNs. After the mice were euthanized, we further collected the blood and organ samples for blood examination and histology analysis. No significant differences in blood biochemical, whole blood test, and tissue slices between PBS and gCMs-MNs were observed (Figure S13, S14 and Table S1), further demonstrating the favorable in vivo compatibility of gCMs-MNs.
In summary, we have rationally developed a core-shell genetically edited cell membrane-coated nanoparticles against both local tumor growth and distant tumor metastasis. The biomimetic gCM shell helps reduce the immune clearance of MNs; in a feedback manner, the MN core delivers the gCM shell into tumor microenvironment under magnetic navigation. The MN core and gCM shell could serve to repolarize TAMs towards M1 phenotype and block the CD47-SIRPα pathway, thus promoting the cytophagy of cancer cells by macrophages. In addition, CD47 blockade by gCMs also trigged robust T cell-mediated devastation of cancer cells owing to the improved antigen presentation by macrophages and dendritic cells. In malignant melanoma and triple negative breast carcinoma mouse models, the gCMs-MNs exhibited significantly prolonged overall survival by controlling both local growth and distant metastasis, encouraging the translation of these biomimetic nanomaterials in cancer immunotherapy.
The employment of genetically edited cell membranes for nanoparticle coating represents an encouraging nanobiotechnology that has superior potential in clinical translation.[29] Benefiting from the membrane-anchored and transmembrane proteins, the cell membrane shell can protect nanoparticle core from immune clearance, ensuring desirable in vivo performance.[45] These biomimetic genetically engineered nanoparticles may also open up an exciting new frontier in personalized medicine. Similar to chimeric antigen receptor-modified T (CAR-T) cell treatment strategy,[46] the cells collected from a patient can be genetically engineered and the membranes can be used for nanoparticle coating before infusing them back into the same patient, which enables the maximization of immune tolerance to nanoparticles.[26, 47, 48] These genetically edited cell membrane nanoparticles represent a significant step forward in nanobiotechnology with great potential to extend the immunotherapeutic arsenal.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), National Natural Science Foundation of China (Grant No. 81972003), and Natural Science Foundation of Guangdong (Grant No. 2017A030313668). All animal studies were approved by the Institutional Review Board of Southern Medical University in accordance with the guidelines for the protection of animal subjects.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
The authors declare no conflict of interest.
Contributor Information
Lang Rao, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, MD 20892, United States..
Shu-Kun Zhao, Biobank, Shenzhen Second People’s Hospital, The First Affiliated Hospital of Shenzhen University, Shenzhen 518035, China.; School of Physics and Technology, Wuhan University, Wuhan 430072, China.
Churan Wen, Breast Center, Department of General Surgery, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China..
Bo Cai, Research Center for Tissue Engineering and Regenerative Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China..
Yue Sun, School of Physics and Technology, Wuhan University, Wuhan 430072, China..
Qian-Fang Meng, School of Physics and Technology, Wuhan University, Wuhan 430072, China..
Guangyu Yao, Breast Center, Department of General Surgery, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China..
Ni Xie, Biobank, Shenzhen Second People’s Hospital, The First Affiliated Hospital of Shenzhen University, Shenzhen 518035, China..
Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, MD 20892, United States..
References
- [1].Pardoll DM, Nat. Rev. Cancer 2012, 12, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Feng M, Jiang W, Kim BYS, Zhang CC, Fu Y-X, Weissman IL, Nat. Rev. Cancer 2019, 19, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E, Nature 2019, 574, 45. [DOI] [PubMed] [Google Scholar]
- [4].Gordon S, Nat. Rev. Immunol 2003, 3, 23. [DOI] [PubMed] [Google Scholar]
- [5].Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, van Rooijen N, Weissman IL, Cell 2009, 138, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kershaw MH, Smyth MJ, Science 2013, 341, 41. [DOI] [PubMed] [Google Scholar]
- [7].Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, Park CY, Zhao FF, Kohrt HE, Malumbres R, Briones J, Gascoyne RD, Lossos IS, Levy R, Weissman IL, Majeti R, Cell 2010, 142, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, Lovelace P, Scheeren FA, Chao MP, Weiskopf K, Tang C, Volkmer AK, Naik TJ, Storm TA, Mosley AR, Edris B, Schmid SM, Sun CK, Chua MS, Murillo O, Rajendran P, Cha AC, Chin RK, Kim D, Adorno M, Raveh T, Tseng D, Jaiswal S, Enger PO, Steinberg GK, Li G, So SK, Majeti R, Harsh GR, van de Rijn M, Teng NNH, Sunwoo JB, Alizadeh AA, Clarke MF, Weissman IL, Proc. Natl. Acad. Sci. USA 2012, 109, 6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, Xu H, Peng H, Fu Y-X, Xu MM, Nat. Med 2015, 21, 1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, Kline J, Roschewski M, LaCasce A, Collins GP, Tran T, Lynn J, Chen JY, Volkmer J-P, Agoram B, Huang J, Majeti R, Weissman IL, Takimoto CH, Chao MP, Smith SM, Engl N. J. Med 2018, 379, 1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Noy R, Pollard Jeffrey W., Immunity 2014, 41, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H, Wang J, Wen D, Zhang Y, Lu Y, Yang G, Jiang C, Wang J, Dotti G, Gu Z, Nat. Nanotechnol 2019, 14, 89. [DOI] [PubMed] [Google Scholar]
- [13].Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, Nat. Rev. Clin. Oncol 2017, 14, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kulkarni A, Chandrasekar V, Natarajan SK, Ramesh A, Pandey P, Nirgud J, Bhatnagar H, Ashok D, Ajay AK, Sengupta S, Nat. Biomed. Eng 2018, 2, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang P, Chen Y, Zeng Y, Shen C, Li R, Guo Z, Li S, Zheng Q, Chu C, Wang Z, Zheng Z, Tian R, Ge S, Zhang X, Xia N-S, Liu G, Chen X, Proc. Natl. Acad. Sci. USA 2015, 112, 6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang J, Li P, Yu Y, Fu Y, Jiang H, Lu M, Sun Z, Jiang S, Lu L, Wu MX, Science 2020, 367, eaau0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pitt JM, André F, Amigorena S, Soria J-C, Eggermont A, Kroemer G, Zitvogel L, The J Clin. Invest 2016, 126, 1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wiklander OPB, Brennan MÁ, Lötvall J, Breakefield XO, EL Andaloussi S, Sci. Transl. Med 2019, 11, eaav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang P, Zhang L, Qin Z, Hua S, Guo Z, Chu C, Lin H, Zhang Y, Li W, Zhang X, Chen X, Liu G, Adv. Mater 2018, 30, 1705350. [DOI] [PubMed] [Google Scholar]
- [20].Koh E, Lee EJ, Nam G-H, Hong Y, Cho E, Yang Y, Kim I-S, Biomaterials 2017, 121, 121. [DOI] [PubMed] [Google Scholar]
- [21].Zhang X, Wang C, Wang J, Hu Q, Langworthy B, Ye Y, Sun W, Lin J, Wang T, Fine J, Cheng H, Dotti G, Huang P, Gu Z, Adv. Mater 2018, 30, 1707112. [DOI] [PubMed] [Google Scholar]
- [22].Han X, Shen S, Fan Q, Chen G, Archibong E, Dotti G, Liu Z, Gu Z, Wang C, Sci. Adv 2019, 5, eaaw6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nature 2015, 526, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rao L, Bu L-L, Cai B, Xu J-H, Li A, Zhang W-F, Sun Z-J, Guo S-S, Liu W, Wang T-H, Zhao X-Z, Adv. Mater 2016, 28, 3460. [DOI] [PubMed] [Google Scholar]
- [25].Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z, Adv. Mater 2015, 27, 7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu W-L, Zou M-Z, Liu T, Zeng J-Y, Li X, Yu W-Y, Li C-X, Ye J-J, Song W, Feng J, Zhang X-Z, Nat. Commun 2019, 10, 3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Proc. Natl. Acad. Sci. USA 2011, 108, 10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rao L, Cai B, Bu L-L, Liao Q-Q, Guo S-S, Zhao X-Z, Dong W-F, Liu W, ACS Nano 2017, 11, 3496. [DOI] [PubMed] [Google Scholar]
- [29].Fang RH, Kroll AV, Gao W, Zhang L, Adv. Mater 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rao L, Wang W, Meng Q-F, Tian M, Cai B, Wang Y, Li A, Zan M, Xiao F, Bu L-L, Li G, Li A, Liu Y, Guo S-S, Zhao X-Z, Wang T-H, Liu W, Wu J, Nano Lett. 2019, 19, 2215. [DOI] [PubMed] [Google Scholar]
- [31].Wang P, Wang H, Huang Q, Peng C, Yao L, Chen H, Qiu Z, Wu Y, Wang L, Chen W, Theranostics 2019, 9, 1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jing L, Qu H, Wu D, Zhu C, Yang Y, Jin X, Zheng J, Shi X, Yan X, Wang Y, Theranostics 2018, 8, 2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Weiskopf K, Ring AM, Ho CCM, Volkmer J-P, Levin AM, Volkmer AK, Özkan E, Fernhoff NB, van de Rijn M, Weissman IL, Garcia KC, Science 2013, 341, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hochmuth R, Evans C, Wiles H, McCown J, Science 1983, 220, 101. [DOI] [PubMed] [Google Scholar]
- [35].Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, Esparza R, Richard C, Ramaswamy V, Remke M, Volkmer AK, Willingham S, Ponnuswami A, McCarty A, Lovelace P, Storm TA, Schubert S, Hutter G, Narayanan C, Chu P, Raabe EH, Harsh G, Taylor MD, Monje M, Cho Y-J, Majeti R, Volkmer JP, Fisher PG, Grant G, Steinberg GK, Vogel H, Edwards M, Weissman IL, Cheshier SH, Sci. Transl. Med 2017, 9, aaf2968. [DOI] [PubMed] [Google Scholar]
- [36].Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M, Nature 2007, 450, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mantovani A, Allavena P, Sica A, Balkwill F, Nature 2008, 454, 436. [DOI] [PubMed] [Google Scholar]
- [38].Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ, Weissleder R, Nat. Biomed. Eng 2018, 2, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ovais M, Guo M, Chen C, Adv. Mater 2019, 31, 1808303. [DOI] [PubMed] [Google Scholar]
- [40].Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, Daldrup-Link HE, Nat. Nanotechnol 2016, 11, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shi C, Liu T, Guo Z, Zhuang R, Zhang X, Chen X, Nano Lett. 2018, 18, 7330. [DOI] [PubMed] [Google Scholar]
- [42].Yu G-T, Rao L, Wu H, Yang L-L, Bu L-L, Deng W-W, Wu L, Nan X, Zhang W-F, Zhao X-Z, Liu W, Sun Z-J, Adv. Funct. Mater 2018, 28, 1801389. [Google Scholar]
- [43].Nel A, Xia T, Mädler L, Li N, Science 2006, 311, 622. [DOI] [PubMed] [Google Scholar]
- [44].Rao L, Bu L-L, Ma L, Wang W, Liu H, Wan D, Liu J-F, Li A, Guo S-S, Zhang L, Zhang W-F, Zhao X-Z, Sun Z-J, Liu W, Angew. Chem. Int. Ed 2018, 57, 986. [DOI] [PubMed] [Google Scholar]
- [45].Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R, Nature 2017, 546, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fesnak AD, June CH, Levine BL, Nat. Rev. Cancer 2016, 16, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang R, Xu J, Xu L, Sun X, Chen Q, Zhao Y, Peng R, Liu Z, ACS Nano 2018, 12, 5121. [DOI] [PubMed] [Google Scholar]
- [48].Chen Z, Zhao P, Luo Z, Zheng M, Tian H, Gong P, Gao G, Pan H, Liu L, Ma A, Cui H, Ma Y, Cai L, ACS Nano 2016, 10, 10049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.