

Abstract
Background
Bevacizumab has promising activity against recurrent glioblastoma (GBM). However, acquired resistance to this agent results in tumor recurrence. We hypothesized that vorinostat, a histone deacetylase (HDAC) inhibitor with anti-angiogenic effects, would prevent acquired resistance to bevacizumab.
Methods
This multicenter phase II trial used a Bayesian adaptive design to randomize patients with recurrent GBM to bevacizumab alone or bevacizumab plus vorinostat with the primary endpoint of progression-free survival (PFS) and secondary endpoints of overall survival (OS) and clinical outcomes assessment (MD Anderson Symptom Inventory Brain Tumor module [MDASI-BT]). Eligible patients were adults (≥18 y) with histologically confirmed GBM recurrent after prior radiation therapy, with adequate organ function, KPS ≥60, and no prior bevacizumab or HDAC inhibitors.
Results
Ninety patients (bevacizumab + vorinostat: 49, bevacizumab: 41) were enrolled, of whom 74 were evaluable for PFS (bevacizumab + vorinostat: 44, bevacizumab: 30). Median PFS (3.7 vs 3.9 mo, P = 0.94, hazard ratio [HR] 0.63 [95% CI: 0.38, 1.06, P = 0.08]), median OS (7.8 vs 9.3 mo, P = 0.64, HR 0.93 [95% CI: 0.5, 1.6, P = 0.79]) and clinical benefit were similar between the 2 arms. Toxicity (grade ≥3) in 85 evaluable patients included hypertension (n = 37), neurological changes (n = 2), anorexia (n = 2), infections (n = 9), wound dehiscence (n = 2), deep vein thrombosis/pulmonary embolism (n = 2), and colonic perforation (n = 1).
Conclusions
Bevacizumab combined with vorinostat did not yield improvement in PFS or OS or clinical benefit compared with bevacizumab alone or a clinical benefit in adults with recurrent GBM. This trial is the first to test a Bayesian adaptive design with adaptive randomization and Bayesian continuous monitoring in patients with primary brain tumor and demonstrates the feasibility of using complex Bayesian adaptive design in a multicenter setting.
Keywords: Bayesian adaptive trial design, bevacizumab, vorinostat, recurrent glioblastoma, progression free survival
Key Points.
Acquired resistance to bevacizumab therapy results in failure of this anti-angiogenic strategy against recurrent glioblastoma, which could be overcome by HDAC inhibition.
Treatment with vorinostat, an HDAC inhibitor, combined with bevacizumab was not superior to bevacizumab against recurrent GBM in clinical or quality of life measures.
This multicenter study establishes the feasibility of using a Bayesian study design in brain tumor trials.
Importance of the Study.
Anti-angiogenic strategies using bevacizumab, a vascular endothelial growth factor inhibitor, improve outcome in only a small subset of patients with recurrent glioblastoma due to intrinsic or adaptive resistance to this agent. This multicenter randomized trial tested the hypothesis that HDAC inhibition using vorinostat, a brain-penetrant HDAC inhibitor, could overcome emergent resistance to the anti-angiogenic effects of bevacizumab. It demonstrates no improvement in PFS or OS for this combination compared with bevacizumab alone. In addition, quality of life measures assessed through the validated MDASI-BT questionnaire showed no improvement in patient-reported symptoms with this combination. This is the first study to utilize a Bayesian adaptive randomized trial against brain tumors and establishes the feasibility of such complex designs for future Bayesian design trials in neuro-oncology.
Treatment with bevacizumab is the current standard of care for adults with recurrent glioblastoma (GBM).1 However, these tumors progress when tumor cells escape their dependence on vascular endothelial growth factor (VEGF)–driven angiogenesis, the target of bevacizumab therapy.2,3 Several studies have demonstrated a role for primary or acquired resistance to agents targeting the VEGF pathway. For instance, genetic ablation of VEGF-A results in increased invasiveness of tumor cells and co-option of existing vessels; such results have also been seen in antibody mediated VEGF inhibition and in human tumors after bevacizumab treatment.4–7 In animal models of malignancy, GBMs which expressed VEGF and basic fibroblast growth factor (bFGF) showed less response compared with rhabdomyosarcomas which were dependent on VEGF alone, suggesting a role for other angiogenic factors in modifying tumor response to VEGF inhibition.8 Further, tumor xenografts which initially responded to VEGF inhibition showed subsequent progression during treatment in association with high expression of angiopoetin-1, platelet-derived growth factor B (PDGF-B), and ephrin B2.9 These data provide strong support for the presence of several intrinsic or emergent mechanisms that mediate resistance to anti-angiogenic therapy.9–12 Agents targeting such alternative pathways have been shown to overcome such adaptive resistance and resensitize resistant tumors to anti-angiogenic strategies.12,13
Histone deacetylase (HDAC) inhibitors target nuclear and cytoplasmic deacetylases and cause pleiotropic effects on various biological pathways relevant to malignancies,14 including downregulation of pro-angiogenic factors such as VEGF, bFGF, angiopoietin, and TIE2 (tunica interna endothelial cell kinase 2)15 and decreased transcriptional activity of hypoxia inducible factor 1α,16,17 a key mediator of resistance to anti-VEGF therapy.18,19 Among several HDAC inhibitors currently in clinical trials, vorinostat, an orally bioavailable small molecular weight hydroxamic acid-based inhibitor, has shown preliminary evidence of clinical activity as a single agent in a phase II trial in patients with recurrent GBM.20 Based on these data, we hypothesized that HDAC inhibition, by blocking signaling pathways that are described in preclinical and clinical settings to be involved in tumor progression in the setting of VEGF pathway inhibition, could prevent adaptive responses to bevacizumab-mediated VEGF inhibition and improve clinical outcome. To test this hypothesis, we conducted a multicenter trial through the Brain Tumor Trials Collaborative (BTTC) using a novel Bayesian adaptive randomized phase II trial design to compare the efficacy of bevacizumab plus vorinostat with bevacizumab alone against recurrent GBM.
Patients and Methods
Eligibility
Key eligibility criteria included age ≥18 years, KPS ≥60, adequate organ function, and unequivocal radiological evidence of disease recurrence after prior radiation therapy (completed at least 12 wk prior to study entry). Patients with histologically confirmed World Health Organization (WHO) grade III or IV glioma were eligible for the phase I portion; those with recurrent supratentorial WHO grade IV glioma with ≤3 relapses were eligible for the phase II trial. Key exclusion criteria included pregnancy or nursing, serious intercurrent illness, inability to consent, and prior treatment with bevacizumab, vorinostat, or other HDAC inhibitors including valproic acid (due to its known HDAC activity).21 Patients were required to be at least 4 weeks from prior investigational therapy, 6 weeks from prior nitrosourea therapy, and 4 weeks from other cytotoxic therapy. Bevacizumab-specific exclusion criteria included uncontrolled hypertension, cardiac insufficiency greater than New York Heart Association grade II, cardiac or cerebrovascular ischemic event or major bleeding event within 6 months before study start, recent surgery, recent trauma, or presence of non-healing wounds. The trial was registered at ClinicalTrials.Gov (NCT01266031) and conducted according to the principles of the Declaration of Helsinki, with informed consent obtained from all participants and with the approval of the institutional review board of each participating center.
Evaluation During Study
On-study evaluation included a history and physical examination at baseline and at the start of each cycle, complete blood counts every 2 weeks, and serum chemistries every 4 weeks. A brain MRI scan and a patient-completed MD Anderson Symptom Inventory‒Brain Tumor (MDASI-BT) survey were obtained at baseline and before every other cycle (approximately every 8 wk). Blood pressure, coagulation tests, and urine protein/creatinine ratio were recorded at baseline and prior to each infusion of bevacizumab.
Treatment Plan
All eligible patients registered with the BTTC Office of Multicenter Clinical Research (OMCR) and initiated treatment within 96 hours after registration.
Phase I study.
—A 3 + 3 design was utilized with the initial cohort treated with bevacizumab at 10 mg/kg on days 1 and 15 intravenously combined with vorinostat at 400 mg daily orally on days 1–7 and 15–21 of a 28-day cycle. Vorinostat starting dose of 400 mg daily was chosen given that higher doses were known to be associated with toxicity; planned dose de-escalation of vorinostat was to 300 mg, 200 mg, or 100 mg daily.
Phase II study.
—Patients were randomized using a Bayesian adaptive randomization (BAR) design to 2 competing treatment arms: bevacizumab alone or the combination of bevacizumab and vorinostat (at maximum tolerated dose [MTD]) in a 28-day cycle. Patients continued treatment until tumor progression or occurrence of unacceptable toxicity. Bevacizumab-related toxicity was managed according to standard practice without dose reductions. Vorinostat dose was reduced for grade ≥3 drug-related toxicity with 2 dose reductions permitted. If treatment hold exceeded 4 weeks, the patients were taken off the study. Patients who discontinued treatment due to progression were followed for survival every 3 months; others who discontinued therapy were followed every 2 months until progression or start of a new therapy.
Endpoints
For the phase I study, primary endpoint was the dose limiting toxicity (DLT) of bevacizumab plus vorinostat. Toxicities were graded according to the Common Terminology Criteria v4.0. DLT was defined as treatment-related adverse events occurring in the first cycle of treatment (4 wk) including grade 4 hematological toxicity (except grade 4 uncomplicated neutropenia or febrile neutropenia of ≤5 days duration) or non-hematologic grade 3 or 4 events except medically manageable toxicities. For the phase II trial, the primary endpoint was progression-free survival (PFS) calculated from the time of randomization until tumor progression or death. Radiological responses and progression were assessed using the MacDonald criteria22 (given that the initiation of the study preceded the establishment of the Response Assessment in Neuro-Oncology criteria); however, anticipating non-enhancing patterns of recurrence, bidimensionally measurable MRI fluid attenuated inversion recovery progression associated with clinical decline was deemed disease progression. PFS was determined by site principal investigators who were provided guidelines for such determination per protocol specifications. The secondary endpoint of overall survival (OS) was calculated from the time of randomization until death from any cause.
Patient-Reported Symptoms
Patient-reported symptoms were assessed using the MDASI-BT self-reporting tool,23 which patients completed approximately every 8 weeks during treatment. Participation was not mandatory. Data were analyzed for completion rates, overall mean symptom and interference scores, and change score from baseline through each treatment cycle. Mean differences in symptom and interference change scores between treatment arms were evaluated with independent-samples t-tests. Significance level was set at P < 0.05. Computations for the patient-reported symptoms were done using IBM SPSS for Windows v23.
Correlative Studies
Plasma samples for the optional biomarker analysis were obtained at pretreatment, on cycle 1, days 1, 2, and 15 (pre- and post-infusion), and cycle 2 (pre-infusion). Approximately 7 cc of venous blood was collected and centrifuged at room temperature and 1 mL of supernatant plasma was stored frozen at −80°C, shipped on dry ice, and maintained frozen until analysis. The samples were analyzed according to the manufacturer’s instructions using commercially available antibody arrays designed to quantitatively detect 60 human angiogenic factors (Quantibody Human Angiogenesis Arrays 1 and 3 [QAH-ANG-1000–1], Ray Biotech).
Study Design and Statistical Analysis
The phase I component was conducted using a 3 + 3 design with DLT assessment within the first 4 weeks of treatment. At each dose level, if 1/3 of the patients experienced DLT, the cohort would be expanded to 3 more patients. If ≤1/6 patients experienced DLT, the MTD was considered to have been reached and this dose would be used for the phase II part of the study. If ≥2/6 patients experienced DLT, the MTD was considered exceeded and the dose would be de-escalated. The starting dose was vorinostat at 400 mg daily combined with bevacizumab at 10 mg/kg. Planned dose de-escalation of vorinostat was 300 mg, 200 mg, or 100 mg daily.
The phase II portion of the trial was conducted using a Bayesian adaptive design with BAR and Bayesian continuous monitoring (BCM), in which patients were randomized fairly between the 2 arms at the start of the trial for the first 20 patients (10 per arm). Thereafter, as the trial progressed and data accrued, the randomization became unbalanced in favor of the treatment that, on average, had longer median PFS. Therefore, each successive patient was more likely to receive the more efficacious treatment. The technical details of the design are summarized in Supplementary data 1. The BAR is based on PFS, rather than OS, because PFS can be observed more quickly to provide sufficient data for making adaptive decisions in a timely fashion. A minimum of 20 and a maximum of 90 patients were planned to be accrued with an anticipated accrual rate between 3 and 5 patients per month. During the trial, the evidence of the relative efficacy between 2 arms was continuously monitored using BCM based on Bayesian posterior probability. The trial would stop early and a treatment arm (experimental or control arm) would be declared as “winner” if the posterior probability of the median PFS of that treatment arm being larger than that of the other arm exceeded 0.995. The BCM could save sample size if the experimental arm was either more effective or ineffective compared with the control. If the trial did not stop early and the maximum 90 patients were accrued, a treatment arm would be selected as “winner” if the probability of the median PFS of that treatment arm being larger than that of the other arm exceeded 0.995. The trial was conducted using a web-based program developed by the Department of Biostatistics and Applied Mathematics at MDACC through which OMCR personnel randomized patients to the 2 arms and updated their current status on an ongoing basis. The PFS and OS were summarized using the Kaplan–Meier method and the comparison between groups was evaluated by the standard log-rank test without adjusting for adaptive randomization, as the latter has little impact on the inference. Both univariate and multivariate Cox regression models were applied to assess the effect of covariates of interest on PFS and OS. All computations were carried out in SAS 9.4 and R 3.2.4.
Results
Phase I Trial
Six patients were enrolled in the phase I portion of the study and all were evaluable. One patient in the first cohort experienced DLT (grade 3 alanine aminotransferase elevation and grade 3 hyperglycemia) possibly related to vorinostat. The cohort was expanded by 3 more patients, with none experiencing a DLT in the first cycle. The starting doses of 10 mg/kg bevacizumab on days 1 and 15 and vorinostat at 400 mg daily on days 1–7 and 15–21 were therefore identified as the phase II combination dose.
Phase II Trial
Patient characteristics.
—The study was conducted through the 16 institutions of the BTTC over a 24-month period. All patients had received radiation therapy and temozolomide previously. Among 90 patients accrued, one did not receive study treatment and four were not evaluable; of the remaining 85 patients, 11 were evaluable for toxicity only given that they did not receive sufficient duration of treatment and 74 were evaluable for both toxicity and response (Figure 1, CONSORT diagram). Descriptive analysis of baseline patient characteristics is summarized in Table 1 and showed no significant difference between the 2 treatment groups.
Fig. 1.

Consolidated Standards of Reporting Trials (CONSORT) diagram.
Table 1.
Summary of patients’ demographics and clinical characteristics
| Covariate | Levels | Total | Treatment Group | P-value | |
|---|---|---|---|---|---|
| Bevacizumab | Bevacizumab +Vorinostat | ||||
| All patients | 85 (100%) | 38 (44.7%) | 47 (55.3%) | ||
| Sex | Female | 27 (31.8%) | 10 (26.3%) | 17 (36.2%) | 0.3319 |
| Male | 58 (68.2%) | 28 (73.7%) | 30 (63.8%) | ||
| Performance status | 100 | 8 (9.4%) | 3 (7.9%) | 5 (10.6%) | 0.7363 |
| 90 | 26 (30.6%) | 10 (26.3%) | 16 (34%) | ||
| 80 | 22 (25.9%) | 12 (31.6%) | 10 (21.3%) | ||
| 70 | 25 (29.4%) | 12 (31.6%) | 13 (27.7%) | ||
| 60 | 4 (4.7%) | 1 (2.6%) | 3 (6.4%) | ||
| Prior radiotherapy | Yes | 85 (100%) | 38 (100%) | 47 (100%) | |
| Prior systemic therapy | No | 2 (2.4%) | 1 (2.6%) | 1 (2.1%) | 1.0000 |
| Yes | 83 (97.6%) | 37 (97.4%) | 46 (97.9%) | ||
| Prior surgery | Yes | 85 (100%) | 38 (100%) | 47 (100%) | |
| Other prior therapy | No | 74 (87.1%) | 35 (92.1%) | 39 (83%) | 0.3313 |
| Yes | 11 (12.9%) | 3 (7.9%) | 8 (17%) | ||
| Prior radiosensitizer therapy | No | 34 (40%) | 17 (44.7%) | 17 (36.2%) | 0.4228 |
| Yes | 51 (60%) | 21 (55.3%) | 30 (63.8%) | ||
| Relapse number | 0 | 3 (3.5%) | 2 (5.3%) | 1 (2.1%) | 0.6579 |
| 1 | 62 (72.9%) | 29 (76.3%) | 33 (70.2%) | ||
| 2 | 19 (22.4%) | 7 (18.4%) | 12 (25.5%) | ||
| 3 | 1 (1.2%) | 0 (0%) | 1 (2.1%) | ||
| Patient evaluable for response | No | 11 (12.9%) | 8 (21.1%) | 3 (6.4%) | 0.0566 |
| Yes | 74 (87.1%) | 30 (78.9%) | 44 (93.6%) | ||
| Primary cause of death | Unknown | 22 | |||
| Attributed to protocol treatment | 1 (1.6%) | 0 (0%) | 1 (2.7%) | 0.1360 | |
| Due to other cause | 4 (6.3%) | 0 (0%) | 4 (10.8%) | ||
| Due to disease | 58 (92.1%) | 26 (100%) | 32 (86.5%) | ||
Toxicity data.
—Among the 85 patients evaluable for toxicity, the most frequent adverse events of any grade or treatment relationship were fatigue, hypertension, anorexia, headache, hyperglycemia, nausea, anemia, cognitive disturbance, and seizures, the majority of which were grade ≤2 in severity. Treatment-related toxicities grade ≥3 were not significantly different between the 2 arms (bevacizumab, 27 events and bevacizumab plus vorinostat, 31 events). Three patients experienced serious adverse events (grade 4 or 5) possibly related to treatment, 1 in the bevacizumab arm (grade 4 decreased ejection fraction) and 2 in the bevacizumab plus vorinostat arm (grade 4 colonic perforation and grade 5 thromboembolic event) (Supplementary Tables 1 and 2).
Patient-reported outcomes.
—Of the 96 patients accrued to the clinical trial, 65 (68%) consented to answering the optional MDASI-BT questionnaire, with 5 not having received treatment. Of these, 83% completed baseline assessment (Supplementary Table 3). Patients in each arm reported similar symptom burden at baseline between the bevacizumab and the bevacizumab plus vorinostat arms, with a mean score (standard deviation [SD]) of 1.3 (1.2) vs 1.7 (1.7), respectively. Patients in both arms reported moderate interference at baseline (mean = 2.2 for both arms). There were no significant differences in change scores for symptom burden between treatment arms. The mean change score from baseline to end of therapy was −0.8 (SD = 0.9) for patients in the bevacizumab arm and −0.4 (SD = 1.3) for patients in the bevacizumab plus vorinostat arm (P < 0.43). Interference scores worsened in both arms during treatment (a change of 1 point or greater), but there were no significant differences in change scores between treatment arms, with a mean (SD) change score from baseline to end of therapy of −1.3 (1.7) for the bevacizumab arm and −1.5 (2.1) for the bevacizumab plus vorinostat arm (P < 0.84) (Table 2). For 12 patients who received at least 6 cycles, no difference between arms was seen for those who remained on therapy.
Table 2.
MDASI-BT symptom burden and interference mean scores and change scores by cycle and treatment arm
| Bevacizumab | Bevacizumab + Vorinostat | P-value | Bevacizumab Change score from baseline | Bevacizumab + Vorinostat Change score from baseline | P-value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean | SD | n | Mean | SD | n | Mean | SD | n | Mean | SD | |||
| Symptom burden | ||||||||||||||
| Baseline | 18 | 1.3 | 1.2 | 32 | 1.7 | 1.7 | 0.38 | – | – | – | – | – | – | – |
| Cycle 2 | 11 | 1.3 | 1.0 | 20 | 1.3 | 1.2 | 0.92 | 10 | 0.0 | 0.5 | 20 | 0.4 | 1.5 | 0.32 |
| Cycle 4 | 9 | 1.7 | 1.2 | 12 | 2.1 | 1.8 | 0.58 | 8 | −0.2 | 0.6 | 12 | −0.1 | 1.5 | 0.90 |
| Cycle 6 | 7 | 1.8 | 1.5 | 6 | 1.6 | 2.0 | 0.84 | 7 | −0.4 | 1.1 | 6 | −0.8 | 2.2 | 0.70 |
| Cycle 8 | 2 | 1.5 | 0.1 | 4 | 2.6 | 2.3 | 0.40 | 2 | 0.2 | 1.0 | 4 | −1.8 | 2.6 | 0.39 |
| Cycle 10 | 2 | 1.5 | 0.3 | 2 | 2.1 | 2.8 | 0.77 | 2 | 0.2 | 0.9 | 2 | −1.1 | 2.2 | 0.55 |
| Cycle 12 | 1 | 1.1 | – | 0 | – | – | – | 1 | −0.3 | – | 0 | – | – | – |
| End of therapy | 11 | 2.1 | 1.3 | 17 | 2.3 | 2.0 | 0.75 | 9 | −0.8 | 0.9 | 17 | −0.4 | 1.3 | 0.43 |
| Interference | ||||||||||||||
| Baseline | 18 | 2.2 | 2.1 | 32 | 2.2 | 2.7 | 0.982 | – | – | – | – | – | – | – |
| Cycle 2 | 11 | 1.5 | 1.5 | 21 | 1.8 | 1.9 | 0.625 | 10 | 0.5 | 1.1 | 21 | −0.1 | 2.2 | 0.47 |
| Cycle 4 | 9 | 2.6 | 1.8 | 12 | 3.0 | 2.8 | 0.716 | 8 | −0.5 | 1.0 | 12 | −1.1 | 2.6 | 0.59 |
| Cycle 6 | 7 | 3.3 | 2.5 | 6 | 1.0 | 1.3 | 0.068 | 7 | −1.5 | 1.7 | 6 | −0.4 | 1.3 | 0.24 |
| Cycle 8 | 2 | 1.4 | 0.6 | 4 | 2.0 | 3.2 | 0.823 | 2 | 1.2 | 1.6 | 4 | −1.3 | 3.2 | 0.37 |
| Cycle 10 | 2 | 1.6 | 0.4 | 2 | 4.1 | 3.7 | 0.437 | 2 | 1.0 | 1.9 | 2 | −3.1 | 3.9 | 0.35 |
| Cycle 12 | 1 | 1.2 | – | 0 | – | – | – | 1 | −0.2 | – | 0 | – | – | – |
| End of therapy | 11 | 3.2 | 2.1 | 18 | 2.7 | 2.9 | 0.639 | 9 | −1.3 | 1.7 | 18 | −1.5 | 2.1 | 0.84 |
Plasma biomarker of angiogenesis.
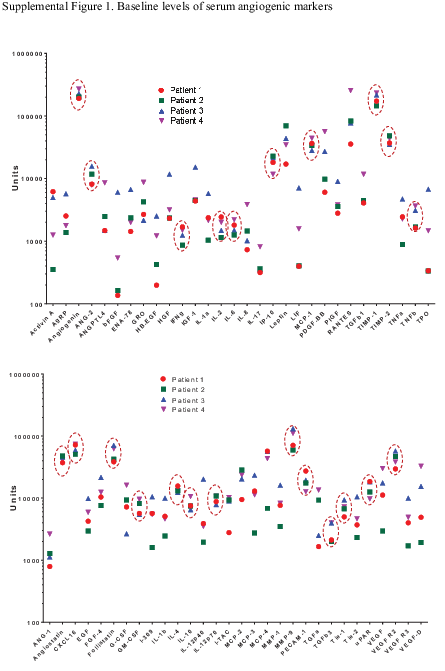
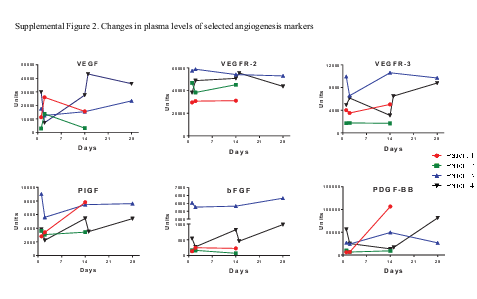
—An exploratory objective of the trial was to assess the changes in plasma biomarkers implicated in angiogenesis. Samples over multiple time points were available from only 4 patients among the subset who participated in this part of the study. Baseline assessment of 60 human angiogenic factors in plasma samples showed interpatient clustering in levels of several factors (Supplementary Figure 1, circled). However, there was no consistent treatment-related change in plasma levels of angiogenic factors related to adaptive resistance including VEGF, VEGF receptor (R)-2, VEGFR-3, bFGF, PDGF with 2 B chains (PDGF-BB), and placenta growth factor (PlGF) (Supplementary Figure 2).
Efficacy.
—At the time of PFS analysis, 70 of the 74 evaluable patients had progressed and/or died. The median PFS time was 3.71 months (95% CI: 2.79, 4.21). The difference between treatment groups was not significant (P = 0.94). The median PFS and PFS rate at 3, 6, and 12 months are as summarized (Table 3). The PFS6 rates were 28% for the bevacizumab arm and 25% for the bevacizumab plus vorinostat arm, which were not significantly different (P = 0.94). In the OS assessment, 60 of the 74 evaluable patients had died at the time of analysis, with a median OS time of 8.11 months (95% CI: 6.18, 9.63) and a median follow-up time of 19.84 months (95% CI: 13.50, 27.30) with no significant difference between treatment groups (P = 0.64). The median OS times and OS rate at 3, 6, and 12 months are as summarized (Table 3). In analysis of time to progression (TTP), 65 of the 74 evaluable patients had disease progression with a median TTP of 3.78 months (95% CI: 2.86, 5.29) and no significant difference between treatment groups (P = 0.71). The median TTP times and TTP rates at 3, 6, and 12 months are as summarized in Table 4 and Supplementary Tables 4 and 5.
Table 3.
Summary of PFS and OS rate at 3, 6, and 12 months
| Covariate | Months | Progression-Free Survival | Overall Survival | ||
|---|---|---|---|---|---|
| PFS Rate (95% CI) | P-value | OS Rate (95% CI) | P-value | ||
| All Patients | 3 | 0.60 (0.48, 0.70) | 0.92 (0.82, 0.96) | ||
| 6 | 0.26 (0.16, 0.37) | 0.64 (0.51, 0.74) | |||
| 12 | 0.10 (0.04, 0.18) | 0.29 (0.18, 0.40) | |||
| Female | 3 | 0.64 (0.42, 0.80) | 0.0434 | 0.88 (0.67, 0.96) | 0.0331 |
| 6 | 0.35 (0.17, 0.54) | 0.67 (0.45, 0.82) | |||
| 12 | 0.26 (0.11, 0.45) | 0.40 (0.21, 0.59) | |||
| Male | 3 | 0.58 (0.42, 0.70) | 0.94 (0.82, 0.98) | ||
| 6 | 0.21 (0.11, 0.34) | 0.62 (0.46, 0.74) | |||
| 12 | 0.02 (0.00, 0.10) | 0.22 (0.11, 0.36) | |||
| Treatment Group | |||||
| Bevacizumab | 3 | 0.59 (0.39, 0.74) | 0.9401 | 0.93 (0.75, 0.98) | 0.6398 |
| 6 | 0.28 (0.13, 0.44) | 0.66 (0.45, 0.80) | |||
| 12 | 0.07 (0.01, 0.20) | 0.28 (0.13, 0.46) | |||
| Bevacizumab + Vorinostat | 3 | 0.61 (0.44, 0.73) | 0.91 (0.77, 0.96) | ||
| 6 | 0.25 (0.13, 0.39) | 0.62 (0.46, 0.75) | |||
| 12 | 0.13 (0.05, 0.24) | 0.29 (0.16, 0.43) | |||
Table 4.
Summary of median OS and PFS times
| Covariate | Level | Median OS Time (mo) | Median PFS Time (mo) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Failed | Censored | Median (95% CI) | P-value | Total | Failed | Censored | Median (95% CI) | P-value | ||
| All patients | 74 | 60 | 14 | 8.11 (6.18, 9.63) | 74 | 70 | 4 | 3.71 (2.79, 4.21) | |||
| Treatment | Bevacizumab | 30 | 23 | 7 | 9.26 (5.88, 11.37) | 0.6398 | 30 | 29 | 1 | 3.94 (1.94, 5.68) | 0.9401 |
| Bevacizumab + vorinostat | 44 | 37 | 7 | 7.79 (5.06, 9.63) | 44 | 41 | 3 | 3.68 (2.33, 3.94) | |||
Efficacy was analyzed in both univariate and multivariate Cox regression models for PFS and OS. Upon multivariate analysis adjusted for performance status (hazard ratio [HR] = 1.64, P = 0.07, 60–80 vs 90–100), current relapse number (HR = 2.32, P = 0.006, 2–3 vs 0–1), and number of cycles administered (HR = 0.68, P < 0.0001), patients treated with bevacizumab and vorinostat had a 37% lower chance of progression, compared with patients treated with bevacizumab alone (HR = 0.63, P = 0.08) (Supplementary Table 6). Similarly, in multivariate analysis adjusted for performance status (HR = 1.60, P = 0.09, 60–80 vs 90–100) and number of cycles administered (HR = 0.73, P < 0.0001), patients treated with bevacizumab plus vorinostat had a 7% lower chance of death compared with patients treated with bevacizumab alone (HR = 0.93, P = 0.79) (Supplementary Table 6). Results of the Kaplan–Meier analysis for PFS and OS are depicted in Figure 2.
Fig. 2.

Kaplan–Meier curves for PFS (left) and OS (right) (E = evaluable, N = total).
Discussion
This trial examined whether targeting of potential resistance mechanisms to bevacizumab therapy by HDAC inhibition using vorinostat could improve outcome in recurrent GBM patients. In a phase I study of the combination of bevacizumab, vorinostat, and irinotecan, Chinnaiyan et al reported poor tolerance of irinotecan and recommended testing bevacizumab with vorinostat only (as in this study) for better tolerance24; our study confirmed that this combination was well tolerated. Previous studies have assessed the role of HDAC inhibition combined with bevacizumab; Lee et al conducted a phase II study combining panobinostat and bevacizumab in patients with recurrent GBM but the study was closed prematurely due to interim results not meeting criteria for continued accrual.25 Ghiaseddin et al reported the results of combining vorinostat and bevacizumab in adults with recurrent GBM which did not show improvement in PFS6 or OS.26 Lastly, Peters et al reported that the combination of bevacizumab, vorinostat, and temozolomide resulted in a PFS6 of 53.8%, which was not significantly better than historical controls.27 These nonrandomized trials with historical controls and heavily pretreated patients limited the ability to determine the true benefit of the combination.
From a patient-reported outcome perspective, overall, patients enrolled in this study had moderate interference of symptoms in daily life, but a low symptom burden. This interference worsened during the course of therapy but was not different between the two arms, including for those receiving at least 6 treatment cycles. These findings indicate limited additional symptom burden for the combination of vorinostat and bevacizumab and highlight the interference of the disease with daily life independent of the toxicities of either therapy.
In the biomarker analysis, certain markers showed interpatient consistency in values, whereas others were disparate from patient to patient. None of the predicted angiogenesis bypass pathway proteins such as PDGF, FGF, or PlGF showed any specific treatment-related changes, possibly because such changes in the tumor microenvironment may not be reflected in plasma protein levels. No definite conclusions can be drawn from these results due to the limited number of patients with sufficient samples for analysis. Larger numbers of samples are needed to determine the relationship of such markers to clinical outcome; however, the analysis demonstrates the feasibility to do multiplexed plasma marker assays in a multicenter study.
The combination of vorinostat and bevacizumab was not superior to bevacizumab alone in improving either PFS or OS in this study. Galanis et al reported that vorinostat can cross the intact blood–brain barrier and inhibit HDACs and has a modest level of antitumor activity against recurrent GBM.20 In addition, “vascular normalization” by bevacizumab was expected to improve tumor delivery of vorinostat.28 However, bevacizumab-induced decreased blood supply could have reduced vorinostat delivery to the tumor as reported for other agents,29 negating the potential benefits of the combination. Alternatively, vorinostat may have been ineffective in overcoming resistance mechanisms to bevacizumab in recurrent GBM. Our primary hypothesis that HDAC inhibition could postpone tumor recurrence by blocking alternative mechanisms of angiogenesis was not substantiated by the current study.
Previous discussions on the Bayesian adaptive design with BAR and BCM in brain tumors have been limited to computer simulation studies.30 To our knowledge, this study is the first prospective brain tumor trial conducted using such a design. The advantage of the Bayesian adaptive design is that it allows allocating more patients to a more effective treatment arm, and if there is strong evidence that the experimental arm is more effective or less effective than the control, the design allows early stopping of the trial with fewer patients accrued. In our trial, we noted that the experimental arm had a similar efficacy as the control arm in terms of PFS; thus, the advantage of the adaptive design was not fully demonstrated by this trial. Given that there was no a priori knowledge whether the trial would be positive or negative, this could not have been predicted when the trial was designed.
The implementation of Bayesian adaptive designs, such as BAR and BCM, is logistically challenging as it requires real-time communication of all events to the coordinating center, real-time decision making based on accrued data, and continuous involvement of the treating physicians, the biostatistician, and the study coordinating team throughout the conduct of the trial. In our experience, the key is to integrate the database with an adaptive decision algorithm such that clinical data can be captured, updated, and fed into the algorithm in real time to facilitate adaptive decisions. For this trial, we developed a web-based program that seamlessly integrated data capture, quality monitoring, storage, and real-time adaptive decision making. Clinical sites could log into the program through a web browser, update the patient data, and obtain the real-time adaptive randomization decision. Developing such a program was a continuous refinement process that requires close collaboration among physicians, biostatistician, and the study coordinating team.
Critics of adaptive designs have raised the issue that such trials require intensive resource allocation, overall collection of more data from patients to run outcome-adaptive allocation, and potentially the need to run slowly enough to allow events to drive the BAR and BCM, thus compromising efficiency.31,32 In addition, concerns have been raised whether patients may mistakenly assume that they will be allocated to the better arm when advised about the nature of the trial design during consenting. However, these concerns have been challenged by advocates of such designs, who contest the existence of any ethical issues in such designs and cite the fact that these trial designs could increase the probability that patients would be randomized to a more efficacious arm (if this exists) while not allocating them to an inferior treatment (if one arm is not more efficacious than the other).33–35 Although the trial requires additional resources, if a treatment is truly better than the control arm, the trial design would allow for a more efficient trial conduct. A complete discussion of these issues is beyond the scope of this manuscript, but we would like to emphasize that the successful completion of this trial demonstrates the feasibility to conduct complex Bayesian adaptive designs in a multicenter setting in the brain tumor population. Although this trial used 2 treatment arms, the same design could be used for multiple arms and could identify the best of such arms. A similar but larger international multicenter adaptive randomized trial platform with multiple treatment arms, the so-called AGILE GBM trial, is currently being planned and, over its course, is expected to test multiple agents against a common control arm.36 Given the potential of Bayesian adaptive designs to improve the efficiency and success of clinical trials,37 this trial provides an example for future trials for implementing novel adaptive designs.
Funding
This investigator-initiated study was supported by National Cancer Institute grant K24CA160777 (to V.P.), the Lasker Clinical Research and Intramural Research Program of the National Institute of Health (to J.W.), research funding from Genentech, and study drugs from Genentech (bevacizumab) and Merck Sharp & Dohme Corp (vorinostat).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We would like to acknowledge the central trial management teams at The MD Anderson Cancer Center and the National Cancer Institute for exceptional multicenter coordination of the trial. We would also like to acknowledge the clinical research teams of each BTTC member institution, the philanthropic support of the BTTC by the Head for the Cure Foundation, and the sponsors at Genentech and Merck Sharp & Dohme Corp, who provided study agents and funding for this study. Lastly, we gratefully acknowledge the participation of all patients and their caregivers who made the trial possible.
Conflict of interest statement.
VKP reports research funding from Abbvie, Bexion, Beigene, Celldex, DNAtrix, Novartis; advisory or data safety monitoring board honoraria from Novocure, SK Lifesciences, Ziopharm, and Orbus Therapeutics, stock ownership in Gilead and Amarin outside the submitted work; PG reports research funding from Cortice, Celldex and Immunocellular Therapeutics; MDG reports employment with Texas Oncology and Abbvie; honoraria and consultancy from Abbvie; research funding from Novocure; HC reports research funding from Newlink Genetics, Plexxicon, Kadmon, Orbus, Merck, DNAtrix, Abbvie, Beigene; and advisory board honoraria from Foundation Medicine, Abbvie, Innocrin, Tactical Therapeutics, Deciphera, Newlink Genetics, Merck, Best Doctors, Karyopharm Therapeutics; TW reports advisory board honoraria from Tocagen, Orbus, travel and accommodation coverage reimbursed by Tocagen and Novocure; JR reports employment with Astellas, equity in Celldex and Agenus, and stock options in Excicure; FI reports honoraria from PrimeOncology and Guidepoint and research funding from Novocure, Bristol Myer Squibb, Celldex, Stemline, Regeneron, Incyte, Northwest Biotherapeutics, and Immunocellular Therapeutics; KF reports research funding from Abbvie, Boston Biomedical, Diffusion Pharmaceuticals, Orbus, Tocagen, Vascular Biogenics, Immunocellular Therapeutics, FivePrime Therapeutics, Cantex, DNATrix, and Stemline Therapeutics, and honoraria and travel from UCB Pharma; NA reports honoraria from Novocure; WKAY reports stock ownership, honoraria and intellectual property from DNAtrix, travel or expenses from Amgen.
Authorship statement.
Experimental design: VKP, JW, YY, TSA, MRG. Implementation and conduct of trial: VKP, JW, TSA, YY, PG, HC, TW, JR, MDG, DT, FI, NA, NP, KF, DP, MC, RM, MPP, WKAY, MRG. Analysis and interpretation of data: VKP, JW, YY, TSA, EV, JW, JX, MRG. Writing of manuscript and approval of final manuscript: All authors.
References
- 1. Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14(11):1131–1138. [DOI] [PubMed] [Google Scholar]
- 2. Lombardi G, Pambuku A, Bellu L, et al. Effectiveness of antiangiogenic drugs in glioblastoma patients: a systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. 2017;111:94–102. [DOI] [PubMed] [Google Scholar]
- 3. Wang N, Jain RK, Batchelor TT. New directions in anti-angiogenic therapy for glioblastoma. Neurotherapeutics. 2017;14(2):321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pàez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rubenstein JL, Kim J, Ozawa T, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2(4):306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hardian RF, Goto T, Kuwabara H, et al. An autopsy case of widespread brain dissemination of glioblastoma unnoticed by magnetic resonance imaging after treatment with bevacizumab. Surg Neurol Int. 2019;10:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Groot JF, Fuller G, Kumar AJ, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12(3):233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362(6423):841–844. [DOI] [PubMed] [Google Scholar]
- 9. Huang J, Soffer SZ, Kim ES, et al. Vascular remodeling marks tumors that recur during chronic suppression of angiogenesis. Mol Cancer Res. 2004;2(1):36–42. [PubMed] [Google Scholar]
- 10. Lu KV, Bergers G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013;2(1):49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int J Mol Sci. 2018;19(4):E1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glade Bender J, Cooney EM, Kandel JJ, Yamashiro DJ. Vascular remodeling and clinical resistance to antiangiogenic cancer therapy. Drug Resist Updat. 2004;7(4-5):289–300. [DOI] [PubMed] [Google Scholar]
- 13. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111(9):1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27(32):5459–5468. [DOI] [PubMed] [Google Scholar]
- 15. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. [DOI] [PubMed] [Google Scholar]
- 16. Kim MS, Kwon HJ, Lee YM, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7(4):437–443. [DOI] [PubMed] [Google Scholar]
- 17. Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15(20):2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu YL, DeLay M, Jahangiri A, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim SH, Jeong JW, Park JA, et al. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol Rep. 2007;17(3):647–651. [PubMed] [Google Scholar]
- 20. Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27(12):2052–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Göttlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–1280. [DOI] [PubMed] [Google Scholar]
- 23. Armstrong TS, Mendoza T, Gning I, et al. Validation of the M.D. Anderson symptom inventory brain tumor module (MDASI-BT). J Neurooncol. 2006;80(1):27–35. [DOI] [PubMed] [Google Scholar]
- 24. Chinnaiyan P, Chowdhary S, Potthast L, et al. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro Oncol. 2012;14(1):93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee EQ, Reardon DA, Schiff D, et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. 2015;17(6):862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghiaseddin A, Reardon D, Massey W, et al. Phase II study of bevacizumab and vorinostat for patients with recurrent world health organization grade 4 malignant glioma. Oncologist. 2018;23(2):157–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peters KB, Lipp ES, Miller E, et al. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J Neurooncol. 2018;137(2):349–356. [DOI] [PubMed] [Google Scholar]
- 28. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–3736. [DOI] [PubMed] [Google Scholar]
- 29. Van der Veldt AA, Lubberink M, Bahce I, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21(1):82–91. [DOI] [PubMed] [Google Scholar]
- 30. Trippa L, Lee EQ, Wen PY, et al. Bayesian adaptive randomized trial design for patients with recurrent glioblastoma. J Clin Oncol. 2012;30(26):3258–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hey SP, Kimmelman J. Are outcome-adaptive allocation trials ethical? Clin Trials. 2015;12(2):102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korn EL, Freidlin B. Outcome-adaptive randomization: is it useful? J Clin Oncol. 2011;29(6):771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Berry DA. Commentary on hey and kimmelman. Clin Trials. 2015;12(2):107–109. [DOI] [PubMed] [Google Scholar]
- 34. Lee JJ. Commentary on Hey and Kimmelman. Clin Trials. 2015;12(2):110–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saxman SB. Commentary on Hey and Kimmelman. Clin Trials. 2015;12(2):113–115. [DOI] [PubMed] [Google Scholar]
- 36. Alexander BM, Ba S, Berger MS, et al. ; GBM AGILE Network Adaptive global innovative learning environment for glioblastoma: GBM AGILE. Clin Cancer Res. 2018;24(4):737–743. [DOI] [PubMed] [Google Scholar]
- 37. Giles FJ, Kantarjian HM, Cortes JE, et al. Adaptive randomized study of idarubicin and cytarabine versus troxacitabine and cytarabine versus troxacitabine and idarubicin in untreated patients 50 years or older with adverse karyotype acute myeloid leukemia. J Clin Oncol. 2003;21(9):1722–1727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.