

Abstract
Background:
Crohn’s disease (CD) may progress from an inflammatory to a stricturing or penetrating disease phenotype. The aim of our study was to identify single nucleotide polymorphisms (SNPs) that predict disease progression in patients of the Swiss IBD Cohort Study (SIBDCS).
Methods:
We applied a multi-state Markov model for progression behavior of CD with three behavioral states according to the Montreal classification. The model considered transition from B1 to B2/B3 or from B2 to B3 stage. Model dynamics were summarized with transition intensities by including the effect of SNPs and calculating transition intensities for each SNP.
Results:
We included 1276 CD patients [669 (52.4%) B1, 248 (19.4%) B2, 359 (28.1%) B3 patients] with a median follow-up of 6.8 (interquartile range = 3.6–9.1; range 0–11.6) years. Probability for a B1 patient to develop a stenosis (B1 to B2, q = 0.033) was twice as much as compared to developing a penetrating complication (B3) during the disease course. In contrast, the probability of entering B3 stage was similar regardless of whether antecedent stricture was present (B2 to B3, q = 0.016) or not (B1 to B3, q = 0.016). We identified SNPs within the gene loci encoding ZMIZ1, LOC105373831 and KSR1 as carrying the highest risk for progression to B3, while the presence of SNPs within gene loci TNFSF15 and CEBPB-PTPN1 protected from progression to B2 or B3.
Conclusion:
We identified new genetic risk factors that can predict disease course in CD patients. A closer understanding on the functional impact of these genetic variations might improve our treatment options finally to prevent disease progression in CD patients.
Keywords: B stage, Crohn’s disease, fistulas, genetic risk factors, Montreal classification, stenosis single-nucleotide polymorphism
Introduction
Crohn’s disease (CD) affects about one out of 200 people in developed countries with rising incidence and prevalence, often involving lifelong debilitating symptoms.1,2 Although at initial diagnosis the majority of inflammatory bowel disease (IBD) patients have a purely inflammatory disease, during their disease course, more than 50% of patients finally develop a stricturing or penetrating disease,3–5 corresponding to a progression in the B stage according to the Montreal classification. 4
At the beginning of the anti-tumor necrosis factor (TNF) era it was found that over a 5-year period 10% of patients progress from an inflammatory to a stricturing disease and 26% of patients progress to a penetrating disease phenotype. 4 However, recent studies estimate the number of patients with a progression to penetrating disease behavior over time to be smaller, while other studies did not detect an effect of biological or immunosuppressive medications on progression to B3 stage.6–8 However, contradictory data from The Netherlands suggested that disease progression (B-stage progression) in CD patients is still unaltered and not affected by biological or immunosuppressive medications. 9
Knowledge about the factors driving the disease progression from an inflammatory to a stricturing or penetrating disease phenotype and the resulting onset of long-term complications is crucial. Clinical parameters, such as the initial CD location at diagnosis, smoking and the age of the patient at diagnosis, have been established as risk factors for a progression to B stage.3,4,10–14
Genetic risk factors for the development of penetrating CD include African American origin and family history. 15 In addition, it has been demonstrated that host genetics and particularly the presence of specific single nucleotide polymorphisms (SNPs) within IBD risk genes critically impact the pathogenesis of CD. Such SNPs have also been associated with the development of penetrating CD. 16 However, these data have not been confirmed so far.
Here, we aimed at identifying the impact of specific genetic risk factors for B-stage progression in CD patients using patients from the Swiss IBD cohort study (SIBDCS).
Patients and methods
Patient data and cohort design
Demographic and clinical data were obtained from the database of the nationwide SIBDCS. The SIBDCS represents a Swiss-wide multicenter prospective observational study and includes IBD patients from all over Switzerland. The SIBDCS study has been continuously funded by the Swiss National Science Foundation (SNSF) since 2006. Repetitively applying a standardized questionnaire, patient data are collected once a year and entered into a central database. Therefore, the location of the disease according to the Montreal classification over time is available for all patients included. Comprehensive inclusion and exclusion criteria and further details on the SIBDCS are described elsewhere.17,18 The SIBDCS has been approved by the respective ethical committees in Switzerland (Ethics Committee of the Canton Zürich: EK-1316). All patients signed an informed consent for data collection and analysis for research purposes. The presented study has been evaluated and approved by the scientific board of the SIBDCS.
Study design
We investigated genetic risk SNPs for the development of penetrating CD (Montreal B3) of SIBDCS patients. Genotyping of SIBDC patients for 160 IBD risk SNPs was performed using Matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOFF) mass spectrometry as described previously. 19 Analyses were performed on those 160 SNPs which were known as IBD risk SNPs at the time of performing our molecular analyses. 20 For our study, we included 1276 CD patients for whom clinical and genetic information was available. The median follow-up was 6.8 years [interquartile range (IQR) = 3.6–9.1 years; range 0–11.6 years]. Each SNP was coded as follows: “0”, if no risk allele was present, “1”, if one risk allele was present (heterozygous), “2”, if two risk alleles were present (homozygous).
Calculation of variant allele frequencies
Allele frequencies of variants were calculated using the BCFtools software package 21 and 1000Genomes populations genotype data 22 downloaded from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/GRCh38_positions/. In addition, allele frequency information estimated from populations of the Haplotype Reference Consortium (HRC) reference panel was extracted from data provided by the Sanger Imputation Server, https://imputation.sanger.ac.uk and http://www.haplotype-reference-consortium.org/home. 23
Association look-ups with other traits and diseases
In order to highlight variants with strong evidence of association with other traits or diseases, we queried 13 variants against the PhenoScanner database version 2 24 and extracted information about known associations with genome-wide significance (p < 5 × 10−8) from published genome wide association studies (GWAS). PhenoScanner is a curated database holding publicly available results from large-scale genetic association studies published through the NHGRI-EBI GWAS catalogue (https://www.ebi.ac.uk/gwas/), NHLBI GRASP (https://grasp.nhlbi.nih.gov/), dbGaP catalogues (https://www.ncbi.nlm.nih.gov/gap/) and UK Biobank pheWAS by Benjamin Neale (http://www.nealelab.is/uk-biobank).
Statistical analysis
We considered a multi-state Markov model for progression behavior of CD with three disease states according to the Montreal classification of CD. 25 State B1 represents inflammatory disease without the presence of stenosis or fistula, B2 comprises stenoses but without fistula and B3 comprises the presence of any kind of fistula with or without additional stenosis.
Our model allowed the following transitions between the different states: a patient in B1 state can either remain in B1, or evolve into either B2 (develop a stenosis) or alternatively straight into B3 (develop a fistula). A patient in B2 state, already having a current stenosis or a history thereof, can either remain in B2, or evolve into B3 (develop a penetrating complication). A patient in B2 cannot go back to B1 as B1 is defined as “no history of complication (fistula or stenosis)”. A patient in B3 has already a history of fistula, and can only stay in B3. This B3 state is therefore called the “absorbing state”, as a patient in B3 will always stay in B3 and can never leave this B3 state any more.
To represent the dynamic of the model better, we computed the following probabilities: probability of a B1 patient staying in B1 state, probability of a B1 patient ending up in B2, probability of a B1 patient ending up in B3 state and probability of a B2 patient ending up in B3 state after 1, 5, 10 or 15 years.
For each scenario, the effect of the number of risk alleles on the transition intensities was summarized with hazard ratios (HRs) with 95% confidence interval (CI; HR > 1 indicating higher intensities, resulting in more complications; HR < 1 indicating lower intensities, resulting in fewer complications). Again, in each case, the dynamics of the system were illustrated with the probabilities of finally having transitioned into (or remained, respectively, in the case of sustained B1 stage) in the different B stages after 1, 5, 10 or 15 years.
We finally performed two different modelings, a linear and a categorical modeling, to demonstrate these SNP effects in more detail. First, we investigated the “linear” effect model. Here, the number of risk alleles was taken as a continuous variable, which forced the effect to be linear. This meant that having two risk alleles exerted twice the effect of having only one, and going from zero to one risk allele was the same effect as going from one allele to two. We then performed the “categorical” effect model: here the effect of the number of risk alleles could be more general. This meant that there was more freedom to define the different effects. We did not “force” any kind of relationship between effects of zero, one or two risk alleles. To this end, we put the SNPs into the model and tested if there was an overall effect on the transition intensities and analyzed the significant SNPs in detail.
Results
Patient characteristics
In total, we included 1276 CD patients in our study. At baseline, 669 (52.4%) patients presented with B1 disease, 248 (19.4%) patients with B2 and 359 (28.1%) patients with B3 according to the Montreal classification. Patient characteristics are given in Table 1.
Table 1.
Characteristics at baseline for the 1276 patients with clinical and genetic information.
| Gender | |
| Male | 628 (49.2%) |
| Female | 648 (50.8%) |
| Age | |
| Median, q25–q75, min–max | 37.8, 26.1–50.3, 16.2–85.5 |
| Stenosis | 425 (33.3%) |
| Non-perianal fistula | 171 (13.4%) |
| Perianal fistula | 245 (19.2%) |
| Behavior | |
| B1 | 669 (52.4%) |
| B2 (stenosis but no fistula) | 248 (19.4%) |
| B3 (fistula) | 359 (28.1%) |
| Follow-up time (years) | |
| Median, q25–q75, min–max | 6.8, 3.6–9.1, 0–11.6 |
Baseline probabilities for B stage migration during disease course
The dynamics of the model were summarized with transition intensities representing the instantaneous probability of switching from state to state. Those “baseline” intensities (with 95% CI) were: q = 0.033 (0.027–0.039) to switch from B1 to B2; q = 0.016 (0.012–0.021) to switch from B1 to B3 and q = 0.016 (0.011–0.023) to switch from B2 to B3. This means that there was twice as much “probability” for a patient to develop a stenosis than a fistula during the disease course. In contrast, the probability of developing a fistula was similar, if already a stenosis was present or not. Detailed probabilities over the entire observation period are given in Table 2. The probability that a patient developed a stenosis is increased in the first year, while the probability that a patient developed a fistula rose after 1 and 5 years of disease, respectively. In other words, the number of patients transforming into B2 and into B3 disease increased over time. After 15 years, about 22% of patients were suffering from penetrating disease and more than 30% suffered from stricturing disease (Figure 1).
Table 2.
“Baseline” probabilities (with 95% CI) to develop a certain disease state.
| Transition | After 1 year | After 5 years | After 10 years |
|---|---|---|---|
| B1 to B1 (stay in B1) | 0.952 (0.943–0.958) | 0.780 (0.747–0.812) | 0.608 (0.561–0.653) |
| B1 to B2 (stenosis but no fistula) | 0.033 (0.027–0.039) | 0.144 (0.119–0.171) | 0.245 (0.206–0.285) |
| B1 to B3 (fistula) | 0.016 (0.012–0.021) | 0.076 (0.060–0.097) | 0.147 (0.118–0.180) |
| B2 to B2 (stay in B2) | 0.984 (0.978–0.990) | 0.925 (0.891–0.948) | 0.855 (0.800–0.896) |
| B2 to B3 (stenosis to fistula) | 0.016 (0.010–0.022) | 0.075 (0.052–0.109) | 0.145 (0.104–0.200) |
CI, confidence interval.
Figure 1.
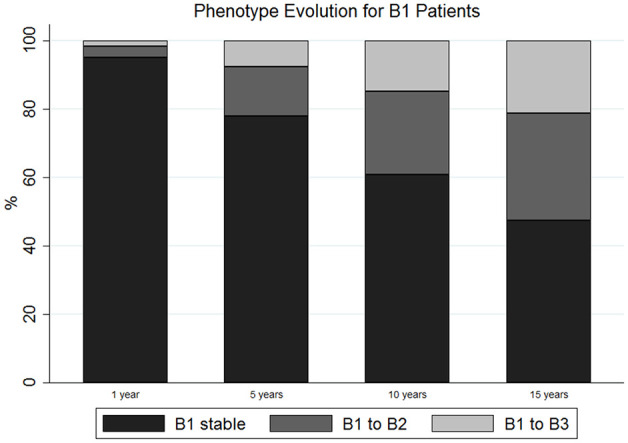
Phenotypic evolution of Crohn’s disease (CD) patients during their disease course.
Specific SNPs promote the transition intensities
In the next step, we analyzed the impact of genetic information/variation by calculating the transition intensities for each SNP. Using our linear modeling approach, we identified nine out of 160 SNPs being significantly associated with B-stage progression (Table 3). The minor allele frequency of these SNPs in the European ancestry population as well as the GWAS significance of those nine SNPs is displayed in Supplemental Tables 1 and 2. We next studied the number of relevant risk alleles identified with the linear modeling being present in each individual patient. We found that out of the 18 possible risk alleles (originating from homozygous or heterozygous presence of the nine significant SNPs), more than 20% of the patients had 11 risk alleles or more, while only a minority had less than eight risk alleles. The median number of risk alleles per patient was 11 (IQR 10–12), the minimum per patients 0 and the maximum number of risk alleles per patient was 17 (Figure 2).
Table 3.
These SNPs have been detected as significant in the linear modeling.
| Linear modeling | Gene symbol | Overall p value |
|---|---|---|
| Rs1250546 | ZMIZ1 | 0.0004 |
| Rs913678 | CEBPB-PTPN1 | 0.0010 |
| Rs11010067 | No gene assigned | 0.0086 |
| Rs17229285 | LOC105373831 | 0.0103 |
| Rs2836878 | No gene assigned | 0.0139 |
| Rs2066847 | NOD2 | 0.0161 |
| Rs17694108 | No gene assigned | 0.0233 |
| Rs2945412 | KSR1 | 0.0280 |
| Rs4246905 | TNFSF15 | 0.0422 |
SNP, single nucleotide polymorphism.
Figure 2.
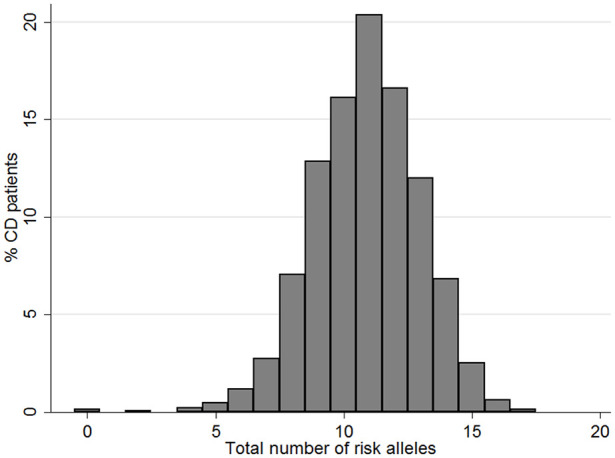
Distribution of the number of relevant risk alleles being present in one patient.
Effects of single SNPs on disease course of CD patients in the linear modeling
We next investigated the impact of the presence of single SNPs for the disease course of CD patients in our linear modeling (Table 4, Supplemental Table 3, Supplemental Figure 1). The presence of SNP rs11010067 is highly unfavorable for CD patients. Patients having one or two risk alleles have a greater risk of proceeding into a state with stenosis or fistula, with patients carrying two risk alleles being at the greatest risk for proceeding into a B3 state. Patients carrying SNP rs1250546 are at particularly high risk of transition from B1 to B3. Of note, the presence of this SNP has no major role for progression to B2 stage, and even makes progression from B2 into B3 stage very unlikely (Table 4). Patients with SNP rs17229285 are protected from the onset of stenosis, but are at high risk of developing fistulas without stenosis. Patients carrying this SNP with stenosis are, however, unlikely to progress further into the B3 stage. The presence of SNP rs17694108 protects the patients from transition from B1 to B3 stage, however, clearly enhances the risk of developing fistula when already having a stenosis. Patients carrying SNP rs2066847 are protected from the onset of stenosis, but are at slightly increased risk of developing fistulas. The presence of SNPs rs2836878, rs4246905 and rs913678 seems to be protective for CD patients, as it prevents progression into B2 and also B3 stage. In contrast, patients carrying the SNP rs2945412 are generally at greater risk of developing stenosis and fistulas and are at particularly high risk of developing fistulas, when a stenosis is already present.
Table 4.
The following SNPs exhibit significant transition intensities in the linear modeling.
| Transition intensities | HR (95% CI) |
|---|---|
| Rs 11010067 | |
| B1 to B2 | 1.338 (1.020–1.754) |
| B1 to B3 | 1.678 (1.132–2.487) |
| B2 to B3 | 1.234 (0.709–2.147) |
| Rs 1250546 | |
| B1 to B2 | 1.020 (0.768–1.356) |
| B1 to B3 | 2.313 (1.413–3.787) |
| B2 to B3 | 0.525 (0.308–0.897) |
| Rs 17229285 | |
| B1 to B2 | 0.729 (0.554–0.958) |
| B1 to B3 | 1.587 (1.049–2.393) |
| B2 to B3 | 0.730 (0.434–1.228) |
| Rs 17694108 | |
| B1 to B2 | 1.026 (0.771–1.364) |
| B1 to B3 | 0.489 (0.289–0.830) |
| B2 to B3 | 1.363 (0.786–2.362) |
| Rs 2066847 | |
| B1 to B2 | 0.513 (0.355–0.741) |
| B1 to B3 | 1.205 (0.506–2.871) |
| B2 to B3 | 1.052 (0.454–2.437) |
| Rs 2836878 | |
| B1 to B2 | 0.792 (0.591–1.063) |
| B1 to B3 | 0.570 (0.378–0.860) |
| B2 to B3 | 0.721 (0.418–1.244) |
| Rs 2945412 | |
| B1 to B2 | 1.271 (0.940–1.719) |
| B1 to B3 | 1.568 (0.991–2.482) |
| B2 to B3 | 1.606 (0.858–3.006) |
| Rs 4246905 | |
| B1 to B2 | 0.691 (0.515–0.928) |
| B1 to B3 | 0.794 (0.509–1.238) |
| B2 to B3 | 0.713 (0.409–1.243) |
| Rs 913678 | |
| B1 to B2 | 0.696 (0.523–0.926) |
| B1 to B3 | 0.557 (0.368–0.842) |
| B2 to B3 | 0.554 (0.388–1.135) |
CI, confidence interval; SNP, single nucleotide polymorphism.
Effects of single SNPs on disease course of CD patients in the categorical modeling
We then performed the “categorical” effect model: The eight out of 160 SNPs that were detected as significantly associated with B-stage progression are presented in Table 5. For the categorical model, the impact of specific SNPs on disease course is given in Table 6 and Supplemental Table 4. The minor allele frequency of these SNPs in the European ancestry population as well as the GWAS significance of those nine SNPs is displayed in Supplemental Tables 1 and 2. Using this model, having two risk alleles of SNP rs11010067 is specifically unfavorable for patients regarding the progression of B1 into B3 stage and also regarding the progression from B2 into B3 stage. With respect to the progression from B1 into B2 stage, there is no difference whether one or two risk alleles are present. Patients carrying SNP rs11150589 are protected from progression from B1 into either B2 or B3 stage. However, carriers of this SNP are at higher risk of developing fistulas, when stenosis is already present. The presence of one or two risk alleles of SNP rs11230563 is highly unfavorable for progression of B1 into B3 stage. In contrast, this SNP does not seem to play a major role for progression from B1 into B2 or from B2 into B3 stage. Patients carrying SNP rs1363907 are generally protected from progression to a stricturing or penetrating disease phenotype. Carriers of SNP rs17229285 are at higher risk of developing fistulas directly from B1 stage, while the risk of developing a stenosis or fistulas when stenoses are present is only mildly elevated. Patients that carry the SNP rs2945412 are at highly elevated risk of developing stenosis, but also fistulas from either B1 or B2 stage. Patients carrying the SNP rs913678 are in general protected from the onset of stenosis or fistulas. With respect to SNP rs917997, patients are at higher risk of developing stenosis, but are protected from the onset of fistulas.
Table 5.
These SNPs have been detected as significant in the categorical modeling.
| Categorical modeling | Gene symbol | Overall p value |
|---|---|---|
| Rs913678 | CEBPB-PTPN1 | 0.0036 |
| Rs11010067 | No gene assigned | 0.0369 |
| Rs17229285 | LOC105373831 | 0.0030 |
| Rs11230563 | CD6 | 0.0356 |
| Rs1363907 | ERAP1, ERAP2 | 0.0193 |
| Rs917997 | IL18RAP | 0.0151 |
| Rs2945412 | KSR1 | 0.0097 |
| Rs11150589 | ITGAL | 0.0353 |
SNP, single nucleotide polymorphism.
Table 6.
The following SNPs exhibit significant transition intensities in the categorical modeling.
| Transition intensities | HR (95% CI) For 1 risk allele |
HR (95% CI) For 2 risk alleles |
|---|---|---|
| Rs 11010067 | ||
| B1 to B2 | 1.662 (1.070–2.581) | 1.665 (0.921–3.010) |
| B1 to B3 | 1.815 (0.913–3.605) | 2.807 (1.257–6.267) |
| B2 to B3 | 1.134 (0.508–2.530) | 1.610 (0.513–5.055) |
| Rs 11150589 | ||
| B1 to B2 | 0.822 (0.516–1.311) | 1.643 (0.998–2.703) |
| B1 to B3 | 0.678 (0.365–1.258) | 0.664 (0.289–1.526) |
| B2 to B3 | 1.143 (0.514–2.544) | 0.401 (0.514–1.456) |
| Rs 11230563 | ||
| B1 to B2 | 1.032 (0.586–1.820) | 0.579 (0.310–1.079) |
| B1 to B3 | 3.977 (0.854–18.506) | 3.029 (0.637–14.407) |
| B2 to B3 | 0.985 (0.327–2.968) | 0.641 (0.197–2.080) |
| Rs 1363907 | ||
| B1 to B2 | 0.822 (0.523–1.292) | 1.645 (0.992–2.727) |
| B1 to B3 | 0.717 (0.378–1.362) | 1.218 (0.570–2.605) |
| B2 to B3 | 0.382 (0.164–0.894) | 0.536 (0.179–1.603) |
| Rs 17229285 | ||
| B1 to B2 | 1.071 (0.685–1.676) | 0.453 (0.238–0.860) |
| B1 to B3 | 2.970 (1.056–8.353) | 3.401 (1.162–9.954) |
| B2 to B3 | 1.014 (0.435–2.366) | 0.458 (0.138–1.520) |
| Rs 2945412 | ||
| B1 to B2 | 3.568 (1.415–9.000) | 2.985 (1.778–7.564) |
| B1 to B3 | 1.525 (0.496–4.685) | 2.418 (0.828–4.685) |
| B2 to B3 | 1.267 (0.274–5.860) | 2.226 (0.509–9.733) |
| Rs 913678 | ||
| B1 to B2 | 0.826 (0.459–1.487) | 0.517 (0.278–0.965) |
| B1 to B3 | 0.328 (0.160–0.672) | 0.286 (0.137–0.599) |
| B2 to B3 | 0.679 (0.245–1.884) | 0.448 (0.150–1.337) |
| Rs 917997 | ||
| B1 to B2 | 1.536 (1.028–2.295) | 1.122 (0.505–2.492) |
| B1 to B3 | 0.691 (0.355–1.345) | 2.654 (1.262–5.580) |
| B2 to B3 | 0.905 (0.402–2.036) | 2.315 (0.763–7.030) |
CI, confidence interval; HR, hazard ratio; SNP, single nucleotide polymorphism.
Discussion
In our study we aimed to identify genetic risk factors that might predict the progression of CD patients from an inflammatory disease state B1 to an either stricturing B2 and/or penetrating disease state B3. Identifying such risk factors at the time of initial diagnosis might help in the decision to start with a more aggressive initial therapy to prevent disease complications. It has already been shown that in CD patients the early administration of anti-TNF antibody treatment can potentially prevent the disease progression towards a penetrating, but not a stricturing phenotype. 9
Our data are in accordance with previous findings regarding the progression of CD patients towards structuring or penetrating complications during their disease course. We found that the majority of CD patients will eventually develop strictures/stenosis or penetrating complications. The percentage of patients that progresses during their life-time are comparable to previous studies.4,26,27 In particular, Cosnes et al. demonstrated in their long-term observational study that the transition of CD patients over a 5-year period from inflammatory B1 to structuring B2, and/or penetrating B3 complications occurs in 10% or, respectively, 26% of CD patients. 4 Nevertheless, although some more recent data have shown that a smaller fraction of CD patients might change their disease behavior during the disease course,6–8,28 further studies are not supporting this observation. 9
We found that from a total of 162 analyzed SNPs there were only a few SNPs associated with a high risk of disease progression from stage B1 into either stage B2 or B3 (e.g. rs11010067, rs1250546, rs2945412, rs11230563, rs917997) or solely into stage B3 (rs17229285) or B2 (rs17694108). This suggests that the disease course in CD patients is not only determined by genetics, but is also influenced by other patient characteristics. In line with this, the genetic impact on disease development in IBD patients has previously been estimated to constitute only about 25%.16,29,30
Stricturing disease was in previous studies found to be associated with ASCA, CBir1 and GM-CSF seropositivity, while penetrating disease was associated also with ASCA and CBir1, but not GM-CSF seropositivity. 31 The three gene loci NOD2, major histocompatibility complex (MHC) and 3p21 (MST1) were previously found to be strongly associated with CD. Moreover, NOD2 is associated with penetrating, but not with stricturing disease. 16 In-depth analysis of the MHC region showed that the strongest signals for disease location were associations with HLA-DRB1*01:03, HLS-DRB1*07:01 and SNP rs77005575. 16
Previous studies already described a role for a number of SNPs located within the TNFSF15 gene locus as being associated with disease progression in CD patients.13,32 In our paper we identified a further SNP within this gene locus, namely SNP rs4246905, to protect from disease progression, confirming the role of TNFSF15 in this regard. On a functional level, TNFSF15, also known as TL1A, and its receptor DR3 have been shown to be widely expressed in inflamed intestinal tissue of IBD patients. 33 TNFSF15 has been shown to exert a dual role in intestinal immunity. Via its involvement in innate immune responses, it plays a protective role for the intestinal mucosa by ameliorating inflammatory responses and promoting wound healing during acute inflammation. However, TNFSF15 also exerts pro-inflammatory effects primarily by modulating T-cell and innate lymphoid cell activation via its role in adaptive immunity. 34 Further data have suggested that TNFSF15 might be a critical mediator of intestinal fibrosis, dependent on the intestinal microbiota, and suggested that targeting TNFSF15 might be a plausible approach for the treatment of CD and other inflammatory diseases.35,36 Further studies are needed to elucidate such potential pathogenetic mechanisms and therapeutic potentials in more detail. Such improved molecular understanding might then finally guide therapy in CD patients at initial CD diagnosis and propose a less aggressive first-line therapy leading to fewer side effects and finally to improved quality of life.
Interestingly, we found that the protective or detrimental effect of certain SNPs depends on the number of risk alleles being present in one patient, for example, rs11150589 located on the gene locus encoding Integrin alpha L (ItgaL) or rs1363907 located on gene locus encoding endoplasmic reticulum aminopeptidases (ERAP) 1 and 2. At a functional level, ItgaL is involved in immune cell adhesion, 37 ERAP1 and 2 are involved in cell surface cleavage of cytokine receptors and antigen processing that suggests an important role in inflammation. 38 This observation demonstrates that also the number of risk alleles is critical for the development of the disease course and shows that the gene–host–disease interplay is highly complex and, to date, still not well understood.
Overall, we tested 160 SNPs available in the SIBDCS and we found more or less 5% of significant ones, as one could expect. This is a higher number of relevant SNPs than, for example, Cleynen et al. detected in their large study from 2016. 16 We are fully aware that genetics cannot explain the disease progression, but it is still interesting to find which SNPs might be the most important ones, according to our data.
It is also interesting to note that we “only” found eight or nine significant SNPs, despite our quite large sample size of 1276 patients. Although we have presented evidence that our findings point towards a possible role for the detected variants in disease progression, we stress that none survives multiple testing correction and hence better powered studies are needed to confirm the bona fide nature of each individual association.
Of note, adjustment for specific factors is important for such a study as we performed. Our results indeed are adjusted for disease duration. In our previous publication studying the very same patient collective from the SIBDCS, 39 we found by multivariate testing, the hazard ratio for migrating from B1 to pB3 (HR 0.27) and from B2 to pB3 (HR 0.12) was lower in patients >40 years compared to patients <17 years. In this study, we additionally found that immunosuppression (HR 0.38) and treatment with anti-TNF for >1 year (HR 0.30) were associated with a decreased likelihood of transitioning from stage B1 to pB3. 39
A limitation of our study is, however, that our SNPs analyzed apply to patients living in Switzerland. While different ethnicities might be present in the cohort, the majority consists of a Caucasian race. It would be important and interesting to expand the study and to examine if our SNP results can be verified in patient collectives including other ethnicities. Further, it must be mentioned that we tried to exemplarily perform a multivariate model with all SNPs mentioned in Table 4. However, the coefficients did not convincingly change, which is due to the fact that correlations between SNPs are quite low.
In summary, within our study we identified novel associations between SNPs in CD risk genes and the disease course in CD patients. Notably, we found genetic variations that confer increased or decreased risk for disease progression. However, we also demonstrated that developing a stenosis is not a prerequisite for progressing to B3 state. The validation of those genetic associations and an improved molecular understanding about the functional role in CD pathogenesis might finally result in the identification of predictive biomarkers that could help in first-line therapy decision-making.
Supplemental Material
Supplemental material, sj-pdf-1-tag-10.1177_1756284820959252 for Genetic risk factors predict disease progression in Crohn’s disease patients of the Swiss inflammatory bowel disease cohort by Felicitas Ditrich, Sena Blümel, Luc Biedermann, Nicolas Fournier, Jean-Benoit Rossel, David Ellinghaus, Andre Franke, Eduard F. Stange, Gerhard Rogler and Michael Scharl in Therapeutic Advances in Gastroenterology
Footnotes
Author contributions: Guarantor of the article: Prof. Dr. Michael Scharl.
Specific author contributions: FD, DE analyzed and interpreted the data. FD wrote the first draft of the manuscript. MS conceived, designed and supervised the research. MS and GR obtained funding. MS, LB, LR, ES, DE, AF and SB interpreted the data and wrote the manuscript. NF and JBR performed statistical analyses. All authors edited the manuscript and approved the final version.
Conflict of interest: The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Stiftung Experimentelle Biomedizin to MS, Swiss National Science Foundation (grant no. 314730-146204, grant no. 314730_166381 and grant no. CRSII3_154488/1) to MS and to GR for the Swiss IBD Cohort (grant no. 3347CO-108792).
ORCID iDs: Sena Blümel  https://orcid.org/0000-0002-0518-5505
https://orcid.org/0000-0002-0518-5505
Luc Biedermann
https://orcid.org/0000-0003-0824-4125
Michael Scharl
https://orcid.org/0000-0002-6729-1469
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Felicitas Ditrich, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland; Department of Internal Medicine, Hospital Zollikerberg, Zollikerberg, Switzerland.
Sena Blümel, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Luc Biedermann, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Nicolas Fournier, Center for Primary Care and Public Health (Unisanté), University of Lausanne, Lausanne, VD, Switzerland.
Jean-Benoit Rossel, Center for Primary Care and Public Health (Unisanté), University of Lausanne, Lausanne, VD, Switzerland.
David Ellinghaus, Institute of Clinical Molecular Biology, Christian-Albrechts-University of Kiel, University Hospital Schleswig Holstein, Kiel, Germany.
Andre Franke, Institute of Clinical Molecular Biology, Christian-Albrechts-University of Kiel, University Hospital Schleswig Holstein, Kiel, Germany.
Eduard F. Stange, Department of Internal Medicine I, University Hospital Tübingen, Tübingen, Germany
Gerhard Rogler, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Michael Scharl, Department of Gastroenterology and Hepatology, University Hospital Zurich, Rämistrasse 100, Zurich, 8091, Switzerland.
References
- 1. Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012; 142: 46–54.e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- 2. Floyd DN, Langham S, Séverac HC, et al. The economic and quality-of-life burden of Crohn’s disease in Europe and the United States, 2000 to 2013: a systematic review. Dig Dis Sci 2015; 60: 299–312. [DOI] [PubMed] [Google Scholar]
- 3. Louis E, Collard A, Oger AF, et al. Behaviour of Crohn’s disease according to the Vienna classification: changing pattern over the course of the disease. Gut 2001; 49: 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cosnes J, Cattan S, Blain A, et al. Long-term evolution of disease behavior of Crohn’s disease. Inflamm Bowel Dis 2002; 8: 244–250. [DOI] [PubMed] [Google Scholar]
- 5. Schwartz DA, Loftus EV, Jr, Tremaine WJ, et al. The natural history of fistulizing Crohn’s disease in Olmsted County, Minnesota. Gastroenterology 2002; 122: 875–880. [DOI] [PubMed] [Google Scholar]
- 6. Nguyen GC, Nugent Z, Shaw S, et al. Outcomes of patients with Crohn’s disease improved from 1988 to 2008 and were associated with increased specialist care. Gastroenterology 2011; 141: 90–97. [DOI] [PubMed] [Google Scholar]
- 7. Gower-Rousseau C, Savoye G, Colombel JF, et al. Are we improving disease outcomes in IBD? A view from the epidemiology side. Gut 2014; 63: 1529–1530. [DOI] [PubMed] [Google Scholar]
- 8. Annese V, Duricova D, Gower-Rousseau C, et al. Impact of new treatments on hospitalisation, surgery, infection, and mortality in IBD: a focus paper by the epidemiology committee of ECCO. J Crohns Colitis 2016; 10: 216–225. [DOI] [PubMed] [Google Scholar]
- 9. Jeuring SFG, van den Heuvel TR, Liu LYL, et al. Improvements in the long-term outcome of Crohn’s disease over the past two decades and the relation to changes in medical management: results from the population-based IBDSL cohort. Am J Gastroenterol 2017; 112: 325–336. [DOI] [PubMed] [Google Scholar]
- 10. Polito JM, II, Childs B, Mellits ED, et al. Crohn’s disease: influence of age at diagnosis on site and clinical type of disease. Gastroenterology 1996; 111: 580–586. [DOI] [PubMed] [Google Scholar]
- 11. Beaugerie L, Seksik P, Nion-Larmurier I, et al. Predictors of Crohn’s disease. Gastroenterology 2006; 130: 650–656. [DOI] [PubMed] [Google Scholar]
- 12. Biedermann L, Fournier N, Misselwitz B, et al. High rates of smoking especially in female Crohn’s disease patients and low use of supportive measures to achieve smoking cessation – data from the Swiss IBD cohort study. J Crohns Colitis 2015; 9: 819–829. [DOI] [PubMed] [Google Scholar]
- 13. Zeitz J, Fournier N, Labenz C, et al. Risk factors for the development of fistulae and stenoses in Crohn disease patients in the Swiss inflammatory bowel disease cohort. Inflamm Intest Dis 2017; 1: 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lang BM, Biedermann L, van Haaften WT, et al. Genetic polymorphisms associated with smoking behaviour predict the risk of surgery in patients with Crohn’s disease. Aliment Pharmacol Ther 2018; 47: 55–66. [DOI] [PubMed] [Google Scholar]
- 15. Rieder F, Zimmermann EM, Remzi FH, et al. Crohn’s disease complicated by strictures: a systematic review. Gut 2013; 62: 1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cleynen I, Boucher G, Jostins L, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet 2016; 387: 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pittet V, Juillerat P, Mottet C, et al. Cohort profile: the Swiss Inflammatory Bowel Disease Cohort Study (SIBDCS). Int J Epidemiol 2009; 38: 922–931. [DOI] [PubMed] [Google Scholar]
- 18. Pittet V, Michetti P, Mueller C, et al. Cohort profile update: the Swiss Inflammatory Bowel Disease Cohort Study (SIBDCS). Int J Epidemiol 2019; 48: 385–386f. [DOI] [PubMed] [Google Scholar]
- 19. Storm N, Darnhofer-Patel B, van den Boom D, et al. MALDI-TOF mass spectrometry-based SNP genotyping. Methods Mol Biol 2003; 212: 241–262. [DOI] [PubMed] [Google Scholar]
- 20. Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danecek P, Mccarthy SA. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics 2017; 33: 2037–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. 1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015; 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016; 48: 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Staley JR, Blackshaw J, Kamat MA, et al. Phenoscanner: a database of human genotype–phenotype associations. Bioinformatics 2016; 32: 3207–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a working party of the 2005 montreal world congress of gastroenterology. Can J Gastroenterol 2005; 19 (Suppl. A): 5A–36A. [DOI] [PubMed] [Google Scholar]
- 26. Cosnes J, Gower-Rousseau C, Seksik P, et al. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011; 140: 1785–1794. [DOI] [PubMed] [Google Scholar]
- 27. Camus M, Seksik P, Bourrier A, et al. Long-term outcome of patients with Crohn’s disease who respond to azathioprine. Clin Gastroenterol Hepatol 2013; 11: 389–394. [DOI] [PubMed] [Google Scholar]
- 28. Gower-Rousseau C, Vasseur F, Fumery M, et al. Epidemiology of inflammatory bowel diseases: new insights from a French population-based registry (EPIMAD). Dig Liver Dis 2013; 45: 89–94. [DOI] [PubMed] [Google Scholar]
- 29. Scharl M, Rogler G. Inflammatory bowel disease pathogenesis: what is new? Curr Opin Gastroenterol 2012; 28: 301–309. [DOI] [PubMed] [Google Scholar]
- 30. Zuk O, Hechter E, Sunyaev SR, et al. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 2012; 109: 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kugathasan S, Denson LA, Walters TD, et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 2017; 389: 1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang D-H, Yang S-K, Song K, et al. TNFSF15 is an independent predictor for the development of Crohn’s disease-related complications in Koreans. J Crohns Colitis 2014; 8: 1315–1326. [DOI] [PubMed] [Google Scholar]
- 33. Bamias G, Martin C, III, Marini M, et al. Expression, localization, and functional activity of TL1A, a novel Th1-Polarizing cytokine in inflammatory bowel disease. J Immunol 2003; 171: 4868–4874. [DOI] [PubMed] [Google Scholar]
- 34. Valatas V, Kolios G, Bamias G. TL1A (TNFSF15) and DR3 (TNFRSF25): a co-stimulatory system of cytokines with diverse functions in gut mucosal immunity. Front Immunol 2019; 10: 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shih DQ, Zheng L, Zhang X, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol 2014; 7: 1492–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jacob N, Jacobs JP, Kumagai K, et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol 2018; 11: 1466–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chidlow JH, Jr, Glawe JD, Alexander JS, et al. VEGF164 differentially regulates neutrophil and T cell adhesion through ItgaL- and ItgaM-dependent mechanisms. Am J Physiol Gastrointest Liver Physiol 2010; 299: G1361–G1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pepelyayeva Y, Amalfitano A. The role of ERAP1 in autoinflammation and autoimmunity. Hum Immunol 2019; 80: 302–309. [DOI] [PubMed] [Google Scholar]
- 39. Cernoch PS, Fournier N, Zeitz J, et al. Lower risk of B1-to-pB3-Stage migration in Crohn’s disease upon immunosuppressive and Anti-TNF treatment in the Swiss IBD cohort study. Dig Dis Sci 2019; 65: 2654–2663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-tag-10.1177_1756284820959252 for Genetic risk factors predict disease progression in Crohn’s disease patients of the Swiss inflammatory bowel disease cohort by Felicitas Ditrich, Sena Blümel, Luc Biedermann, Nicolas Fournier, Jean-Benoit Rossel, David Ellinghaus, Andre Franke, Eduard F. Stange, Gerhard Rogler and Michael Scharl in Therapeutic Advances in Gastroenterology