

Abstract
Purpose:
Small molecule inhibitors have had a major impact on cancer care. While treatments have demonstrated clinically promising results, they suffer from dose-limiting toxicities and the emergence of refractory disease. Considerable efforts made to address these issues have more recently focused on strategies implementing particle-based probes that improve drug delivery and accumulation at target sites, while reducing off-target effects.
Experimental Design:
Ultrasmall (<8 nm) core-shell silica nanoparticles, C’ dots, were molecularly engineered to function as multivalent drug delivery vehicles for significantly improving key in vivo biological and therapeutic properties of a prototype epidermal growth factor (EGFR) tyrosine kinase inhibitor, gefitinib. Novel surface chemical components were used to conjugate gefitinib-dipeptide drug linkers and deferoxamine (DFO) chelators for therapeutic delivery and PET imaging labels, respectively.
Results:
Gefitinib-bound C’ dots (DFO-Gef-C’ dots), synthesized using the gefitinib analog, APdMG, at a range of drug-to-particle ratios (DPR; DPR=11–56), demonstrated high stability for DPR values≤ 40, bulk renal clearance, and enhanced in vitro cytotoxicity relative to gefitinib (LD50=6.21 nM versus 3 μM, respectively). In human non-small cell lung cancer mice, efficacious Gef-C’ dot doses were at least 200-fold lower than that needed for gefitinib (360 nmoles versus 78 μmoles, respectively), noting fairly equivalent tumor growth inhibition and prolonged survival. Gef-C’ dot-treated tumors also exhibited low phosphorylated EFGR levels, with no appreciable wild-type EGFR target inhibition, unlike free drug.
Conclusions:
Results underscore the clinical potential of DFO-Gef-C’ dots to effectively manage disease and minimize off-target effects at a fraction of the native drug dose.
Keywords: ultrasmall, nanoparticle, gefitinib, drug delivery, non-small cell lung cancer, small molecular inhibitor, therapeutic window, silica, survival
Introduction
Since the 2001 FDA approval of the first small molecule inhibitor (SMI), imatinib, for treatment of chronic myelogenous leukemia, numerous small molecule drugs have been developed, sparking a revolution in cancer treatment (1,2). Owing to their precise inhibition of molecular targets, which are often overexpressed or aberrantly activated in cancer, SMIs have shown robust responses in the clinic while reducing off-target, dose-limiting toxicities (3). To date, the number of FDA-approved small molecule kinase inhibitors number 52 with numerous other candidates in clinical trials (1,4), including the reversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), gefitinib (Iressa) (5) for the treatment of advanced non-small cell lung cancer (NSCLC) (6,7). TKIs gain access and bind to the receptor’s adenosine triphosphate (ATP) binding site, inhibiting receptor autophosphorylation and downstream signaling cascades (7,8); this mechanism of action is ideally suited to treating NSCLC cases that harbor activating receptor mutations (e.g., L858R, delE746-A750) (9,10). In addition to using gefitinib to treat NSCLC, its effectiveness has also been evaluated in glioblastoma multiforme (GBM). EGFR gene amplification is observed in approximately 40% of all GBM cases, with about 50% of those tumors also expressing the constitutively active EGFRvIII mutations (11). EGFRvIII-mutated GBM lacks the extracellular EGF binding domain normally necessary for receptor activation (12,13). Despite the loss of extracellular exons 2–7, EGFRvIII mutants demonstrate continuous, albeit low-level, EGFR signaling that is further compounded by limited receptor internalization and downregulation (14). Studies investigating the use of gefitinib for patients with the EGFRvIII mutation, however, have demonstrated less than adequate outcomes. Researchers cite the possibility of poor transport across the blood-brain-barrier and the presence of drug efflux pumps as mechanisms for its failure (15,16).
The treatment of NSCLC with gefitinib, by comparison, has demonstrated good response rates and prolonged disease-free survival in patients. However, resistance to this therapy inevitably occurs after an average treatment time of 10 months (17). Explanations for the emergence of a resistant phenotype can be attributed to the appearance of a secondary receptor mutation (T790M), compensatory pathway activation, and low levels of intracellular drug accumulation (18,19). Given that ~90% of the bioavailable gefitinib exists in a plasma protein bound state (17), it is not surprising that only a small percentage accumulates within the cellular compartment of tumors. Estimates of the percentage of free drug (i.e. unbound to plasma protein) amount to no more than 6% of the total administered dose (20). This is of concern given that the unbound portion of drug is believed to be a reflection of its level of intracellular uptake at target sites (20). Furthermore, studies evaluating the pharmacokinetic (PK) profile of 18F-labeled gefitinib have demonstrated rapid hepatobiliary clearance and low blood activity measurements within 20 minutes of injection in both vervet monkey and mouse models (21). Interestingly, biodistribution analysis of gefitinib-treated mice 2 hours post-injection demonstrate very low levels of activity present across multiple tumor models (Average Standard Uptake Values, SUV= 0.16) (21). Comparing tumor uptake values to those observed in liver (SUV = 3.28 ± 0.62) demonstrates a shockingly low tumor-to-liver ratio of 0.049 (21). These results present two important points that require addressing. The first being that very low levels of gefitinib accumulate within tumors, as this is a postulated mechanism by which resistance may develop (17). Published results of poor tumor uptake make it clear that there exists a wide berth for improving the delivery of gefitinib, and other small molecular drugs, to sites of disease. The second point worth noting is that >10-fold increased accumulations occur in off target sites (i.e. liver) when compared with tumor tissue (21). Efforts to amass more drug at the site of disease, while reducing its unwanted deposition in non-target organs, will undoubtedly expand the therapeutic index of these treatments and provide better efficacy and improved patient outcomes.
The use of particle-based delivery systems has served as one approach towards improving drug delivery and limiting off-target accumulations (22). To date, at least 51 nanomedicines have garnered FDA approval for use in the clinic (23). These include both imaging agents, as well as nanoparticle-based therapies that increase drug accumulation and efficacy, while reducing dose-limiting toxicities (23). Although nanoparticles exhibit a broad range of sizes, typically a few nanometers to 1000 nm, and span a number of classes based on the material from which they are derived (24), particles in the sub-100 nm range are preferable as drug delivery vehicles, in part due to their ability to cross physiological barriers with relative ease in comparison to larger constructs (24). Furthermore, the smallest subset of particle probes (i.e. <8 nm) allow for efficient transport within the circulation, increased tumor accumulation via the enhanced permeability and retention effect (EPR) (25), and dominant renal excretion (26). By contrast, larger nanoparticle delivery platforms (>10 nm) are at a greater risk of recognition and opsonization by the reticuloendothelial system (RES), as evidenced by their high accumulation in liver, spleen, and bone marrow (26,27), thereby leading to their rapid elimination from the bloodstream and limiting their availability for accumulation at target sites (28).
In an effort to harness the unique characteristics of nanoparticles for drug delivery, attempts have been made to synthesize gefitinib-loaded nanoparticle constructs (Supplemental Table 1; 29–35). Unfortunately, these reports largely focus solely on in vitro cytotoxic effects, without investigation into their therapeutic potential in vivo (29–32,35). Nanoparticle delivery platforms whose physical properties, surface chemistry, and stoichiometry can be precisely tuned to maximize targeted uptake and diffusion of drug throughout areas of disease while evading uptake and elimination by the RES are critically important design features. Such optimized particle probes should be able to strike a better balance between their high drug loading capacity and their ability to maintain favorable PK and clearance properties. Combining these properties into a single delivery platform will undoubtably improve efficacy and product safety by reducing dose-limiting toxicities.
Herein, we investigate the feasibility of utilizing a clinically-translated ultrasmall (<8 nm) core-shell silica nanoparticle, Cornell prime dots (C’ dots) (36–38), as a drug delivery vehicle offering improved therapeutic properties over the corresponding native drug in a patient-derived model of NSCLC. Our prior work has shown that dye-encapsulated C’ dots can be surface modified with an array of functional groups (37–43) and radiotracers (40,44) for mapping disease in both surgical (45) and non-surgical settings (39,41), as well as for investigating biological properties and targeted inhibition in a high grade glioma model (46). In addition, we have demonstrated the successful conjugation and in vitro therapeutic activity of a protease-sensitive gefitinib-linker construct to an earlier generation silica nanoparticle, which was found to inhibit EGFR signaling in vitro (47). We improve upon these earlier particle surface chemical designs in this work through the addition of dibenzocyclooctyne (DBCO) click chemistry and DFO for zirconium-89 chelation and positron emission tomography (PET) imaging – all while maintaining a renally-clearable platform. The newer click chemistry design has led to significantly higher drug loading than reported previously, with improved precision and surface chemical control (47). Imparted with the ability to tailor drug-to-particle (DPR) ratios, we investigated the alteration of PK and biodistribution as a function of DPR. A candidate DFO-Gef-C’ dot displaying low liver uptake and favorable biodistribution was identified and investigated as a prototype NDC in both in vitro and in vivo studies. DFO-Gef-C’ dots (DPR = 40) were actively taken into cells and displayed low-nanomolar cytotoxicity (LD50 = 6.21 nM). In vivo, we observed that DFO-Gef-C’ dots inhibited tumor growth and prolonged survival to the same degree as that found with gefitinib alone. However, mice treated with the DFO-Gef-C’ dot construct received a fraction of the total drug dose administered to mice treated with gefitinib (360 nmoles vs 78 μmoles, respectively). In addition, evaluation of off-target EGFR effects demonstrated minimal suppression of wild-type EGFR levels in the DFO-Gef-C’ dot treated mice, as compared to those treated with the free drug. Together, these data demonstrate that sub 8-nm C’ dots can be molecularly engineered to function as clinically-promising drug delivery vehicles, offering distinct advantages over larger-size probes in terms of their key biological properties, as well as their potential to abrogate dose-limiting toxicities and improve disease control and patient outcomes.
Materials and Methods
Synthesis of azido-dPEG8-Phe-Lys-PABC-APdMG Drug-Linker.
The drug-linker was prepared as previously described (47).This is a multi-step synthesis wherein the final coupling step with HS-dPEG8 was replaced with an azido-dPEG8-OH spacer (QuantaBiodesign) to accommodate azide-alkyne 1–3 dipolar cycloadditions. ESI-HRMS (m/z) for C60H81ClFN11O15: [M+H]+ calc 1250.5659, obs 1250.5662.
Materials, synthesis, and characterization of DFO-DBCO-PEG-Cy5-C’ dots.
Superdex 200 resin for GPC, and vivaspin 30k MWCO spin filters were purchased from GE Healthcare Life Sciences. 5 M NaCl in water solution, dimethyl sulfoxide (DMSO), (3-aminopropyl)trimethoxysilane (APTMS), tetramethyl orthosilicate (TMOS), (3-mercaptopropyl)trimethoxysilane (MPTMS), and 2.0 M ammonia in ethanol were all purchased from Sigma-Aldrich. Methoxy-PEG(6–9)-silane (~500 g/mol) was purchased from Gelest. Cy5(+)-maleimide with net positive charge was purchased from Lumiprobe. DI water was generated using a Millipore IQ7000 system (18.2 MΩ·cm). DBCO-PEG4-NHS was purchased from Click Chemistry Tools. P-SCN-Bn-Deferoxamine (98%) (DFO-NCS) was purchased from Northernchem Inc. All chemicals were used as received.
C’ dots with positively charged Cy5(+) dyes were synthesized as reported previously (37,38,48). Briefly, for a 10 mL batch, 0.367 μmol Cy5(+) dye with maleimide mono functionality was dissolved in DMSO overnight in a glovebox. Mercaptopropyl-trimethoxysilane (MPTMS) was mixed with the dissolved dye (23:1 ratio) by pipette and allowed to react overnight in the glove box. The next day, 1 mL of 0.02 M ammonium hydroxide was added into 9 mL deionized water in a round-bottom flask. 68 μL tetramethylorthosilicate (TMOS) and the prepared Cy5(+) dye-silane conjugate were added into the flask drop-wise and allowed to react overnight under stirring. The following day, 100 μL of mPEG(6–9EO)-silane was added into the flask drop-wise under stirring and allowed to react overnight. The next day, stirring of the solution was stopped and the flask was heated at 80°C for 24 hours. Following the heat treatment step, the particles were syringe filtered with a 200 nm membrane to remove any large aggregates in the solution. Additional DBCO and DFO groups were further attached to the C’ dot particle surface using a previously reported PPSMI protocol and employing APTMS as the functional silica surface modifier for subsequent reactions with DBCO and DFO derivatives (DBCO-PEG4-NHS and DFO-NCS), respectively (43). The resulting reaction solution of C’ dots was purified by centrifuging with a 30K MWCO PES membrane spin filter, and finally by GPC purification through Superdex 200 resin on a Bio-Rad FPLC. The purified particles were characterized using fluorescence correlation spectroscopy (FCS) on a home-built setup, equipped with a 633-nm solid-state laser as the excitation source for Cy5(+) dye, and UV/Vis spectroscopy on a Cary 5000 spectrometer as described previously (43,49). TEM images were taken using a FEI Tecnai T12 Spirit microscope operated at an acceleration voltage of 120kV.
Synthesis and characterization of DFO-Gef-PEG-Cy5-C’ dots (or DFO-Gef-C’ dots).
For a typical synthesis of DFO-Gef-C’ dots having a DPR of 40, 37.5 nmols of Gef-linker-Azide were added to 100 μL PBS solution of DFO-DBCO-PEG-Cy5-C’ dots (7.5 μM). The mixture was continuously shaken at RT overnight. Free Gef-linker-azide was removed by PD-10 column purification (PBS buffer as the mobile phase). As-synthesized DFO-Gef-C’ dots were then subjected to morphology, size and surface payload density characterizations using a combination of TEM, FCS, and UV/Vis spectroscopy. The hydrodynamic particle size, particle brightness, and concentration were obtained by fitting the FCS auto-correlation curves (43,49). The number of Cy5 dyes (or functional groups of DFO, DBCO) per DFO-Gef-C’ dot was calculated using a similar method we reported previously (43,49). Surface payload density (or DPR) of DFO-Gef-C’ dot was estimated by measuring and comparing the absorption peak intensity of Gefitinib at 346 nm and Cy5 dye at 651 nm. Absorbance spectra of DFO-Gef-C’ dots with varied DPRs were obtained using the HPLC UV detector.
Enzymatic cleavage of DFO-Gef-C’ dots.
To study the enzymatic cleaving capability of DFO-Gef-C’ dots, 50 μL (15 μM) of DFO-Gef-C’ dots (DPR 40) in PBS was mixed with (or without) 50 μL of 0.25% Trypsin-EDTA (ThermoFisher). The mixture was kept on a shaking platform (650 rpm) at 37 °C for 30 min. To quantify the efficacy of DFO-Gef-C’ dot cleavage, both samples were analyzed using HPLC to obtain the area under the curve (AUC) ratio between gefitinib (AUC346nm) and C’ dots (represented by Cy5 dye, AUC651nm). The percent cleaved was calculated using the following equation:
Payload stability of DFO-Gef-C’ dots in serum.
For serum stability studies, 100 μL (15 μM) of DFO-Gef-C’ dots (DPR 40) in PBS was mixed with 100 μL of human or mouse serum. The mixtures were kept on a shaking platform (650 rpm) at 37 °C for 24 hours. The AUC ratio between gefitinib (AUC346nm) and C’ dots (AUC651nm) was acquired using the same HPLC method described previously (47). The percentage (%) of retained gefitinib in the NDC was calculated using the following equation:
89Zr-oxalate production.
89Zr was produced at Memorial Sloan Kettering Cancer Center on a TR19/9 cyclotron (Ebco Industries Inc.) via the 89Y(p,n)89Zr reaction, and purified to yield 89Zr with a specific activity of 5.28–13.43 mCi/μg (470–1195 Ci/mmol) of zirconium(50). Activity measurements were performed using a CRC-15R Dose Calibrator (Capintec). For the quantification of activities, experimental samples were counted on an Automatic Wizard2 γ-Counter (PerkinElmer). All in vivo experiments were performed according to protocols approved by the Memorial Sloan Kettering Institutional Animal Care and Use Committee. A purity of greater than 95% was confirmed using radio-TLC for all of the 89Zr-labeled DFO-Gef-C’ dots.
89Zr radiolabeling of DFO-Gef-C’ dots.
For 89Zr labeling, 0.75 nmol of DFO-Gef-C’ dots was mixed with 1 mCi of 89Zr-oxalate in HEPES buffer (pH 8) at 37 °C for 60 min; final labeling pH was kept at 7–7.5(44). An EDTA challenge process was introduced to remove any non-specifically bound 89Zr by incubating the mixture at 37 °C for additional 30–60 min. The final 89Zr labeling yield was in the range of 70 to 80% (n>5). As synthesized 89Zr-DFO-Gef-C’ dots were then purified using a PD-10 column. The final radiochemical purity was estimated to be greater than 99% (measured by using Radio-TLC).
Cell culture.
The human NSCLC cell line, ECLC26, was received as a generous gift from the laboratory of Dr. Charles Rudin. This cell line demonstrates a typical EGFR activating mutation, L858R (c.2573 T>G) for lung adenocarcinoma. No concurrent mutations were seen in AKT1, BRAF, ERBB2, KRAS, MEK1, NRAS, or PIK3CA. Cells were cultured in Roswell Park Memorial Institute 1640 media (RPMI 1640) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Media Preparation Facility, Memorial Sloan Kettering). Cultures were maintained in a humidified incubator with 5% CO2 at 37°C.
Western blot.
ECLC26 cells were plated at a density of 1×106 cells/well in a 6-well plate and allowed to attach overnight. The following day, culture media was replaced with normal growth media supplemented with either free gefitinib or DFO-Gef-C’ dots at the described concentrations for 18 hours. Cells were then washed 2X with PBS and collected via trypsinization. Collected cell suspensions were centrifuged and resulting pellets were lysed using RIPA lysis buffer (ThermoFisher Scientific) containing phosphatase inhibitor (Roche). Total protein concentrations were determined using the BCA assay (ThermoFisher Scientific). A total of 20 μg of protein per sample was loaded into a NuPAGE 4–12% Bis-Tris gel (Invitrogen) and run according to standard Western blot protocol. Expression and phosphorylation levels of EGFR were visualized using anti-EGFR (#4267S, Cell Signaling; 1:2000) and anti-phospho-EGFR (#3777S, Cell Signaling, 1:1000) antibodies. β-actin (sc-47778, Santa Cruz, 1:5000) expression was used as a loading control.
Concentration and temperature-dependent uptake.
For concentration-dependent uptake studies, ECLC26 cells were first seeded at a density of 0.5×106 cells per well in a 6-well plate and allowed to attach overnight. The following morning, growth media was removed and replaced with media containing the indicated concentrations of DFO-Gef-C’ dots and allowed to incubate at 37°C for 24 hours prior to analysis. Determination of cellular uptake was demonstrated using flow cytometry. Briefly, cells were washed 2x with PBS and detached from the plate surface using trypsin. Detached cells were collected, and trypsin neutralized with excess growth media prior to centrifugation. Cells were then resuspended in flow buffer containing 0.05 mg/ml DAPI for live/dead determination. Triplicate samples were analyzed on a LSRFortessa flow cytometer (BD Biosciences). Results were displayed as both median fluorescence intensities (MFI), as well as a percentage of DFO-Gef-C’ dots positively staining cells. Graphs were generated using Prism 7 software (GraphPad).
For temperature-dependent uptake studies, three 6-well plates were seeded a day prior to study initiation in a manner analogous to the methods described above. Seeded cells were then treated with 0, 10, 100, or 500 nM DFO-Gef-C’ dots in full growth media and placed at either 4°C, 25°C, or 37°C for an incubation period of 4 hours. Cells were collected and analyzed using flow cytometry. Graphical depiction of median fluorescence intensities and DFO-Gef-Cy5-C’ dots positively staining cell percentages were created using Prism 7 software (GraphPad).
Time-dependent viability and proliferation assays.
ECLC26 cells were plated in opaque 96-well plates at a density of 5×103 cells per well and allowed to attach overnight. The following day, cells were treated with free gefitinib and gefitinib-linker at a concentration of 500 nM in full media. DFO-Gef-C’ dots were evaluated at particle concentrations of 500 nM and 11.11 nM. Plates were collected at the respective time points and cell viability assessed using the CellTiter-Glo assay (Promega) according to manufacturer’s instructions. The percent of viable cells was calculated by first normalizing the luminescence values for each treatment time point to their respective untreated control values. These values were then divided by the treatment group’s mean value at 0 hours and multiplied by 100. Additionally, a proliferation index was calculated by dividing each treatment group’s initial (0 hour) mean luminescence value into all collected timepoint values. Data for both viability and proliferation were plotted using Prism7 software (GraphPad).
Super-resolution Confocal Microscopy.
To visualize the dose-dependent uptake of DFO-Gef-C’ dots, ECLC26 cells were plated on Poly-D-Lysine coated coverslips in 24-well plates at a density of 1×105 cells per well. The cells were allowed to attach overnight, prior to the addition of full growth media supplemented with 0.1 mg/ml FITC-Dextran (Fisher Scientific) and DFO-Gef-C’ dots at the described concentrations for 24 hours. At the conclusion of the incubation, coverslips were washed 2X with PBS and then treated with 0.05 mg/ml DAPI in flow buffer for 15 minutes. Coverslips were washed 2X with PBS prior to mounting on glass microscope slides using Prolong Gold mounting media (ThermoFisher). Samples were then imaged using a Zeiss LSM880 point-scanning confocal microscope equipped with an Airyscan, super-resolution detector (Molecular Cytology Core, Memorial Sloan Kettering Cancer Center). Images were processed using Zen software (Zeiss) and displayed using Imaris Image Analysis Software (Bitplane). Quantification of the number of foci and their intensities was performed by the manual thresholding of images and particle analysis using ImageJ.
Determination of LD50.
LD50 values were determined by incubating increasing concentrations of each treatment with ECLC26 cells in opaque 96-well plates. Briefly, cells were plated at a density of 5×103 cells per well and allowed to attach overnight in full growth media. Next, cells were treated with free gefitinib, gefitinib-linker, or DFO-Gef-Cy5-C’ dot containing media for 7 days. At the conclusion of treatment, cell viability was assessed using the CellTiter-Glo assay (Promega). LD50 values were calculated in a manner identical to previously reported methods(41) and displayed using Prism7 software (GraphPad).
In-vivo PET and ex vivo biodistribution studies.
For in vivo PET imaging, mice with or without ECLC26 tumors were i.v.-injected with 200–300 μCi (7.4–11.1 MBq) 89Zr-DFO-Gef-C’ dots. Approximately 5 min prior to PET imaging, mice were anesthetized by inhalation of 2% isoflurane (Baxter Healthcare, Deerfield, IL)/oxygen gas mixture, and placed on the scanner bed; anesthesia was maintained using 1% isoflurane/gas mixture. PET imaging, performed using a small-animal PET scanner (Focus 120 microPET; Concorde Microsystems, or Inveon PET/CT, Siemens), was performed in normal, healthy mice at 0.5, 5, 24, 72, 120 and 168 h p.i. 89Zr-DFO-Gef-C’ dots of varying DPR (Supplemental Figure 3), as well as in ECLC26 xenografted mice at 0.5, 24, 48 and 72 h p.i. (Supplemental Figure 4). An energy window of 350–700 keV and a coincidence timing window of 6 ns were used. Data were sorted into 2D histograms by Fourier rebinning, and transverse images were reconstructed by filtered back-projection into a 128 × 128 × 63 (0.72 × 0.72 × 1.3 mm3) matrix. The PET imaging data were normalized to correct for non-uniformity of response, dead-time count losses, positron branching ratio, and physical decay to the time of injection; no attenuation, scatter, or partial-volume averaging corrections were applied. The counting rates in the reconstructed images were converted to activity concentrations (percentage injected dose per gram of tissue, %ID/g) by use of a system calibration factor derived from the imaging of a mouse-sized water-equivalent phantom containing 89Zr. Region-of-interest (ROI) analyses of the PET data were performed using Inveon Research Workplace (IRW) software, with results presented as %ID/g values. At the last post-injection time point (72 h or 168 h), mice were sacrificed after PET scanning, and tumor and major organs harvested for ex vivo radioassay analysis. Mouse organs were wet-weighted, counted in a Wizard2 γ-Counter (PerkinElmer), and converted to %ID/g (mean ± SD).
Dosimetry.
Time-activity curves, derived for each tissue, were analytically integrated, accounting for radioactive decay, to yield the corresponding cumulative activity. Organ absorbed doses were then calculated by multiplying the cumulative activity by the 89Zr equilibrium dose constant for non-penetrating radiations (positrons), assuming complete local absorption of such radiation, and ignoring the contribution of penetrating radiations (i.e., γ-rays). Mouse normal organ cumulated activities were converted to human normal organ cumulated activities by taking into account differences in total-body and organ masses between mice and humans (assuming 70-kg standard human). Calculated human normal-organ cumulated activities were entered into the OLINDA dosimetry program to compute standard human organ absorbed doses using the formalism of the Medical Internal Dosimetry Committee of the Society of Nuclear Medicine(51). This human dosimetry model is a “normal” (i.e., tumor-free) anatomic model.
In vivo tumor growth inhibition and survival.
ECLC26 flank xenografts were established by injecting 5×106 cells (100 μl of a 1:1 mixture of Matrigel:PBS) into the right flank of female nu/nu (nude) mice (Abico). Tumor growth was monitored over time via caliper measurements and recorded. Once tumors reached ~200 mm3, treatments were initiated. Briefly, free gefitinib was administered daily in mice by orally gavaging 150 mg/kg gefitinib in a solution of 1% Tween-80/10% kolliphor/dH2O (200 μl). Mice treated with saline vehicle or DFO-Gef-C’ dots (15 μM) received i.v.-injections (200 μl) of their respective treatments on Days 0, 3, and 6. Tumor volumes were monitored daily by caliper measurements. Tumor volumes were calculated using the formula: Tumor Volume = (long axis x short axis2)/2. Tumor volume growth curves were generated using Prism7 software (GraphPad).
For survival studies, ECLC26 xenografted mice (n=10/group) underwent identical treatment protocols to those described above. However, tumors were measured daily until they reached the endpoint for tumor size (1 cm3), which concluded the study. Once a mouse met the established endpoint, it was recorded as an event in a Kaplan-Meier survival curve and displayed using Prism7 software (GraphPad). All studies were performed in accordance with protocols approved by the MSKCC Institutional Animal Care and Use Committee, and conformed to the NIH guidelines for animal welfare.
Statistical Analysis.
Group means and SDs were calculated for time-dependent changes in the cellular proliferation index and tumor volume, and Kaplan-Meier analyses were applied to survival data. Statistical comparisons between the experimental groups were performed by Student’s t-test (unpaired, two-tailed), and considered statistically significant if P < 0.05. No less than three replicates were generated per group (*P < .05; **P < .01; ***P < .001), unless otherwise noted. All graphs were constructed and analysed using GraphPad Prism 7 (GraphPad Software, San Diego, CA).
Results
Synthesis and Characterization of DFO-DBCO-PEG-Cy5-C’ dots, Gef-linker-azide, and DFO-Gef-C’ dots.
Ultrasmall fluorescent silica nanoparticles (C’ dots) were synthesized in aqueous solution as described previously (Figure 1a) (37,38,48). Briefly, tetramethyl orthosilicate (TMOS), a silica precursor, and positively charged silane/cyanine 5 (Cy5+) dye conjugates were combined in deionized water at slightly basic conditions under stirring. TMOS, upon the hydrolysis and condensation reactions, form negatively charged ~2 nm sized silica clusters, which aggregate over time by sandwiching the positively charged fluorescent Cy5 dyes in between. Upon silica cluster/fluorescent dye formations reaching an electrostatically stable size around 6 nm, particle growth stops, and the silica particle surfaces are further coated by PEG-silane molecules for steric stabilization. As shown previously, this chemistry gives final silica particles a homogeneous surface chemistry (i.e. fully encapsulated Cy5 dyes in the silica matrix and fully PEGylated outer particle surface without dyes creating hydrophobic patches in the PEG layer) (48,52). After the synthesis of the base C’ dot particle platform, amine-silane molecules were added to the solution in order to functionalize the C’ dot surface with primary amines. This was achieved via the post-PEGylation surface modification by insertion (PPSMI) method allowing for the insertion of small silane derivatives, here amine-silane molecules, between the interstitial space of PEG molecules on the C’ dot surface (43). Subsequently, the available primary amines on the C’ dot surface were terminated via click chemistry addition of DBCO and DFO molecules (DBCO-PEG4-NHS, and DFO-NCS).
Figure 1. Synthesis and characterization of DFO-Gef-C’ dots.
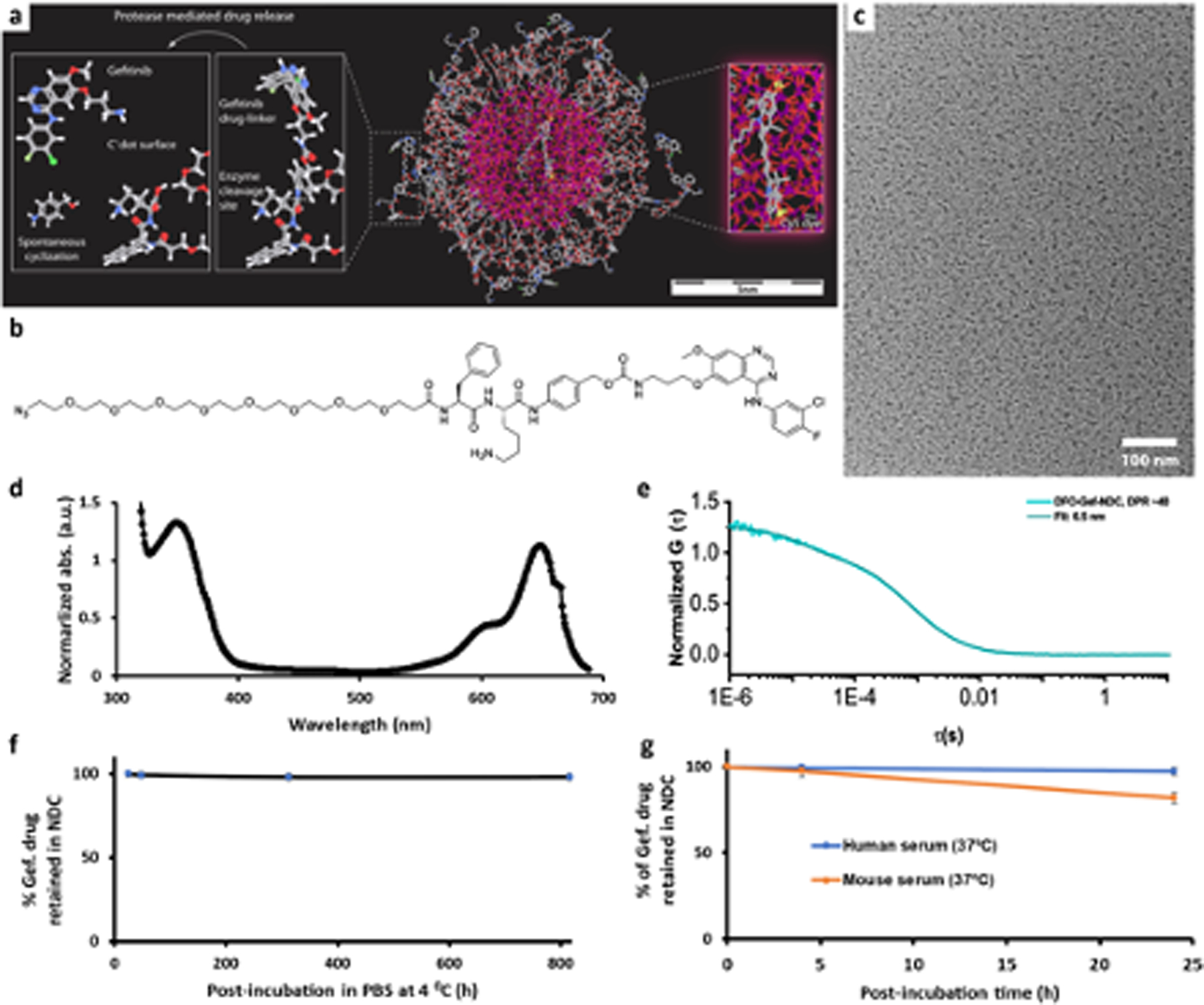
a) Molecular rendering of Gef-C’ dots. b) Chemical structure of azido-dPEG8-Phe-Lys-PABC-APdMG Drug-Linker. c) Representative TEM image of DFO-Gef-C’ dots. d) A representative UV-Vis absorbance spectrum of DFO-Gef-C’ dots (DPR 40). e) FCS correlation curve with fit of DFO-Gef-C’ dots (DPR 40). f) Payload stability monitoring of DFO-Gef-C’ dots in PBS over one month at 4 °C. g) Payload stability of DFO-Gef-C’ dots in mouse and human serum at 37 °C under shaking (650 rpm).
Synthesis of the azido-dPEG8-Phe-Lys-PABC-APdMG drug-linker (Gef-linker-azide) was carried out in a manner analogous to previously reported methods (47) with a minor modification to the addition of the terminal group. Briefly, we utilized the previously described multistep approach (Supplemental Figure 1a) to synthesize a modified gefitinib derivative (APdMG) that retains its activity and is capable of undergoing conjugation with the designed PEG spacer. For this work, we have substituted the terminal HS-dPEG8 with an azido-dPEG8-OH spacer (Figure 1b and Supplemental Figure 1b). The addition of the terminal azido group now enables the addition of Gef-linker-azide through strain-promoted azide-alkyne click chemistry reactions (SPAAC) with the accessible DBCO on the DFO-PEG-Cy5-C’ dot surface. DFO was reacted with C’ dot surface DBCO in a similar manner to APdMG-drug linkers, and served as a 89Zr chelator for use in positron emission tomography (PET) imaging. As described elsewhere (43) the proper nomenclature for this final C’ dot product with the attached gefitinib analogue would be: C’dot(Cy5+)-PEG6–9_amine-NCS-DFO_amine-NHS-PEG4-DBCO-azide-dPEG8-Phe-Lys-PABC-APdMG (beyond the first ligand of the base particle, here PEG6–9, underscores denote ligands directly connected to the silica core particle surface). For brevity, it is herein referred to as DFO-Gef-C’ dots (Figure 1a, 1c).
Efforts to accurately assess and demonstrate the attachment of gef-linker-azide to the nanoparticle surface were carried out using HPLC techniques. Acquired UV-Vis spectra of DFO-Gef-C’ dots (drug-to-particle ratio, or DPR, of 40) illustrate well-distinguished absorption peaks at 346 nm (gefitinib) and 651 nm (C’ dots), respectively (Figure 1d). We further found that variations in DPR from 15 to 50 can be precisely controlled by simply changing the reaction ratio between the Gef-linker-azide and DFO-DBCO-PEG-Cy5-C’ dot (Supplemental Figure 2a). In addition, area under the curve (AUC) ratios between AUC346nm and AUC651nm were found to increase linearly (R2>0.99) with DPR (Supplemental Figure 2a, inset). FCS measurements and quantitative analysis of the FCS correlation curves obtained for DFO-Gef-C’ dots suggested no significant increase in their size over the DPR range of 15 to 50, likely due to backfolding of the hydrophobic drugs in-between PEG chains (Figure 1e and Supplemental Figure 2b); a DPR of 40 corresponded to a diameter of 6.5 nm. To demonstrate trypsin-responsive drug release, DFO-Gef-C’ dots (suspended in PBS) were mixed with trypsin-EDTA and kept on a shaking platform at 37 °C. After 30 min, the AUC ratios between AUC346nm and AUC651nm of DFO-Gef-C’ dots with or without the presence of trypsin-EDTA were obtained using HPLC. Significantly reduced AUC346nm/AUC651nm from 1.92 to 0.26 clearly indicated the enzyme-sensitive release of gefitinib from the DFO-Gef-C’ dots (Supplemental Figure 3). Next, we evaluated payload stability of DFO-Gef-C’ dots while stored in PBS (4 °C, pH 7.4) or during incubation with serum (mouse or human). As shown in Figure 1f, <2% pre-release of the drug from DFO-Gef-C’ dots (DPR 40) was found after storing in PBS for >800 hours (or >1 month). Higher payload release (i.e., 3–20%), however, was observed upon incubation of the same NDC with human or mouse serum for 24 hours (Figure 1g), which was felt to be due to the presence of proteases capable of inducing drug-linker cleavage.
Biodistribution, clearance profiles, and in vivo tumor targeting of 89Zr-DFO-Gef-C’ dots.
Although no obvious increase in particle size was observed for DFO-Gef-C’ dots of varying DPR, we conducted screening biodistribution and PET imaging studies to investigate DPR-dependent tissue uptake of DFO-Gef-C’ dots, given that PK is governed by properties other than particle size. NDCs displaying a DPR ranging from ~10 to >50 were selected for radiolabeling with 89Zr using previous protocols (44). Naive female mice, i.v.-injected with representative 89Zr-DFO-Gef-C’ dots, showed similar biodistribution profiles. Specifically, high blood and urinary activities were observed at 0.5 h post-injection, indicated by the high radioactivity overlying the hearts and bladders of injected mice (Supplemental Figure 4). Serial PET imaging over time showed clearance of 89Zr-DFO-Gef-C’ dots from the blood pool and whole body, but with an increase in non-specific liver accumulation with rising DPR. Nearly a 4-fold increase in liver uptake was found over a 1-week period as the DPR increased from ~10 to >50 (Supplemental Figure 4a–d, respectively); these findings were confirmed on ex vivo biodistribution studies (Supplemental Figure 4e). Analysis of these studies led to the selection of a DFO-Gef-C’ dots bearing a DPR of ~40 for all subsequent studies, as it displayed an optimal DPR and an acceptable pharmacokinetic profile, with around 5% or less accumulating in off-target organs, including the RES.
To quantitatively assess the renal and hepatic clearance rates of 89Zr-DFO-Gef-C’ dots (DPR 40), a metabolic-cage study was later performed in normal healthy mice. Each mouse was initially injected systemically with ~50 μCi of 89Zr-DFO-Gef-C’ dots and housed in a metabolic cage. Urine and fecal specimens were collected separately at 6, 24, 48, 72, 120 and 168 h post-injection, and their activities assayed using a CRC®−55tR dose calibrator. Resulting renal and hepatic clearances of 89Zr- DFO-Gef-C’ dots were measured over a 1-week duration (Figure 2a and Supplemental Table 2). Urinary and fecal clearance at 6 h p.i. was found to be 25.0±2.0 percent of the injected dose (%ID) and 0.6±0.9 %ID, respectively, yielding a urinary-to-fecal clearance ratio close to 40, and thus clearly indicating dominant renal clearance at early post-injection time points. Cumulative clearance in both urine and feces increased to 32.2±2.1 %ID and 10.2±2.5 %ID by 24 h post-injection, respectively. As shown in Figure 2a, renal clearance of 89Zr-DFO-Gef-C’ dots continued to increase over time, with the total clearance estimated to be 45.2±1.5 %ID on Day 7. Total hepatic clearance of 89Zr-DFO-Gef-C’ dots was 16.4±3.7 %ID on Day 7. The final renal-to-hepatic-clearance ratio was ~3 (Supplemental Table 2).
Figure 2. Renal/hepatic clearance and biodistribution of 89Zr-DFO-Gef-C’ dots (DPR 40) in healthy mice after i.v. administration.
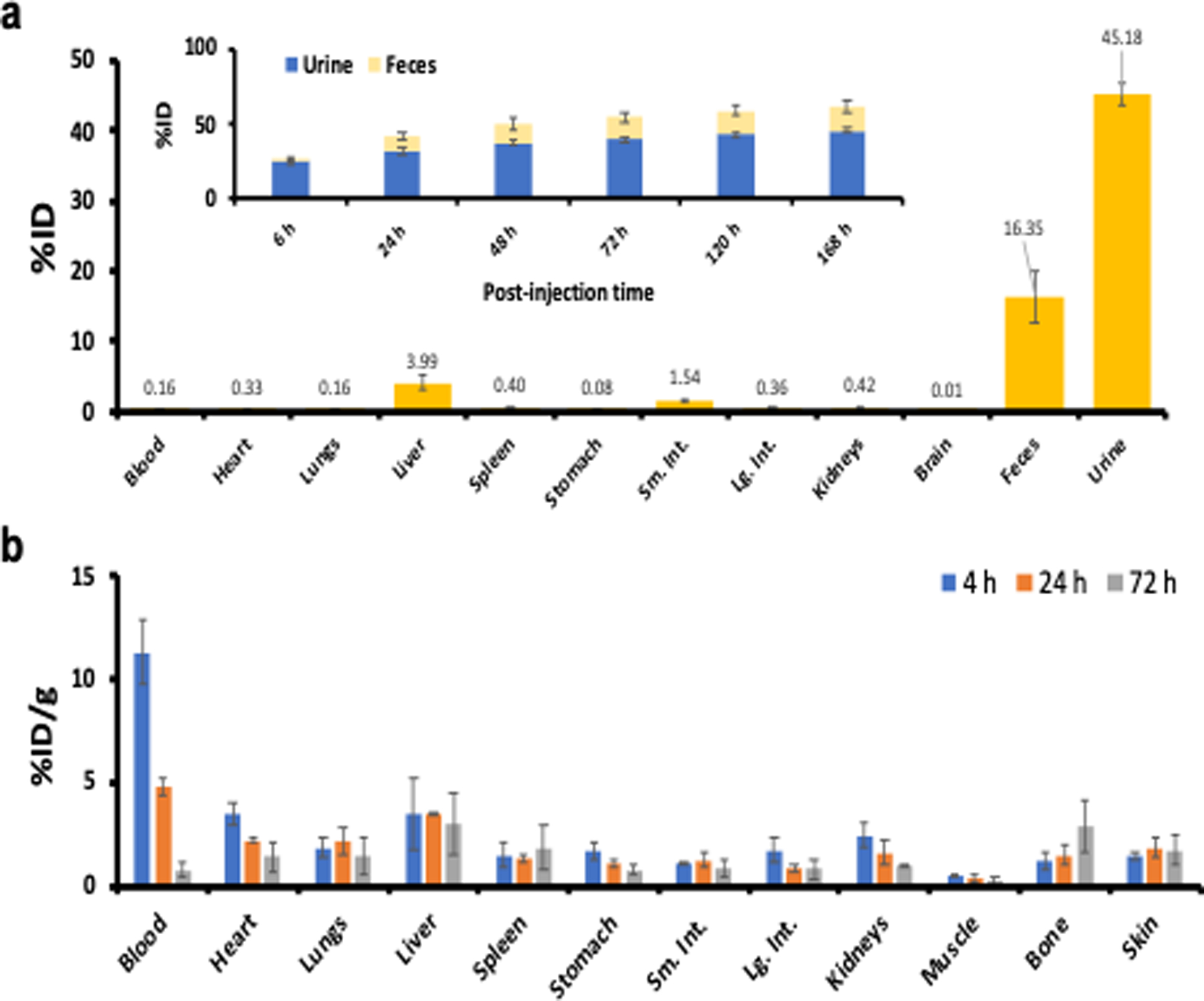
a) Biodistribution of 89Zr-DFO-Gef-C’ dots in major organs and tissues on day 7, expressed in terms of %ID (n=3; error bars: mean±SD). Inset shows the cumulative clearance measurements of 89Zr-DFO-Gef-C’ dots in urine and feces at various p.i. time points (n=3; error bars: mean±SD). b) Biodistribution of 89Zr-DFO-Gef-C’ dots in healthy mice (n=3) at 4, 24 and 72 h post-injection.
To evaluate time-dependent changes in particle distributions, mice were sacrificed and all major organs (Supplemental Table 2) were collected, wet-weighed, and counted with a gamma probe 7 days post-injection. The ex vivo biodistribution data are displayed as the %ID of 89Zr-DFO-Gef-C’ dots in healthy female mice (Figure 2a and Supplemental Table 3). In addition to dominant accumulations of 89Zr-DFO-Gef-C’ dots found in the urine and feces, respective particle uptake values in the liver, spleen and kidneys were found to be ~4 %ID or less. A more complete biodistribution study, presented as the percentage of the injected dose per gram (%ID/g), was performed in a separate cohort of mice over a range of post-injection time points. Radioactivity was predominantly found in both mouse blood and urine at early post-injection time points (Figure 2b and Supplemental Table 4). As expected, the uptake of 89Zr-DFO-Gef-C’ dots in all major organs, including liver and kidney, was found to be ~5 %ID/g or less at 24 h post-injection. Favorable radiation dosimetry of 89Zr-DFO-Gef-C’ dots (DPR 40) in a 70-kg standard man (Supplemental Table 5), estimated based on the biodistribution data, using the OLINDA dosimetry program (51), was also achieved.
Next, we investigated in vivo passive targeting, largely attributable to the EPR effect, of i.v.-injected 89Zr-DFO-Gef-C’ dots in mice xenografted with a patient-derived model of NSCLC, ECLC26. Mice were serially imaged using a Focus 120 microPET scanner. Significant bladder activity was observed 0.5 h post-injection for all tumor-bearing mice, while cardiac uptake was estimated to be ~20 %ID/g, clearly indicating predominant renal clearance and circulation of particles in the bloodstream (Supplemental Figure 5). ECLC26 tumor uptake values increased with time; at 0.5 h post-injection, uptake was roughly 3.2±1.2 %ID/g, while after 24 h, it peaked at 4.7±0.5 %ID/g (Supplemental Figure 6a). 89Zr-DFO-Gef-C’ dots demonstrated similar clearance profiles as other 89Zr-labeled C’ dots reported previously (41,42,44), with time-dependent tumor-to-blood and tumor-to-muscle ratios increasing over time (Supplemental Figure 6b & d). Liver uptake was found to be as high as the ECLC26 tumor uptake with the tumor-to-liver ratio calculated to be ~1 after 24 h post-injection (Supplemental Figure 6c). Ex vivo biodistribution findings further confirmed that ECLC26 tumor was indeed the organ with the highest uptake (Supplemental Figure 6e, Supplemental Table 6). Taken together, we demonstrated the dominant renal clearance, low RES accumulation, and passive targeting of 89Zr-DFO-Gef-C’ dots in ECLC26 NSCLC tumor-bearing mice, paving the way for the following in vitro and in vivo therapeutic efficacy evaluations.
In vitro analysis of DFO-Gef-Cy5-C’ dot functionality, uptake, and cytotoxicity.
Initially, we chose to examine the ability of DFO-Gef-C’ dots, bearing varying DPRs, to inhibit phosphorylation of the epidermal growth factor receptor (pEGFR) in vitro. Given the high liver uptake of 89Zr-DFO-Gef-C’ dots with a DPR of 55, we excluded this construct from these studies and focused on DFO-Gef-C’ dots with liver uptake values <6% ID/g. We chose to evaluate the potency of DFO-Gef-C’ dots in both a DPR- and concentration-dependent manner using ECLC26 cells. Cells were exposed to gefitinib or DFO-Gef-C’ dots (DPRs = 11, 18, 40) for 18 hours, and subsequently examined for inhibition of pEGFR. Western blot analysis demonstrated reduced levels of pEGFR across all treatment groups that occurred in both a DPR- and dose-dependent manner (Supplemental Figure 7a). Combining the results of our screening PK and biodistribution studies (Supplemental Figure 4a–e) with these target inhibition data (Supplemental Figure 7a), we observed that DFO-Gef-C’ dots having a DPR of 40 provided the greatest efficacy while also retaining low levels of liver uptake and primary renal clearance. Given these observations, we chose to advance DFO-Gef-C’ dots exhibiting a DPR of 40 as the lead candidate for all subsequent experiments.
Inhibition of EGFR signaling relies on the internalization of gefitinib and subsequent binding to the intracellular kinase domain (7,8); therefore, we sought to demonstrate that DFO-Gef-C’ dots are capable of internalization via a number of in vitro assays. We performed a dose-dependent (0–500 nM) uptake study of DFO-Gef-C’ dots co-incubated with ECLC26 cells for 24 hours, followed by flow cytometric analysis. The results show a strong increase in particle intensity (Cy5 fluorescence) within cells as a result of increased incubation concentration, where 500 nM of DFO-Gef-C’ dots demonstrated the highest median fluorescence intensity (MFI; 5000.8 a.u.) and total percent of DFO-Gef-C’ dots positive cells (99.35%). (Figure 3a; Supplemental Figure 7b). The increase in particle uptake, corresponding to increased incubation concentrations, was then further confirmed via super-resolution confocal microscopy (Figure 3a (insert); Supplemental Figure 8). Internalized particles appear to localize within the endolysosomal compartment, as demonstrated by its co-localization with 70 kDa FITC-dextran (53). Increases in intracellular particle accumulation were observed as both increases in the quantity of DFO-Gef-C’ dot foci (Cy5; red) per cell, as well as focal signal intensities (Supplemental Figure 8e). We next sought to understand whether the intracellular accumulation of DFO-Gef-C’ dots was occurring via an active or passive process. This was determined by incubating DFO-Gef-C’ dots with ECLC26 cells for 4 hours across a range of temperatures (4°C, 25°C, and 37°C). The acquired data revealed a temperature-dependent increase in both the cellular MFI values (Figure 3b), as well as the percentage of cells positive for DFO-Gef-C’ dot uptake (Supplemental Figure 7). Given that this approach is a well-established readout of active endocytosis (39), we concluded that DFO-Gef-C’ dot internalization occurs via an energy-dependent, active mechanism.
Figure 3. Cellular uptake and cytotoxicity of DFO-Gef-C’ dots in ECLC26 Non-Small Cell Lung Cancer Cells.
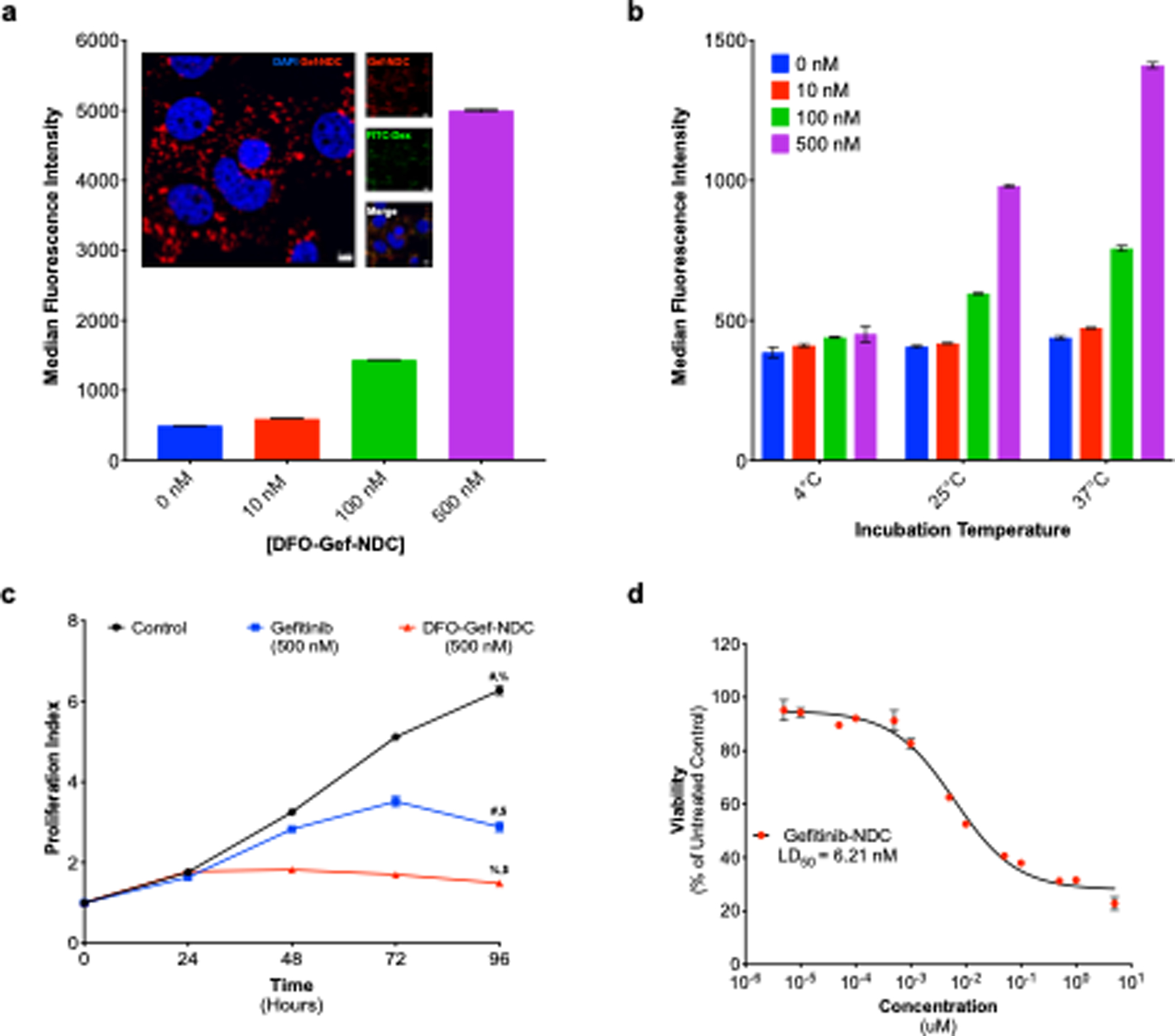
a) Dose-dependent (0–500 nM) uptake of DFO-Gef-C’ dots in ECLC26 non-small cell lung cancer cells following a 24-hour incubation. Internalization of DFO-Gef-C’ dots was confirmed using super-resolution confocal microscopy (insert, scale bar=5 μm). b) Temperature-dependent uptake of DFO-Gef-C’ dots across doses tested in a, demonstrating that internalization occurs through an active process. c) Effect of DFO-Gef-C’dot administration on the proliferation of ECLC26 cells. Cells were incubated with 500 nM concentrations of gefitinib or DFO-Gef-C’ dots over time, and cell numbers were evaluated using the CellTiter-glo assay (# p < 0.0001; % p <0.0001; $ p = 0.0004). d) LD50 curves generated following exposure of ECLC26 to DFO-Gef-C’ dots for 72 hrs. The LD50 of DFO-Gef-C’ dots was calculated to be 6.21 nM.
Lastly, we evaluated the cytotoxicity of DFO-Gef-C’ dots (DPR = 40) versus free gefitinib in ECLC26 cells. We began by examining the anti-proliferative effect of DFO-Gef-C’ dots versus that of free gefitinib at a concentration of 500 nM. We observed an approximate 2-fold reduction in the proliferative capacity of cells treated with DFO-Gef-C’ dots when compared to those exposed to free gefitinib at 96 hours (p = 0.0004; Figure 3c). This was expected given that the concentration of DFO-Gef-C’ dots was based on the concentration of the particle, and would therefore equate to an ~20 μM concentration of gefitinib. To correct for this, we also treated ECLC26 cells with particle concentrations that would equate to the molar equivalent of the administered drug. These calculations were based on the nanoparticle concentration and DPR of the DFO-Gef-C’ dots. At identical drug dosages, DFO-Gef-C’ dots demonstrated modest improvement (~15%; p = 0.0386) over free gefitinib in its ability to inhibit cell growth (Supplemental Figure 7d). Further, the gefitinib-linker construct also demonstrated an improved efficacy (~19%; p = 0.0242) over native gefitinib, a result likely due to an increase in the solubility of the drug (Supplemental Figure 7d).
We next calculated the median lethal dose, or LD50, for DFO-Gef-C’ dots by incubating with ECLC26 cells across a range of increasing concentration (10−11 M–10−5 M) for 72 hours, and subsequently assessing cell viability. Using this data, we demonstrated an LD50 value for the DFO-Gef-C’ dots to be 6.21 nM (Figure 3d). By comparison, the LD50 values for free gefitinib and the gefitinib-linker were 1946 nM and 76 nM, respectively (Supplemental Figure 7e). The LD50 value calculated for the DFO-Gef-C’ dots was based on the concentration of the nanoparticle (pLD50). Therefore, to determine the total amount of drug administered, the pLD50 was multiplied by the DPR (DPR = 40) to account for drug loading. Implementing this calculation, the theoretical LD50 of gefitinib was estimated to be ~248.4 nM. This value is nearly identical to the experimental LD50 (LD50 = 251.6 nM) achieved when particle dosing was adjusted for drug content (Supplemental Figure 7e). These results suggest that the attachment of gefitinib to the particle platform increases drug lethality by a factor of ~7.7 over that of free gefitinib, in addition to providing an enhanced cytotoxic effect.
DFO-Gef-C’ dot inhibits ECLC26 tumor growth and prolongs survival
We next sought to demonstrate the in vivo therapeutic capabilities of DFO-Gef-C’ dots in ECLC26 tumor-bearing mice. We chose to evaluate the efficacy of DFO-Gef-C’ dots in comparison to gefitinib, as well as a saline vehicle control. Gefitinib was administered via oral gavage and dosed at 150 mg/kg/day, while DFO-Gef-C’ dots were administered as a 200 μl tail vein injection of a 15 μM solution every third day. Similarly, saline vehicle control treated mice received 200 ul of intravenous saline every third day. Over the course of a 10-day period, we observed clear inhibition of tumor growth in both the gefitinib and DFO-Gef-C’ dot treated groups, in comparison to vehicle controls. At the conclusion of the study, mice in the gefitinib treatment group had a 92% (p = 0.023) reduction in tumor volume when compared to vehicle control treated animals (Figure 4a). Similarly, mice treated with DFO-Gef-C’ dots showed an 87.2% (p = 0.027) reduction in overall tumor volume (Figure 4a). While these results appear similar, if the total administered dose of gefitinib is taken into consideration, the results imply that DFO-Gef-C’ dots are more efficacious than gefitinib is in its native form. Calculations to determine the total dose of administered gefitinib revealed DFO-Gef-C’ dots required only 360 nmoles of conjugated drug-linker to elicit an anti-tumor response similar to the 78 μmoles of orally administered gefitinib (Figure 4b). Calculating the difference in dosages equates to an over 200-fold reduction of administered gefitinib in DFO-Gef-C’ dot treated animals. Together, the reduction in administered drug dose and nearly equivalent treatment response observed in DFO-Gef-C’ dot treated mice indicate that attachment of gefitinib to the C’ dot surface enhances the therapeutic efficacy of the drug through possible mechanisms such as improved pharmacokinetics, increased bioavailability, enhanced internalization, superior solubility, and better tumor tissue distribution.
Figure 4. Treatment of ECLC26 tumors using a 200-fold smaller DFO-Gef-NDC dose than gefitinib results in nearly equivalent growth inhibition and overall survival.
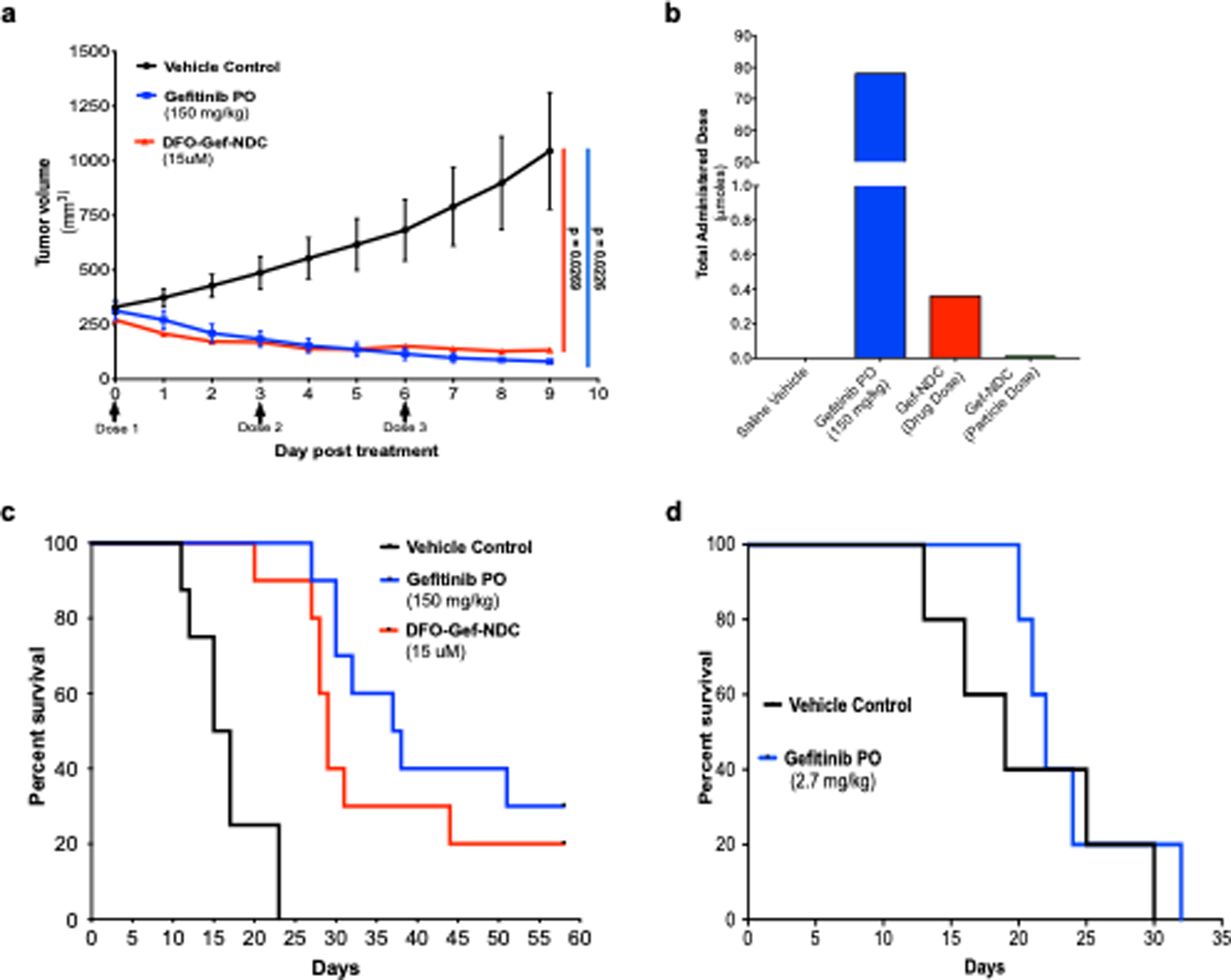
a) Tumor growth inhibition curves of ECLC26 tumor-bearing mice (n = 3/group) treated with saline vehicle, free gefitinib (150 mg/kg/day; oral gavage), or DFO-Gef-C’ dots (Days 0, 3, 6; 200 μl of 15 μM; i.v.). b) Total administered dose of gefitinib across all treatment groups described in (a). Mouse receiving free gefitinib were administered a total dose of 78 μmoles, where DFO-Gef-C’ dot treated mice received a total dose of gefitinib equal to 360 nmoles. c) Kaplan-Meier curves depicting the overall survival of mice following an identical treatment paradigm to (a) (n = 10/group). Median survival times of vehicle, gefitinib, and DFO-Gef-C’ dot treated mice were 16, 37.5, and 29 days, respectively (Vehicle:Gefitinib PO p < 0.0001; Vehicle:DFO-Gef-C’ dots p < 0.0001; Gefitinib PO:DFO-Gef-NDC p = 0.2627). d) Kaplan-Meier survival curve for ECLC26 tumor-bearing mice treated with a DFO-Gef-C’ dot equivalent dose (i.e. 360 nmoles) of gefitinib versus vehicle control treated mice (p = 0.4944).
In addition to tumor inhibition studies, we also evaluated the ability of gefitinib and DFO-Gef-C’ dots to prolong survival in comparison to vehicle control treated mice. ECLC26 tumor bearing mice were randomized to one of three groups (n = 10/group), and a dosing strategy identical to that used for the previous treatment study was employed. Mice receiving the vehicle control treatment demonstrated a median survival time of 16 days. By comparison, mice treated with daily gefitinib or DFO-Gef-C’ dots demonstrated median survival times of 37.5 (p < 0.001) and 29 (p < 0.001) days, respectively (Figure 4c). Notably, treatment with gefitinib led to a total of 3 apparent cures, while 2 DFO-Gef-C’ dot treated mice also lacked any discernable tumor at the conclusion of the study. While gefitinib treatment did slightly improve the median survival time, albeit not significantly (p = 0.2627), it also required the administration of nearly 3 orders of magnitude more drug. To demonstrate the effectiveness of gefitinib at a dose equal to that administered in the DFO-Gef-C’ dot treated group, we conducted a second survival study in which gefitinib was administered at a dose of 2.7 mg/kg (instead of 150mg/kg) on days 0, 3, and 6 of the study as was done with DFO-Gef-C’ dots. When comparing gefitinib-treated mice to those administered vehicle control, the median survival times were nearly identical (19 vs 22 days, respectively; p = 0.4944; Figure 4d).
Together, these data demonstrate that DFO-Gef-C’ dots are capable of providing robust tumor growth inhibition and prolong survival at substantially reduced dosages of gefitinib relative to the free drug. While the effectiveness of the DFO-Gef-C’ dots does closely align with that of gefitinib for the evaluated dosing strategies, gefitinib itself lacks the capacity to prolong survival when administered at identical doses to those used for DFO-Gef-C’ dots. Therefore, attachment of gefitinib to the C’ dot surface improves the therapeutic efficacy of the drug and, in turn, requires only a fraction of the dose to illicit a similar anti-tumor effect. These results indicate that optimization of DFO-Gef-C’ dot dosing may lead to dramatic improvements in treatment response that are superior to free gefitinib alone.
Ex vivo histological analyses of ECLC26 tumor and non-target tissues.
The effectiveness of gefitinib relies on its ability to inhibit the phosphorylation of EGFR, ultimately leading to a suppression of the receptor’s tumor-driving signaling cascade and reduced tumor growth (54). In an effort to demonstrate successful inhibition of EGFR signaling, ECLC26 tumor-bearing mice were treated with a single dose of either the saline vehicle (200 μl), free gefitinib (150 mg/kg), or DFO-Gef-C’ dots (15 μM; 200 μl). Tumor tissue samples were then collected 18 hours post-injection, and the level of phosphorylated EGFR (p-EGFR) was visualized using immunohistochemistry. Results in mice treated with the vehicle alone demonstrate robust p-EGFR staining (brown) throughout the bulk of the tumor area, while mice treated with either gefitinib or DFO-Gef-C’ dots show markedly reduced levels of EGFR phosphorylation across the majority of the tumor area as demonstrated by H&E staining of an adjacent tissue section (Figure 5a).
Figure 5. Gef-C’ dots inhibit p-EGFR levels in a tumor-specific manner sparing normal tissue inhibition and organ toxicity.
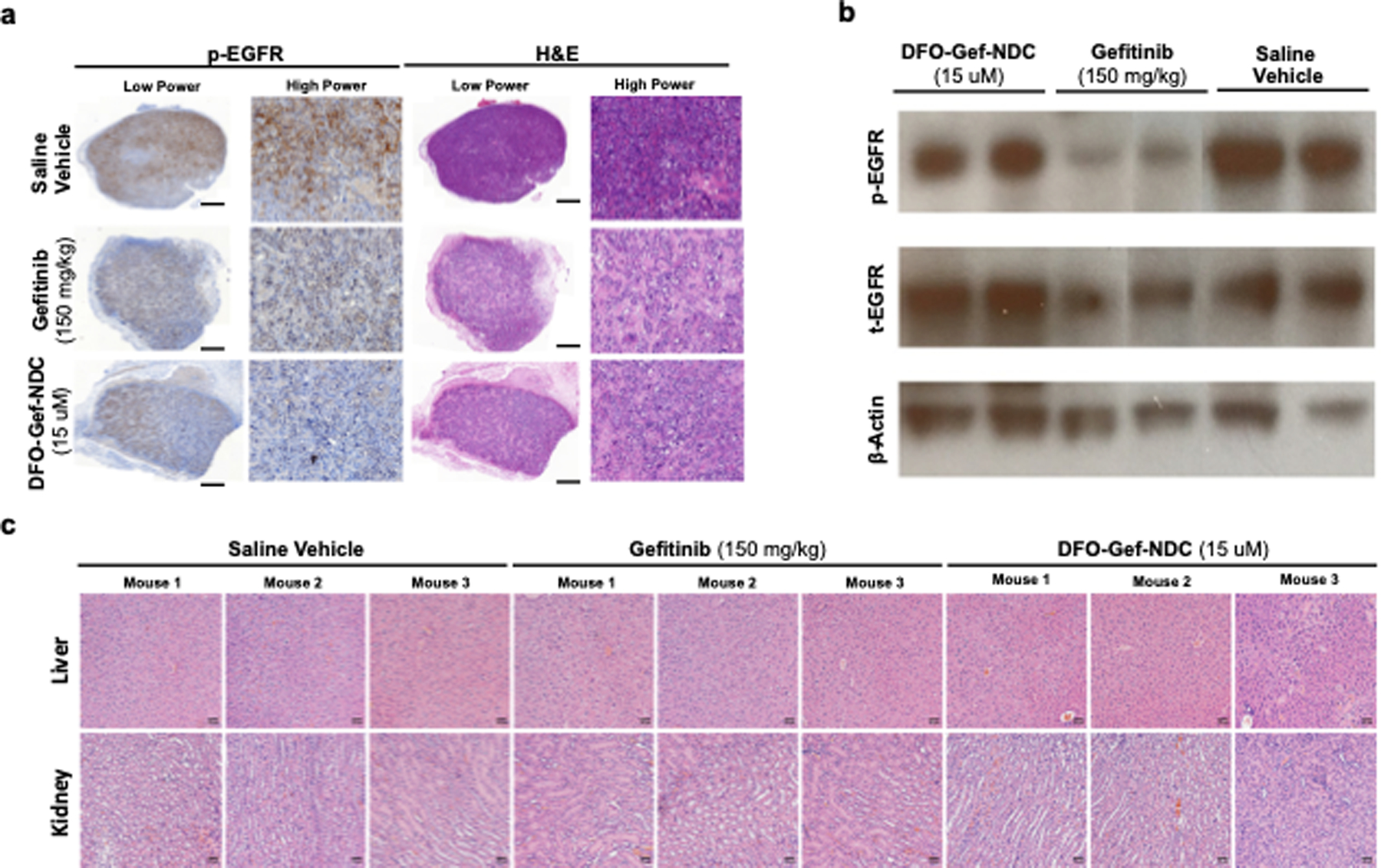
a) ECLC26 tumors were treated and collected 18-hours post-injection of saline vehicle (200 μl), free gefitinib (150 mg/kg; gavage), or Gef-C’ dots (200 μl; 15 μM). Expression of p-EGFR was evaluated using immunohistochemistry. H&E staining of an adjacent section is also displayed (scale bars = 1 mm). b) Western blot analysis of ear clippings collected at the termination of treatment studies shown in Figure 4. Expression levels of p-EGFR, total EGFR, and β-actin were evaluated in each group (n=2 mice/group). c) Liver and kidney samples were also collected following the treatment studies conducted in Figure 4 (scale bars = 50 μm). Tissue sections from each mouse (n=3 mice/treatment) were histologically assessed by an independent pathologist using H&E staining.
One of the most prevalent issues with the cancer drug therapies is their propensity to induce off-target effects and toxicities. We hypothesized that given the altered pharmacokinetics of gefitinib following its attachment to the C’ dot surface, we would be able to reduce drug accumulation in non-target tissues and limit the effect of the drug at these anatomical sites. Given that skin rashes are a common side effect of gefitinib administration, we chose to evaluate ear clippings from mice treated in the aforementioned growth inhibition study to serve as a surrogate of wild-type EGFR inhibition. We examined the expression of total EGFR, p-EGFR, and β-actin (loading control) using Western blot to determine the effect of our treatments (Figure 5b). Comparison of total and phosphorylated EGFR levels in the saline vehicle treated group to both gefitinib and DFO-Gef-C’ dot treatments illustrated a clear and strong suppression of p-EGFR in mice treated with gefitinib, a result not observed in mice treated with the DFO-Gef-C’ dots. Interestingly, the levels of total EGFR expression also show marked reductions in the ear clippings of mice treated with gefitinib when compared to the DFO-Gef-C’ dots and vehicle controls. These data indicate that DFO-Gef-C’ dots, while capable of similar tumor growth inhibition, result in less suppression of non-target, wild-type EGFR, and that gefitinib may elicit unwanted side effects, not only through the reduction of p-EGFR levels, but also from the inhibition of EGFR expression in general.
Lastly, we performed limited toxicology studies on mice from each treatment arm (n=3/arm) in our tumor growth inhibition studies. The toxicology reports included the examination of tissue histology in both the liver and kidneys of mice that had been treated with gefitinib, DFO-Gef-C’ dots, or vehicle control (Figure 5c). Analysis performed by an independent pathologist showed no abnormalities or obvious tissue damage in the treatment groups based on their gross histological examination. In addition to histopathology, complete blood counts (Supplemental Table 7–8) and serum chemistries (Supplemental Table 9–10) were also performed on these mice. Complete blood count results look similar across all evaluated groups and remain largely within normal limits. Two mice treated with DFO-Gef-C’ dots, and one mouse treated with gefitinib, showed mildly elevated white blood cell and lymphocyte numbers that is likely attributed to a B cell response, as nude mice lack T lymphocytes. The observed increases are minor and, given that increased lymphocyte counts by themselves are not considered an adverse event in preclinical toxicology, we consider these treatments to be well-tolerated in this model. Evaluation of serum chemistry values revealed no significant abnormalities or differences between mice treated with gefitinib or DFO-Gef-C’ dots when compared to saline vehicle treated mice. Together, these findings demonstrate DFO-Gef-C’ dots are capable of inhibiting tumor-associated EGFR signaling while limiting the suppression of the wild-type receptors. According to the assayed histological and hematological parameters, DFO-Gef-C’ dots are well-tolerated when administered to mice at the assessed dosages, indicating that DFO-Gef-C’ dots function as a safe and novel drug delivery platform for the administration of small molecule inhibitors.
Discussion
The implementation of SMIs in the clinic for the treatment of oncological malignancies has had a significant impact on patient care outcomes. Exploting the overly expressed or aberrantly activated pro-tumor signaling pathways found in cancers, a number of SMIs have been developed that effectively inhibit tumor growth through suppression of these driving signal cascades. Despite such advances in drug design, these small targeted therapeutics still suffer from a number of setbacks that preclude them from functioning at their ultimate potential. Issues such as poor solubility, limited bioavailability, serum binding, short circulation half-lives, off-target accumulation, and unwanted side effects limit their potential in the clinic. While, from a practical standpoint, these drugs would be expected to perform well in a living system, the reality is that they still require improvements to increase their overall effectiveness.
Considerable efforts have been made to improve upon the performance of these molecular drugs by combining them with a potential carrier in the hopes of increasing circulation times and overall targeted accumulations, while limiting their off-target effects. One such approach has involved the use of nanoparticle carriers that either encapsulate drugs or covalently attach them to the particle surface. Herein, we synthesized a nanoparticle drug conjugate using a gefitinib analog (APdMG) attached to the surface of ultrasmall (i.e., 6.5 nm hydrodynamic diameter) core-shell silica nanoparticles, termed C’ dots, via a protease sensitive drug linker. In the current study, we were able to precisely control drug loading on the particle surface and investigated the effects of altering the number of drugs per particle on the corresponding PK profile. This approach enabled the selection of an optimal construct exhibiting bulk renal clearance, DFO-Gef-C’ dots (DPR=40), for next-stage pre-clinical studies. DFO-Gef-C’ dots were found to undergo active endocytosis into ECLC26 NSCLC cells where, upon release of the therapeutic payload, a potent cytotoxic response was elicited. In vivo, DFO-Gef-C’ dots displayed favorable PK and biodistribution profiles that resulted in high tumor accumulation and low levels of wild-type EGFR signaling inhibition. These results offer promise for the use of the ultrasmall C’ dot platform as a useful drug delivery vehicle for a variety of therapeutics.
While the outcomes of this study are promising, we do acknowledge a few practical issues. The first being our choice to use a first-generation prototype inhibitor in place of newer generation EGFR inhibitors. While we understand current generation EGFR kinase inhibitors are more potent and selective than gefitinib, the work presented here provides foundational proof-of-concept data by which the biological and therapeutic properties of toxic small molecule drugs might potentially be improved by their attachment to C’ dots. While we do not suggest that DFO-Gef-C’ dots are more effective than current therapies, we do show that the attachment of gefitinib to the C’ dot surface enhances its efficacy and reduces non-tumor EGFR pathway inhibition over the free drug alone. It should also be noted that gefitinib is highly insoluble in aqueous environments and, therefore, renders intravenous delivery challenging without the use of organic solvents. Previous attempts to deliver gefitinib via intravenous injection, when dissolved in the minimally necessary amount of DMSO, quickly led to respiratory distress and animal mortality. It is for these reasons that the studies presented here administered gefitinib through oral gavage, following standard oncology care.
A second issue may be the lack of tumor-specific targeting. Previously, we have demonstrated the benefits of tumor targeting to enhance delivery of a SMI into high-grade gliomas (46). While we can’t discount the possibility that the addition of a targeting moiety to the particle surface (i.e. anti-EGFR antibody fragment) would have increased NDC accumulation and effectiveness, the scope of this project was centered around demonstrating a therapeutic and survival benefit following attachment of a prototype SMI, gefitinib, to the particle surface. This omission allows for future investigations into the use of targeting ligands, in conjugation with more toxic payloads, in efforts to further improve NDC tumor accumulation while minimizing normal tissue deposition.
Lastly, we acknowledge the use of a non-orthotopic, single tumor model. We chose to utilize the ECLC26 tumor cell line as it is directly derived from a patient who presented with an EGFR activating mutation, L858R (c.2573 T>G) and is, therefore, an ideal candidate for EGFR inhibitor therapy. In order to implement an orthotopic lung model for use in demonstrating tumor control and overall survival, introduction of bioluminescent reporters (i.e. luciferase) would have been necessary to monitor treatment changes. We chose not to pursue this avenue for our initial proof-of-concept study, but instead focused on the use of a pristine, patient-derived tumor model that allowed direct, accurate visualization and serial assessment of treatment response in subcutaneous tumors. Tumor models that better recapitulate the complexity of the tumor microenvironment are a focus of ongoing studies.
In this study, we present the first demonstration of therapeutic responsiveness and survival benefit following the administration of DFO-Gef-C’ dots in a xenograft model of NSCLC. Systemic administration of DFO-Gef-C’ dots resulted in robust downregulation of p-EGFR levels in ECLC26 xenografts, which closely resembled the downregulation observed by treatment with gefitinib alone. However, unlike gefitinib, DFO-Gef-C’ dots do not inhibit wild-type EGFR signaling, nor total receptor expression, as evidenced by preserved p-EGFR levels in tissue samples collected from mouse ear clippings. Additionally, the total drug dose given as part of the DFO-Gef-C’ dot platform was over 2 orders of magnitude less than the dose received by mice in the gefitinib treatment arm. This result, together with limited EGFR inhibition in normal tissues, indicates that attachment of gefitinib to the C’ dot surface increases the efficacy of gefitinib when compared to the free drug formulation. While tumor growth inhibition studies in this work were not adjusted for dose and timing, we believe that optimization of these parameters may lead to even more impressive disease control while continuing to limit side effects and, in turn, expand upon the therapeutic window of gefitinib. This work elucidates the utility of the ultrasmall C’ dot as an extremely promising drug delivery vehicle for small molecule drugs, and opens its possible use to other, less well-tolerated, toxins as a mechanism to increase drug dosing, accumulation, and efficacy while lessening dose-limiting toxicities.
Supplementary Material
Statement of Translational Relevance.
Targeted molecular drugs have revolutionized the cancer therapy landscape; however, these therapies are often plagued by poor solubility, bioavailability, tumor accumulation, and dose-limiting toxicity. We demonstrate that a clinically-translatable ultrasmall silica nanoparticle, C’-dots, functionalized with new surface chemical components and the prototype epidermal growth factor receptor tyrosine kinase inhibitor, gefitinib, led to order of magnitude increases in drug loading, enhanced therapeutic efficacy, and limited non-target receptor inhibition at a fraction of the native drug dose in a non-small cell lung cancer model. Findings highlight the potential utility of therapeutic C’ dots as an efficacious drug delivery vehicle, laying the foundation for attaching highly toxic compounds as a means of improving delivery/uptake, pharmacokinetics, and therapeutic index.
Conflicts of Interest and Source of Funding:
FC, MZT, KM, PC, MSB, and UW hold interest in Elucida Technologies, Inc., which has licensed IP from Cornell and MSKCC on C’ dots and their application in oncology. No other authors declare a conflict of interest. This study was funded by grants from the National Institutes of Health (1U54 CA199081-01 to M.B. and U.W.) and Sloan Kettering Institute (Core Grant P30 CA008748CCSG).
References
- 1.Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends in pharmacological sciences 2015;36(7):422–39 doi 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 2002;8(5):935–42. [PubMed] [Google Scholar]
- 3.Chabner BA, Roberts TG Jr. Chemotherapy and the war on cancer. Nature Reviews Cancer 2005;5:65 doi 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 4.Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacological research 2019;152:104609 doi 10.1016/j.phrs.2019.104609. [DOI] [PubMed] [Google Scholar]
- 5.Cohen MH, Williams GA, Sridhara R, Chen G, McGuinn WD Jr., Morse D, et al. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839; Iressa) tablets. Clinical cancer research : an official journal of the American Association for Cancer Research 2004;10(4):1212–8. [DOI] [PubMed] [Google Scholar]
- 6.Tiseo M, Bartolotti M, Gelsomino F, Bordi P. Emerging role of gefitinib in the treatment of non-small-cell lung cancer (NSCLC). Drug design, development and therapy 2010;4:81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muhsin M, Graham J, Kirkpatrick P. Gefitinib. Nature Reviews Drug Discovery 2003;2:515 doi 10.1038/nrd1136. [DOI] [PubMed] [Google Scholar]
- 8.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature reviews Molecular cell biology 2001;2(2):127–37 doi 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 9.Hirsch FR, Varella-Garcia M, Bunn PA Jr., Franklin WA, Dziadziuszko R, Thatcher N, et al. Molecular predictors of outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2006;24(31):5034–42 doi 10.1200/jco.2006.06.3958. [DOI] [PubMed] [Google Scholar]
- 10.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science (New York, NY) 2004;305(5687):1163–7 doi 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 11.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia (New York, NY) 2010;12(9):675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proceedings of the National Academy of Sciences of the United States of America 1992;89(7):2965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Struve N, Riedel M, Schulte A, Rieckmann T, Grob TJ, Gal A, et al. EGFRvIII does not affect radiosensitivity with or without gefitinib treatment in glioblastoma cells. Oncotarget 2015;6(32):33867–77 doi 10.18632/oncotarget.5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. The FEBS journal 2013;280(21):5350–70 doi 10.1111/febs.12393. [DOI] [PubMed] [Google Scholar]
- 15.Padfield E, Ellis HP, Kurian KM. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Frontiers in oncology 2015;5:5 doi 10.3389/fonc.2015.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ayrton A, Morgan P. Role of transport proteins in drug absorption, distribution and excretion. Xenobiotica; the fate of foreign compounds in biological systems 2001;31(8–9):469–97 doi 10.1080/00498250110060969. [DOI] [PubMed] [Google Scholar]
- 17.Zhao C, Han SY, Li PP. Pharmacokinetics of Gefitinib: Roles of Drug Metabolizing Enzymes and Transporters. Current drug delivery 2017;14(2):282–8 doi 10.2174/1567201813666160709021605. [DOI] [PubMed] [Google Scholar]
- 18.Kosaka T, Yamaki E, Mogi A, Kuwano H. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. Journal of biomedicine & biotechnology 2011;2011:165214 doi 10.1155/2011/165214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galetti M, Alfieri RR, Cavazzoni A, La Monica S, Bonelli M, Fumarola C, et al. Functional characterization of gefitinib uptake in non-small cell lung cancer cell lines. Biochemical pharmacology 2010;80(2):179–87 doi 10.1016/j.bcp.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Brahmer J, Messersmith W, Hidalgo M, Baker SD. Binding of gefitinib, an inhibitor of epidermal growth factor receptor-tyrosine kinase, to plasma proteins and blood cells: in vitro and in cancer patients. Investigational new drugs 2006;24(4):291–7 doi 10.1007/s10637-006-5269-2. [DOI] [PubMed] [Google Scholar]
- 21.Su H, Seimbille Y, Ferl GZ, Bodenstein C, Fueger B, Kim KJ, et al. Evaluation of [(18)F]gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. European journal of nuclear medicine and molecular imaging 2008;35(6):1089–99 doi 10.1007/s00259-007-0636-6. [DOI] [PubMed] [Google Scholar]
- 22.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annual review of medicine 2012;63:185–98 doi 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 23.Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharmaceutical research 2016;33(10):2373–87 doi 10.1007/s11095-016-1958-5. [DOI] [PubMed] [Google Scholar]
- 24.Singh R, Lillard JW Jr., Nanoparticle-based targeted drug delivery. Experimental and molecular pathology 2009;86(3):215–23 doi 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bregoli L, Movia D, Gavigan-Imedio JD, Lysaght J, Reynolds J, Prina-Mello A. Nanomedicine applied to translational oncology: A future perspective on cancer treatment. Nanomedicine : nanotechnology, biology, and medicine 2016;12(1):81–103 doi 10.1016/j.nano.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nature materials 2014;13(2):125–38 doi 10.1038/nmat3780. [DOI] [PubMed] [Google Scholar]
- 27.Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, et al. Design considerations for tumour-targeted nanoparticles. Nature nanotechnology 2010;5(1):42–7 doi 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle Uptake: The Phagocyte Problem. Nano today 2015;10(4):487–510 doi 10.1016/j.nantod.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuruppu AI, Zhang L, Collins H, Turyanska L, Thomas NR, Bradshaw TD. An Apoferritin-based Drug Delivery System for the Tyrosine Kinase Inhibitor Gefitinib. Advanced healthcare materials 2015;4(18):2816–21 doi 10.1002/adhm.201500389. [DOI] [PubMed] [Google Scholar]
- 30.Zhao L, Yang G, Shi Y, Su C, Chang J. Co-delivery of Gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. Journal of nanobiotechnology 2015;13:57 doi 10.1186/s12951-015-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lam AT, Yoon J, Ganbold EO, Singh DK, Kim D, Cho KH, et al. Colloidal gold nanoparticle conjugates of gefitinib. Colloids and surfaces B, Biointerfaces 2014;123:61–7 doi 10.1016/j.colsurfb.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 32.Shi Y, Su C, Cui W, Li H, Liu L, Feng B, et al. Gefitinib loaded folate decorated bovine serum albumin conjugated carboxymethyl-beta-cyclodextrin nanoparticles enhance drug delivery and attenuate autophagy in folate receptor-positive cancer cells. Journal of nanobiotechnology 2014;12:43 doi 10.1186/s12951-014-0043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni XL, Chen LX, Zhang H, Yang B, Xu S, Wu M, et al. In vitro and in vivo antitumor effect of gefitinib nanoparticles on human lung cancer. Drug delivery 2017;24(1):1501–12 doi 10.1080/10717544.2017.1384862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han W, Shi L, Ren L, Zhou L, Li T, Qiao Y, et al. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal transduction and targeted therapy 2018;3:16 doi 10.1038/s41392-018-0019-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kola Srinivas NS, Verma R, Pai Kulyadi G, Kumar L. A quality by design approach on polymeric nanocarrier delivery of gefitinib: formulation, in vitro, and in vivo characterization. International journal of nanomedicine 2017;12:15–28 doi 10.2147/ijn.S122729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Science translational medicine 2014;6(260):260ra149 doi 10.1126/scitranslmed.3009524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma K, Mendoza C, Hanson M, Werner-Zwanziger U, Zwanziger J, Wiesner U. Control of Ultrasmall Sub-10 nm Ligand-Functionalized Fluorescent Core–Shell Silica Nanoparticle Growth in Water. Chemistry of Materials 2015;27(11):4119–33 doi 10.1021/acs.chemmater.5b01222. [DOI] [Google Scholar]
- 38.Ma K, Zhang D, Cong Y, Wiesner U. Elucidating the Mechanism of Silica Nanoparticle PEGylation Processes Using Fluorescence Correlation Spectroscopies. Chemistry of Materials 2016;28(5):1537–45 doi 10.1021/acs.chemmater.6b00030. [DOI] [Google Scholar]
- 39.Benezra M, Phillips E, Overholtzer M, Zanzonico PB, Tuominen E, Wiesner U, et al. Ultrasmall integrin-targeted silica nanoparticles modulate signaling events and cellular processes in a concentration-dependent manner. Small (Weinheim an der Bergstrasse, Germany) 2015;11(14):1721–32 doi 10.1002/smll.201402331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen F, Ma K, Benezra M, Zhang L, Cheal SM, Phillips E, et al. Cancer-Targeting Ultrasmall Silica Nanoparticles for Clinical Translation: Physicochemical Structure and Biological Property Correlations. Chemistry of materials : a publication of the American Chemical Society 2017;29(20):8766–79 doi 10.1021/acs.chemmater.7b03033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen F, Ma K, Madajewski B, Zhuang L, Zhang L, Rickert K, et al. Ultrasmall targeted nanoparticles with engineered antibody fragments for imaging detection of HER2-overexpressing breast cancer. Nature communications 2018;9(1):4141 doi 10.1038/s41467-018-06271-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen F, Zhang X, Ma K, Madajewski B, Benezra M, Zhang L, et al. Melanocortin-1 Receptor-Targeting Ultrasmall Silica Nanoparticles for Dual-Modality Human Melanoma Imaging. ACS applied materials & interfaces 2018;10(5):4379–93 doi 10.1021/acsami.7b14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma K, Wiesner U. Modular and Orthogonal Post-PEGylation Surface Modifications by Insertion Enabling Penta-Functional Ultrasmall Organic-Silica Hybrid Nanoparticles. Chemistry of Materials 2017;29(16):6840–55 doi 10.1021/acs.chemmater.7b02009. [DOI] [Google Scholar]
- 44.Chen F, Ma K, Zhang L, Madajewski B, Zanzonico P, Sequeira S, et al. Target-or-Clear Zirconium-89 Labeled Silica Nanoparticles for Enhanced Cancer-Directed Uptake in Melanoma: A Comparison of Radiolabeling Strategies. Chemistry of materials : a publication of the American Chemical Society 2017;29(19):8269–81 doi 10.1021/acs.chemmater.7b02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen F, Madajewski B, Ma K, Karassawa Zanoni D, Stambuk H, Turker MZ, et al. Molecular phenotyping and image-guided surgical treatment of melanoma using spectrally distinct ultrasmall core-shell silica nanoparticles. Sci Adv 2019;5(12):eaax5208 doi 10.1126/sciadv.aax5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Juthani R, Madajewski B, Yoo B, Zhang L, Chen PM, Chen F, et al. Ultrasmall Core-Shell Silica Nanoparticles for Precision Drug Delivery in a High-Grade Malignant Brain Tumor Model. Clinical cancer research : an official journal of the American Association for Cancer Research 2019. doi 10.1158/1078-0432.Ccr-19-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoo B, Ma K, Zhang L, Burns A, Sequeira S, Mellinghoff I, et al. Ultrasmall dual-modality silica nanoparticle drug conjugates: Design, synthesis, and characterization. Bioorganic & medicinal chemistry 2015;23(22):7119–30 doi 10.1016/j.bmc.2015.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gardinier TC, Turker MZ, Hinckley JA, Katt WP, DomNwachukwu N, Woodruff F, et al. Controlling Surface Chemical Heterogeneities of Ultrasmall Fluorescent Core–Shell Silica Nanoparticles as Revealed by High-Performance Liquid Chromatography. The Journal of Physical Chemistry C 2019;123(37):23246–54 doi 10.1021/acs.jpcc.9b06905. [DOI] [Google Scholar]
- 49.Kohle FFE, Hinckley JA, Wiesner UB. Dye Encapsulation in Fluorescent Core–Shell Silica Nanoparticles as Probed by Fluorescence Correlation Spectroscopy. The Journal of Physical Chemistry C 2019;123(15):9813–23 doi 10.1021/acs.jpcc.9b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nuclear medicine and biology 2009;36(7):729–39 doi 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stabin MG, Sparks RB, Crowe E. OLINDA/EXM: the second-generation personal computer software for internal dose assessment in nuclear medicine. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2005;46(6):1023–7. [PubMed] [Google Scholar]
- 52.Gardinier TC, Kohle FFE, Peerless JS, Ma K, Turker MZ, Hinckley JA, et al. High-Performance Chromatographic Characterization of Surface Chemical Heterogeneities of Fluorescent Organic–Inorganic Hybrid Core–Shell Silica Nanoparticles. ACS Nano 2019;13(2):1795–804 doi 10.1021/acsnano.8b07876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Humphries WHt, Szymanski CJ, Payne CK. Endo-lysosomal vesicles positive for Rab7 and LAMP1 are terminal vesicles for the transport of dextran. PloS one 2011;6(10):e26626 doi 10.1371/journal.pone.0026626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res 2002;62(20):5749–54. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.