

Abstract
TGFβ induces the differentiation of hepatic stellate cells (HSCs) into tumor-promoting myofibroblasts but underlying mechanisms remain incompletely understood. Because endocytosis of TGFβ receptor II (TβRII), in response to TGFβ stimulation, is a prerequisite for TGF signaling, we investigated the role of protein diaphanous homolog 1 (known as Diaph1 or mDia1) for the myofibroblastic activation of HSCs. Using shRNA to knockdown Diaph1 or SMIFH2 to target Diaph1 activity of HSCs, we found that the inactivation of Diaph1 blocked internalization and intracellular trafficking of TβRII and reduced SMAD3 phosphorylation induced by TGFβ1. Mechanistic studies revealed that the N-terminal portion of Diaph1 interacted with both TβRII and Rab5a directly and that Rab5a activity of HSCs was increased by Diaph1 overexpression and decreased by Diaph1 knockdown. Additionally, expression of Rab5aQ79L (active Rab5a mutant) increased whereas the expression of Rab5aS34N (inactive mutant) reduced the endosomal localization of TβRII in HSCs compared to the expression of wild-type Rab5a. Functionally, TGFβ stimulation promoted HSCs to express tumor-promoting factors, and α-smooth muscle actin, fibronection, and CTGF, markers of myofibroblastic activation of HSCs. Targeting Diaph1 or Rab5a suppressed HSC activation and limited tumor growth in a tumor implantation mouse model. Thus, Dipah1 and Rab5a represent targets for inhibiting HSC activation and the hepatic tumor microenvironment.
Keywords: cancer-associated fibroblasts, colorectal cancer, endosomal trafficking of receptors, IncuCyte Live-Cell Analysis, biotinylation
1 ∣. INTRODUCTION
In response to tumor growth in the liver, hepatic stellate cells (HSCs), which are liver-specific pericytes, are activated and differentiated into myofibroblasts. Activated-HSC/myofibroblasts, in turn, promote tumor growth by paracrine mechanisms, including the release of growth factors, cytokines, extracellular matrix proteins, and matrix metalloproteinases.1-3 TGFβ is the most potent factor that induces the myofibroblastic activation of HSCs and its effect is mediated by a series of intracellular signaling events, including the formation of a complex of TGFβ receptor II (TβRII) and TGFβ receptor I (TβRI) at the plasma membrane, endocytosis of the receptor complex, phosphorylation of SMAD2/3, and gene transcription in the nucleus.4,5 Understanding how each signaling event is regulated may lead to novel strategies and targets to inhibit HSC activation and the hepatic tumor microenvironment.
Protein diaphanous homolog 1 (commonly known as Diaph1 or mDia1) is a member of the family of formin proteins. It is an effector of Rho small GTPases that regulates actin polymerization and the shape and motility of the cell at the downstream of Rho.6-8 Through Formin Homology 2 (FH2) domain, Diaph1 nucleates actin filaments by binding to spontaneously formed actin dimers. It also facilitates the elongation of the filaments by preventing the binding of capping proteins to actin filament barbed ends and aid the addition of the actin monomers (G-actin) to the filaments.7,9,10 Through the FH1 domain, Diaph1 binds to profilin bound G-actin to bring G-actin into the close proximity to the barbered ends for filament elongation.11 Additionally, Rho small GTPases activate Diaph1 by binding to its GTPase-binding domain (GBD) and releasing Diaph1 from its auto-inhibition.7,12
In patients, genetic mutations of Diaph1 were associated with deafness and macrothrombocytopenia13,14 and the expression level of Diaph1 in cancer cells correlated with the clinical stage and metastasis of the diseases.15,16 In Drosophila embryos, protein diaphanous (Dia) regulated the endocytosis of E-cadherin and morphogenesis of epithelium17 and in mammalian cells, Diaph1 was targeted to endosomes by a RhoB-dependent mechanism.18,19 mDia1 was recruited to the phagocytic cup of murine macrophage where it promoted the phagocytosis of CD11bCD18 (αMβ2 integrins).20-22 Additionally, mDia1 knockout mice developed neutropenia because of the attenuated internalization of CD11 of neutrophils and enhanced neutrophil adhesion to the endothelium,23 supporting mDia1 contributed to phagocytosis although the mechanisms involved in were not well understood.
Because TGFβ stimulation leads to the endocytosis of TβRI/TβRII complexes and subsequent phosphorylation of SMAD2/3 at early endosomes,24,25 we analyzed the role of Diaph1 in intracellular trafficking of TβRII and TGFβ-mediated HSC activation in this study. Using Diaph1 shRNA to knockdown Diaph1 or small molecule SMIFH226 to inhibit Diaph1 function, we demonstrated that the inactivation of Diaph1 suppressed TGFβ-mediated intracellular trafficking of TβRII and SMAD3 phosphorylation through modulating Rab5a, a key regulator for the biogenesis of endocytic vesicles. Functionally, TGFβ1 induced the activation of HSCs into myofibroblasts and stimulated HSCs to produce paracrine tumor-promoting factors. Inactivation of Diaph1 or Rab5a suppressed the tumor-promoting effect of HSCs in vitro and in a tumor implantation mouse model. Diaph1 and Rab5a thus represent targets for inhibiting HSC activation and the hepatic tumor microenvironment.
2 ∣. MATERIALS AND METHODS
2.1 ∣. Cell lines, antibodies, and reagents
Primary human HSCs were purchased from ScienCell Research Laboratories (Carlsbad, CA).1,27,28 They were cultured in DMEM (supplemented with 10% fetal bovine serum [FBS], penicillin and streptomycin), and used up to nine passages. HT29 human colorectal cancer cells were from ATCC (HTB38, Manassas, VA) and authenticated by short tandem repeat (STR) DNA profiling by Genetica. Mycoplasma contamination was regularly monitored by a MycoAlert detection kit (Lonza, Basel, Switzerland). Cells free of infection were used for experiments.
Antibodies for Western blot: anti-TβRII (184948; Abcam, Cambridge, UK; 1:1000), anti-Diaph1 (E-4) (373807; Santa Cruz Biotechnology, Dallas, TX; 1:500), anti-α-SMA (A5228; Millipore Sigma, Burlington, MA; 1:5000), anti-fibronectin (610077; BD Transduction Laboratory, Franklin Lakes, NJ; 1:500), anti-P-SMAD3 (S423/425) (9520; Cell Signaling, Danvers, MA; 1:500), anti-SMAD2/3 (sc-133098; Santa Cruz Biotechnology; 1:5000), anti-HA (c29F4) (3724; Cell Signaling; 1:1000), anti-GAPDH (G8140; US Biological, Salem, MA; 1:5000), anti-GAPDH (G8795; Millipore Sigma; 1:2000), anti-Rab5 (D-11) (46692; Santa Cruz Biotechnology; 1:500), anti-GFP (G1544; Millipore Sigma; 1:4000), anti-DYKDDDDK (FLAD tag) (2368; Cell Signaling technology; 1:1000), anti-tenascin C (sc-20932; Santa Cruz Biotechnology; 1:10 000), anti-PD-L1 ( 4059; ProSci, San Diego, CA; 1:500), anti-IGF1 (H-9) (518040; Santa Cruz Biotechnology; 1:200), anti-CTGF (209780; Abcam; 1:1000), and anti-CTGF (sc-14939; Santa Cruz Biotechnology; 1:500).
Antibodies for immunofluorescence: anti-EEA1 (610456; BD Transduction Laboratory; 1:100), anti-LAMP1 (H4A3) (20011; Santa Cruz Biotechnology; 1:50), anti-HA (c29F4) (3724; Cell Signaling; 1:1000), anti-α-SMA (A5228; Millipore Sigma, Burlington, MA; 1:4000), OctA-Probe (G-8) (FLAG tag) (166384; Santa Cruz Biotechnology; 1:500), anti-tenascin C (sc-20932; Santa Cruz Biotechnology; 1:2000), anti-CTGF (209780; Abcam; 1:800).
SMIFH2 (4401) was purchased from Tocris Bioscience, Bristol, UK; TGFβ1 (240-B) was purchased from R & D Systems Inc, Minneapolis, MN; Nocodazole (M1404) was purchased from Millipore Sigma; Bafilomycin A1 (1334) was purchased from Tocris, Minneapolis, MN.
2.2 ∣. Plasmids and subcloning
A retroviral vector encoding TβRII-HA cDNA was generated by a prior study.1 Diaph1-Emerald cDNA (54157; Addgene, Watertown, MA) and Rab5Q79L cDNA (28046; Addgene) were inserted into the pMMP retroviral vector, respectively, by standard PCR-based subcloning techniques. A FLAG-tag was added to the N-terminus of the constructs so that pMMP-FLAG-Rab5Q79L, pMMP-FLAG-Rab5wt, and pMMP-FLAG-Rab5S34N were subsequently generated using a Q5 Site-Directed Mutagenesis kit (E0554; New England Biolabs, Ipswich MA). pGEX6p-1βRII was generated by a prior study.1 To generate additional bacterial expression vectors, Rab5a cDNA, full-length Diaph1 cDNA, and an N-terminal fragment of Diaph1 encoding a.a. 1-453 were inserted into the pGEX4T1 vector individually using a Gibson Assembly Cloning Kit (E5510; New England Biolabs). The PCR primers used for subcloning were:
pGEX4T1 Forward: TTCCCGGGTCGACTCGAGCGGCCG
pGEX4T1 Reverse: TTCCGGGGATCCACGCGGAACCAG
Diaph1 Forward: CTGGTTCCGCGTGGATCCCCGGAAATGGAGCCGCCCGGC
Diaph1 (1-453) Reverse: CGGCCGCTCGAGTCGACCCGGGAATTATCCCTCAATCTCAATCTGG
Full-length Diaph1 Reverse: CGGCCGCTCGAGTCGACCCGGGAATTAGCTTGCACGGCC
Rab5a forward: CTGGTTCCGCGTGGATCCCCGGAAATGGCTAGTCGAGGC
Rab5a reverse: CGGCCGCTCGAGTCGACCCGGGAATTAGTTACTACAACAC
2.3 ∣. Retroviral and lentiviral transduction of HSCs
Lentiviral constructs encoding Diaph1 shRNA (TRCN0000118678 and TRCN0000118679) or Rab5a shRNA (TRCN0000007974 and TRCN0000007975) were bought from the MISSION shRNA library (Millipore Sigma) through the University of Minnesota Genomic Center. A construct encoding non-targeting shRNA was used as a control (SHC202; Millipore Sigma). Lentiviruses and retroviruses were generated by transfecting 293T cells with three plasmids using the Effectene Transfection Reagent (301425; Qiagen, Hilden, Germany), as we previously described.1,29-31 Virus-containing supernatant was collected at 48 hours or 72 hours after plasmid transfection. The transduction of HSCs was performed by incubating cells with 25%-50% viral supernatant supplemented with 8 μg/mL of polybrene at 37°C overnight. About 72 hours later, HSCs were collected for further experiments.
2.4 ∣. Immunofluorescence (IF) and image quantification
HSCs or murine tissue sections were fixed with 4% paraformaldehyde followed by permeabilization by 0.2% Triton X-100 and a primary antibody was applied. After incubation with a primary antibody for 2 hours at room temperature or overnight at 4°C, an Alexa Fluor-conjugated second antibody (Thermo Fisher Scientific, Waltham, MA) was used for IF labeling and detection. Cell nuclei were counterstained with DAPI (D1306; Thermo Fisher Scientific). IF signals were captured by an Axio observer (Zeiss, Oberkochen, Germany) with or without ApoTome function.32
To analyze the sorting of TβRII to early endosomes or lysosomes induced by TGFβ1, HSCs expressing TβRII-HA were serum-starved and pretreated with 40 αg/mL of cycloheximide for 1 hour to block the synthesis of proteins including TβRII-HA. TGFβ1 was added in and TGFβ1/receptor binding was permitted by incubating the cells for 30 minutes at 4°C. After washing off free TGFβ1, cells were incubated at 37°C for various times and harvested for double IF for EEA-1/TβRII-HA or LAMP1/TβRII-HA. IF was captured by an Axio observer using a 63x lens with ApoTome function. The rate of TβRII-HA/EEA-1 and TβRII-HA/LAMP1 colocalization was analyzed as we previously described.1
2.5 ∣. Western blot (WB) and coimmunoprecipitation (coIP)
RIPA lysis buffer containing 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS was used for WB, and a lysis buffer containing 1% Nonidet P-40 and 5% glycerol was used for coIP. Both buffers were supplemented with PMSF, NaF, Na3VO4, and protease inhibitors (A32965; Thermo Fisher Scientific). For coIP, 300-500 μg of the total protein, 1-2 μg of a primary antibody, and 30 μL of slurry of protein G Sepharose 4 Fast Flow (GE Healthcare Life Sciences, Pittsburgh, PA) were mixed and rotated at 4°C for overnight. After washing the Sepharose beads for four times, Laemmli sample buffer (Bio-Rad, Hercules, CA) was added into elute the precipitated proteins for WB. WB was performed as previously described1,28,33 and densitometric analysis was performed using the Image J software (NIH).
2.6 ∣. Quantification of plasma membrane TβRII and TβRII internalization by biotinylation-based approaches
To quantitate TβRII at the plasma membrane, cell surface proteins were first labeled with biotin (EZ-Link Sulfo-NHS-Biotin, 21217; Thermo Fisher Scientific) for 30 minutes at 4°C. After free biotin was washed off, HSCs were lysed and the lysates were incubated with streptavidin agarose beads (S1638; Millipore Sigma) so that the biotinylated cell surface proteins were pulled down by the beads.1,28 After the biotinylated proteins were eluted and loaded onto a PAGE gel for electrophoresis, TβRII at the cell surface was detected and quantitated by WB using anti-TβRII.
To analyze TβRII internalization induced by TGFβ1, serum-starved HSCs were first labeled with cleavable biotin, EZ-Link Sulfo-NHS-SS-Biotin (A39258; Thermo Fisher Scientific), and stimulated with TGFβ1.28 HSCs without TGFβ1 stimulation were harvested and the biotinylated TβRII detected from this group represented the level of TβRII at the plasma membrane. For the group with TGFβ1 stimulation, cells were first incubated with TGFβ1 for 10 minute at 37°C to allow the internalization of TGFβ receptors and GSH stripping buffer (50 mM glutathione, 75 mM NaCl, 10 mM EDTA, 1% BSA, 0.075 N NaOH) was added in to cleave the biotin from the proteins at the plasma membrane. Cells were then lysed and streptavidin agarose beads were used to pull down the biotinylated proteins. The biotinylated TβRII detected from this group represented internalized TβRII by TGFβ1. After densitometric analysis, an equation was used for calculation: TβRII internalization rate = internalized TβRII/ plasma membrane TβRII × 100%.
2.7 ∣. Quantification of TβRII degradation induced by TGFβ
Serum-starved HSCs were pretreated with 40 μg/mL of cycloheximide for 1 hour to block protein synthesis. TGFβ1 was added in and TGFβ1/receptor binding was allowed by incubating the cells at 4°C for 30 minutes. After washing off the free TGFβ1, cells were incubated at 37°C for indicated times and harvested for WB to quantitate the protein level of TβRII in HSCs.33 Densitometry was performed by the ImageJ software and TβRII degradation curves were generated by the GraphPad Prism 5 software (GraphPad Software, Inc, La Jolla, CA). To test if Bafilomycin A1 prevented TβRII degradation, Bafilomycin A1 (10 nM) was added into one sample and cells were incubated with Bafilomycin A1 for 6 hours prior to TGFβ1 stimulation.
2.8 ∣. Rab5 activity assay
The activity of Rab5 was assessed by a Rab5 activation assay kit (83701; NewEast Biosciences, King of Prussia, PA), according to the manufacturer’s recommended protocol. In brief, cell lysates containing equal amounts of proteins in equal volumes were incubated with a mouse monoclonal antibody specifically recognizing GTP-bound Rab5, and protein A/G agarose beads were later added into pull down GTP-bound Rab5. After the agarose beads were washed, a sample buffer was added in to elute active Rab5 for WB using a rabbit polyclonal anti-Rab5 antibody.
2.9 ∣. GST fusion proteins
Bacterial expression vectors were transformed into E coli BL21 (DE3) individually. Expression of Rab5a, Diaph1 (1-453), or full-length Diaph1 (Diaph1 FL) was induced by 100 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) and culture of cells at 16°C, while the expression of TβRII was induced by 50 μM IPTG and culture of cells at 15°C. A lysis buffer, containing 25 mM Tris-HCl pH7.6, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1mg/mL lysosome, 1% NP-40, 1% TritonX-100, was then added to lyse the cells. Cells were incubated on ice for 20 minutes and subjected to sonication until the lysates turned clear. After centrifugation at 15 000 rpm for 15 minutes, the supernatants were collected and GST-fused proteins were purified by Glutathione Sepharose 4B beads (17075601; GE Healthcare). Removal of the GST tag from GST-TβRII fusion protein was performed by PreScission Protease (GE27-0843-01; Millipore Sigma) and removal of the GST tag from GST-Rab5a was performed by Thrombin (10602400001; Millipore Sigma).
2.10 ∣. In vitro protein binding assay
To study if Diaph1 bound to Rab5a or TβRII and TβRII bound to Rab5a in vitro, GST pull down assays were performed.1 GST pull down assays were setup by mixing a detagged protein with 2nd protein conjugated to Glutathione Sepharose 4B beads (molar ratio 2:1) in a binding buffer (25 mM Tris-HCl pH7.6, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1% NP-40, 0.1 mM PMSF, protease inhibitors) in a reaction volume of 200 μL. Protein binding was allowed by incubating the mixture at 10°C for 4 hours followed by centrifugation at 500 ×g for 5 minutes to precipitate the Glutathione Sepharose 4B beads. After washing the beads two times with binding buffer, the protein pulled-down was eluted by Laemmli sample buffer and loaded onto an SDS-PAGE gel for WB. Anti-Rab5 (D-11) (46692; Santa Cruz technology) and anti-TβRII (184948; Abcam) were used to detect the pulled-down proteins. Last, the blots were subjected to Ponceau S staining to visualize the GST-fused proteins used for pull down assays.
2.11 ∣. Real-time tracking of tumor/HSC coculture by IncuCyte live-cell analysis
To analyze the role of HSCs in tumor cell proliferation in vitro, we setup HT29/HSC coculture and used the IncuCyte S3 Live-Cell Analysis system to track HT29/HSC coculture. HT29 cells were tagged by emerald by lentiviral transduction. HSCs expressing control or Diaph1 shRNA were first seeded into a 96-plate culture plate (5000 cells/well) and on 2nd day, HT29 expressing emerald was seeded on the top of HSCs (10 000 cells/well) for HT29/HSC coculture. The cells were then put in the IncuCyte S3 Live-Cell Analysis system for real-time analysis of the green fluorescence of HT29 for 72 hours. Data were analyzed by the software provided and the total area of green fluorescent cells per picture was calculated, which represented the density of HT29 cells.
2.12 ∣. In Vitro cell proliferation assay
Conditioned media (CMs) collected from HSCs were used as stimulants for HT29 cell proliferation in an in vitro assay. To collect CMs, HSCs were first transduced with lentiviruses encoding NT shRNA or Diaph1 shRNA, or retroviruses encoding LacZ or Rab5S34N, 48 hours later their culture media were replaced by serum-free DMEM medium. After an additional 48 hours, the culture media were collected and filtered through a 0.45 μm syringe filter so that the cells and cell debris were efficiently removed. Filtered CMs were then aliquoted and stored at −80°C until use. To assess the role of each CM in promoting HT29 cell proliferation, 5000 HT29 cells, suspended in 100 μL of each CM, were seeded into a well of 96-well plate. Quantification of HT29 cells was performed at 0, 24, 48, and 72 hours with a CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay kit (Promega, Madison, WI), according to the manufacturer’s recommended protocol.
2.13 ∣. Subcutaneous coinjection of HSC/HT29 into mice
Animal studies were approved by the Institutional Animal Care and Use Committee of the University of Minnesota. To evaluate the effect of HSCs on HT29 tumor growth in vivo, 0.5 × 106 HT29 cells mixed with HSCs (0.5 × 106) were injected into 8-week-old male nude mice (553; Charles River, Wilmington, MA) subcutaneously. Tumor sizes were measured with a caliper at different days and mice were killed at Day 14. Tumor nodules were calculated by an equation: tumor volume = (width)2 × length/2. The GraphPad Prism 5 software (GraphPad Software, Inc, La Jolla, CA) was used to generate tumor growth curves and perform statistical analyses.28,32
2.14 ∣. Statistical analysis
Data were expressed as mean ± SD and subjected to statistical analysis using the two-tailed Student’s t test or ANOVA (GraphPad Software, Inc, La Jolla, CA). P < .05 was considered statistically different.
3 ∣. RESULTS
3.1 ∣. Inactivation of Diaph1 inhibits TGFβ1 signaling of HSCs and induces TβRII accumulation at the plasma membrane
To investigate if Diaph1 regulates TGFβ signaling of HSCs, we first collected control and Diaph1 knockdown HSCs for Western blot (WB) to quantitate SMAD3 phosphorylation (p-SMAD3) induced by TGFβ1. Two lentiviruses each encoding a distinct Diaph1 shRNA were used to transduce HSCs to knockdown Diaph1. Lentiviruses encoding non-targeting shRNA (NT shRNA) were used as controls. In control HSCs, TGFβ1 stimulation led to a time-dependent increase of p-SMAD3 and this effect of TGFβ1 was inhibited by the knockdown of Diaph1 (P < .05, Figure 1A). Consistently, SMIFH2 (25 μM), a small molecule targeting the FH2 domain of Diaph1, inhibited p-SMAD3 induced by TGFβ1 (P < .05, Figure 1B). These data suggested that the inactivation of Diaph1 affected the TGFβ signaling at the upstream of the TGFβ signaling, possibly at the TGFβ receptor level.
FIGURE 1.
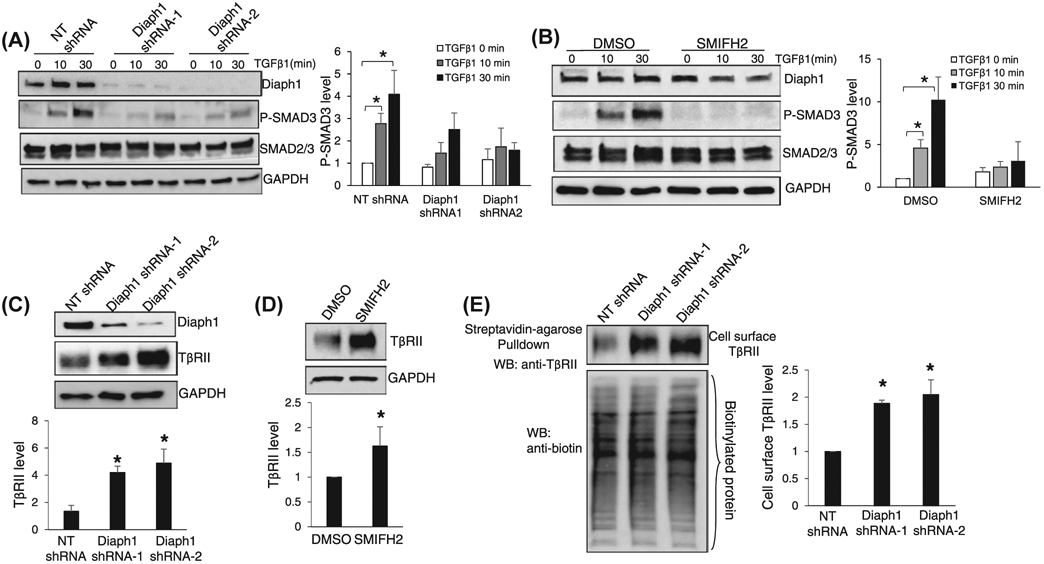
Inactivation of Diaph1 leads to TβRII accumulation at the plasma membrane and inhibits SMAD3 phosphorylation induced by TGFβ1. A and B, HSCs expressing NT shRNA (control) or Diaph1 shRNA, HSCs preincubated with DMSO (control) or SMIFH2 (25 μM) were serum-starved and stimulated with TGFβ1 (5 ng/mL) for indicated times. Cells were collected for WB for p-SMAD3. Knockdown of Diaph1 or SMIFH2 suppressed the phosphorylation of SMAD3 induced by TGFβ1. *P < .05 by ANOVA. n = 4 repeats. C and D, HSCs expressing NT shRNA or Diaph1 shRNA, HSCs preincubated DMSO or SMIFH2 were subjected to WB for TβRII. Knockdown of Diaph1 or SMIFH2 increased the protein level of TβRII of HSCs. *P < .05 by ANOVA for C. and t test for D, n = 3 repeats. E, TβRII at the plasma membrane was quantitated by biotinylation of cell surface proteins followed by streptavidin-agarose pull down and WB for TβRII. Knockdown of Diaph1 significantly increased the cell surface TβRII of HSCs. *P < .05 by ANOVA, n = 3 repeats
Because TβRII is a better model than TβRI for studying receptor endocytosis and turnover,34 we focused on the role of Diaph1 for intracellular trafficking of TβRII in this study. WB revealed that knockdown of Diaph1 or incubation with SMIFH2 increased the protein level of TβRII of HSCs (P < .05, Figure 1C,D). Biotinylation of cell surface proteins followed by streptavidin agarose pull down assay demonstrated that the protein level of TβRII at the plasma membrane of HSCs was higher in Diaph1 knockdown cells than in control HSCs (P < .05, Figure 1E). Because TβRII undergoes endocytosis constitutively in the presence or absence of TGFβ1 stimulation,35 these data suggested that the inactivation of Diaph1 may block the internalization of TβRII so that TβRII was accumulated at the plasma membrane and the TGFβ1 signaling was inhibited. This hypothesis was tested by the experiments below.
3.2 ∣. Inactivation of Diaph1 blocks the endocytosis of TβRII induced by TGFβ1
We first performed immunofluorescence (IF) to test if Diaph1 influenced the sorting of TβRII into early endosomes under TGFβ1 stimulation. Because commercial anti-TβRII antibodies were not reliable for IF, HSCs transduced with retroviruses encoding TβRII-HA were used and TβRII-HA fusion protein was detected by IF using anti-HA.1,28,33 Cycloheximide (40 μg/mL) was added in cell culture to block the translation of TβRII during TGFβ1 stimulation and HSCs were collected for double IF for TβRII-HA and early endosome antigen 1 (EEA-1),1 marker of early endosomes. IF data revealed that the stimulation of HSCs with TGFβ1 for 10 minutes indeed led to the increase of colocalization of TβRII-HA and EEA-1 in HSCs and this effect of TGFβ1 was reduced by Diaph1 shRNA or SMIFH2 (P < .05, Figure 2A,B). As quantitated by biotinylation-based assay, 72% ± 11% of cell surface TβRII of control HSCs was internalization whereas only 25% ± 4% of that of Diaph1 knockdown HSCs was internalized in response to TGFβ1 stimulation (P < .05, Figure 2C). Thus, targeting Diaph1 of HSCs blocked internalization and endosomal sorting of TβRII induced by TGFβ1. To test the role of microtubule for TGFβ1 signaling and endocytosis of TβRII, we added nocodazole so cells were incubated with nocodazole for 1 hour prior to TGFβ1 stimulation. Nocodazole at 200 nM or 500 nM significantly reduced the phosphorylation of SMAD3 induced by TGFβ1 (P < .05, Supplementary Figure 1A). Consistently, it blocked TGFβ1-induced endocytosis of TβRII-HA (P < .05, Supplementary Figure 1B). The phenotypes induced by nocodazole incubation recapitulated those induced Diaph1 inactivation, indicating that Diaph1 may promoted TβRII endocytosis and TGFβ1 signaling through microtubules.
FIGURE 2.

Inactivation of Diaph1 inhibits the endocytosis of TβRII induced by TGFβ1. A and B, HSCs expressing TβRII-HA by retroviral transduction were transduced with NT shRNA or Diaph1 shRNA lentiviruses (A), or incubated with SMIFH2 (B). Cells serum-starved and pretreated with cycloheximide (40 μg/mL) were stimulated with TGFβ1 for 10 minutes and collected for double IF for HA (green) and EEA-1(red). TGFβ1 increased the colocalization of these two proteins (yellow) and this effect of TGFβ1 was reduced by Diaph1 shRNA or SMIFH2. Quantitative data were shown on the bottom. *P < .05 by ANOVA, n = 12 cells per group in A and n = 10 cells per group in B. Bar, 20 μm. C, HSCs were subjected to biotinylation using cleavable biotin followed by stimulation with TGFβ1 for 10 minutes. Cells were lysed for streptavidin-agarose pull down and WB to quantitate internalization of TβRII mediated by TGFβ1. Diaph1 shRNA reduced the rate of TβRII internalization induced by TGFβ1. *P < .05 by t test, n = 3 repeats
3.3 ∣. Inactivation of Diaph1 blocks lysosomal degradation of TβRII mediated by TGFβ1
In response to TGFβ1 stimulation, TβRII was internalized and a fraction of it was sorted from early endosomes to lysosomes for degradation.35 Therefore, TGFβ1 stimulation led to a time-dependent downregulation of TβRII.1,28,33 So we next tested the role of Diaph1 in lysosomal degradation of TβRII induced by TGFβ1. HSCs incubated with cycloheximide and TGFβ1 for 30 or 60 minutes were collected for double IF for TβRII-HA and lysosomal-associated membrane protein 1 (LAMP1),1 marker of late endosome/lysosomes. As shown by double IF, incubation of HSCs with TGFβ1 for 30 or 60 minutes indeed led to an increase of the colocalization of TβRII-HA and LAMP1 in control cells and this effect of TGFβ1 was inhibited by Diaph1 shRNA or SMIFH2 (P < .05, Figure 3A,C). Additionally, HSCs incubated with TGFβ1 for different times were collected for WB, which revealed that TβRII degraded much faster in control HSCs than in Diaph1 knockdown or SMIFH2-incubated HSCs (P < .05, Figure 3B,D). The half-life of TβRII was 48-51 minutes for control cells, 102.9 minutes for Diaph1 knockdown HSCs, and 102.6 minutes for SMIFH2-incubated cells, respectively (Figure 3B,D). Consistently, preincubation of HSCs with Bafilomycin A1, an inhibitor of lysosomes, effectively prevented the downregulation of TβRII induced by TGFβ1 (P < .05, Supplementary Figure 2). Together, these data supported that Diaph1 was required for lysosomal sorting and degradation of TβRII induced by TGFβ1.
FIGURE 3.

Inactivation of Diaph1 inhibits the lysosomal degradation of TβRII induced by TGFβ1. A, HSCs expressing TβRII-HA were transduced by NT shRNA or Diaph1 shRNA lentiviruses. Cells pretreated with cycloheximide were stimulated with TGFβ1 for indicated times and collected for double IF for HA (green) and LAMP1 (red). TGFβ1 increased the colocalization of these two proteins (yellow) and this effect of TGFβ1 was reduced by Diaph1 shRNA. *P < .05 by ANOVA, n = 10 cells each group. Bar, 20 μm. B, HSCs treated as described in A were collected for WB for TβRII with quantitative data shown on the bottom. Diaph1 shRNA inhibited the degradation of TβRII induced by TGFβ1. *P < .05 by ANOVA, n = 3 repeats. C, HSCs expressing TβRII-HA were incubated with SMIFH2. Cells preincubated with cycloheximide were stimulated with TGFβ1 and collected for double IF. TGFβ1 increased the colocalization of these two proteins (yellow) and this effect of TGFβ1 was reduced by SMIFH2. *P < .05 by ANOVA, n = 10 cells each group. Bar, 20 μm. D, HSCs treated as described in C were collected for WB for TβRII. SMIFH2 inhibited the degradation of TβRII induced by TGFβ1. *P < .05 by ANOVA, n = 3 repeats
3.4 ∣. Targeting Diaph1 suppresses the myofibroblastic activation of HSCs induced by TGFβ1
To test the role of Diaph1 for the activation of HSCs into myofibroblasts, control and Diaph1 knockdown HSCs stimulated with TGFβ1 for 24 hours were collected for WB and IF. TGFβ1 indeed stimulated control HSCs to express α-smooth muscle actin (α-SMA) and fibronectin, markers of activated-HSC/myofibroblasts, and this effect of TGFβ1 was inhibited by Diaph1 knockdown (P < .05, Figure 4A). α-SMA IF also showed that the knockdown of Diaph1 by two different shRNAs consistently reduced the percentage of activated-HSC/myofibroblasts in response to TGFβ1 stimulation (P < .05, Figure 4B). Consistently, SMIFH2 suppressed the activation of HSCs into myofibroblasts as determined by both WB and IF (P < .05, Figure 4C,D). Thus, Diaph1 is required for TGFβ1-stimulated myofibroblastic activation of HSCs.
FIGURE 4.

Inactivation of Diaph1 suppresses TGFβ-mediated activation of HSCs into myofibroblasts. A, HSCs transduced with lentiviruses encoding NT shRNA (control) or Diaph1 shRNA were serum-starved and stimulated with TGFβ1 for 24 hours. Cell lysates were subjected to WB for α-SMA and fibronectin. Diaph1 shRNA suppressed TGFβ1-mediated HSC activation. Densitometry data were shown on the bottom. *P < .05 by ANOVA, n = 3 repeats. B, HSCs treated as described in A were subjected to IF for α-SMA (red). Diaph1 knockdown suppressed the formation of stress fibers in HSCs induced by TGFβ1. Bar, 50 μm. *P < .05 by ANOVA, n = 6 randomly picked microscopic fields, each containing 100-200 cells. C, DMSO (control) and SMIFH2-incubated HSCs were stimulated with TGFβ1 for 24 hours. WB revealed that TGFβ1-mediated HSC activation was inhibited by SMIFH2. Densitometry data were shown on the right. *P < .05 by ANOVA. n = 3 repeats. D, TGFβ1 induced the formation of α-SMA-positive stress fibers and SMIFH2 suppressed this effect of TGFβ1 on HSCs. Bar, 50 μm. *P < .05 by ANOVA, n = 6 randomly picked microscopic fields, each containing 100-200 cells
3.5 ∣. Diaph1 binds to Rab5 and promotes its activity
To understand how Diaph1 promoted the internalization and trafficking of TβRII, we focused on Rab5 small GTPase, a key regulator for the formation, movement, tethering, and fusion of endocytic vesicles.36-40 Because the effect of Rab5 on membrane fusion is mediated by EEA-1,41 we performed IF for EEA-1 and found that Diaph1 knockdown indeed reduced the EEA-1 IF density of each endosome in HSCs (P < .05, Figure 5A). We next generated three FLAG-tagged Rab5a constructs, FLAG-Rab5awt (wild-type Rab5a), FLAG-Rab5aQ79L (GTPase-deficient mutant, constitutively active), and FLAG-Rab5aS34N (bound to GDP, dominant negative).41 HSCs expressing each Rab5a construct by retroviral transduction were collected for IF for EEA-1, which revealed that the expression of FLAG-Rab5aQ79L led to the expansion of EEA-1-positive endosomes whereas expression of FLAG-Rab5aS34N led to shrinkage of EEA-1-positive endosomes, compared to FLAG-Rab5awt (P < .05, Figure 5B). We next collected HSCs expressing FLAG-Rab5awt for IF for FLAG and found that the knockdown of Dipah1 indeed reduced Rab5a IF density of each endosome (P < .05, Figure 5C). Together, these data suggested that the inactivation of Diaph1 may reduce the activity of Rab5a and this hypothesis was tested by additional experiments below.
FIGURE 5.

Diaph1 interacts with Rab5 and promotes Rab5 activity. A, HSCs expressing NT or Diaph1 shRNA were subjected to IF for EEA-1. Quantitative data are shown on the right. Diaph1 shRNA reduced the EEA-1 IF density of each endosome. *P < .05 by t test, n = 6 cells each group. Bar, 20 μm. B, HSCs expressing FLAG-Rab5awt, FLAG-Rab5S34N (inactive), or FLAG-Rab5aQ79L (constitutively active) by retroviral transduction were subjected to IF for EEA-1. FLAG-Rab5aQ79L increased whereas FLAG-Rab5S34N reduced EEA-1 IF density of each endosome, compared to FLAG-Rab5awt. *P < .05 by t test, n = 6 cells each group. Bar, 20 μm. C, HSCs expressing FLAG-Rab5awt were transduced by NT shRNA or Diaph1 shRNA lentiviruses and cells were subjected to IF for FLAG. Diaph1 shRNA reduced Rab5a IF density of each endosome. *P < .05 by t test, n = 6 cells each group. Bar, 20 μm. D, HSCs expressing NT, Diaph1 shRNA, GFP, or Diaph1-Emerald were collected and subjected to Rab5 activity measurement using a Rab5 activation assay kit. Diaph1 shRNA reduced (upper) whereas overexpression of Diaph1 (lower) increased the level of GTP bound Rab5a. *P < .05 by t test. n = 3. E, Left, HSCs expressing FLAG-Rab5awt were transduced by LacZ or Diaph1-Emerald retroviruses and cells were subjected to IP. Anti-FLAG was used to pull down FLAG-Rab5a and coprecipitated Diaph1-Emerald was detected by WB using anti-GFP antibody. Diaph1 and Rab5a interacted in HSCs. Right, GST-fused Diaph1 proteins were purified from bacteria and used for GST pull down assay. Diaph1-bound Rab5a was quantitated by WB. GST fusion proteins were shown by Ponceau S staining. F, HSCs expressing Diaph1-Emerald were transduced with retroviruses encoding FLAG-Rab5awt or FLAG-Rab5aQ79L and cells were subjected to IF for FLAG (red) and GFP (green). Diaph1-Emerald colocalized with FLAG-Rab5awt and FLAG-Rab5aQ79L (arrows) in HSCs. Bar, 20 μm
Because Rab5 cycles from GDP-bound (inactivate) to GTP-bound form (active), we used a Rab5 activation assay kit to quantitate GTP-bound Rab5 in control HSCs, Diaph1 knockdown HSCs, and HSCs overexpressing Diaph1-Emerald. While the knockdown of Diaph1 significantly reduced the level of GTP-bound Rab5, overexpression of Diaph1-Emerald increased it (P < .05, Figure 5D), confirming that Diaph1 indeed promoted the Rab5 activity of HSCs. We next collected HSCs expressing both FLAG-Rab5awt and Diaph1-Emerald by retroviral transduction for coimmunoprecipitation (coIP) to test if both proteins interacted in HSCs. Anti-FLAG was used to pull down FLAG-tagged Rab5awt and WB for GFP was used to detect precipitated Diaph1-Emerald. The fact that FLAG-Rab5awt and Diaph1-Emerald were coprecipitated by anti-FLAG (Figure 5E, left) suggested that Diaph1 and Rab5a interacted in HSCs. To further test Diaph1/Rab5a binding, we used the Gibson Assembly to create three bacterial expression vectors, pGEX4T1-Diaph FL (full-length Diaph1), pGEX4T1-Diaph (1-453) (a.a. 1-453 of Diaph1), and pGEX4T1-Rab5a. GST-fused proteins were purified from bacteria cultures for GST pull down assays, which revealed that Rab5a bound to both Diaph1 (1-453) and Diaph1 FL in vitro (Figure 5E, right). Double IF confirmed that Diaph1-Emerald colocalized with either FLAG-Rab5awt or FLAG-Rab5aQ79L in HSCs (Figure 5F). Thus, Diaph1 bound to Rab5a directly to promote its activity in HSCs.
3.6 ∣. TβRII interacts with Diaph1 and active Rab5a at endosomes
We next investigated if TβRII interacted with both Diaph1 and Rab5a in HSCs. We first collected HSCs expressing TβRII-HA and Diaph1-Emerald by retroviral transduction for coIP. Anti-HA was used to pull down TβRII-HA and WB was used to detect coprecipitated Diaph1-Emerald, which revealed coprecipitation of Diaph1-Emerald and TβRII-HA by anti-HA pull down (Figure 6A, left). Double IF confirmed that Diaph1-Emerald and TβRII-HA colocalized at the plasma membrane and endosomes of HSCs (arrows, Figure 6A). We next collected HSCs expressing FLAG-Rab5awt and TβRII-HA for coIP and found that FLAG-Rab5awt and TβRII-HA were indeed coprecipitated by anti-HA (Figure 6B, left). To test if the binding of TβRII to Diaph1 or Rab5a was direct or not, we transformed bacteria with pGEX6P1-TβRII, a vector that we previously constructed.1 TβRII was purified from bacteria and GST pull down assays revealed that TβRII indeed bound to Diaph1 (1-453) and Diaph1 FL (Supplementary Figure 3A), and additionally, Rab5a in vitro (Supplementary Figure 3B). Together, these data confirmed that TβRII directly bound to Diaph1 and Rab5a. In addition, we found that the expression of FLAG-Rab5awt led to prominent endocytic vesicles in HSCs where TβRII-HA and FLAG-Rab5awt colocalized to (arrows, Figure 6B), Moreover, expression of FLAG-Rab5aQ79L increased whereas the expression of FLAG-Rab5aS34N decreased TβRab5a colocalization at the endosomes (P < .05, Figure 6B, right). Together, these data supported that TβRII bound to Diaph1 and active Rab5a directly and that endosomal localization of TβRII was promoted by the Diaph1-Rab5a axis.
FIGURE 6.

TβRII interacts with Diaph1 and Rab5a and inactivation of Rab5a suppresses TGFβ1-stimulated HSCs activation. A, Left, HSCs expressing TβRII-HA and Diaph1-Emerald were collected for IP and HSCs expressing TβRII-HA and GFP were used as a control. Anti-HA was used to pull down TβRII-HA and coprecipitated Diaph1-Emerald was detected by WB using anti-Diaph1. TβRII-HA interacted with Diaph1-Emerald in HSCs. Right, double IF revealed that TβRII-HA and Diaph1-Emerald colocalized at the plasma membrane and endocytic vesicles (arrows). Bar, 20 μm. B, Left, HSCs expressing TβRII-HA and FLAG-Rab5awt were collected for IP and HSCs expressing TβRII-HA and LacZ were used as a control. Anti-HA was used to pull down TβRII-HA and coprecipitated FLAG-Rab5awt was detected by WB using anti-FLAG. TβRII-HA interacted with FLAG-Rab5awt in HSCs. Right, HSCs coexpressing TβRII-HA and FLAG-tagged wild-type Rab5a or a mutant were subjected to double IF. FLAG-Rab5aQ79L increased whereas FLAG-Rab5aS34N reduced TβRII-HA/Rab5a colocalization, compared to FLAG-Rab5awt (arrows). *P < .05 by ANOVA, n = 6 cells per group. Bar, 20 μm. C and D, HSCs expressing LacZ (control) or FLAG-Rab5aS34N were stimulated with TGFβ1 for indicated times and collected for WB for p-SMAD3, α-SMA, and CTGF. Expression of Rab5aS34N suppressed TGFβ1-mediated phosphorylation of SMAD3 and HSC activation. *P < .05 by ANOVA, n = 3. E, HSCs expressing NT or Rab5a shRNA were stimulated with TGFβ1 for 24 hours and collected for WB for α-SMA and CTGF. Knockdown of Rab5a inhibited TGFβ1-mediated HSC activation. *P < .05 by ANOVA, n = 3 repeats
3.7 ∣. Inactivation of Rab5a suppressed HSC activation
The Rab5aS34N mutant significantly reduced the endosomal targeting of TβRII (Figure 6B, right), so we tested if it inhibited TGFβ signaling and myofibroblastic activation of HSCs. To this end, HSCs expressing LacZ (control) or FLAG-Rab5aS34N by retroviral transduction were stimulated with TGFβ1 and cells were collected for WB for p-SMAD3 and stellate cell activation markers, α-SMA and CTGF. WB revealed that FLAG-Rab5aS34N indeed reduced the level of p-SMAD3, α-SMA, and CTGF of HSCs induced by TGFβ1 (P < .05, Figure 6C,D). α-SMA IF confirmed that more than 70% of control HSCs were differentiated into myofibroblasts whereas less than 20% of FLAG-Rab5aS34N-expressing HSCs were differentiated by TGFβ1 (P < .05, Supplementary Figure 4A). Consistently, knockdown of Rab5a by two different shRNAs suppressed SMAD3 phosphorylation and HSC activation induced by TGFβ1 (P < .05, Supplementary Figures 4B and 6E). Thus, similar to Diaph1, Rab5a promoted TGFβ1 signaling and myofibroblastic activation of HSCs.
3.8 ∣. Targeting Diaph1 or Rab5a suppresses HSC-derived tumor-promoting factors
Our RNA sequencing data revealed that TGFβ1 stimulation of HSCs increased the transcripts of genes encoding tumor-promoting factors, such as tenascin C, IGF1, CTGF, and PD-L1 (Supplementary Figure 5A) (GSE127964). To analyze if TGFβ1 promoted these HSC-derived paracrine factors through Diaph1 or Rab5a, we collected control HSCs, Diaph1 knockdown, and Rab5a knockdown HSCs for WB. In control HSCs, TGFβ1 stimulation indeed increased the protein level of tenascin C, IGF1, CTGF, and PD-L1 and this effect of TGFβ1 was inhibited by the knockdown of Diaph1 (P < .05, Figure 7A) or knockdown of Rab5a (P < .05, Supplementary Figures 5B and 6E). Thus, Diaph1 and Rab5a represented targets for inhibiting HSC activation and paracrine tumor-promoting effect of HSCs. This hypothesis was further tested by in vitro and animal studies below.
FIGURE 7.

Knockdown of Diaph1 or Rab5a suppresses tumor-promotion effect of HSCs in vitro and in mice. A, HSCs expressing NT shRNA or Diaph1 shRNA were stimulated with TGFβ1 and collected for WB. TGFβ1 stimulated HSC to produce tenascin C, IGF1, PD-L1, and CTGF and this effect of TGFβ1 was abolished by Diaph1 shRNA. *P < .05 by ANOVA, n = 3 repeats. B, HT29 human colorectal cancer cells tagged by emerald were added to control or Diaph1 knockdown HSCs for HT29/HSC coculture. Real-time IncuCyte Live-Cell Analysis showed that the knockdown of Diaph1 reduced the effect of HSCs on promoting HT29 proliferation in vitro. *P < .05 by ANOVA, n = 8. C and D, HT29 cells (0.5 × 106) were mixed with different HSCs (0.5 × 106) in vitro and they were coinjected into nude mice by subcutaneous injection. Tumor sizes were measured by a caliber at indicated times and mice were killed on Day 14 after coinjection. Diaph1 knockdown HSCs and Rab5a knockdown HSCs were less effective than control HSCs at promoting HT29 growth in mice. *P < .05 by ANOVA, n = 8, 10 tumors
3.9 ∣. Targeting Diaph1 or Rab5a of HSCs limits tumor growth in vitro and in a HSC/tumor coimplantation mouse model
The liver is a common site for colorectal cancer to metastasize, so we selected HT29 human colorectal cancer cells to study how Diaph1-Rab5a axis of HSCs influenced HT29 proliferation. We first tagged HT29 cells with green fluorescence by transducing the cells with lentiviruses encoding emerald. HT29 cells were then added into a 96-well-plate coated with either control or Diaph1 knockdown HSCs previously for HT29/HSC coculture. Using real-time tracking the green fluorescence of HT29 by IncuCyte S3 Live-Cell Analysis, we found that coculture of HT29 with control HSCs promoted HT29 proliferation compared to coculture with Diaph1 knockdown HSCs (P < .05, Figure 7B). To test if Diaph1 or Rab5a promoted HT29 proliferation through the release of soluble factors, conditioned media (CMs) were collected from control HSCs, Diaph1 knockdown HSCs, HSCs expressing LacZ, or HSCs expressing Rab5aS34N, and they were used as stimulants for HT29 proliferation in a MTS-based cell proliferation assay.1 As expected, CMs of control HSCs (HSCs expressing NT shRNA or LacZ) promoted HT29 proliferation compared to the basal medium (Supplementary Figure 6A,B). Importantly, this effect of CM on HT29 proliferation was significantly reduced by the knockdown of Diaph1 or expression of the Rab5aS34N mutant in HSCs (P < .05, Supplementary Figure 6A,B). Thus, targeting Diaph1 or Rab5a of HSCs inhibited the tumor-promoting effect of HSCs in vitro.
We next performed HSC/HT29 coinjection28,32 to test the role of Diaph1 or Rab5a of HSCs for tumor growth in vivo. About 0.5 × 106 HT29 cells were mixed with different HSCs (0.5 × 106 cells) in vitro and they were coinjected into nude mice subcutaneously. HT29 cells were allowed to grow in mice for 14 days and tumor sizes were measured by a caliber at different days. Consistent with the in vitro experiments, Diaph1 knockdown or Rab5a knockdown HSCs were less effective at promoting HT29 growth in mice, compared to control HSCs (P < .05, Figure 7C,D). WB and IF showed that myofibroblastic densities in tumors arising from HT29/HSC-Diaph1s0hRNA coinjections or HT29/HSC-Rab5ashRNA coinjections were significantly reduced compared to the tumors arising from control coinjections (P < .05, Figure 8A,B, Supplementary Figure 6C). Consistently, the protein levels of CTGF, PD-L1, IGF-1, and tenascin C were also reduced in the tumors arising from HT29/HSC-Diaph1shRNA coinjections or HT29/HSC-Rab5ashRNA coinjections (P < .05, Figure 8A,B, Supplementary Figure 6C). Although PD-L1 was not important for this study as athymic mice were used for tumor implantation, it was clinically relevant to cancer treatment. Thus, targeting Diaph1 or Rab5a of HSCs suppressed HSC-derived tumor-promoting factors and limited tumor growth in mice.
FIGURE 8.

Targeting Diaph1 or Rab5a suppresses myofibroblastic activation of HSCs and HSC-derived tumor-promoting factors in vivo. A,B, Tumor nodules, as described in Figure 7C, were subjected to WB. In the tumors arising from HT29/HSC-Diaph1shRNA or HT29/HSC-Rab5ashRNA coinjections, the protein levels of α-SMA, CTGF, PD-L1, IGF-1, and tenascin C were significantly reduced, compared to those in the tumors arising from control coinjections. *P < .05 by ANOVA, n = 5, 6. IF for α-SMA and tenascin C were also shown in A, lower panels. *P < .05 by t test, n = 4. Bar, 50 μm. C, Schematic presentation of the finding of this study that the Diaph1-Rab5a axis promotes the internalization of TGFβ receptors and myofibroblastic activation of HSCs
4 ∣. DISCUSSION
We used Diaph1 shRNA to knockdown Diaph1 and small molecule SMIFH2 to inhibit the activity of Diaph1 in this study to investigate if Diaph1 regulated the endocytosis of TβRII and TGFβ signaling of HSCs. Our data demonstrated that Diaph1 was indeed required for the TGFβ-stimulated internalization of TβRII and intracellular trafficking of TβRII in HSCs (Figure 8C). Mechanistically, the N-terminal portion of Diaph1 bound to both TβRII and Rab5a directly and promoted the activity of Rab5a, a key regulator for the formation, movement, tethering, and fusion of endocytic vesicles. The knockdown of Diaph1 reduced Rab5a activity, Rab5a and EEA-1 IF density of endosomes, and endosomal sorting of TβRII of HSCs, thereby blocking TGFβ1-mediated internalization and intracellular trafficking of TβRII. Functionally, the inactivation of Diaph1 or Rab5a suppressed HSC activation and HSC-derived tumor-promoting factors, and reduced the tumor-promoting effect of HSCs in vitro and in a HSC/tumor coimplantation mouse model. Diaph1 and Rab5a thus represent targets for suppressing HSC activation and the hepatic tumor microenvironment.
We showed that Diaph1 promoted the internalization and intracellular trafficking of TβRII by activating Rab5a but it is unclear if endocytic pit formation and endocytosis of TβRII were dependent upon Diaph1-mediated actin bundling or not. This study also showed that small molecule SMIFH2, targeting the actin polymerization activity of Diaph1, suppressed the intracellular trafficking of TβRII and SMAD3 phosphorylation induced by TGFβ1. SMIFH2 was not an inhibitor specific for Diaph1 and may have broader roles than inhibiting the actin polymerization activity of formins. Additionally, clathrin-mediated endocytosis (receptor-mediated endocytosis) was known to be the least reliant on local actin polymerization.42 So we tested the role of microtubule for endocytosis of TβRII and TGFβ signaling using a microtubule poison nocodazole. We demonstrated that the incubation of HSCs with nocodazole prior to TGFβ1 stimulation indeed inhibited TβRII internalization and TGFβ signaling (Supplementary Figure 1A,B), indicating that Diaph1 may promote Rab5a activation and TβRII internalization through mechanisms dependent upon microtubules.
Myofibroblastic activation of HSCs is controlled by a series of TGFβ signaling events and the downstream biological responses, such as the formation of α-SMA-positive stress fibers to define the phenotypes of activated-HSC/myofibroblasts. Because the development of α-SMA positive stress fibers is known to be regulated by RhoA43,44 and Diaph1 is an effector of RhoA, it is possible that RhoA-Diaph1 signaling axis may contribute to HSC activation through modulating the formation of SMA-positive-stress fibers. Moreover, the movement of endocytic vesicles within a cell occurs along microtubules18 and Diaph1 is known to stabilize microtubules so as to target β1-integrin and membrane type1-matrix metalloproteinase (MT1-MMP) to the plasma membrane.15,45 Thus, in addition to promoting the endocytosis of TβRII at the upstream of the TGFβ signaling, Diaph1 may also participate in and regulate multiple steps of the TGFβ signaling cascade for HSC activation.
Diaph1 has been characterized as an oncogenic protein in cancer cells. The gene expression level of Diaph1 was elevated in multiple cancers, including cancer of prostate, breast, lung and lymphocyte,45 and it correlated with the clinical stage of laryngeal squamous cell carcinoma,16 metastasis of colorectal cancer,15 and poor overall survival of patients with high grade (class G3) head and neck cancer.16 Diaph1 inhibited the apoptosis of cancer cells16 and promoted cancer cell adhesion, invasion, and metastasis in vitro.15,45 Additionally, the knockdown of Diaph1 in HCT116 colorectal cancer cells reduced lung metastasis in a metastasis mouse model.15 Here we demonstrated that targeting Diaph1 of HSCs suppressed TGFβ-stimulated HSC activation, the tumor-promoting microenvironment, and tumor growth in mice. Together, our findings and others highlight Diaph1 as a therapeutic target for both cancer cells and the tumor microenvironment and that the development of inhibitors specific for Diaph1 may be promising to target cancer progression and metastasis.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank the support from the Hormel Institute and Hormel Foundation.
Funding information
NIH grant R01 CA160069 to N. Kang.
Abbreviations:
- α-SMA
alpha-smooth muscle actin
- CTGF
connective tissue growth factor
- DAPI
4′,6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- ECM
extracellular matrix
- EEA-1
early endosomal antigen-1
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GST
glutathione S-transferase
- HA
hemagglutinin
- H & E
hematoxylin and Eosin staining
- HSC
hepatic stellate cell
- IF
immunofluorescence
- IGF-1
insulin growth factor-1
- IP
immunoprecipitation
- LAMP1
lysosome-associated membrane glycoprotein 1
- min
minute
- NT shRNA
non-targeting short hairpin RNA
- P-SMAD
phosphorylated SMAD
- PD-L1
programmed death-ligand 1
- SMAD
mothers against decapentaplegic homolog
- TβRI
transforming growth factor-beta receptor I
- TGFβ
transforming growth factor-beta
- TβRII
transforming growth factor-beta receptor II
- WB
western blot analysis
Footnotes
COMPETING INTERESTS
All authors have declared that no conflict of interest exists.
DATA AND MATERIALS AVAILABILITY
Gene Expression Omnibus GSE127964.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Liu C, Billadeau DD, Abdelhakim H, et al. IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. J Clin Invest. 2013;123:1138–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang N, Gores GJ, Shah VH. Hepatic stellate cells: partners in crime for liver metastases? Hepatology. 2011;54:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang N, Shah VH, Urrutia R. Membrane-to-nucleus signals and epigenetic mechanisms for myofibroblastic activation and desmoplastic stroma: potential therapeutic targets for liver metastasis? Mol Cancer Res. 2015;13:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 5.Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. J Cell Biochem. 2007;102:593–608. [DOI] [PubMed] [Google Scholar]
- 6.Narumiya S, Ishizaki T, Watanabe N. Rho effectors and reorganization of actin cytoskeleton. FEBS Lett. 1997;410:68–72. [DOI] [PubMed] [Google Scholar]
- 7.Li F, Higgs HN. The mouse formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003;13:1335–1340. [DOI] [PubMed] [Google Scholar]
- 8.Zuidscherwoude M, Green HLH, Thomas SG. Formin proteins in megakaryocytes and platelets: regulation of actin and microtubule dynamics. Platelets. 2019;30:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copeland JW, Copeland SJ, Treisman R. Homo-oligomerization is essential for F-actin assembly by the formin family FH2 domain. J Biol Chem. 2004;279:50250–50256. [DOI] [PubMed] [Google Scholar]
- 10.Kozlov MM, Bershadsky AD. Processive capping by formin suggests a force-driven mechanism of actin polymerization. J Cell Biol. 2004;167:1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paul AS, Pollard TD. The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr Biol. 2008;18:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallar BJ, Stropich BN, Schoenherr JA, Holman HA, Kitchen SM, Alberts AS. The basic region of the diaphanous-autoregulatory domain (DAD) is required for autoregulatory interactions with the diaphanous-related formin inhibitory domain. J Biol Chem. 2006;281:4300–4307. [DOI] [PubMed] [Google Scholar]
- 13.Lynch ED, Lee MK, Morrow JE, Welcsh PL, Leon PE, King MC. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science. 1997;278:1315–1318. [PubMed] [Google Scholar]
- 14.Stritt S, Nurden P, Turro E et al. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood. 2016;127:2903–2914. [DOI] [PubMed] [Google Scholar]
- 15.Lin YN, Bhuwania R, Gromova K et al. Drosophila homologue of Diaphanous 1 (DIAPH1) controls the metastatic potential of colon cancer cells by regulating microtubule-dependent adhesion. Oncotarget. 2015;6:18577–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Zhou L, Zhang Y, et al. DIAPH1 Is upregulated and inhibits cell apoptosis through ATR/p53/Caspase-3 signaling pathway in laryngeal squamous cell carcinoma. Dis Markers. 2019;2019:6716472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levayer R, Pelissier-Monier A, Lecuit T. Spatial regulation of Dia and Myosin-II by RhoGEF2 controls initiation of E-cadherin endocytosis during epithelial morphogenesis. Nat Cell Biol. 2011;13:529–540. [DOI] [PubMed] [Google Scholar]
- 18.Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts AS. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol Cell. 2000;5:13–25. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez-Borja M, Janssen L, Verwoerd D, Hordijk P, Neefjes J. RhoB regulates endosome transport by promoting actin assembly on endosomal membranes through Dia1. J Cell Sci. 2005;118:2661–2670. [DOI] [PubMed] [Google Scholar]
- 20.Lewkowicz E, Herit F, Le Clainche C, Bourdoncle P, Perez F, Niedergang F. The microtubule-binding protein CLIP-170 coordinates mDia1 and actin reorganization during CR3-mediated phagocytosis. J Cell Biol. 2008;183:1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brandt DT, Marion S, Griffiths G, Watanabe T, Kaibuchi K, Grosse R. Dia1 and IQGAP1 interact in cell migration and phagocytic cup formation. J Cell Biol. 2007;178:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colucci-Guyon E, Niedergang F, Wallar BJ, Peng J, Alberts AS, Chavrier P. A role for mammalian diaphanous-related formins in complement receptor (CR3)-mediated phagocytosis in macrophages. Curr Biol. 2005;15:2007–2012. [DOI] [PubMed] [Google Scholar]
- 23.Mei Y, Feng G, Rahimi N et al. Loss of mDia1 causes neutropenia via attenuated CD11b endocytosis and increased neutrophil adhesion to the endothelium. Blood Adv. 2017;1:1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayes S, Chawla A, Corvera S. TGF beta receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anders RA, Dore JJ Jr, Arline SL, Garamszegi N, Leof EB. Differential requirement for type I and type II transforming growth factor beta receptor kinase activity in ligand-mediated receptor endocytosis. J Biol Chem. 1998;273:23118–23125. [DOI] [PubMed] [Google Scholar]
- 26.Rizvi SA, Neidt EM, Cui J et al. Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem Biol. 2009;16:1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dou C, Liu Z, Tu K et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology. 2018;154(2209-2221):e2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu K, Li J, Verma VK et al. VASP promotes TGF-β activation of hepatic stellate cells by regulating Rab11 dependent plasma membrane targeting of TGF-β receptors. Hepatology (Baltimore, MD). 2015;61:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decker NK, Abdelmoneim SS, Yaqoob U, et al. Nitric oxide regulates tumor cell cross-talk with stromal cells in the tumor microenvironment of the liver. Am J Pathol. 2008;173:1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang N, Yaqoob U, Geng Z, et al. Focal adhesion assembly in myofibroblasts fosters a microenvironment that promotes tumor growth. Am J Pathol. 2010;177:1888–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Routray C, Liu C, Yaqoob U, et al. Protein kinase G signaling disrupts Rac1-dependent focal adhesion assembly in liver specific pericytes. Am J Physiol Cell Physiol. 2011;301:C66–C74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Tu K, Liu D et al. p300 acetyltransferase is a cytoplasm-to-nucleus shuttle for SMAD2/3 and TAZ nuclear transport in transforming growth factor beta-stimulated hepatic stellate cells. Hepatology. 2019;70:1409–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu C, Li J, Xiang X, et al. PDGF receptor alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and post transcriptional regulation of TGF-beta receptors. Am J Physiol Gastrointest Liver Physiol. 2014;307:G749–G759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koli KM, Arteaga CL. Processing of the transforming growth factor beta type I and II receptors. Biosynthesis and ligand-induced regulation. J Biol Chem. 1997;272:6423–6427. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell H, Choudhury A, Pagano RE, Leof EB. Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell. 2004;15:4166–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bucci C, Parton RG, Mather IH et al. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. [DOI] [PubMed] [Google Scholar]
- 37.Barbieri MA, Roberts RL, Gumusboga A et al. Epidermal growth factor and membrane trafficking. EGF receptor activation of endocytosis requires Rab5a. J Cell Biol. 2000;151:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu G, Zhai P, Liu J, Terzyan S, Li G, Zhang XC. Structural basis of Rab5-Rabaptin5 interaction in endocytosis. Nat Struct Mol Biol. 2004;11:975–983. [DOI] [PubMed] [Google Scholar]
- 39.Fouraux MA, Deneka M, Ivan V et al. Rabip4' is an effector of rab5 and rab4 and regulates transport through early endosomes. Mol Biol Cell. 2004;15:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffenberg S, Liu X, Nikolova L et al. A novel membrane-anchored Rab5 interacting protein required for homotypic endosome fusion. J Biol Chem. 2000;275:24661–24669. [DOI] [PubMed] [Google Scholar]
- 41.Simonsen A, Lippe R, Christoforidis S et al. EEA1 links PI(3) K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. [DOI] [PubMed] [Google Scholar]
- 42.Hinze C, Boucrot E. Local actin polymerization during endocytic carrier formation. Biochem Soc Trans. 2018;46:565–576. [DOI] [PubMed] [Google Scholar]
- 43.Edlund S, Landstrom M, Heldin CH, Aspenstrom P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002;13:902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandbo N, Lau A, Kach J, Ngam C, Yau D, Dulin NO. Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-beta. Am J Physiol Lung Cell Mol Physiol. 2011;301:L656–L666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim D, Jung J, You E, Ko P, Oh S, Rhee S. mDia1 regulates breast cancer invasion by controlling membrane type 1-matrix metalloproteinase localization. Oncotarget. 2016;7:17829–17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.