

Abstract
Combination therapies that target multiple pathways involved in immune rejection of transplants hold promise for patients in need of restorative surgery. Herein, a non-interacting multiphase molecular assembly approach is developed to crystallize tofacitinib, a potent JAK1/3 inhibitor, within a shear-thinning self-assembled fibrillar peptide hydrogel network. The resulting Microcrystalline Tofacitinib Hydrogel (MTH) can be syringe-injected directly to the grafting site during surgery to locally deliver the small molecule. The rate of drug delivered from MTH is largely controlled by the dissolution of the encapsulated microcrystals. A single application of MTH, in combination with systemically delivered CTLA4-Ig, a co-stimulation inhibitor, affords significant graft survival in mice receiving heterotopic heart transplants. Locoregional studies indicate that the local delivery of tofacitinib at the graft site enabled by MTH is required for the observed enhanced graft survival.
Keywords: self-assembly, peptide, hydrogel, transplant, immunotherapy
Graphical Abstract
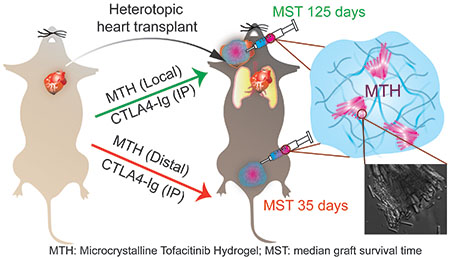
Combination therapies targeting multiple pathways involved in immune rejection of transplants hold promise for restorative surgery patients. We developed Microcrystalline Tofacitinib Hydrogel (MTH), which can be syringe-injected directly to the grafting site to locally deliver tofacitinib, a potent JAK1/3 inhibitor. A single application of MTH, in combination with systemic CTLA4-Ig, a co-stimulation inhibitor, affords significant graft survival in mice receiving heterotopic heart transplants.
1. Introduction
Transplantation is the only life-saving procedure available to patients with end-stage organ disease. In parallel, Vascularized Composite Allotransplantation (VCA) provides a restorative option for patients requiring the transplantation of multiple tissues as a single functional unit like hand, knee or face. 1–3 Successful patient outcome after transplantation correlates with effective life-long application of immunosuppressive drugs. While calcineurin inhibitors such as cyclosporine A combined with antiproliferative agents like mycophenolate mofetil can effectively inhibit allograft rejection, the chronic high doses of calcineurin inhibitors given systemically cause deleterious side effects including hypertension, hyperglycemia, nephrotoxicity, diabetes or even lymphoma. 4, 5 These effects often impose the need to use lower doses of immunosuppressants that do not properly control rejection, resulting in graft loss in the long term (chronic rejection). This post-transplant conundrum warrants the need to develop safer and more effective therapeutic strategies, a result that we posit will require innovative methods of delivering drugs.
Rejection is facilitated by alloreactive T lymphocytes that are activated by a series of tightly regulated processes.6 Donor antigens bound to major histocompatibility complex (MHC) presented from Antigen Presenting Cells (APC) provides the initial stimulus to antigen-specific T cells through T cell receptor (TCR) binding which mediates consequent signaling cascades. However, co-stimulation is required for T cell activation. An important co-stimulatory T cell receptor is CD28, which binds to two partners, CD80 and CD86 expressed on the surface of APCs. Without co-stimulation, TCR-engaged T cells become anergic. Further, at distinct stages of T cell response, signaling through cytokines generated from dendritic cells and macrophages is necessary. For example, it’s been shown that CD4+ T cells can condition dendritic cells to produce IL-12, which in turn, is required for the development of CD8+ T cell effector function and graft rejection.7 Thus, TCR signaling, co-stimulation and cytokine signaling facilitate graft rejection and represent opportunities for therapeutic intervention. CTLA4-Ig is a co-stimulation inhibitor that binds to CD80 and CD86 that showed promise in primate studies8–12 and Belatacept, a second-generation of CTLA4-Ig, is being used in humans. Although Belatacept can prove beneficial, the rate of acute rejection episodes often exceeds those of calcineurin inhibitors.13, 14 Emerging evidence is showing that the effectiveness of co-stimulation inhibitors can be hampered by inflammatory cytokines, some of which can support alloreactive T cell activation in a CD28-independent manner.15–24
We envision that a combination therapy involving both the inhibition of co-stimulation and inflammatory cytokine signaling could prolong graft survival.25 The JAK/STAT pathway is prominent in inflammatory cytokine signaling and the small molecule tofacitinib is a potent inhibitor of Janus kinase 1 and 3, two of the four tyrosine kinases (JAK1,2,3 and TYK2) in the pathway.26 Tofacitinib has shown promising anti-rejection efficacy in non-human primate transplant models and in humans receiving kidney transplantation.27–29 Unpublished data from our lab suggest that a combination therapy of tofacitinib delivered via repeated oral gavages synergizes with CTLA4-Ig to enhance the survival of mice receiving heterotopic heart transplant grafts. Although promising, systemic and prolonged administration of Tofacitinib can be problematic. In kidney transplant clinical trials, although the drug did lead to lower incidence of rejection and better renal function as compared to calcineurin inhibitors, it also caused higher rates of infection and occurrence of post-transplant lymphoproliferative disorders. Retrospective analysis indicated that these deleterious side-effects correlated with high plasma concentrations of drug and ubiquitous biodistribution, most likely a result of frequent systemic dosing to maintain therapeutic concentrations of the short-lived (3 h) drug.14, 30 Thus, we hypothesized that a sustained localized delivery strategy where tofacitinib was released proximal to the transplanted tissue would avoid systemic distribution, and in combination with systemically delivered CTLA4-Ig might be ideal to prolong graft survival.4, 31–34
There has been considerable recent interest in designing local delivery platforms for applications in immunotherapy.35–38 For the local delivery of tofacitinib, we initially targeted a hydrogel delivery system that could be shear-thin injected directly onto tissue during the transplant surgery. Directly encapsulating soluble tofacitinib into such a material would seem the most straight-forward approach. However, tofacitinib is quite hydrophilic with a measured logP ~1.1539 and prone to rapid burst release, especially from materials composed primarily of water. To overcome this problem, we developed a material that contains microcrystalline deposits of drug encapsulated within an injectable hydrogel matrix where the release profile is largely dependent on the rate limiting step of crystal dissolution, Figure 1a. As will be shown, application of this material to transplanted heart in mice in combination with systemically delivered CTLA4-Ig significantly prolonged median graft survival, Figure 1b. Parallel to the development of our material, Langer et al. recently showed that preformed crystals of GW2580 colony stimulating factor 1 receptor inhibitor embedded within an alginate gel can prevent fibrosis of medical implants, further exemplifying the utility of crystals in drug delivery.40 In general, if the gel network minimally influences the drug of interest then the rate of dissolution of crystals within the network will define the release profile of the therapy. In turn, the rate of dissolution depends on crystal morphology, surface energies and defect-related microtopology.41, 42 These features are difficult to engineer a priori but can be reliably reproduced by faithfully recapitulating crystal growth conditions. We developed a unique method of formulation in which small molecule crystallization and peptide assembly are non-interacting processes. Thus, both the initial crystallization of the drug and its subsequent delivery by dissolution is not influenced by its accompanying gel network, whose role is to deliver the microcrystalline cargo to a tissue of interest by simple syringe injection.
Figure 1.
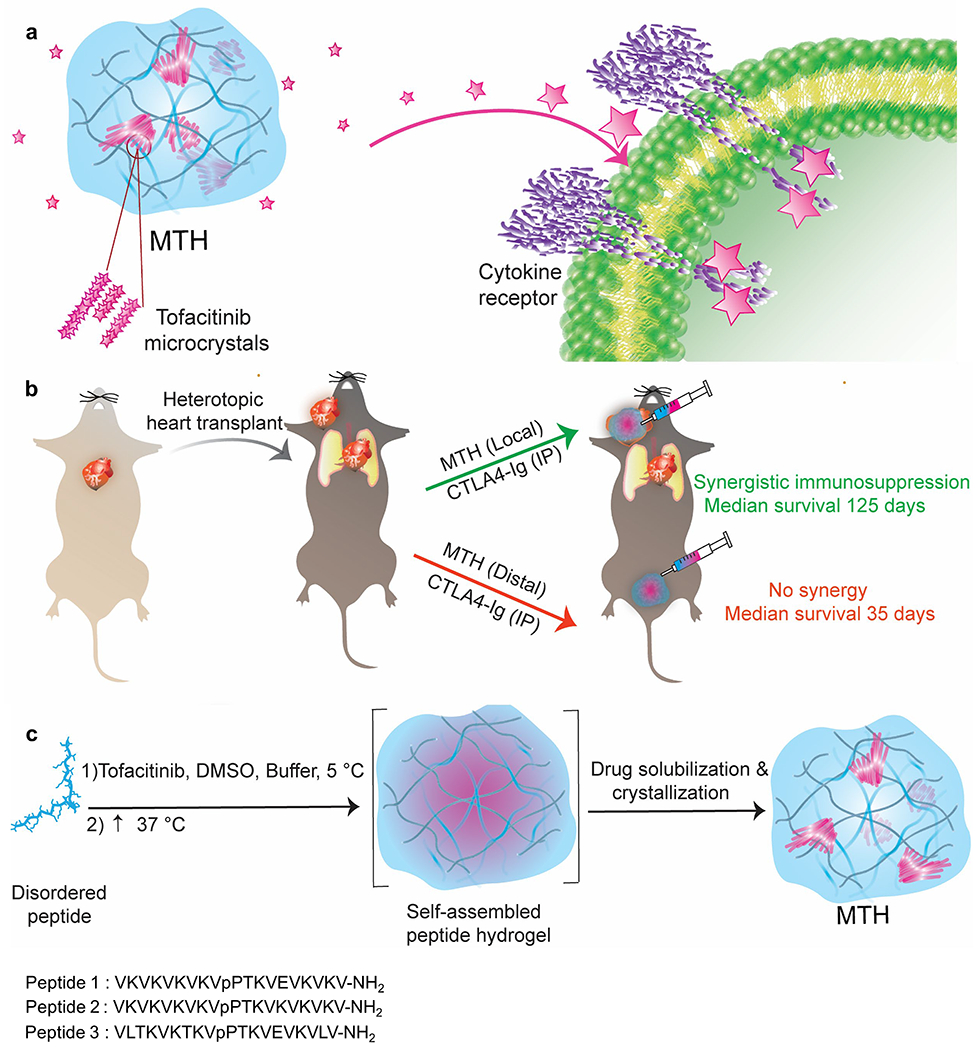
(a) Schematic of MTH comprised of microcrystalline deposits of tofacitinib encapsulated within a fibrillar hydrogel network of self-assembled peptide. Crystal dissolution governs the release of the drug which inhibits JAK1/3 in the JAK/STAT pathway (b) MTH applied locally to a heterotopic heart transplant along with the intraperitoneal (IP) systemic administration of CTLA4-Ig significantly inhibits rejection. Distal application of MTH in the same model has no affect. (c) Temporal molecular events leading to the formations of MTH. Triggered peptide self-assembly occurs concurrently with rapid Tofacitinib (Tofa) aggregation. As the gel’s fibril network nears completion, Tofa aggregates solubilize and the drug subsequently crystallizes. The sequences of the gelating peptides used in this study are shown.
2. Results and Discussion
2.1. Formulation of Microcrystalline Tofacitinib Hydrogel (MTH)
MTH is prepared by taking advantage of multiple non-interacting molecular assembly processes that occur over different time regimes in the same pot. Peptide assembly takes place first leading to fibril formation and gelation, which is concurrent with the rapid aggregation of tofacitinib, which forms amorphous precipitate. Over time, the tofacitinib solubilizes enabling its subsequent crystallization within the hydrogel’s fibril network, Figure 1c. The hydrogel network is formed using a class of previously reported self-assembling peptide designed to undergo triggered assembly in response to environmental cues.43–45 For example, Peptides 1, 2 and 3 can be triggered to assemble by the addition of buffer to increase the solution pH and ionic strength, which decreases intramolecular charge allowing self-association to take place. Increasing the solution temperature also facilitates assembly by driving the hydrophobic effect.46 Ultimately, a physically crosslinked network of fibers is formed constituting the formation of the gel. Within each fiber, peptides assemble into a bilayered cross β-structure, individually adopting a β-hairpin conformation.47 Figure 2a visually shows the time-dependent process of MTH formulation, which is initiated by the addition of a tofacitinib buffered solution containing DMSO to an aqueous solution of unfolded peptide 1 at 5 °C. The sample is then allowed to warm to 37 °C. Although most crystalline deposits are micron in size, some larger clusters can be visually observed. This process can be monitored by following the change in transmittance at 700 nm of the sample as a function of time, Figure 2b. Tofacitinib precipitation is immediate leading to gross light scattering, which resolves over about 25 minutes as the amorphous drug aggregates solubilize. Crystallization then occurs over the next 4-5 h finalizing MTH preparation. Transmittance during the formation of the gel network alone (minus tofacitinib) shows that the gel phase is nearly transparent. Differential Interference Contrast (DIC) microscopy clearly shows the formation of micron-sized bundled crystals of tofacitinib (Figure 2c) whose needle-like morphology is similar to crystals of the drug grown in the absence of hydrogel, Figure S1. The tofacitinib deposits are birefractive under polarized light and generate second harmonics indicating that the chiral molecule has formed non-centrosymmetric crystals, Figures 2d,e. The encapsulation efficiency using this procedure is 100%.
Figure 2.
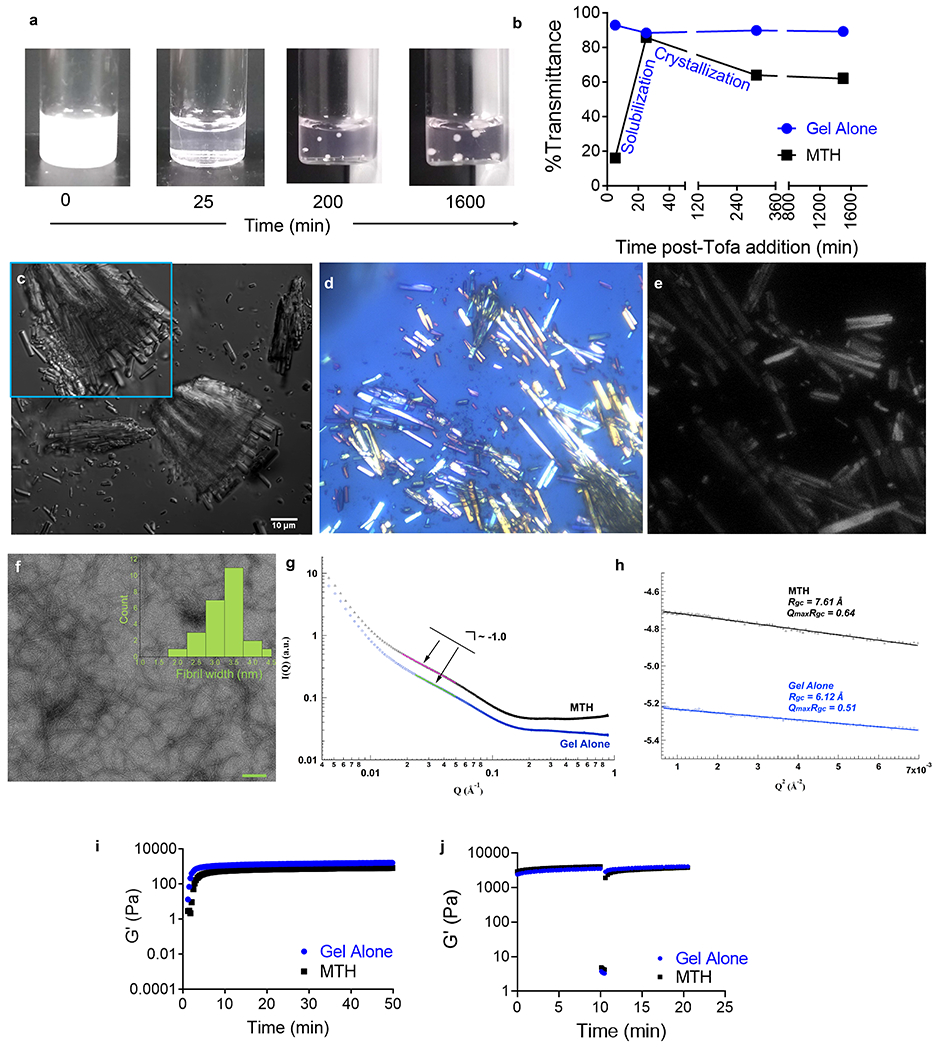
(a) Visual change in phase behavior during the formation of MTH. (b) Transmittance as a function of time monitored during MTH formation compared to peptide 1 gel alone. (c) DIC micrograph of tofacitinib microcrystals within MTH (magnified in inset). Polarized microscopy (d) and second harmonic generation (e) of MTH showing crystalline nature of deposits. (f) TEM of peptide 1 fibrils isolated from MTH, scale bar (100 nm). Inset shows measured fibril widths (n = 25). (g) Power law fit of SAXs data for MTH and gel alone and (h) modified Guinier analysis deriving similar cross-sectional radii of gyration values. (i) Time-sweep oscillatory rheology of MTH comprising 16 mM tofacitinib, 5% (v/v) DMSO and 1% (w/v) peptide 1 compared to peptide 1 gel alone (1% w/v), frequency = 6 rad/s, strain = 0.2%. (j) Time-sweep shear thin-recovery oscillatory rheology of MTH and peptide 1 gel at 24 h. MTH in this Figure comprises 16 mM tofacitinib, 5% (v/v) DMSO and 1% (w/v) peptide 1.
A combination of microscopy, scattering and rheology experiments were conducted to further investigate the time-dependent assembly events underlying the formation of MTH. We were especially interested in whether the presence of tofacitinib influences the formation of peptide fibrils and/or the formation of the fibrillar hydrogel network. Figure 2f shows TEM of peptide fibrils isolated from MTH. Their local morphology, including measured fibril widths (~3-3.5 nm) were similar to those of fibrils formed in the absence of drug (Figure S2) and consistent with the previously determined solid state NMR structure of fibrils formed by another member of this class of self-assembling peptide.47 Figure 2g shows Small Angle X-ray Scattering (SAXS) profiles for both peptide 1 gel alone and MTH containing tofacitinib as well as their corresponding power law fits in the intermediate q region. Both have similar slopes of ~-1 indicating the formation of rod-like objects (fibrils) and a modified Guinier analysis (Figure 2h) provide similar values of cross-sectional radii of gyration (Rgc = 6.1 and 7.6 Å). Close examination of the higher Q region shows a broad peak for both samples at q = 0.282 Å−1 and sharper peaks at 0.564, 0.846, and 1.128 Å−1 in a ratio of 1:2:3:4, indicating a periodic spacing of 2.2 nm which corresponds to the height of a bilayer of β-hairpins that have assembled into an extended cross β-structure that has laminated, Figure S3. SAXS and Wide Angle X-ray Scattering (WAXS) show a peak at 1.334 Å−1 for both samples, corresponding to a d-spacing of 4.7 Å, the distance between the β-strands of the sheet-rich fibrils, Figure S3 and S4. The evolution of the fibril network of MTH was next examined by oscillatory rheology. Figure 2i shows time-sweep data monitoring the formation of MTH over the initial 50 minutes of its formation. Within this time regime, the fibril network of the gel has developed, but tofacitinib has yet to crystallize (Figure 2b). MTH initially forms a moderately stiff gel (~500 Pa) within 10 minutes whose rate of formation and modulus closely resembles peptide 1 gel alone. This indicates that the presence of tofacitinib does not influence the formation of the fibril network defining the gel. Over ~24 h both MTH and the control gel stiffen considerably (~4000 Pa, Figure S5). Figure 2j shows time-sweep shear thin-recovery data for MTH at 24 h. After the initial 10 minutes, a large strain (1000%) is applied for 30 seconds to thin the material, which is subsequently allowed to recover. The data show that MTH demonstrates shear-thin recovery behavior to nearly the same extent as peptide 1 gel alone, indicating that the crystalline tofacitinib minimally influences the recovery properties of MTH. Thus, as will be shown, MTH has rheological properties suitable for syringe delivery. Taken together, the TEM, scattering, rheological data and similar morphologies of crystals grown in the absence or presence of gel indicate that tofacitinib crystallization does not influence peptide self-assembly, the local morphology of the resulting fibrils, nor the evolution of the fibril network and vice versa.
2.2. Ex Vivo Tofacitinib Release From MTH
The rate of tofacitinib release from MTH is designed to be largely governed by the rate of dissolution of the microcrystalline deposits encapsulated within the fibril network. We tested this assertion by studying the influence of drug loading, the composition of the gel-forming peptide, and the formulation process in the context of tofacitinib release. Figure 3a shows that the release profile is profoundly dependent on the final concentration of drug loaded. At concentrations below 3.2 mM, tofacitinib is unable to crystallize, remaining soluble during formulation. As a result, it is burst-released from the gel network within several hours. In contrast, at concentrations > 6.4 mM, crystalline deposits are formed, and the release is slowed in a concentration-dependent manner, consistent with crystal dissolution being rate-determining. The highest loading concentration studied (16 mM) provides a sustained release of its payload over a week. The final volume percent of DMSO used in preparing MTH also influences the release rate, Figure 3b. At values < 10% DMSO, there is little effect of solvent. However, at greater values, drug is released more quickly suggesting that DMSO greatly aids in the solubilization of the microcrystalline deposits hastening its diffusion out of the gel. Rate values derived over the first 4 h show a similar trend, Figure S6. Next, the effect of the composition of the self-assembling peptide was studied. Peptides 1, 2, and 3 differ both in their overall amphiphilicity and charge state at physiological pH, characteristics imbibed to the fibril networks that they form. Peptides 1, 2 and 3 carry formal charges of +7, +9, and +5 per monomer at neutral pH. These electrostatic differences are greatly magnified in the gel state, which is composed of charged fibrils containing millimolar amounts of self-assembled peptide. With respect to amphiphilicity, peptides 1 and 2 are quite similar, but peptide 3, which incorporates two leucine residues at positions 2 and 19 at the expense of lysine differs considerably. Gels formed by peptide 3 carry significantly less charge and are more hydrophobic.
Figure 3.
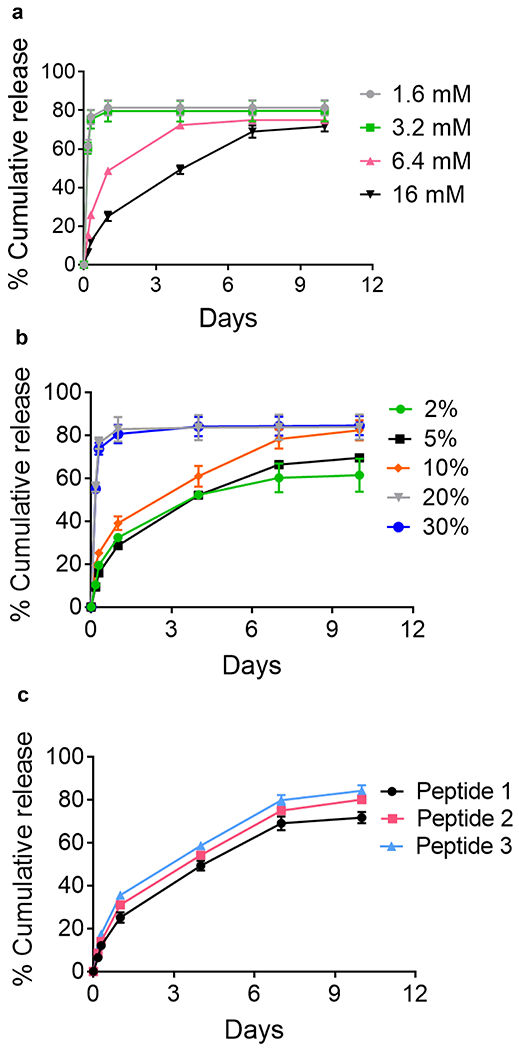
(a) Cumulative release of tofacitinib from 1% (w/v) peptide 1 gel containing 5% (v/v) DMSO as a function of loading concentration. Only at [Tofacitinib]≥ 6.4 mM are microcrystalline deposits formed. (b) Cumulative release of tofacitinib from MTH composed of 16 mM tofacitinib, 1% (w/v) peptide 1, and varying amounts of DMSO. (c) Cumulative release of tofacitinib from gels comprising 16 mM tofacitinib, 5% (v/v) DMSO and 1% (w/v) peptide 1, 2 or 3. Release into an infinite sink (25 mM HEPES, 150 mM NaCl, pH 7.4) at 37 °C was monitored for each experiment. Lines are provided to guide the eye.
Thus, if tofacitinib was interacting with the network to any great extent hydrophobically or electrostatically, either in an attractive or repulsive manner, then a deviation in release profile should be observed amongst the different peptide gels. Figure 3c shows very similar release profiles for the three gels indicating that the drug, in fact, does not interact significantly with the gel network. Finally, we measured drug release as a function of the formulation process. Three distinct MTH gels were prepared where the drug was added before, during and after peptide self-assembly, Figure S7. The release profiles of the resulting materials were nearly identical suggesting that again, peptide self-assembly nor the existence of the resulting fibrillar network influences the process of tofacitinib crystallization to any great extent since the subsequent rate of crystal dissolution is not affected. The similar rates also highlight the flexibility of the formulation process in that tofacitinib crystallization can be induced at any time during the production of MTH. Taken together, these experiments show that tofacitinib release is largely dependent on the rate of crystal dissolution and not the exact nature of the peptide gel nor its formulation history. Although defining the exact mechanism of crystal dissolution is beyond the scope of this paper, it is most likely a combination of surface-kinetic and mass-transport controlled processes as is the case with most small molecule and inorganic crystals.48, 49
Lastly, although the formulation process of MTH is quite flexible, the exact molecular form of tofacitinib is very important. All of the properties of MTH discussed thus far are dependent on using the free base of the drug for material preparation. For example, transmittance spectroscopy and interference contrast microscopy show that the commercially available citrate salt of tofacitinib is unable to form microcrystalline deposits within the gel, Figure S8a,b.TEM shows that the citrate analog results in fibril coarsening and SAXS shows that the gel network structure is altered, providing a power law fit of -1.4, which is more consistent with forming mass fractals than rods, Figure S8c,d. It is likely that the negatively charged citrate counter ions partition to the positively charged fibrils to induce fibril clustering. Finally, because the citrate salt of tofacitinib does not form microcrystals, it is rapidly released from the gel, Figure S8e. Considering that the adaptive immune response peaks for allotransplant rejection at around 3-10 days, a final formulation of MTH comprising 16 mM tofacitinib, 5% (v/v) DMSO and 1% (w/v) peptide 1 was chosen, which can provide delivery of drug over this time regime. Peptide 1, as opposed to peptides 2 or 3, was chosen for further study because it affords the most mechanically rigid hydrogel with encapsulated tofacitinib, Figure S9.
2.3. In vitro immune modulatory properties of MTH
In vitro experiments were first conducted prior to animal studies to ensure that MTH could release biologically active tofacitinib. Non-selective JAK inhibitors are known to affect both the development of monocyte-derived dendritic cells and their activation.50, 51 Figure 4 shows results from an experiment in which the activation of bone marrow-derive dendritic cells (BMDCs) was monitored as a function of material composition. After 5 days of culture, cells are exposed to different amounts of MTH or gel alone for 24 h followed by lipopolysaccharide, a Toll-like receptor 4 ligand and stimulant. BMDC activation was assessed by following CD80, CD86, and MHCII expression. LPS exposure resulted in the upregulation of all the markers, which were markedly countered by the presence of MTH. CD80 and CD86 levels returned to nearly basal levels. MHCII levels were also affected but to a lesser extent, an observation consistent with recent findings showing that soluble tofacitinib only moderately influences MHCII expression (Raimondi, manuscript in preparation). In contrast to MTH, the application of gel alone had little effect on BMDC activation. Thus, MTH is capable of delivering enough tofacitinib at both applied volumes studied here to inhibit the stimulatory capacity of mouse antigen presenting cells.
Figure 4.
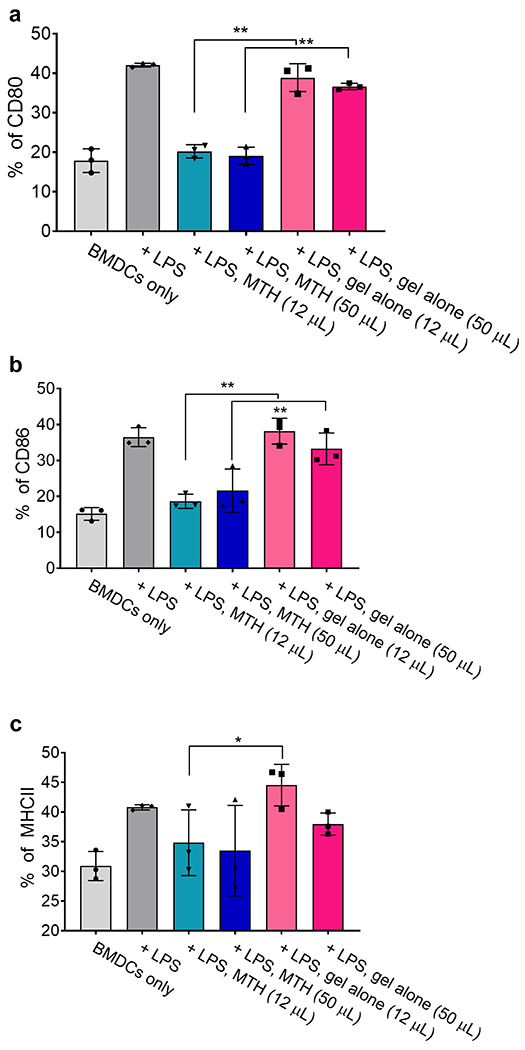
MTH-released tofacitinib inhibits BMDC activation. The indicated volume of MTH or gel alone was added via transwell inserts to the culture plates containing BMDCs followed by LPS. Cell activation was measured via (a) CD80, (b) CD86, and (c) MHCII expression levels.
2.4. In vivo properties of MTH and potential to modulate transplant rejection
We first assessed the bioresorptive capacity of MTH by ultrasound after subcutaneous injection into immune competent C57BL/6J mice. Figures 5a,b show that the implanted material is degraded over time with about 50% of the gel remaining after two weeks. Importantly, this time frame is compatible with the targeted time regime for tofacitinib delivery (~ 10 days), Figure 3a. Again, immune response peaks at around 3-10 days for allotransplant rejection. The data also show that the gel alone (no tofacitinib) degrades in a similar time frame as does MTH in mice receiving an IP regimen of CTLA4-Ig.This indicates that neither tofacitinib nor CTLA4-Ig influence the resorptive processes leading to MTH’s degradation. Immune cell inflation was monitored histologically at the implant site to gain insight into the mechanism of degradation and to establish a baseline for the immune response to MTH in the absence of an allotransplant. Figure 5c shows sections taken through the implanted MTH and surrounding normal tissue. MTH stains light pink in all the images. At day 1 (d1), there is a detectable level of neutrophils that have infiltrated the periphery of the implanted materials of all three groups: MTH, gel alone and MTH + CTLA4-Ig. However, in mice receiving MTH there are fewer neutrophils than the gel alone group and strikingly few neutrophils in the MTH+CTLA4-Ig group. At d10, the striking difference in immune cell infiltrates is lost. Neutrophils can be found nearly throughout all of the implanted materials with macrophages localized mainly at the periphery of each of the material implants. At d30, all the groups appear histologically similar with scattered small clusters of neutrophils accompanying large numbers of macrophages that are found throughout each of the injection sites. Some of the macrophages appear enlarged, presumably from phagocytosing the peptide gel. There were no signs of significant B cell nor T cell infiltration into the material and no signs of toxicity in the surrounding tissue. Taken together, the ultrasound and histology show that MTH is biocompatible with degradation taking about a month by phagocytosis. Locally delivered tofacitinib, alone or in combination with CTLA4-Ig dampens initial neutrophil response, but not macrophage activity.
Figure 5.
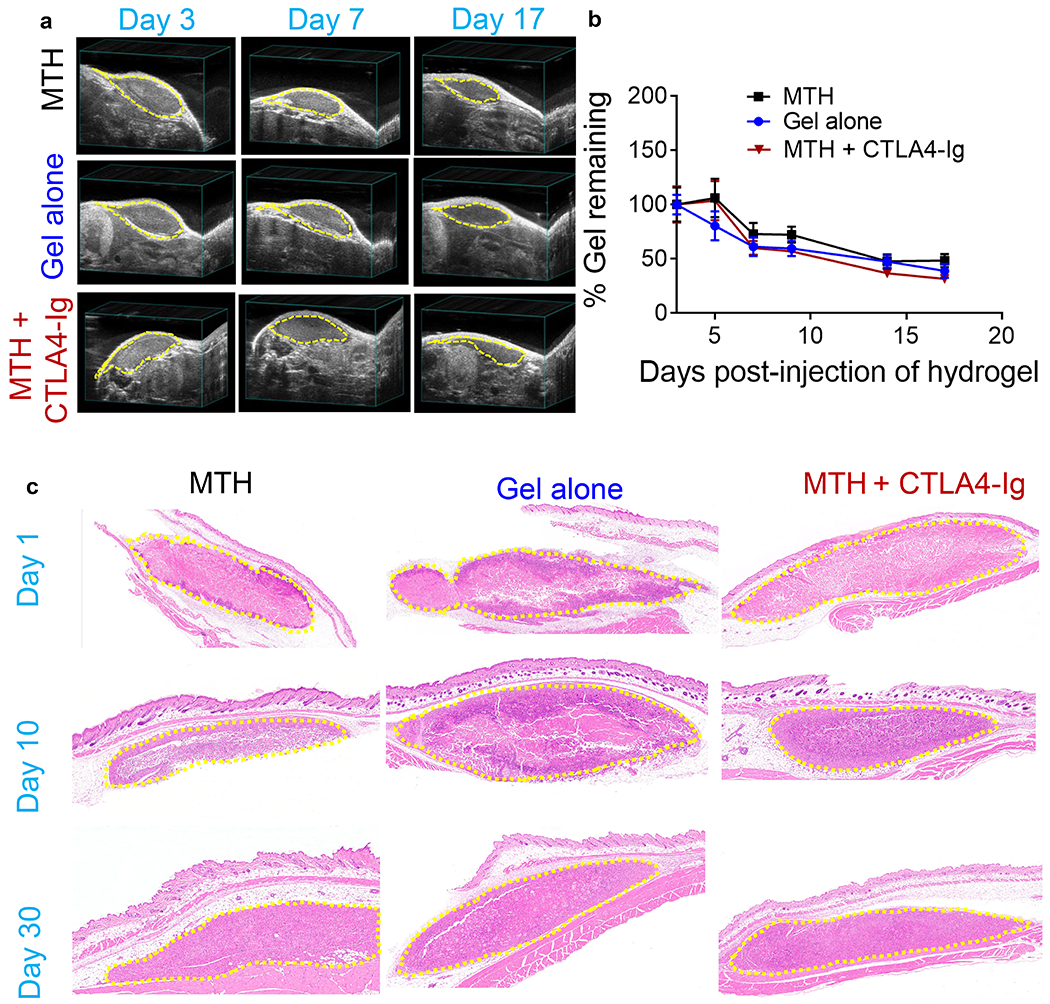
(a) Bioresorption of subcutaneously injected MTH (16 mM tofacitinib, 5% (v/v) DMSO and 1% (w/v) peptide 1), 1% (w/v) peptide 1 gel alone, and MTH co-administered with CTLA4-Ig delivered IP monitored by ultrasound at d3, 7, 17. (b) Quantitation of echograms at d3, 5, 7, 9, 14, and 17 (n = 5). (c) H&E staining of tissue sections taken at d1, 10, and 30 post injection from C57BL/6J mice that received a single subcutaneous injection of MTH, gel alone, and MTH co-administered with CTLA4-Ig delivered IP. The injection sites are outlined by yellow dash.
We next assessed the efficacy of the combination therapy in a mouse heterotopic heart transplant model, Figure 6a–c. Here, the heart of a BALB/c mouse is transplanted into the neck of a C57BL/6J recipient, a full MHC mismatched combination where the graft is typically rejected on post-operative day 7 (POD 7) if left untreated. Surgical dissection of the lateral neck of the recipient mouse exposes the common carotid artery and external jugular vein, and cuff- based anastomosis of the heart transplant follows. After the vessel clamps are removed and the heart perfused, MTH or control gel alone is applied to fill the entire graft pocket before fully closing the animal. As expected, animals receiving monotherapy MTH or gel alone are characterized by a median graft survival time (MST) of 7 days, Figure 6d. Intraperitoneal (IP) administration of monotherapy CTLA4-Ig on POD 0, 2, 4 and 6 prolongs graft survival to an MST of 35 days. Gratifyingly, combination therapy of MTH applied once locally to the graft along with CTLA4-Ig given IP at the same schedule as the monotherapy resulted in significant extension of graft survival to 125 days. The local application of MTH is necessary to delay rejection. If the combination therapy is given where MTH is administered to a location distant from the graft, for example at the base of the tail, the MST drops back down to 35 days, the value of CTLA4-Ig monotherapy. This suggests that tofacitinib stays localized to the implantation site of MTH. We tested this assertion by measuring the amount of tofacitinib in plasma by LCMSn as a function of time after MTH was introduced subcutaneously. Figure 6e shows that only a very small amount (<1%) of the administered tofacitinib partitions to the plasma at early times, which decreases to a steady state near the detection limits (~ 2 ng/mL) of the analysis. Thus, nearly all of the drug remains at or near the injection site. Lastly, histological analysis of transplanted heart isolated from animals on POD21 receiving combination therapy (MTH +CTLA4-Ig, Figure 6f/g) versus CTLA4-Ig only (Figure 6h/i) was performed. Grafts that received combination therapy contained a large number of pericardial acidophilic engorged macrophages, similar to that observed in Figure 5c, most likely due to the presence of the MTH gel. Importantly, mice receiving combination therapy presented with less inflammation with respect to lymphoid/mononuclear infiltration of the epicardium and myocardium compared to the CTLA4-Ig monotherapy group. The combination therapy group also presented less pericardial collagen. Since MTH does not elicit a lymphocytic response on its own, the relative number of infiltrating lymphocytes at the graft is a good indicator of the efficacy of the combination therapy at limiting inflammation, which is consistent with the observed enhanced graft survival in Figure 6d.
Figure 6.
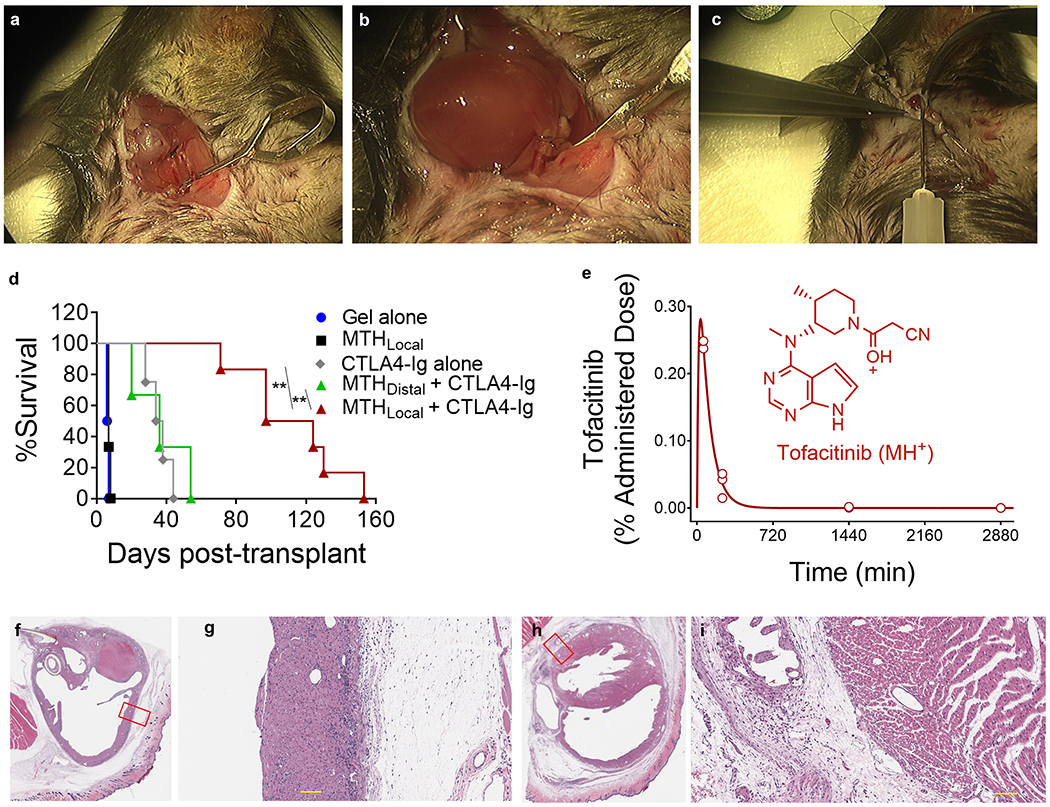
(a) Dissection of the lateral neck of the recipient mouse to isolate the common carotid artery and external jugular vein. (b) Cuff-based anastomosis of the carotid artery to the donor aortic arch and jugular vein to the pulmonary trunk of the transplanted heart. (c) 150 µL of MTH containing 750 µg (16 mM) tofacitinib or control gel is injected into the surgical pocket pericardially prior to final closure by purse string suture. (d) Transplant survival curves of C57BL/6J mice receiving BALB/c heart transplants under different treatment regimes. Mice were syringe-injected at either the graft site or a distal location (base of the tail) with MTH on POD 0. The gel alone group was injected only at the graft site. Where indicated, CTLA4-Ig was IP administered on POD 0, 2, 4, and 6 (500 µg/injection). n = 3-4 for each group. ** indicates statistically significance, p <0.01. (e) Percent of tofacitinib from MTH found in the plasma as a function of time. 150 µL of MTH having a total of 750 µg of tofacitinib was injected subcutaneously and blood drawn at indicated times. The MS1 (M+H)+ ion (C16H21N6O) was used to calculate tofacitinib concentration using ruxolitinib as an internal standard. MS2 analysis using the collision-induced secondary ion (C13H20N5) showed similar results, Supporting Figure S10. H&E staining of tissue sections taking from a BALB/c heart harvested on POD21 that had been transplanted into a C57BL/6J mouse. Heart section of mouse treated with MTH/CTLA4-Ig combination therapy (f, g) or CTLA4-Ig monotherapy (h, i). Scale bar is 100 µm on panels g and i.
3. Conclusion
A combination therapy of CTLA4-Ig delivered by IP and tofacitinib delivered locally by a single application of MTH significantly enhances graft survival in a murine heterotopic heart transplant model. Targeting two essential pathways in graft rejection, co-stimulation and cytokine signaling, proved effective and was enabled by a new microcrystalline formulation of tofacitinib. MTH is shear-thin injectable facilitating its application directly to grafts during surgery, is biocompatible, non-inflammatory and biodegradable. Importantly, the self-assembled, peptide-based gel accommodates the crystallization of tofacitinib within its network during material formulation and subsequent crystal dissolution enabling retarded release. Iťs important that the hydrogel and drug be non-interacting so that the rate of drug release is largely dependent on crystal dissolution. Using other injectable materials such as PLGA particle formulations may be complicated since their hydrolytic degradation and release profiles are heavily dependent on the identity of the encapsulated drug.52 Since the release rate of tofacitinib is largely dependent on the rate of crystal dissolution there is an opportunity to utilize crystal engineering to further control drug release. Although not explored in this current work, developing crystal growth conditions within the gel network to control final crystal morphologies, underlying surface energies, and defects that impact dissolution and thus control drug release is an exciting possibility.53 Additionally, inclusion of the biologic CTLA4-Ig in a shear-thin injectable MTH represents an exciting parallel area of investigation – at both the engineering and immunological levels – that could potentially maximize therapeutic efficacy while minimizing the manifestation of side effects. Lastly, this material platform can be extended to other small molecule immune modulators that specifically affect other players in graft rejection.
4. Experimental Section
4.1. Synthesis
Peptides were synthesized using Fmoc-based solid phase peptide synthesis strategy with RINK amide resin and HCTU activation. Resin-bound dry peptide was cleaved and side chain protecting groups were removed using cleavage cocktail of TFA/thioanisole/ethanedithiol/anisole (90:5:3:2) for 2 h under Argon atmosphere. After diethyl ether precipitation, crude peptides were purified via RP-HPLC using a preparative Vydac C18 peptide column with a flow rate of 8 mL/min. Standard A (0.1% TFA in water) and Standard B (90% acetonitrile, 9.9% water, 0.1% TFA) were used as elutants. Analytical HPLC chromatograms and ESIMS spectra of the purified peptides are provided in supplementary data (Figures S11 and S12, respectively).
4.2. Preparation of MTH and tracking the crystallization process
Peptide stock solutions (2%, w/v) were prepared in glass vials by dissolving 1 mg of peptide in 50 µL of chilled sterile water. Tofacitinib free base (LC Laboratories) stock solution was prepared in spectrophotometric grade DMSO with a final concentration of 100 µg/µL. Required volume of drug stock solution was added into a chilled solution of HEPES Buffered Saline (2X HBS, 50 mM HEPES, 300 mM NaCl, pH 7.4) to make the final volume as 50 µL. At this point, mixing of the resulting suspension was deliberately avoided. Peptide solution was transferred into the suspension, and samples were mixed to initiate hydrogelation. This resulted into MTH (Microcrystalline Tofacitinib Hydrogel) consisting of 1% (w/v) peptide in a final total volume of 100 µL. Tofacitinib was loaded to the final concentrations of 1.6, 3.2, 6.4 and 16 mM in MTH. MTH samples were placed in an incubator at 37 °C.
To spectroscopically monitor the crystallization process within the MTH, the samples were prepared as described above. 180 µL of the suspension was transferred to disposable ultra-micro cuvettes (BrandTech Scientific, Inc.) immediately after the addition of the peptide solution. The absorbance of the MTH samples was measured at 700 nm using a UV-VIS spectrophotometer (Agilent 8453) at 5, 25, 300 and 1500 minutes after the initiation of hydrogelation. %Transmittance (%T) was derived from the absorbance values using the equation: log(%T) = 2 - Absorbance.
4.3. Characterization of MTH
MTH samples were stored at 37 °C after preparation. The samples were characterized with confocal microscopy, polarized microscopy, second harmonic generation and transmission electron microscopy after one day.
4.3.1. Microscopy
For Confocal Microscopy, 10 µL of the MTH was placed on a glass slide, covered with a cover slip and sealed with nail polish. Differential Interference Contrast (DIC) channel of a confocal microscope (Zeiss LSM 710) was used to characterize the tofacitinib crystals suspended in the MTH matrix as well as the crystals formed in the absence of the MTH hydrogel.
For Polarized Light Microscopy, MTH samples were dispensed into the sample well of a crystallization plate (Swissci/MRC UV-XPO) and imaged using a Rock Imager (Formulatrix) by cross-polarized visible light. Second Harmonic Generation (SHG) images were also acquired similarly.
For Transmission Electron Microscopy (TEM), MTH samples were diluted 100X with water and 5 µL the solution was applied to 200 mesh carbon-coated copper grids (Electron Microscopy Sciences). The sample was applied for 30 s and blotted with a filter paper. Aqueous uranyl formate (0.75%, w/v) was used for negative staining. Images of the fibril nanostructure were acquired on a Hitachi 7650 transmission electron microscope at 80 kv accelerating voltage.
4.3.2. Small-Angle X-ray Scattering and Wide-Angle X-ray Scattering
The hierarchical structures of the hydrogels were studied by simultaneous small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) techniques at beamline 12ID-B of Advanced Photon Source, Argonne National Laboratory. Photon energy was 13.3-KeV and sample-to-detector distance was set in a way that q ranges in SAXS and WAXS are overlapping and achieve a total range of 0.0045 < q < 2.66 Å-1, where q = (4π/λ)sinθ, and 2θ is the scattering angle. The 2D scattering patterns were recorded with Pilatus 2M detector for SAXS and Pilatus 300K detector for WAXS with exposure time of 0.1s while the samples were scanned vertically and horizontally at 100 μm increments. Twenty-one 2D scattering images were collected for each sample in vertical and horizontal scans, respectively. The 2D images were reduced to 1D scattering profiles using the Matlab software package at the beamline. The 1D SAXS and WAXS profiles were grouped by samples and averaged.
4.3.3. Dynamic Oscillatory Rheology
The gelation process of the MTH and Peptide 1 gel alone was monitored using dynamic oscillatory rheology. 150 µL of a 2% (w/v) peptide 1 solution was first prepared by dissolving Peptide 1 in chilled sterile water. Required volume of Tofacitinib stock solution in DMSO was added into a chilled solution of HEPES Buffered Saline (2X HBS, 50 mM HEPES, 300 mM NaCl, pH 7.4) to make the final volume as 150 µL. The peptide solution was then added to the drug suspension, mixed together and transferred quickly to the rheometer (AR G2, TA instruments) plate, which was pre-equilibrated at 5 °C. The parallel plate tool (25 mm diameter) was then lowered to a gap height of 0.5 mm and the temperature was increased linearly to 37 °C to initiate gelation.
Shear-thinning and recovery ability of the samples were measured on the MTH and the Peptide 1 gel alone, 24 h after preparation. 100 μL of the samples were prepared in flexiPERM® chambers (Greiner Bio-One) and after 24 h, the samples were transferred to the center of the rheometer plate (pre-equilibrated at 37 ºC). The upper geometry (8 mm diameter) was lowered to a gap height of 0.5 mm. A dynamic time sweep was first performed to measure the storage (G’) modulus at a frequency of 6 rad/s and 0.2% strain as a function of time for 10 minutes. This was immediately followed by the application of 1000% strain at a frequency of 6 rad/s for 30 s. To monitor the recovery ability of the samples after shear, another dynamic time sweep (6 rad/s, 0.2% strain) was performed for 10 minutes. The linear viscoelastic range for the time sweep experiment was verified by performing a dynamic frequency sweep (0.1 to 100 rad/sec at 0.2% strain) which was followed by a dynamic strain sweep (0.1 to 1000% strain at 6 rad/sec) step.
4.4. Ex vivo drug release
For determining the release of tofacitinib from the MTH as a function of the drug concentration, DMSO stock solutions of tofacitinib were prepared at a range of concentrations so that the ratio of DMSO and 2X HBS could be maintained at 5% (v/v) during MTH preparation while allowing for varying the concentrations of the drug within the MTH. To determine the release of tofacitinib as a function of DMSO volume, volume ratio of DMSO and 2X HBS during MTH preparation was varied while keeping the final total volume of the MTH at 100 µL. In each case, concentrations of DMSO stock solutions of tofacitinib was adjusted in a way that would allow for maintaining a final tofacitinib concentration of 16 mM within MTH. MTH samples composed of 16 mM tofacitinib and 1% (w/v) peptides were prepared using Peptide 1, 2 and 3 to determine the influence of the peptide on the release rate of tofacitinib. In each case, MTH samples were prepared in glass vials, tightly wrapped with parafilm and stored at a 37 ºC incubator. 1 mL of 1X HBS (25 mM HEPES, 150 mM NaCl, pH 7.4) was added to the top of gel after 24 h and the vials were agitated at 37 ºC (70 rpm). At scheduled time points, the entire volume of buffer above the gel was removed and replaced with fresh buffer. Drug concentration was determined for each removed aliquot as a function of time by measuring absorbance at 290 nm in a UV-VIS spectrophotometer (Agilent 8453) with a standard curve constructed under similar conditions.
4.5. In vitro immune modulatory properties of MTH
Peptide 1 gel alone and MTH comprising 16 mM tofacitinib, 5% (v/v) DMSO and 1% (w/v) peptide 1 was prepared in syringes equipped with 27G needles. MTH and gel alone were syringe-injected into Corning® Transwell® microporous membrane inserts (8 µm) for transferring 12 µL and 50 µL gels in each group. The inserts were placed onto a 24-well plate seeded with murine bone marrow-derived dendritic cells (BMDCs). The cells were seeded in 24-well plate (2X105 cells/mL per plate) on Day 0, with the addition of the MTH and Peptide 1 gel alone onto transwell inserts on Day 6. BMDCs were stimulated with LPS (Enzo, Lot # 02261813) at 200 ng/mL on Day 7 for overnight, collected on Day 8, and stained with anti-CD11c-FITC (N418, eBioScience), anti-CD80-APC (16-10A1, BioLegend), anti-CD86-eF450 (GL1, eBioscience), and anti-MHCII-eF710 (M5/114.15.2, eBioScience) antibodies. The expression levels of these maturation markers were measured on a BD LSRII flow cytometer and the data were analyzed using FlowJo software (version 10.5.3, TreeStar) by gating on CD11c+ single cells.
4.6. In vivo studies
All animal protocols were approved by the institutional animal ethics committees at NCI Frederick and Johns Hopkins University School of Medicine. 6-8 weeks old C57BL/6J and BALB/c mice were raised in a specific pathogen-free environment.
For animal experiments, Peptide 1 was first decontaminated of any possible endotoxin by loading 5-6 mL of the peptide solution (at 10mg/mL) prepared in sterile water to an endotoxin removal spin column (Pierce, Rockford, IL). The peptide solution was retrieved by centrifugation after shaking for 2 h at 4 °C. The resin was repeated washed and the solution was lyophilized to collect the peptide. Endotoxin levels were tested using a LAL chromogenic endotoxin quantification kit (Pierce, Rockford, IL). Endotoxin levels across different batches of the peptide were routinely found to be below detection level. For preparing MTH, an equal volume of Tofacitinib suspension in 2X HBS was mixed thoroughly with an aqueous solution of a 2% (w/v) Peptide 1 in a glass vial and the suspension was quickly drawn into a sterile 27-gauge syringe. The resulting MTH was stored at 37 °C for 24 h before administration.
4.6.1. Ultrasound imaging
All Animal studies were performed according to the Frederick National Laboratory for Cancer Research (Frederick, MD) Animal Care and Use Committee guidelines. 150 μL of MTH composed of 16 mM tofacitinib and 1% (w/v) of Peptide 1 was subcutaneously administered on the lower back of 6-8 weeks old C57BL/6J mice (n = 5), Charles River Laboratories International, Inc., Frederick, MD) on Day 0. A separate group of mice was similarly administered with Peptide 1 gel alone (n = 5) at the same time. The mice in the third group (n = 5) were intraperitoneally administered with CTLA4-Ig on Day 0, 2, 4 and 6 (500 µg per injection) in addition to receiving subcutaneously administered MTH (150 μL, composed of 16 mM tofacitinib and 1% (w/v) of Peptide 1) on the flank on Day 0. Hydrogel degradation was evaluated by performing serial B-mode ultrasound imaging using the Vevo2100 preclinical scanner (VisualSonics Inc., Toronto, CA) at days 3, 5, 7, 9, 14 and 17 post injection. 3D ultrasound volumes were acquired with the MS 550S (40 MHz; 40 µm axial image resolution) linear array transducer with a step size of 0.076 mm while keeping the animal in the prone position. Prior to imaging, fur was removed around the imaging area by manual shaving followed by the application of depilatory cream. Ultrasound gel was then applied on the skin for impedance matching during imaging. Mice were anesthetized in the induction chamber with 3% isoflurane with filtered (0.2 µm) air at 1 liter/minute flow rate for 3-4 minutes and then modified for imaging to 2% with O2 as a carrier with a flow rate 1 liter/minute. Animal body temperature was maintained at 37 ºC (heated platforms) during all phases of the experiment; anesthesia induction, imaging, and post procedure recovery. The hydrogel 3D volumes were calculated for each time-point using the parallel contour algorithm (Vevo LAB software ver. 1.7.1, Visual Sonics Inc., Toronto, CA).
4.6.2. H & E staining
After ultrasound imaging at specific time points, mice were euthanized by CO2 asphyxiation and skin tissue were collected from area at the vicinity of hydrogel injection. Collected tissues were fixed in 10% neutral buffered formalin for 24 h and processed to paraffin in graded alcohols. Sections of 5 μm thickness were generated from paraffin-embedded tissues which were then stained with hematoxylin and eosin for histopathological examination.
4.6.3. Heterotopic heart transplant
C57BL/6J mice were transplanted with a heart from BALB/c donors. Prior to final closure of the surgical dissection, 150 µL of Peptide 1 gel alone or MTH (composed of 16 mM tofacitinib) was syringe-injected to fill the entire graft containing pocket. A control group of transplanted animals was prepared by injecting 150 µL of the MTH at a satellite location (base of the tail). Specific groups of mice (with or without the MTH) received intraperitoneal administration of CTLA4-Ig on POD (post-operative day) 0, 2, 4, and 6 (500 µg per injection). Graft survival was then assessed over time via manual palpation, and rejection was defined as complete cessation of cardiac contractility.
4.6.4. Pharmacokinetics
Mice were injected with 150 µL of the MTH in the same location as describe above while the mice did not bear any transplant. Blood was collected at 2, 4, 24 and 48 h after administration. Ruxolitinib internal standard was added with thorough mixing to either a 100-µL aliquot of plasma, or to 50 µL plasma diluted with an equal volume of LC/MS-grade water, to achieve a final concentration of 100 ng/mL. Plasma proteins were removed by precipitation with 4X volumes of LC/MS-grade acetonitrile. After vortexing, the plasma-CH3CN mixture was centrifuged at 14,000 X g for 15 min, and the supernatant solution transferred to a clean Eppendorf tube. The deproteinized extract was evaporated to dryness under UHP nitrogen while maintaining the sample at 30°C. The resultant residue was dissolved in 100 µL LC-MS grade H2O with vortexing and sonication followed by centrifuging for 5 min. The aqueous extract was then transferred to a 2-mL autosampler vial equipped with a low volume glass insert and analyzed as described above. After determination of plasma drug concentrations, pharmacokinetic modelling and data analysis was carried out using GraphPad Prism (Version 7.01).
Tofacitinib concentrations in mouse plasma were measured using a high-resolution LC/MS/MS method. Plasma extract components were separated on an Accela 600 HPLC system equipped with an autosampler using a narrow-bore column system consisting of a 2.1 X 100 mm, 3.5-µm Zorbax SB-C18 analytical column preceded by a 2.1 X 12.5 mm guard cartridge of the same type. This column system was operated in an 18-min cycle using elution at 250 µL/min with a 12-min 2-90% acetonitrile/water/0.1% formic acid gradient followed by a 6-min mobile phase reset and column equilibration. In this analytical HPLC system, tofacitinib and ruxolitinib internal standard had retention times of 3.25 ± 0.05 min and 5.05 ± 0.05 min, respectively. Following an initial 2-min eluent divert to waste, the entire column eluant was directed into the mass spectrometer ion source.
An LTQ-Orbitrap-XL LC/MSn system equipped with an Ion Max electrospray ionization ion source was operated in scanning mode for sequential MS and MS/MS analysis at resolutions of 15,000 and 7,500, respectively. Ion trap helium collision-induced-dissociation (CID) for MS/MS was carried out on the MH+ of tofacitinib and ruxolitinib internal standard using a mass selection window set to encompass the full isotopic profile of these compounds. This approach generated a full scan (m/z 100 – 750), high-resolution record of the primary MS and a full, high-resolution MS/MS (MS2) spectrum of the CID fragmentation of the selected MS1 ions. Instrument operation and control as well as data extraction and analysis were carried out using Xcalibur software (version 2.1.0, SP1). The tofacitinib MH+ (m/z 313.1771) and its CID fragmentation to C13H20N5+ (m/z 246.1713) and to C7H9N4+ (m/z 149.0821) as well as the ruxolitinib MH+ (m/z 307.1666) and its decomposition to C9H8N5+ (m/z 186.0774) were chosen as ions to use for quantitative measurements. High-resolution extracted ion chromatograms were generated for ions of interest using an m/z window of 10 ppm (typically less than ± 3 mDa). Tofacitinib and internal standard peak areas were integrated, and MS1 and MS2 peak area ratio calibration curves were constructed for the appropriate concentration ranges to allow determination of unknowns. For the lowest concentration range (0-100 ng/mL), the limit of quantitation for tofacitinib in 100 µL plasma was 2 ng/mL. Under the same analysis conditions, the limit of detection exceeded 0.3 ng/mL.
4.7. Statistical Analyses
Mice were randomly divided into groups. Data are expressed as mean ± SD except for the ultrasound imaging experiment where the data represents mean ± SEM. Statistical analysis to compare in vitro data between two groups was performed using two-tailed Student’s t-tests, and log-rank test analysis determined differences in transplant survival. GraphPad Prism 7.0 or 8.0. software was used for data analysis and p < 0.05 was considered statistically significant. Significance is denoted as * for p < 0.05, ** for p < 0.01.
Supplementary Material
Acknowledgements
This research was supported by the Center for Cancer Research Intramural Research Program of the National Cancer Institute, National Institutes of Health, by the Department of Plastic and Reconstructive Surgery at Johns Hopkins School of Medicine, and by the USAMRAA through the FY17 Reconstructive Transplant Research Program under Award No. W81XWH1810789. The authors acknowledge Dr. Cory Brayton (Director, Phenotypic Core facility, Johns Hopkins Medicine) for histological evaluation of transplanted heart tissues, Dr. Galit Fichman (Chemical Biology Laboratory, NCI Frederick) and Dr. Ziqiu Wang (Electron Microscopy Laboratory, Leidos Biomedical Research Inc.) for help with Transmission Electron Microscopy. Dr. Joseph D. Kalen and Ms Lisa Riffle (Small Animal Imaging Program, Leidos Biomedical Research Inc.) are acknowledged for help with ultrasound imaging. For the SAXS experiments, we acknowledge the SAXS core facility at the Center for Cancer Research, National Cancer Institute (NCI). The shared scattering beamline 12ID-B resource is allocated under the PUP-24152 agreement between the National Cancer Institute and Argonne National Laboratory (ANL). Use of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, was operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Bueno EM; Diaz-Siso JR; Sisk GC; Chandawarkar A; Kiwanuka H; Lamparello B; Caterson EJ; Pomahac B, Vascularized composite allotransplantation and tissue engineering. J. Craniofac. Surg 2013, 24, 256–63. [DOI] [PubMed] [Google Scholar]
- 2.Diaz-Siso JR; Bueno EM; Sisk GC; Marty FM; Pomahac B; Tullius SG, Vascularized composite tissue allotransplantation--state of the art. Clin. Transplant 2013, 27, 330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman CL; Ouseph R; Marvin MR; Manon-Matos Y; Blair B; Kutz JE, Monitoring and long-term outcomes in vascularized composite allotransplantation. Curr. Opin. Organ Transplant 2013, 18, 652–8. [DOI] [PubMed] [Google Scholar]
- 4.Gajanayake T; Olariu R; Leclere FM; Dhayani A; Yang Z; Bongoni AK; Banz Y; Constantinescu MA; Karp JM; Vemula PK; Rieben R; Vogelin E, A single localized dose of enzyme-responsive hydrogel improves long-term survival of a vascularized composite allograft. Sci. Transl. Med 2014, 6, 249ra110. [DOI] [PubMed] [Google Scholar]
- 5.Platz KP; Mueller AR; Blumhardt G; Bachmann S; Bechstein WO; Kahl A; Neuhaus P, Nephrotoxicity following orthotopic liver transplantation. A comparison between cyclosporine and FK506. Transplantation 1994, 58, 170–8. [PubMed] [Google Scholar]
- 6.Goral S, The Three-Signal Hypothesis of Lymphocyte Activation/Targets for Immunosuppression. Dial. Transplant 2011, 40, 14–16. [Google Scholar]
- 7.Filatenkov AA; Jacovetty EL; Fischer UB; Curtsinger JM; Mescher MF; Ingulli E, CD4 T cell-dependent conditioning of dendritic cells to produce IL-12 results in CD8-mediated graft rejection and avoidance of tolerance. J. Immunol 2005, 174, 6909–17. [DOI] [PubMed] [Google Scholar]
- 8.Kirk AD; Harlan DM; Armstrong NN; Davis TA; Dong Y; Gray GS; Hong X; Thomas D; Fechner JH Jr.; Knechtle SJ, CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 8789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenschow DJ; Zeng YJ; Thistlethwaite JR; Montag A; Brady W; Gibson MG; Linsley PS; Bluestone JA, Long-Term Survival of Xenogeneic Pancreatic-Islet Grafts Induced by Ctla4ig. Science 1992, 257, 789–792. [DOI] [PubMed] [Google Scholar]
- 10.Kremer JM; Westhovens R; Leon M; Di Giorgio E; Alten R; Steinfeld S; Russell A; Dougados M; Emery P; Nuamah IF; Williams GR; Becker JC; Hagerty DT; Moreland LW, Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N. Engl. J. Med 2003, 349, 1907–15. [DOI] [PubMed] [Google Scholar]
- 11.Levisetti MG; Padrid PA; Szot GL; Mittal N; Meehan SM; Wardrip CL; Gray GS; Bruce DS; Thistlethwaite JR Jr.; Bluestone JA, Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J. Immunol 1997, 159, 5187–91. [PubMed] [Google Scholar]
- 12.Bluestone JA; St Clair EW; Turka LA, CTLA4Ig: bridging the basic immunology with clinical application. Immunity 2006, 24, 233–8. [DOI] [PubMed] [Google Scholar]
- 13.Riella LV; Sayegh MH, T-cell co-stimulatory blockade in transplantation: two steps forward one step back! Expert Opin. Biol. Ther 2013, 13, 1557–68. [DOI] [PubMed] [Google Scholar]
- 14.Webber AB; Vincenti F, An Update on Calcineurin Inhibitor-Free Regimens: The Need Persists, but the Landscape has Changed. Transplantation 2016, 100, 836–43. [DOI] [PubMed] [Google Scholar]
- 15.Chong AS; Alegre ML, The impact of infection and tissue damage in solid-organ transplantation. Nat. Rev. Immunol 2012, 12, 459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lichtman AH; Chin J; Schmidt JA; Abbas AK, Role of interleukin 1 in the activation of T lymphocytes. Proc. Natl. Acad. Sci. U. S. A 1988, 85, 9699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rochman I; Paul WE; Ben-Sasson SZ, IL-6 increases primed cell expansion and survival. J. Immunol 2005, 174, 4761–7. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Sasson SZ; Hu-Li J; Quiel J; Cauchetaux S; Ratner M; Shapira I; Dinarello CA; Paul WE, IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 7119–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao DA; Eid RE; Qin L; Yi T; Kirkiles-Smith NC; Tellides G; Pober JS, Interleukin (IL)-1 promotes allogeneic T cell intimal infiltration and IL-17 production in a model of human artery rejection. J. Exp. Med 2008, 205, 3145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SA; Scheller J; Rose-John S, Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Invest 2011, 121, 3375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKay D; Shigeoka A; Rubinstein M; Surh C; Sprent J, Simultaneous deletion of MyD88 and Trif delays major histocompatibility and minor antigen mismatch allograft rejection. Eur. J. Immunol 2006, 36, 1994–2002. [DOI] [PubMed] [Google Scholar]
- 22.Thornley TB; Phillips NE; Beaudette-Zlatanova BC; Markees TG; Bahl K; Brehm MA; Shultz LD; Kurt-Jones EA; Mordes JP; Welsh RM, Type 1 IFN mediates cross-talk between innate and adaptive immunity that abrogates transplantation tolerance. J. Immunol 2007, 179, 6620–6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker WE; Nasr IW; Camirand G; Tesar BM; Booth CJ; Goldstein DR, Absence of innate MyD88 signaling promotes inducible allograft acceptance. J. Immunol 2006, 177, 5307–16. [DOI] [PubMed] [Google Scholar]
- 24.Shen H; Goldstein DR, IL-6 and TNF-alpha synergistically inhibit allograft acceptance. J. Am. Soc. Nephrol 2009, 20, 1032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khalifian S; Raimondi G; Lee WA; Brandacher G, Taming inflammation by targeting cytokine signaling: new perspectives in the induction of transplantation tolerance. Immunotherapy 2014, 6, 637–53. [DOI] [PubMed] [Google Scholar]
- 26.Ghoreschi K; Jesson MI; Li X; Lee JL; Ghosh S; Alsup JW; Warner JD; Tanaka M; Steward-Tharp SM; Gadina M; Thomas CJ; Minnerly JC; Storer CE; LaBranche TP; Radi ZA; Dowty ME; Head RD; Meyer DM; Kishore N; O'Shea JJ, Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol 2011, 186, 4234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rousvoal G; Si MS; Lau M; Zhang S; Berry GJ; Flores MG; Changelian PS; Reitz BA; Borie DC, Janus kinase 3 inhibition with CP-690,550 prevents allograft vasculopathy. Transplant Int. 2006, 19, 1014–21. [DOI] [PubMed] [Google Scholar]
- 28.Changelian PS; Flanagan ME; Ball DJ; Kent CR; Magnuson KS; Martin WH; Rizzuti BJ; Sawyer PS; Perry BD; Brissette WH; McCurdy SP; Kudlacz EM; Conklyn MJ; Elliott EA; Koslov ER; Fisher MB; Strelevitz TJ; Yoon K; Whipple DA; Sun J; Munchhof MJ; Doty JL; Casavant JM; Blumenkopf TA; Hines M; Brown MF; Lillie BM; Subramanyam C; Shang-Poa C; Milici AJ; Beckius GE; Moyer JD; Su C; Woodworth TG; Gaweco AS; Beals CR; Littman BH; Fisher DA; Smith JF; Zagouras P; Magna HA; Saltarelli MJ; Johnson KS; Nelms LF; Des Etages SG; Hayes LS; Kawabata TT; Finco-Kent D; Baker DL; Larson M; Si MS; Paniagua R; Higgins J; Holm B; Reitz B; Zhou YJ; Morris RE; O'Shea JJ; Borie DC, Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003, 302, 875–8. [DOI] [PubMed] [Google Scholar]
- 29.Busque S; Vincenti FG; Tedesco Silva H; O'Connell PJ; Yoshida A; Friedewald JJ; Steinberg SM; Budde K; Broeders EN; Kim YS; Hahn CM; Li H; Chan G, Efficacy and Safety of a Tofacitinib-based Immunosuppressive Regimen After Kidney Transplantation: Results From a Long-term Extension Trial. Transplant. Direct 2018, 4, e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojciechowski D; Vincenti F, Tofacitinib in kidney transplantation. Expert Opin. Invest. Drugs 2013, 22, 1193–9. [DOI] [PubMed] [Google Scholar]
- 31.Fujita T; Takahashi S; Yagihashi A; Jimbow K; Sato N, Prolonged survival of rat skin allograft by treatment with FK506 ointment. Transplantation 1997, 64, 922–5. [DOI] [PubMed] [Google Scholar]
- 32.Shi W; Liu T; Xie L; Wang S, FK506 in a biodegradable glycolide-co-clatide-co-caprolactone polymer for prolongation of corneal allograft survival. Curr. Eye Res 2005, 30, 969–76. [DOI] [PubMed] [Google Scholar]
- 33.Park CG; Hartl CA; Schmid D; Carmona EM; Kim HJ; Goldberg MS, Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci. Transl. Med 2018, 10. [DOI] [PubMed] [Google Scholar]
- 34.Fisher JD; Acharya AP; Little SR, Micro and nanoparticle drug delivery systems for preventing allotransplant rejection. Clin. Immunol 2015, 160, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J; Zheng Z; Chong Y; Li X; Pu L; Tang Q; Yang L; Wang X; Wang F; Liang G, Immune Responsive Release of Tacrolimus to Overcome Organ Transplant Rejection. Adv. Mater 2018, 30, e1805018. [DOI] [PubMed] [Google Scholar]
- 36.Leach DG; Dharmaraj N; Piotrowski SL; Lopez-Silva TL; Lei YL; Sikora AG; Young S; Hartgerink JD, STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials 2018, 163, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soranno DE; Lu HD; Weber HM; Rai R; Burdick JA, Immunotherapy with injectable hydrogels to treat obstructive nephropathy. J. Biomed. Mater. Res., Part A 2014, 102, 2173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Q; Wang C; Zhang X; Chen G; Hu Q; Li H; Wang J; Wen D; Zhang Y; Lu Y; Yang G; Jiang C; Wang J; Dotti G; Gu Z, In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol 2019, 14, 89–97. [DOI] [PubMed] [Google Scholar]
- 39.Pfizer, NDA203214, Application number: 203214Orig1s000. Clinical Pharmacology Review 2011, 3149379, 11. [Google Scholar]
- 40.Farah S; Doloff JC; Muller P; Sadraei A; Han HJ; Olafson K; Vyas K; Tam HH; Hollister-Lock J; Kowalski PS; Griffin M; Meng A; McAvoy M; Graham AC; McGarrigle J; Oberholzer J; Weir GC; Greiner DL; Langer R; Anderson DG, Long-term implant fibrosis prevention in rodents and non-human primates using crystallized drug formulations. Nat. Mater 2019, 18, 892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adobes-Vidal M; Maddar FM; Momotenko D; Hughes LP; Wren SA; Poloni LN; Ward MD; Unwin PR, Face-discriminating dissolution kinetics of furosemide single crystals: in situ three-dimensional multi-microscopy and modeling. Cryst. Growth Des 2016, 16, 4421–4429. [Google Scholar]
- 42.Maddar FM; Adobes-Vidal M; Hughes LP; Wren SA; Unwin PR, Dissolution of Bicalutamide Single Crystals in Aqueous Solution: Significance of Evolving Topography in Accelerating Face-Specific Kinetics. Cryst. Growth Des 2017, 17, 5108–5116. [Google Scholar]
- 43.Smith DJ; Brat GA; Medina SH; Tong D; Huang Y; Grahammer J; Furtmüller GJ; Oh BC; Nagy-Smith KJ; Walczak P, A multiphase transitioning peptide hydrogel for suturing ultrasmall vessels. Nat. Nanotechnol 2016, 11, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Majumder P; Baxa U; Walsh ST; Schneider JP, Design of a multicompartment hydrogel that facilitates time‐resolved delivery of combination therapy and synergized killing of glioblastoma. Angew. Chem., Int. Ed. Engl 2018, 57, 15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller SE; Yamada Y; Patel N; Suárez E; Andrews C; Tau S; Luke BT; Cachau RE; Schneider JP, Electrostatically Driven Guanidinium Interaction Domains that Control Hydrogel-Mediated Protein Delivery In Vivo. ACS Cent. Sci 2019, 5, 1750–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagy-Smith K; Beltramo PJ; Moore E; Tycko R; Furst EM; Schneider JP, Molecular, local, and network-level basis for the enhanced stiffness of hydrogel networks formed from coassembled racemic peptides: Predictions from Pauling and Corey. ACS Cent. Sci 2017, 3, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagy-Smith K; Moore E; Schneider J; Tycko R, Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 9816–9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adobes-Vidal M; Shtukenberg AG; Ward MD; Unwin PR, Multiscale visualization and quantitative analysis of L-cystine crystal dissolution. Cryst. Growth Des 2017, 17, 1766–1774. [Google Scholar]
- 49.Adobes-Vidal M; Pearce H; Unwin PR, Tracking the dissolution of calcite single crystals in acid waters: a simple method for measuring fast surface kinetics. Phys. Chem. Chem. Phys 2017, 19, 17827–17833. [DOI] [PubMed] [Google Scholar]
- 50.Kubo S; Yamaoka K; Kondo M; Yamagata K; Zhao J; Iwata S; Tanaka Y, The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann. Rheum. Dis 2014, 73, 2192–8. [DOI] [PubMed] [Google Scholar]
- 51.Heine A; Held SA; Daecke SN; Wallner S; Yajnanarayana SP; Kurts C; Wolf D; Brossart P, The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122, 1192–202. [DOI] [PubMed] [Google Scholar]
- 52.Siegel SJ; Kahn JB; Metzger K; Winey KI; Werner K; Dan N, Effect of drug type on the degradation rate of PLGA matrices. Eur. J. Pharm. Biopharm 2006, 64, 287–93. [DOI] [PubMed] [Google Scholar]
- 53.Vidal MA Quantitative and Holistic Views of Crystal Dissolution Processes University of Warwick, 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.