

Abstract
Selective elimination of microglia from the brain was shown to dysregulate neuronal Ca2+ signaling and to reduce the incidence of spreading depolarization (SD) during cerebral ischemia. However, the mechanisms through which microglia interfere with SD remained unexplored. Here, we identify microglia as essential modulators of the induction and evolution of SD in the physiologically intact brain in vivo. Confocal- and super-resolution microscopy revealed that a series of SDs induced rapid morphological changes in microglia, facilitated microglial process recruitment to neurons and increased the density of P2Y12 receptors (P2Y12R) on recruited microglial processes. In line with this, depolarization and hyperpolarization during SD were microglia- and P2Y12R-dependent. An absence of microglia was associated with altered potassium uptake after SD and increased the number of c-fos-positive neurons, independently of P2Y12R. Thus, the presence of microglia is likely to be essential to maintain the electrical elicitation threshold and to support the full evolution of SD, conceivably by interfering with the extracellular potassium homeostasis of the brain through sustaining [K+]e re-uptake mechanisms.
Keywords: Microglia, spreading depolarization, P2Y12 receptor, extracellular potassium clearance, inflammation
Introduction
Microglia play many roles in the brain beyond their primary immune function, including the removal of terminally injured neurons and the support of tissue repair through their interactions with neurons or glial cells.1–3 In addition, microglia are critical for the consolidation of neuronal networks. Microglia monitor synaptic function, prune redundant boutons in a complement-mediated manner and assist synapse formation by secreting brain-derived neurotrophic factor.4,5 In the healthy adult brain, surveilling microglia are characterized by motile, ramified processes, which form contacts with different compartments of neurons and respond to increased neuronal depolarization. In turn, the spontaneous and evoked activity of the contacted neurons is suppressed.6 Microglia–neuron interactions may be partially independent of astrocytes, because blockade of astrocyte function by fluoroacetate did not alter glutamate-induced microglial process recruitment to neurons.7
Microglia respond to a wide range of stimuli via purinergic signaling. For example, P2Y12R, which is specifically expressed by microglia in the brain,8 is essential for ATP-induced chemotaxis, process outgrowth to injury, and enhanced surveillance activity.9,10 In turn, microglia can release a wide range of signaling molecules including pro-inflammatory cytokines (IL-1b, IL-18, TNF-α) or K+, which can modulate neuronal excitability.9,11–14
We have previously found that selective elimination of microglia with a colony-stimulating factor 1 receptor (CSF1R) kinase inhibitor15 triggered slow neuronal calcium oscillations (0.1 Hz) in the acute phase of focal cerebral ischemia in mice, while the spontaneous occurrence of cortical spreading depolarization (SD) was hampered.16 SD is a self-propagating wave of abrupt, transient, mass depolarization that reflects a near-complete breakdown of neuronal transmembrane ion gradients and travels slowly (3–5 mm/min) over the cerebrocortical gray matter.17,18 The hallmark of SD is a negative shift in the local field potential filtered in direct current (DC) mode, and an immense elevation of extracellular potassium ([K+]e) and glutamate concentration, accompanied by cellular swelling and the initiation of inflammatory cascades.19–22 SD has been recognized as a potentially harmful event promoting secondary injury in the metabolically compromised ischemic brain, and has been recognized to be the cellular counterpart of migraine aura.23
Microglia react to SD by increased IL-1β release,24,25 and enhanced outward potassium conductivity.26 They have also been shown to remain reactive for days after series of SDs triggered in rats27 and to modulate SD initiation ex vivo.28 However, the mechanisms through which microglia may tune the susceptibility of the nervous tissue to SD are yet to be explored in vivo. Neuronal hyperexcitability and impaired K+ clearance have long been accepted as key contributors to the evolution of SD.29,30 Thus, we hypothesized that surveilling or activated microglia may sustain SD susceptibility by interfering with either of these mechanisms. Accordingly, we investigated whether the presence of microglia is critical for the elicitation or the propagation of an SD triggered in the nervous tissue at resting state (i.e. no prior SDs occurred), and for recurrent SDs in vivo. To understand the mechanisms involved, we evaluated [K+]e changes associated with SDs using ion-selective microelectrodes in microglia-depleted and P2Y12R knock out (P2Y12R KO) mice, and analyzed nanoscale changes in the expression of microglial P2Y12R using super-resolution microscopy.
Materials and methods
Mice
Experiments were carried out in 12–14-weeks-old adult male C57BL/6J (n = 28) and P2Y12R KO (#TF1881, Taconic) (n = 7) mice (male, C57BL/6J background), bred in the SPF unit of the Institute of Experimental Medicine (Budapest, Hungary). The animals were housed under controlled temperature, humidity and lightning conditions (23°C, 12:12-h light/dark cycle, lights on at 7 a.m.), with ad libitum access to food and water.
Selective elimination of microglia was performed in C57BL/6J mice (n = 12) by feeding a chow diet containing CSF1R inhibitor, PLX5622 for three weeks.15 PLX5622 was provided by Plexxikon Inc. and formulated in AIN-76A standard chow by Research Diets (1200 p.p.m.; 1200 mg PLX5622 in 1 kg chow). Body temperature was recorded for every mouse before surgery or electrophysiological recording. No mice were excluded from these studies due to fever, weight loss, infection or behavioral alterations as a result of PLX5622 treatment. P2Y12R KO mice (n = 7) and reference groups (n = 14) were on control diet (Supplementary Table 1).
Surgical procedures
All procedures were approved by the National Food Chain Safety and Animal Health Directorate of Csongrád County, Hungary, conforming to the guidelines of the Scientific Committee of Animal Experimentation of the Hungarian Academy of Sciences (updated Law and Regulations on Animal Protection: 40/2013. (II. 14.) Gov. of Hungary), following the EU Directive 2010/63/EU on the protection of animals used for scientific purposes, and reported in compliance with the ARRIVE guidelines.
Mice were anesthetized with 1.5–2% isoflurane in N2O:O2 (3:2) and were allowed to breathe spontaneously through a head mask. Atropine (0.1%, 0.01 ml) was injected intramuscularly to avoid the production of airway mucus. Body temperature was maintained at 37°C with a servo-controlled heating pad (507222 F, Harvard Apparatus, USA). The animal was fixed in a stereotaxic frame, and two craniotomies (3 mm lateral from sagittal suture, −1 and −3 caudal from bregma) were created with a dental drill (ProLab Basic, Bien Air, Switzerland) on the right parietal bone. The dura mater in each craniotomy was left intact. The rostral window was used for SD elicitation, while electrophysiological variables were monitored in the caudal window. To assess whether any focal inflammatory reaction developed at the site of electrode insertion, microglial activation, leukocyte recruitment and vascular activation were investigated by Iba1, CD45 and ICAM-1 immunofluorescence, respectively. Only minor changes in microglial morphology were noticed no deeper than 100 µm below the meninges, while no increases in CD45 or ICAM-1 immunofluorescence were found. Quantitative measurements took place remotely from the site of electrode insertion (Figure 1).
Figure 1.

Spreading depolarization (SD) attracts microglial processes to neuronal somata and selective elimination of microglia alters the electrical threshold of SD elicitation. (a) Schematic illustration of the experimental setting in series 1, and areas of quantification for the immunofluorescent analysis shown in Panels (b) and (c). Area size (420 µm × 320 µm). (b) Changes in microglial cell body area and the number of branches as obtained from 2D morphological analysis of Iba1-positive microglia in the cerebral cortex (the region used for analysis is identical to Area 1 shown in panel (a)). Mann–Whitney U test (p < 0.05*). Scale bar, 10 μm. (c) Representative images demonstrating microglial (P2Y12R, red) process recruitment to Kv2.1 labeled neuronal soma (green) in the neocortex, contralateral and ipsilateral to SD elicitation. Microglial process density is increased around neurons in the ipsilateral cortex relative to the contralateral cortex in Area 1 (near SD induction) depicted in Panel A. Data are expressed as mean±stdev. Control vs. ipsi p < 0.01, two-way ANOVA followed by Sidak’s multiple comparison test (p < 0.05 for Area 1). Scale bar, 10 μm. (d) Representative images and quantitative analysis confirming elimination of microglia (P2Y12 receptors, green) after feeding mice a PLX5622 (1200 ppm)-containing diet for three weeks. Data are expressed as mean±stdev. Unpaired t-test (p < 0.01**). Scale bar, 50 μm. (e) Microglia depletion increased the electric threshold of SD elicitation for each consecutive SD in a train (Series 1). Data are shown as median and interquartile ranges. Friedman ANOVA for time (p < 0.05* vs. 1st SD, in the depleted group), and a Mann–Whitney U test for group comparison (no significant differences between groups). (f) Microglia depletion altered the electric threshold of SD elicitation (series 1). First SDs (circles) are shown apart from pooled, recurrent SDs (triangles). Black circle and error bars stand for mean ± 95% CI. Mann–Whitney U test (p < 0.05* and p < 0.01** vs. respective Control). (g) The induction of SD required a KCl concentration higher than 1 M in some of the microglia-depleted and P2Y12R KO animals (series 2). Each colored sphere in the graph stands for the induction of an individual SD.
Experimental protocols
Two series of experiments were designed (Supplementary Table 1). In series 1, the electrical threshold of SD induction was determined, whereas in series 2, [K+]e was assessed during SD induced with a topical application of 1 M KCl in the absence of microglia or P2Y12R.
Electrophysiology
In series 1, a glass capillary electrode (outer tip diameter = 20 µm) filled with physiological saline was inserted into the cortex. An Ag/AgCl reference electrode was implanted under the skin of the animal’s neck. DC potential was recorded via a high input impedance pre-amplifier (NL100AK, Digitimer Ltd, UK), connected to a differential amplifier with associate filter and conditioner systems (NL107, NL125 and NL530, Digitimer Ltd, UK). Potential line frequency noise (50 Hz) was removed by a noise eliminator (HumBug, Quest Scientific Instruments, Canada). The resulting signal was then digitized and continuously acquired at a sampling frequency of 500 Hz with a dedicated analog-to-digital (A/D) converter (NI USB-6008/6009, National Instruments, USA) controlled through a custom-made software in Labview (National Instruments, USA).
In series 2, ion-sensitive microelectrodes were prepared according to Viitanen et al.31 Glass capillary microelectrode tips (outer tip diameter: 10–12 µm) were filled with a liquid K+-ion exchanger (Potassium ionophore I – cocktail A; Sigma-Aldrich, Germany),31,32 and the shank of the microelectrode was backfilled with 100 mM KCl. Each K+-selective microelectrode was calibrated in standard solutions of known K+ concentrations (1, 3, 5, 10, 30, 50 and 100 mM) (Figure 3(a)).33 In each experiment, a K+-sensitive microelectrode was lowered into the cortex, together with another microelectrode (outer tip diameter = 20 µm) filled with 150 mM NaCl and 1 mM HEPES to serve as reference and acquire DC potential (<1Hz). An Ag/AgCl electrode implanted under the skin of the animal’s neck was used as common ground. Microelectrodes were connected to a custom-made dual-channel high input impedance electrometer (including AD549LH, Analog Devices, USA) via Ag/AgCl leads. The voltage signal recorded by the reference electrode was subtracted from that of the K+-sensitive microelectrode by dedicated differential amplifiers and associated filter modules (NL106 and NL125, Digitimer Ltd, UK), which yielded potential variations related to changes in [K+]e. The recorded signals were then forwarded to an A/D converter (MP 150, BIOPAC Systems, USA) and digitized by a sample rate of 1 kHz using the software AcqKnowledge 4.2.0 (BIOPAC Systems, USA). The completed preparations were enclosed in a Faraday cage. K+ signals were displayed together with the DC potential essentially as described above.
Figure 3.

Selective elimination of microglia shortens the duration of spreading depolarization (SD)-related potassium elevation. (a) Calibration curve for the K+-selective microelectrodes using K+ solution standards (1, 3, 5, 10, 30, 50, and 100 mM KCl). Microelectrodes were calibrated before and after each in vivo measurement. (b) Baseline level of extracellular potassium concentration ([K+]e) (mean of 60 s) immediately before the induction of each SD. Data are given as mean±stdev. Repeated measures ANOVA for within group variation, and one-way ANOVA followed by Tukey’s multiple comparison for group evaluation (p < 0.05$ and p < 0.01$$ P2Y12R KO vs. depleted, p < 0.05# P2Y12R KO vs. control). (c) The transient increase of [K+]e with recurrent SDs (rSDs) (each trace is the mean of rSDs in each experimental group, series 2). Capital letters indicate variables quantitated in the respective Panels. (d) Peak elevation of the [K+]e shift with rSDs. The base of each bar in the chart is set at [K+]e immediately prior to rSDs. (e) Magnitude of the [K+]e shift with rSDs, expressed as area under the curve (AUC). (f) Duration of the [K+]e shift with rSDs, taken at half amplitude. (g) [K+]e clearance, calculated by an exponential regression at half-time recovery of [K+]e to baseline. In Panels (d)–(f), data are given as mean±stdev. Sample size is indicated in each bar. In Panel (g), triangles denote individual values, while black circle and error bars stand for mean±CI. Note, that the y-axis for control and depleted (left) is an expanded segment of the y-axis (right) for P2Y12R KO (grey arrows). One-way ANOVA followed by Tukey’s multiple comparison for equal variances, or Games-Howell for unequal variances (p < 0.01** Depleted vs. control, p < 0.05$ and p < 0.01$$ P2Y12R KO vs. depleted, p < 0.05# P2Y12R KO vs. Control).
Induction of SD
In each animal, four SDs were triggered at a 15-min of inter-SD interval. In series 1, SDs were triggered as reported earlier.34 Briefly, a concentric bipolar needle electrode with a tip diameter of 40 µm (Neuronelektród KFT, Hungary) was placed upon the dura. It was connected to an opto-coupled stimulus isolator with a constant current output (NL 800, Digitimer Ltd, UK), a pulse generator (NL301), a with-delay panel (NL405), and a pulse buffer (NL510), which enabled the adjustment of amplitude and duration of the stimuli at will. Stimulation was implemented with a single bipolar constant current stimulation. The charge delivered was quantified as Q(µC) = I(mA) × t(ms), and it was raised stepwise with an interstimulus interval of 1 min until SD was observed. Whenever necessary, the position of the needle electrode was adjusted to optimize the contact between the electrode tip and the tissue. Successful elicitation of SD was confirmed by a negative DC-shift of an amplitude greater than 5 mV at the recording electrode.
In series 2, a cotton ball soaked in 1 M KCl was placed into the rostral cranial window. The cotton ball was removed, and the cranial window rinsed with artificial cerebrospinal fluid (aCSF; mM concentrations: 126.6 NaCl, 3 KCl, 1.5 CaCl2, 1.2 MgCl2, 24.5 NaHCO3, 6.7 urea, 3.7 glucose bubbled with 95% O2 and 5% CO2 to achieve a constant pH of 7.4) immediately after each successful elicitation.
Tissue processing and immunostaining
Following in vivo measurements, mice were deeply anesthetized with an overdose of chlorale hydrate (i.p.). For immunohistochemistry, animals were transcardially perfused with ice-cold saline followed by 4% paraformaldehyde 1.5 h after the induction of the first SD. Subsequently, the brains were removed, post-fixed and cryoprotected overnight in 10% sucrose/4%PFA/PBS and were stored in 10% sucrose/PBS at 4°C until 25 µm thick coronal sections were prepared using a sliding microtome (Leica SM 2010 R).
Immunofluorescence
Thick free-floating brain sections (25 µm) were blocked with 5–10% normal donkey serum for 1 h and incubated with different primary antibodies at 4°C, overnight: rabbit anti-Iba-1 (1:1000, #019–19741, Wako Chemicals), goat anti-Iba1 (1:500, NB100-1028, Novusbio), rabbit anti-P2Y12R (1:1000, #55043 A, AnaSpec), rabbit anti-c-fos (1:500, #sc-52, Santa Cruz Biotechnology), guinea pig anti-c-fos (1:500, #226 004, Synaptic Systems), chicken anti-GFAP (1:500, #173 006, Synaptic Systems), guinea pig anti-glutamine synthetase (1:500, #367 005, Synaptic Systems), rat anti-CD45 (1:250, #MCA1388, Bio-Rad), goat anti-ICAM-1 (1:250, #AF796, R&D Systems) and mouse anti-Kv2.1 (1:500, #75-014 NeuroMab). Sections were washed several times with TBS and incubated with the corresponding secondary antibody (Jackson ImmunoResearch, unless specified below) for 2 h: donkey anti-rabbit Alexa 488 (1:1000, #711-546-152), donkey anti-mouse Alexa 488 (1:1000, #715-006-151), donkey anti-chicken Alexa 488 (1:500, #703-546-155), donkey anti-goat Alexa488 (1:500, #705-546-147), donkey anti-rabbit CF568 (1:1000, #20098-1mg, Biotium), donkey anti-rabbit Alexa 594 (1:5000, #A21207, Invitrogen), donkey anti-mouse Alexa 594 (1:500, #715-586-151), donkey anti-rabbit Alexa 647 (1:1000, #711-605-152), donkey anti-mouse Alexa 647 (1:1000, #715-605-150), donkey anti-guinea pig Alexa647 (1:500, #706-606-148). After washing, sections were mounted on glass slides and coverslipped (Fluoromount-G, Southern Biotech).
STORM super-resolution microscopy
To examine the exact localization of P2Y12R, STORM imaging was performed. Immunolabeled (rabbit anti-P2Y12R, 1:1000, #55043 A, AnaSpec, mouse anti-Kv2.1 1:500, #75-014 NeuroMab) sections were covered with imaging medium35 immediately before imaging. Correlated confocal and super-resolution imaging was performed with VividSTORM (donkey anti-rabbit Alexa 647, 1:1000, #711-605-152, Jackson ImmunoResearch, donkey anti-mouse Alexa 488, 1:1000, #715-006-151, Jackson ImmunoResearch, donkey anti-rabbit CF568, 1:1000, #20098-1mg, Biotium). Images were captured with a Nikon N-STORM C2+ super-resolution system based on the platform of a Nikon Ti-E inverted microscope, equipped with a Nikon C2 confocal scan head and an Andor iXon Ultra 897 EMCCD camera and a CFI Apo TIRF 100× objective. Image acquisition and processing were performed using Nikon NIS-Elements AR software with N-STORM module.
Quantitative analysis
In the PLX5622-treated groups, microglia depletion was validated based on P2Y12R immunofluorescence (Figure 1(d)). Two mice were excluded from the analysis pre hoc due to insufficient microglia depletion (less than 85%).
All physiological variables (i.e. DC potential and [K+]e) were first downsampled to 1 Hz, then either analyzed with the inbuilt tools of AcqKnowledge 4.2.0 (BIOPAC System, USA) software, or were transferred into a MATLAB environment (MathWorks, USA). The transient negative DC shift indicative of SD was analyzed to assess amplitude of depolarization, duration of depolarization at half amplitude and area under the curve of post-SD hyperpolarization. Raw [K+]e data expressed in mV were translated into mM concentration using polynomial cubic regression (R2=1) on calibration solutions of data range (1, 3, 10, 30 mM).36 Polynomial regression was used to determine subtle changes in the lower range of the [K+]e concentration accurately. Baseline level of [K+]e was determined by sampling a 1-min average before induction of each SD. Changes in [K+]e during SD were analyzed to determine: peak [K+]e, and duration at half maximum and AUC of potassium elevation. The rate of [K+]e release was determined by linear regression, while half-time recovery of [K+]e to baseline ([K+]e clearance) was calculated by exponential regression as described previously.33
Immunofluorescent quantification
To assess the level of microglia depletion after PLX5622 treatment, images were captured with Zeiss Axiovert 200 M epi-fluorescent microscope and the number of P2Y12R-positive cells counted in three randomly selected ROIs at five different coronal sections from the cortex, hippocampus and striatum.
The number of activated neurons was counted based on c-fos immunostaining on three different coronal sections. Two ROIs (200 µm × 300 µm) were placed near to, and one ROI was placed distant from the stimulating electrode and the number of c-fos positive cells was averaged in each ROI.
Astrocyte reaction to SD was investigated by staining of glial fibrillary acidic protein (GFAP) and glutamine synthetase (GS) on free floating coronal brain sections and images were taken with a 20× objective by Nikon A1R confocal system guided by NIS-Elements Microscope Imaging Software. Integrated density values were measured in three ipsilateral and three contralateral ROIs (250 µm × 80 µm).
For precise microglial morphology analysis, sections were analyzed and only those included that had the best signal-to-noise ratio (crisp microglia-specific staining with low background). Confocal images were taken with a 60× objective by Nikon A1R confocal system guided by NIS-Elements Microscope Imaging Software with a Z-step of 1 µm. Two-dimensional morphological analysis was performed on maximum intensity projection images of the confocal stacks. Data were analyzed using GraphPad Prism 8.0. software.
STORM super-resolution microscopy analysis
The 3D STORM localization points were filtered for photon number, z position (within −300; 300 nm axial distances from the center plane) and local density using VividSTORM software.35 The clusters of selected localization points were determined by the density-based spatial clustering of applications with noise (DBSCAN) algorithm.
Statistical analysis
The statistical approach used has been specified in each figure legend. The software SPSS (SPSS Statistics, Version 22.0, IBM, USA) or the inbuilt statistical functions of MATLAB (MathWorks, USA) were used for statistical analysis. The distribution of the data was tested with Shapiro–Wilk normality test. Outliers were filtered with Grubbs test. Homogeneity of the variances was checked by Levene’s test. A Friedman analysis of variance (ANOVA) and Mann–Whitney U test were used to evaluate electrical threshold level differences of SD induction. A one-way ANOVA model followed by Tukey’s HSD or Games-Howell post hoc test was applied for the analysis of variables derived from the DC potential signature of SD, [K+]e shift with SD, whereas data from series 1 were evaluated by independent t-test or Welch-t test, dependent on the type of data set. A repeated measures paradigm was used to evaluate baseline [K+]e changes. Mann–Whitney U test was used to assess morphological changes (cell body area, number of branches) in microglia labelled with Iba1. Data from P2Y12R, P2Y12R/Kv2.1 immunofluorescence and combined P2Y12R confocal and STORM labeling were analyzed with unpaired-t test, while data from GFAP, GS, Kv2.1/c-fos immunofluorescence were analyzed using two-way ANOVA followed by Sidak’s multiple comparison. The level of significance was defined as p < 0.05* and p < 0.01**.
Data and software availability
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data. All reagents and protocols used in this study are available for sharing upon reasonable request to the authors.
Results
SD facilitates microglia-to-neuron interactions, while microglia depletion alters the electrical threshold of SD elicitation
SD travels across the cerebral cortex restricted to the ipsilateral hemisphere,37 and microglia are supposed to detect changes in neuronal activity induced by SD.26,38 To test the possible effect of repeated SDs on microglia, we first performed 2D morphology analysis on representative microglial cells sampled randomly from the ipsi- and the corresponding contralateral insular cortex of mice (Figure1(a) and (b)). We found a small, but significant decrease in the area of microglial cell bodies (ipsilateral: 36.14 µm2 (median, 32.2–42.2 interquartile range), n = 66 cells; contralateral: 38.84 µm2 (33.6–44.36), n = 79 cells from three mice), and the number of microglial processes originating from the soma was reduced by 17% (n = 79 cells from three mice). SD did not affect microglial numbers in the ipsilateral hemisphere (not shown). Next, we assessed the relationship between microglial processes and neurons in two representative areas in the cerebral cortex (Figure 1(c)). P2Y12R-positive microglial processes were recruited to the vicinity of neurons in the cerebral cortex after repeated SD induction leading to a 25% increase in microglial process density around the neuronal soma visualized by Kv2.1 immunostaining, which was most apparent in the ROI closer to SD induction (Figure 1(c)). This was in line with our earlier observation made under ischemic conditions by using in vivo two-photon imaging.16
To investigate the functional contribution of microglia to neuronal responses following a series of SDs, we selectively eliminated microglia from the brain (Figure 1(d)).8 The electric threshold of SD elicitation was expressed as the lowest electric charge sufficient to trigger SD (Figure 1(e) and (f)). The initiation of the first SD (SD1) required lower electric charge in the microglia depleted group (41 ± 30 vs. 88 ± 28 µC; depleted vs. control), whereas the electric threshold of elicitation for recurrent SDs (rSDs) increased considerably in the microglia depleted group (182 ± 120 vs. 85 ± 30 µC; microglia depleted vs. control) (Figure 1(f)). Statistical analysis confirmed that the elicitation of a series of subsequent rSDs at the absence of microglia required increasingly greater electric charge (from 41 ± 30 to 210 ± 120 µC, corresponding to SD1 and SD4 in the microglia-depleted group). In contrast, the charge necessary for SD elicitation did not change during a series of SDs in the control group (Figure 1(e)). Similarly, during the chemical elicitation (series 2), 1 M KCl proved to be sufficient to trigger SDs repeatedly in the control group, yet higher concentrations of KCl were required to induce SDs in the microglia depleted and P2Y12R KO groups (1.25 ± 0.57 and 1.81 ± 1.6 vs. 1.0 M KCl; microglia depleted and P2Y12R KO vs. control, Figure 1(g)). Thus, microglia appear to be substantially involved in the induction of SDs in the otherwise healthy brain.
The DC potential signature of SD is altered by selective microglia elimination or in the absence of functional P2Y12R signaling
P2Y12R signaling is essential for microglial process chemotaxis towards sites of tissue damage or neuronal hyperactivity in response to the release of purinergic metabolites such as ATP, ADP or adenosine,39 while adenosine is released to the extracellular space upon the development of SD.40 We found that a series of SDs resulted in increased P2Y12R immunopositivity (by 27%) in microglia in the ipsilateral side (Figure 2(a)). Next, we visualized P2Y12 receptors at 20 nm lateral resolution by using STORM super-resolution microscopy as established earlier,16 which are specific for microglia in the brain.8 We found that a series of SDs increased the density of P2Y12R on microglial processes in the vicinity of the neuronal soma (278 ± 29 and 404 ± 44 clustered NLP/cluster in the contralateral side vs. ipsilaterally after SD, Figure 2(b)), suggesting that recruited microglial processes upregulate and/or redistribute P2Y12R in response to SD. The absence of microglial P2Y12Rs also resulted in altered microglial response to repeated SDs: opposite to the decrease in cell body area and number of branches observed in wild type mice ipsilaterally to SDs, P2Y12R KO microglial cells showed an increase in these parameters after SDs (P2Y12R KO somatic area – ipsi: 1.11 of contralateral value (0.97–1.24), n = 66 cells; contra: 1 (0.88–1.25), n = 86 cells from three mice; P2Y12R KO number of branches – ipsi: 1.2 (1–1.4) of contralateral value; contra: 1 (0.8–1.2); Figure 2(c)).
Figure 2.

Selective elimination of microglia or absence of P2Y12R curbs SD and supports hyperpolarization after SD. (a) Confocal analysis revealed increased microglial P2Y12R mean fluorescence intensity (MFI) in response to a series of SDs (assessed in Area 1 according to Figure 1(a)). Unpaired t-test, n = 40 randomly selected microglia from the contralateral hemisphere and n = 38 microglia from the ipsilateral hemisphere from seven mice per group. (b) Representative images depict the enrichment of P2Y12R (STORM, cyan) on microglial processes (P2Y12R, confocal, red) recruited to the Kv2.1 (green) labeled neurons in the neocortex, contralateral and ipsilateral to SD elicitation. Scale bar, 1000 nm. STORM super-resolution microscopy reveals increased P2Y12R densities on microglial processes recruited to neurons, in response to SD. Mean±stdev values of the number of localization points (NLP) are shown normalized to P2Y12R clusters determined by the density-based spatial clustering of applications with noise (DBSCAN) algorithm (Local density filter: 10 neighbours within 150 nm Z-filter: ±300 nm from focal plane). Unpaired t test, p < 0.05*, n = 20 neurons randomly selected from seven mice per group. (c) Changes in microglial cell body area and the number of branches in P2Y12R KO mice as obtained from 2D morphological analysis of Iba1-positive microglia in the cerebral cortex (the region used for analysis is identical to Area 1 shown on Figure 1 Panel (a)). Values normalized to the contralateral side are shown as median±interquartile range. Mann–Whitney U test, (p < 0.05*). Scale bar, 10 μm. (d) The direct current (DC) potential signature of recurrent SDs (rSDs) (each trace is the mean of rSDs in each experimental group, series 2). Capital letters indicate variables quantitated in the respective Panels. (e) Amplitude of the negative DC potential shift of rSDs (series 1 and 2). (f) Duration of the negative DC potential shift of rSDs (series 1 and 2). (g) Area under the curve of the hyperpolarization after rSDs (series 1 and 2). In Panels (d)–(f), data are given as mean±stdev. Sample size is indicated in each bar. Statistical analysis of data in series 1 relied on an independent t-test or a Welch-t test. Data in series 2 were evaluated by a one-way ANOVA paradigm followed by a multiple comparison of Tukey for equal variances, or Games-Howell for unequal variances (p < 0.01** depleted vs. respective control, p < 0.05# and p < 0.01## P2Y12R KO vs. respective control).
Subsequently, we studied whether P2Y12R-mediated actions in microglia contribute to the evolution of SD. In agreement with the results that microglia depletion elevates the elicitation threshold of rSDs, both microglia depletion and the absence of P2Y12R decreased the amplitude of rSDs consistent for both electrical (series 1) and chemical (i.e. 1 M KCl, series 2) SD elicitation (−15.5 ± 2.8 vs. −18.4 ± 2.3 mV, microglia depleted vs. control, series 1; −18 ± 1.3 and −18.7 ± 1.5 vs. −21.4 ± 2.5 mV, microglia depleted and P2Y12R KO vs. control, series 2) (Figure 2(d) and (e)). However, the duration of depolarization was shorter only in the microglia-depleted groups, but not in P2Y12R KO mice, with respect to the control (23.3 ± 8.5 vs. 35.7 ± 6.2 s, microglia depleted vs. control, series 1; 20.9 ± 3.6 vs. 29 ± 9.3 and 25.6 ± 7.8 s, microglia depleted vs. control and P2Y12R KO, series 2) (Figure 2(f)). Finally, the AUC of post-SD hyperpolarization increased in both microglia-depleted and P2Y12R KO mice (166.8 ± 96 vs. 68.8 ± 49 mV × s, microglia depleted vs. control, series 1; 182.8 ± 54.3 vs. 138.6 ± 87.9 vs. 68.9 ± 41.1 mV × s, microglia depleted vs. P2Y12R KO vs. control, series 2) (Figure 2(g)). Thus, microglia appear to contribute to SD propagation – in part – via P2Y12R.
The absence of microglia but not of P2Y12R limits the duration of [K+]e shift during SD
The quantitative assessment of [K+]e variation by potassium-selective microelectrodes was conducted in series 2 (Figure 3(a), Supplementary Table 1). Baseline [K+]e (i.e. the value taken prior to the elicitation of each SD in a train) proved to be consistently higher in P2Y12R KO mice compared to controls or the microglia-depleted group (Figure 3(b)), although the higher concentration remained within the physiological range throughout (i.e. below 4–5 mM) and is, therefore, not expected to have significant pathophysiological consequences. In line with this, microglia depletion did not alter baseline [K+]e or physiological interstitial potassium levels in the cortex (Figure 3(b)).
The analysis of the [K+]e shift with rSDs did not reveal any notable difference in magnitude characterized by the peak amplitude or the AUC across different groups of mice (Figure 3(c) to (e)). However, the duration of [K+]e shift was considerably shorter in the absence of microglia compared to the other two groups (22.4 ± 4 vs. 33.8 ± 10.2 and 41.3 ± 10.6 s, microglia depleted vs. control and P2Y12R KO) (Figure 3(f)), which is consistent with the shorter duration of the DC potential deflection in the same mice (Figure 2(f)). At last, a more rapid clearance of [K+]e was revealed in microglia-depleted mice compared to the other groups. At the same time, [K+]e clearance in the P2Y12R KO mice was hindered with respect to both other groups (16.2 ± 3 vs. 21.8 ± 3.5 and 43.6 ± 21.1 s, microglia depleted vs. control and P2Y12R KO, Figure 3(g)). Taken together, activated microglia seem not only to elongate K+ release but also to decelerate [K+]e uptake, whereas P2Y12 receptor signaling seems not to be involved in these processes.
The absence of microglia alters neuronal activation in response to SD
Since the effects of microglia depletion and P2Y12R-deficiency on potassium uptake after SDs were different and astrocytes are known to contribute to K+ clearance in the brain,41 we investigated whether SD induction resulted in changes in astrocyte GFAP and glutamine synthetase (GS) levels. No significant changes in the integrated density of GFAP and GS immunofluorescence were seen in response to SD (Figure 4(a)), and levels were not altered in P2Y12R KO or microglia-depleted mice (Figure 4(b)). We next investigated whether the absence of microglia or P2Y12R-deficiency resulted in any changes in neuronal activation in response to SD. The transcription of c-fos is controlled by an increase in the intracellular Ca2+ concentration,42 which occurs in neurons in response to SD.43,44 We found that recurrent SDs markedly increased neuronal c-fos expression in the ipsilateral hemisphere (by 54% in Area 1, 88% in Area 2 and 124% in Area 3), compared to the contralateral side (Figure 4(c)). Importantly, the number of c-fos-positive neurons markedly increased in the microglia-depleted group, which was most apparent in the insular cortex and less obvious in the cingulum, in line with the area of SD induction and propagation (Figure 4(c)). These differences were not seen in the insular cortex of P2Y12R KO mice (Figure 4(d)).
Figure 4.

The absence of microglia is associated with augmented neuronal activation after spreading depolarization (SD). (a) Schematic showing the areas used for quantitative analysis relative to the site of SD induction. (b) Representative images showing glial fibrillary acidic protein (GFAP) and glutamine synthetase (GS) immunofluorescence in the ipsilateral hemisphere 1.5 h after SD induction (Area 1 is displayed). Graphs showing integrated density values of GFAP and GS signal normalized to the corresponding areas of the contralateral hemisphere as mean±stdev. Two-way ANOVA followed by Sidak’s multiple comparison. (c) Representative images demonstrate activated, c-fos labeled (green) neurons (Kv2.1, a voltage-dependent K+ channel; magenta) in the cerebral cortex ipsilateral to SD elicitation (mean±stdev). Mice were sacrificed 1.5 h after the induction of the first SD in a train of four events, to allow the assessment of c-fos protein expression. Scale bar, 10 µm. Quantification of c-fos protein expression was performed in areas indicated in the schematic coronal brain section in Panel (a) (ROI: 300 µm × 200 µm). p < 0.01**, control vs. depleted (ipsilaterally), two-way ANOVA followed by Sidak’s multiple comparison. (d) Representative images demonstrate activated, c-fos labeled (green) neurons (Kv2.1, magenta) in the cerebral cortex ipsilateral to SD elicitation in control and P2Y12R KO mice (Area 1 on panel A is shown). Quantification of c-fos protein expression was performed in areas indicated in the schematic coronal brain section in Panel (a) (ROI: 300 µm × 200 µm, only Area 1 and Area 2 assessed based on the results of microglia depletion studies). n = 4–7, data are expressed mean±stdev.
Discussion
Here, we identify microglia as essential modulators of the induction of SD in the physiologically intact brain, in vivo. Moreover, we show that SD alters microglial responses that include enhanced interaction between microglia and neurons, which is associated with sustained potassium uptake and altered neuronal activity after SD. Indeed, c-fos expression induced by SD is attenuated by the inhibition of NMDA receptors or voltage-gated calcium channels,44,45 both involved in physiological neuronal signaling,46 SD evolution47 and excitotoxic injury.48 Our results also provide mechanistic insight into how microglial responses are altered by SD, in part via P2Y12R.
First, we sought to determine whether microglia react to SD in the physiologically intact mouse brain in vivo. Our histological findings that activated microglial processes are recruited to neurons in response to SD, while P2Y12Rs are upregulated and redistributed at this interface extrapolate that microglia sense SD and are ideally positioned to modulate neuronal activity in the non-ischemic brain. In line with our findings, the upregulation of P2Y12R on spinal cord microglia was found to promote hypersensitivity of the nociceptive network in the development of neuropathic pain after peripheral nerve injury.49
Subsequently, we set out to investigate whether selective elimination of microglia alters SD induction in the mouse brain. With constant current stimulation 1 mm rostral to the implanted microelectrodes, we reliably induced SD with no interference with the acquired electrophysiological parameters,34 while the sensibility of the nervous tissue for SD elicitation could be precisely determined. Our data indicate that microglia are essential to maintain the susceptibility of the cerebral cortex for SD. Interestingly, the absence of microglia may render the otherwise intact brain more sensitive to the initiation of an SD, whereupon subsequent SDs are impeded. Microglia have been shown to downregulate both spontaneous and evoked neuronal activities in the optic tectum of larval zebrafish, while injury-induced microglial recruitment augmented neuronal firing.6 The increased neuronal activity state may make it easier to simultaneously discharge a sufficient number of neurons in order to generate an SD via electrical stimulation.47 This suggests that microglial actions may also shape SD induction or propagation differently in different forms of neuropathologies. For example, microglia activated by SD or other stimuli may induce a more proinflammatory milieu making the brain prone to recurrent SDs,26,28 while an absence of microglia was found to reduce SD incidence in the evolving penumbra after experimental stroke in mice.16 Thus, these observations provide evidence for the first time that microglia effectively modulate SD threshold in the non-injured brain in vivo, similar to that seen during brain ischemia,16 indicating that SD susceptibility seems to depend on the activity status of microglia (Figure 5).
Figure 5.
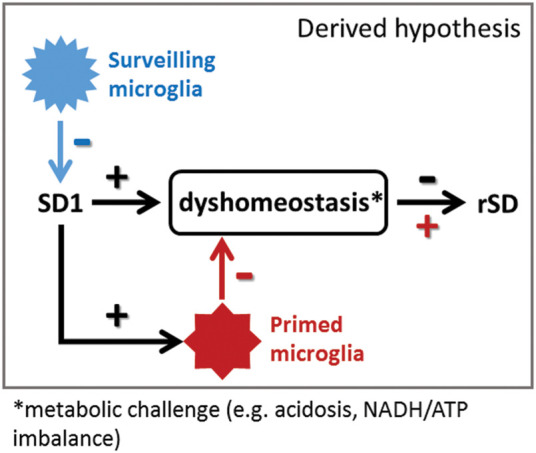
Surveilling microglia may favor lower tissue susceptibility to SD, while primed microglia could support SD occurrence. We propose that resting microglia balance out neuronal overactivation and make SD occurrence less likely. Once SD occurs, the homeostasis of the nervous tissue is challenged, microglia transform to a primed state, which together preserve or increase the susceptibility of the nervous tissue to sustain subsequent SDs.
Surveilling microglia may not only respond rapidly to SD elicitation, but are also expected to change their activity and produce inflammatory mediators to shape the excitability of the nervous tissue during recurrent SD events. This is in agreement with the in vitro observations that pro-inflammatory cytokines, especially TNFα released from activated microglia lowered SD threshold,28 and reduced SD amplitude.50 Besides, there is an existing continuous outward potassium current on surveilling microglia through a recently identified two-pore domain K+ channel,9 which can be potentiated by P2Y12R activation.10 Microglial slow outward potassium currents therefore could curtail the neuronal transmembrane potassium leakage, shifting neuronal membrane potential to more positive values and altering the excitability of neurons.51
We also found that the SD-related transient negative shift of DC potential was markedly attenuated by microglia depletion, and in mice lacking the P2Y12R. Although there is no ultimate consensus on the conditions that govern SD amplitude, a causal link has recently been established between the amplitude of the negative DC shift and the total number of synchronously discharged cortical neurons.52 Considering this association, the reduced SD amplitude observed in our studies may infer the involvement of a possibly smaller cell mass locally depolarized, thus reduced neuronal excitability in the absence of activated microglia, in line with increased SD threshold for recurring SDs. Our results confirm that the excitability of the nervous tissue after SD occurrence is modulated by microglial responses implicating P2Y12R signaling in this process. The repolarization after SD is achieved by the ATP-consuming Na+/K+ pump and astrocytic buffering of K+ and glutamate,20,47,53,54 the efficacy of which is subject to the metabolic status of the tissue.55 Our data indicate that the presence of activated microglia decelerates the uptake of the accumulated [K+]e or glutamate. This process, however, may not require P2Y12R signaling, suggesting that additional microglia-dependent mechanisms also contribute to the restoration of extracellular ion homeostasis after SD. The hyperpolarization that follows an SD is thought to be a transient overshoot of extracellular Cl−, which accompanies cationic changes due to an increased pumping activity of the Na+/K+ ATPase.20 This seems to be consistent with the finding that the absence of activated microglia curbs the peak of depolarization. The smaller depolarization and the more enhanced hyperpolarization observed in the microglia-depleted mice all together signify that microglia shift the balance from hyperpolarization to depolarization, possibly by restraining Na+/K+ ATPase activity.
The evolution of SD is dependent on a massive efflux of potassium through tetraethylammonium-sensitive, ATP-sensitive or large-conductance Ca2+-activated potassium channels,56,57 all of which are also expressed by microglia.58,59 Accordingly, we hypothesized that activated microglia may tune the susceptibility of the cortex to SD by modulating transmembrane potassium currents. Thus, we set out to evaluate [K+]e dynamics with SD at the absence of microglia or P2Y12R. We observed shorter [K+]e-related changes during SDs and facilitated K+ uptake following SDs only in the microglia-depleted group. In contrast to microglia depletion, K+ reuptake was found hindered in the P2Y12R KO animals. Even though the activation of P2Y12R enhances microglial outward potassium currents, which was shown to be countered by pharmacological blockade of the microglial P2Y12R,9–11 and thus P2Y12R appears to be implicated in K+ homeostasis, the mechanism behind the low rate of [K+]e clearance in the absence of P2Y12R cannot be identified with certainty. In line with this, while astrocytes are known for their contribution to the uptake of extracellular potassium, levels of astrocyte markers did not differ between control, microglia-depleted and P2Y12R KO mice. However, astrocyte-microglia interactions may still be altered in microglia-depleted and P2Y12R KO mice, which are not reflected by the markers assessed within this time frame upon SD induction. Similarly, the potential interactions between microglial P2Y12R and neuronal potassium channels such as Kv2.1 will need to be investigated in future studies. Here, Kv2.1 was merely used to visualize neuronal membranes for microglia–neuron interactions and its functional contribution to SD propagation or microglial activity was not investigated. The duration of [K+]e shift is determined by a competing force of opposing potassium currents. Activated microglia may contribute to the potassium outflow through inward rectifying, calcium-activated or ATP-sensitive K+ channels,58–60 independent of P2Y12R signaling. Likewise, increased barium sensitive K+ conductance of microglial cells was shown in response to SD in vitro.26 As the recovery from SD is critically dependent on the activity of the Na+/K+-ATPase, as well as the buffering capacity of the astrocyte network,54 our results indicate that microglia modulate the sustenance or termination of SD by interacting with either of these mechanisms. Here we identified potassium homeostasis as a novel microglia-associated SD-regulatory pathway, in addition to the previously proposed cytokine signalization.28 Furthermore, activated microglia have been known to alter glutamate homeostasis61 and are critical source of the pro-inflammatory IL-1β in the brain,11 both of which have been shown to modulate neuronal sensitivity12,14 and influence either the induction or the evolution of SD.21,47,62 Thus, it is conceivable that these additional, microglia-linked pathways are also implicated in the modulation of SD evolution.
To investigate how selective elimination of microglia affects neuronal activation in the cerebral cortex after SD, we turned to c-fos immunofluorescence. Spreading depolarization induces the expression of the early proto-oncogene c-fos, which is a widely used marker of neuronal activation.45,49 We observed elevated c-fos expression ipsilateral to the SD induction. Based on the known contribution of NMDA receptors and voltage-gated calcium channels to c-fos expression during SD,45,63 microglia are proposed to suppress calcium influx through these channels, and thus protect neurons against SD-related hyperactivity or potential injury. The lack of a major effect of P2Y12R-deficiency on potassium clearance and neuronal c-fos expression collectively suggests that microglia shape neuronal activity via different mechanisms after SD, which is only partially dependent on microglial P2Y12R.
In conclusion, the presence of microglia is likely to be essential to maintain the electrical elicitation threshold and to support the full evolution of SD, partly by sustaining the release or the cellular clearance of [K+]e. While microglial P2Y12R signaling is suggested to take part in sensitizing the tissue to SD, it appears that activated microglia decelerate the recovery from SD independent of their P2Y12 receptors. Altogether, our findings suggest that surveilling microglia may favor SD suppression, while in turn, SD-induced microglial activation preserves the susceptibility of the nervous tissue to sustain subsequent, recurrent SDs.
Supplemental Material
Supplemental material, JCB900097 Supplemental Figure for Microglia alter the threshold of spreading depolarization and related potassium uptake in the mouse brain by Dániel P Varga, Ákos Menyhárt, Balázs Pósfai, Eszter Császár, Nikolett Lénárt, Csaba Cserép, Barbara Orsolits, Bernadett Martinecz, Tamás Szlepák, Ferenc Bari, Eszter Farkas and Ádám Dénes in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
The authors are grateful for the technical assistance provided by Orsolya Ivánkovitsné Kiss. We thank Plexxikon Inc. (Berkeley, USA) for the generous contribution of PLX5622 diet.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from National Research, Development and Innovation Office (NKFIH: K120358, K111923, PD128821), the Ministry for National Economy of Hungary (GINOP-2.3.2-15-2016-00048), the Ministry of Human Capacities of Hungary (EFOP-3.6.1-16- 2016-00008) the Momentum Program of the Hungarian Academy of Sciences (LP2016-4/2016), by ERC-CoG 724994, by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences, and by the Ministry for Innovation and Technology (ÚNKP-19-3-I and ÚNKP-19-4).
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions: Dániel P. Varga, Conceptualization, Methodology, Investigation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing; Ákos Menyhárt, Methodology, Investigation, Formal analysis, Writing – review and editing; Eszter Császár, Investigation, Formal analysis, Visualization; Bernadett Martinecz, Investigation, Visualization; Tamás Szlepák, Investigation, Visualization; Balázs Pósfai, Methodology, Investigation, Formal analysis, Writing – review and editing; Nikolett Lénárt, Methodology, Investigation, Writing – review and editing; Barbara Orsolits, Methodology, Investigation; Csaba Cserép, Methodology, Investigation, Formal analysis, Writing – review and editing; Ferenc Bari, Writing – review and editing; Eszter Farkas, Conceptualization, Supervision, Writing – original draft; Writing – review and editing, Funding acquisition; Ádám Dénes, Conceptualization, Supervision, Methodology, Writing – review and editing, Funding acquisition, Project administration.
Supplemental material: Supplemental material for this article is available online.
ORCID iDs
Balázs Pósfai https://orcid.org/0000-0003-1035-565X
Csaba Cserép https://orcid.org/0000-0001-5513-2471
References
- 1.Shinozaki Y, Shibata K, Yoshida K, et al. Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y1receptor downregulation. Cell Rep 2017; 19: 1151–1164. [DOI] [PubMed] [Google Scholar]
- 2.Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in toll-like receptor-mediated neuronal injury. Glia 2010; 58: 253–263. [DOI] [PubMed] [Google Scholar]
- 3.Neumann H, Kotter MR, Franklin RJM. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 2009; 132: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011; 333: 1456–1458. [DOI] [PubMed] [Google Scholar]
- 5.Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013; 155: 1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Du XF, Liu CS, et al. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell 2012; 23: 1189–1202. [DOI] [PubMed] [Google Scholar]
- 7.Eyo UB, Peng J, Swiatkowski P, et al. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci 2014; 34: 10528–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fekete R, Cserép C, Lénárt N, et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol 2018; 136: 461–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madry C, Kyrargyri V, Arancibia-Cárcamo IL, et al. Microglial ramification, surveillance, and interleukin-1β release are regulated by the two-pore domain K + channel THIK-1. Neuron 2018; 97: 299–312.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swiatkowski P, Murugan M, Eyo UB, et al. Activation of microglial P2Y12 receptor is required for outward potassium currents in response to neuronal injury. Neuroscience 2016; 318: 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kettenmann H, Hanisch U-K, Noda M, et al. Physiology of microglia. Physiol Rev 2011; 91: 461–553. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi H, Jin S, Wang J, et al. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 2006; 281: 21362–21368. [DOI] [PubMed] [Google Scholar]
- 13.Carmen J, Rothstein JD, Kerr DA. Tumor necrosis factor-α modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res 2009; 1263: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klapal L, Igelhorst BA, Dietzel-Meyer ID. Changes in neuronal excitability by activated microglia: differential Na+ current upregulation in pyramid-shaped and bipolar neurons by TNF-α and IL-18. Front Neurol 2016; 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elmore MRP, Najafi AR, Koike MA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014; 82: 380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szalay G, Martinecz B, Lénárt N, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016; 7: 11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 2014; 15: 379–393. [DOI] [PubMed] [Google Scholar]
- 18.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011; 17: 439–447. [DOI] [PubMed] [Google Scholar]
- 19.Chen S-P, Qin T, Seidel JL, et al. Inhibition of the P2X7–PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain 2017; 140: 1643–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand 1981; 113: 437–45. [DOI] [PubMed] [Google Scholar]
- 21.Zhou N, Rungta RL, Malik A, et al. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J Cereb Blood Flow Metab 2013; 33: 1582–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steffensen AB, Sword J, Croom D, et al. Chloride cotransporters as a molecular mechanism underlying spreading depolarization-induced dendritic beading. J Neurosci 2015; 35: 12172–12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron 2015; 86: 902–922. [DOI] [PubMed] [Google Scholar]
- 24.Jander S, Schroeter M, Peters O, et al. Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J Cereb Blood Flow Metab 2001; 21: 218–225. [DOI] [PubMed] [Google Scholar]
- 25.Takizawa T, Qin T, Lopes de Morais A, et al. Non-invasively triggered spreading depolarizations induce a rapid pro-inflammatory response in cerebral cortex. J Cereb Blood Flow Metab. Epub ahead of print 26 June 2019. DOI: 10.1177/0271678X19859381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wendt S, Wogram E, Korvers L, et al. Experimental cortical spreading depression induces NMDA receptor dependent potassium currents in microglia. J Neurosci 2016; 36: 6165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caggiano AO, Kraig RP. Eicosanoids and nitric oxide influence induction of reactive gliosis from spreading depression in microglia but not astrocytes. J Comp Neurol 1996; 369: 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pusic KM, Pusic AD, Kemme J, et al. Spreading depression requires microglia and is decreased by their M2a polarization from environmental enrichment. Glia 2014; 62: 1176–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuura T, Bureš J. The minimum volume of depolarized neural tissue required for triggering cortical spreading depression in rat. Exp Brain Res 1971; 12: 238–249. [DOI] [PubMed] [Google Scholar]
- 30.Grafstein B. Mechanism of spreading cortical depression. J Neurophysiol 1956; 19: 154–71. [DOI] [PubMed] [Google Scholar]
- 31.Viitanen T, Ruusuvuori E, Kaila K, et al. The K+-Cl- cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol 2010; 588: 1527–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bazzigaluppi P, Dufour S, Carlen PL. Wide field fluorescent imaging of extracellular spatiotemporal potassium dynamics in vivo. Neuroimage 2015; 104: 110–116. [DOI] [PubMed] [Google Scholar]
- 33.Yao X, Smith AJ, Jin BJ, et al. Aquaporin-4 regulates the velocity and frequency of cortical spreading depression in mice. Glia 2015; 63: 1860–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hertelendy P, Menyhart A, Makra P, et al. Advancing age and ischemia elevate the electric threshold to elicit spreading depolarization in the cerebral cortex of young adult rats. J Cereb Blood Flow Metab 2017; 37: 1763–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barna L, Dudok B, Miczán V, et al. Correlated confocal and super-resolution imaging by VividSTORM. Nat Protoc 2016; 11: 163–83. [DOI] [PubMed] [Google Scholar]
- 36.Hansen AJ. Extracellular potassium concentration in juvenile and adult rat brain cortex during anoxia. Acta Physiol Scand 1977; 99: 412–420. [DOI] [PubMed] [Google Scholar]
- 37.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grinberg YY, Milton JG, Kraig RP. Spreading depression sends microglia on Lévy flights. PLoS One 2011; 6: e19294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 2006; 9: 1512–1519. [DOI] [PubMed] [Google Scholar]
- 40.Kaku T, Hada J, Hayashi Y. Endogenous adenosine exerts inhibitory effects upon the development of spreading depression and glutamate release induced by microdialysis with high K+ in rat hippocampus. Brain Res 1994; 658: 39–48. [DOI] [PubMed] [Google Scholar]
- 41.Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience 2004; 129: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaczmarek L. c-Fos in learning: beyond the mapping of neuronal activity In: Kaczmarek L, Robertson HA. (eds) Handbook of chemical neuroanatomy. Stockholm: Elsevier, 2002, pp.189–215. [Google Scholar]
- 43.Siesjo BK, Bengtsson F. Calcium fluxes, calcium antagonists, and calcium-related pathology in brain ischemia, hypoglycemia, and spreading depression: a unifying hypothesis. J Cereb Blood Flow Metab 1989; 9: 127–140. [DOI] [PubMed] [Google Scholar]
- 44.Reinhart KM, Shuttleworth CW. Ketamine reduces deleterious consequences of spreading depolarizations. Exp Neurol 2018; 305: 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrera DG, Robertson HA. Application of potassium chloride to the brain surface induces the c-fos proto-oncogene: reversal by MK-801. Brain Res 1990; 510: 166–170. [DOI] [PubMed] [Google Scholar]
- 46.Mayford M, Siegelbaum SA, Kandel ER. Synapses and memory storage. Cold Spring Harb Perspect Biol 2012; 4: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hertelendy P, Varga DP, Menyhárt Á, et al. Susceptibility of the cerebral cortex to spreading depolarization in neurological disease states: the impact of aging. Neurochem Int 2019;127: 125–136. [DOI] [PubMed] [Google Scholar]
- 48.Brouns R, De Deyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clin Neurol Neurosurg 2009; 111: 483–95. [DOI] [PubMed] [Google Scholar]
- 49.Kobayashi K, Yamanaka H, Fukuoka T, et al. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci 2008; 28: 2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richter F, Lütz W, Eitner A, et al. Tumor necrosis factor reduces the amplitude of rat cortical spreading depression in vivo. Ann Neurol 2014; 76: 43–53. [DOI] [PubMed] [Google Scholar]
- 51.Kato G, Inada H, Wake H, et al. Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity. eNeuro 2016; 3: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Speckmann E-J, Elger CE and Gorji A. Neurophysiologic basis of EEG and DC potentials. In: Schomer DL, da Silva LF (eds) Niedermeyer’s Electroencephalography: Basic Principles, Clinical Applications, and Related Fields. 6th ed. Philadelphia: Lippincott Williams and Wilkins, 2011, pp.17–32.
- 53.Walz W. Role of astrocytes in the spreading depression signal between ischemic core and penumbra. Neurosci Biobehav Rev 1997; 21: 135–142. [DOI] [PubMed] [Google Scholar]
- 54.Seidel JL, Escartin C, Ayata C, et al. Multifaceted roles for astrocytes in spreading depolarization: a target for limiting spreading depolarization in acute brain injury? Glia 2016; 64: 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feuerstein D, Backes H, Gramer M, et al. Regulation of cerebral metabolism during cortical spreading depression. J Cereb Blood Flow Metab 2016; 36: 1965–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menyhárt Á, Farkas AE, Varga DP, et al. Large-conductance Ca2+-activated potassium channels are potently involved in the inverse neurovascular response to spreading depolarization. Neurobiol Dis 2018; 119: 41–52. [DOI] [PubMed] [Google Scholar]
- 57.Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 2001; 81: 1065–1096. [DOI] [PubMed] [Google Scholar]
- 58.Cocozza G, di Castro MA, Carbonari L, et al. Ca2+-activated K+ channels modulate microglia affecting motor neuron survival in hSOD1G93A mice. Brain Behav Immun 2018; 73: 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du RH, Sun HB, Hu ZL, et al. Kir6.1/K-ATP channel modulates microglia phenotypes: implication in Parkinson’s disease. Cell Death Dis 2018; 9: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Lucente J, Nguyen HM, Wulff H, et al. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia 2018; 66: 1881–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takaki J, Fujimori K, Miura M, et al. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflammation 2012; 9: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richter F, Eitner A, Leuchtweis J, et al. Effects of interleukin-1ß on cortical spreading depolarization and cerebral vasculature. J Cereb Blood Flow Metab 2017; 37: 1791–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shimazawa M, Hara H, Watano T, et al. Effects of Ca2+channel blockers on cortical hypoperfusion and expression of c‐Fos‐like immunoreactivity after cortical spreading depression in rats. Br J Pharmacol 1995; 115: 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, JCB900097 Supplemental Figure for Microglia alter the threshold of spreading depolarization and related potassium uptake in the mouse brain by Dániel P Varga, Ákos Menyhárt, Balázs Pósfai, Eszter Császár, Nikolett Lénárt, Csaba Cserép, Barbara Orsolits, Bernadett Martinecz, Tamás Szlepák, Ferenc Bari, Eszter Farkas and Ádám Dénes in Journal of Cerebral Blood Flow & Metabolism
Data Availability Statement
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data. All reagents and protocols used in this study are available for sharing upon reasonable request to the authors.