

Abstract
Aralar/AGC1/Slc25a12, the mitochondrial aspartate-glutamate carrier expressed in neurons, is the regulatory component of the NADH malate-aspartate shuttle. AGC1 deficiency is a neuropediatric rare disease characterized by hypomyelination, hypotonia, developmental arrest, and epilepsy. We have investigated whether β-hydroxybutyrate (βOHB), the main ketone body (KB) produced in ketogenic diet (KD), is neuroprotective in aralar-knock-out (KO) neurons and mice. We report that βOHB efficiently recovers aralar-KO neurons from deficits in basal-stimulated and glutamate-stimulated respiration, effects requiring βOHB entry into the neuron, and protects from glutamate excitotoxicity. Aralar-deficient mice were fed a KD to investigate its therapeutic potential early in development, but this approach was unfeasible. Therefore, aralar-KO pups were treated without distinction of gender with daily intraperitoneal injections of βOHB during 5 d. This treatment resulted in a recovery of striatal markers of the dopaminergic system including dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC)/DA ratio, and vesicular monoamine transporter 2 (VMAT2) protein. Regarding postnatal myelination, myelin basic protein (MBP) and myelin-associated glycoprotein (MAG) myelin proteins were markedly increased in the cortices of βOHB-treated aralar-KO mice. Although brain Asp and NAA levels did not change by βOHB administration, a 4-d βOHB treatment to aralar-KO, but not to control, neurons led to a substantial increase in Asp (3-fold) and NAA (4-fold) levels. These results suggest that the lack of increase in brain Asp and NAA is possibly because of its active utilization by the aralar-KO brain and the likely involvement of neuronal NAA in postnatal myelination in these mice. The effectiveness of βOHB as a therapeutic treatment in AGC1 deficiency deserves further investigation.
SIGNIFICANCE STATEMENT Aralar deficiency induces a fatal phenotype in humans and mice and is associated with impaired neurodevelopment, epilepsy, and hypomyelination. In neurons, highly expressing aralar, its deficiency causes a metabolic blockade hampering mitochondrial energetics and respiration. Here, we find that βOHB, the main metabolic product in KD, recovers defective mitochondrial respiration bypassing the metabolic failure in aralar-deficient neurons. βOHB oxidation in mitochondria boosts the synthesis of cytosolic aspartate (Asp) and NAA, which is impeded by aralar deficiency, presumably through citrate-malate shuttle. In aralar-knock-out (KO) mice, βOHB recovers from the drastic drop in specific dopaminergic and myelin markers. The βOHB-induced myelin synthesis occurring together with the marked increment in neuronal NAA synthesis supports the role of NAA as a lipid precursor during postnatal myelination.
Keywords: ARALAR/AGC1 deficiency, β-hydroxybutyrate, ketogenic diet, malate-aspartate shuttle, mitochondrial aspartate-glutamate carrier, mitochondrial disorders
Introduction
Aralar/AGC1 is an electrogenic mitochondrial transporter that extrudes aspartate (Asp) from the mitochondria to the cytosol in exchange for glutamate, and, as the regulatory component of the malate-aspartate shuttle (MAS), plays a crucial role in the transfer of NADH from the cytosol into the mitochondria. AGC1 is expressed in all excitable tissues and, in the CNS, is mainly or exclusively restricted to neurons (Ramos et al., 2003; Berkich et al., 2007; Xu et al., 2007; Pardo et al., 2011). The absence of Aralar-MAS induces a decrease in mitochondrial respiration in neurons but not in astrocytes (Gómez-Galán et al., 2012; Llorente-Folch et al., 2013a; Juaristi et al., 2017). Aralar-knock-out (KO) mice have a short life expectancy [as they die around postnatal day (PND)20–PND22] and develop motor-coordination deficits from PND12 onward, prominent hypomyelination throughout the CNS, epilepsy, and pronounced deficits in Asp and NAA levels in brain and in cultured neurons (Jalil et al., 2005). Besides, aralar-KO mice show hyperactivity, anxiety-like behavior, hyperreactivity, and a failure in the nigrostriatal dopaminergic system (Llorente-Folch et al., 2013b).
In humans, Aralar/AGC1 deficiency causes the rare disease “global cerebral hypomyelination” (OMIM #612949, also named early infantile epileptic encephalopathy 39), a neurodevelopmental disease that matches aralar-KO mice phenotype, as it is characterized by severe hypomyelination, hypotonia, neurodevelopmental arrest, and seizures (Wibom et al., 2009; Falk et al., 2014; Kavanaugh et al., 2019; Pfeiffer et al., 2020). The first patient described for Aralar/AGC1 deficiency initiated a treatment with ketogenic diet (KD) at the age of 6 and for at least 19 months. At this time, the response of this affected patient to the treatment was reported to be dramatic (Dahlin et al., 2015), presenting a clear improvement in psychomotor development and resumed myelination.
KD has a high fat content (80–90%) with little but sufficient protein, and a drastic reduction in carbohydrates content that leads to a switch from glucose to ketogenic metabolism. KD contains both long-chain fatty acids (LCFA) and medium-chain fatty acids (MCFA), which give rise to ketone bodies (KBs) in the liver, increasing the KB: glucose ratio in circulation. KD has been proved to be beneficial in patients with pharmaco-resistant epilepsy (Klepper et al., 2007; Kossoff et al., 2009; Villeneuve et al., 2009; Kessler et al., 2011) and, interestingly, in several metabolic disorders like GLUT-1 deficiency (Alter et al., 2015) and MPC-1 deficiency (Vanderperre et al., 2016). Indeed, it has also been proposed as a therapeutic diet in several neurologic diseases as Alzheimer's disease (Van der Auwera et al., 2005), amyotrophic lateral sclerosis (Zhao et al., 2006), Huntington's disease (Ruskin et al., 2011), autism (Ruskin et al., 2013), Parkinson's disease (VanItallie et al., 2005), and as previously mentioned in two patients with Aralar/AGC1 deficiency (Dahlin et al., 2015; Pfeiffer et al., 2020).
Similarly, the administration in vivo of β-hydroxybutyrate (βOHB), the main metabolic product of KD, has been shown to have the same anticonvulsant and neuroprotective properties as KD (for review, see Maalouf et al., 2009). Understanding the specific role of βOHB in the effects of KD has a special interest in Aralar/AGC1 deficiency because only KB, but not KD lipids, are metabolized by neurons (Thevenet et al., 2016). Therefore, this work is focused on the study of the therapeutic potential of both KD and βOHB administered early in development to aralar-deficient mice. However, it was not possible to complete a KD treatment on aralar-KO mice. On the other hand, our study demonstrates βOHB to be highly effective to rescue reduced basal and agonist-stimulated mitochondrial respiration in aralar-KO neurons bypassing the metabolic failure imposed by Aralar/MAS deficiency. Importantly, βOHB treatment to aralar-KO mice recovered deficiencies both in specific dopaminergic markers and in postnatal myelin synthesis in this mouse model.
Materials and Methods
Animals
Mice with a mixed SVJ129 x C57BL/6 genetic background carrying a decifiency for Aralar/AGC1/Slc25a12 expression [aralar wild-type (WT); heterozygous, aralar+/−; and KO], were obtained from Lexicon Pharmaceuticals Inc (Jalil et al., 2005). All mice used were aged-matched littermates from aralar+/− breeding pairs, without distinction of gender. Mice were housed in a humidity-controlled and temperature-controlled room on a 12/12 h light/dark cycle, receiving water and food ad libitum. All animal procedures were approved by the corresponding institutional ethical committee (Center of Molecular Biology Severo Ochoa) and Autónoma University (CEEA-CBMSO-23/159), and were performed in accordance with Spanish regulations (BOE 67/8509–12, 1988) and European regulations (EU directive 86/609, EU decree 2001–486), reporting followed the ARRIVE Guidelines. All efforts were made to minimize the number of animals used and their suffering.
Neuronal cell culture
Neuronal cultures were obtained from embryonic day (E)15–E16 mouse embryos as previously described (Ramos et al., 2003; Pardo et al., 2006), from crosses between SVJ129 x C57BL/6 aralar+/− mice, and embryonic tissue samples were preserved for genotype determination of each embryo as previously described for aralar (Jalil et al., 2005). Neurons were maintained in a serum-free B27, glutaMAX, and antibiotics supplemented neurobasal medium (NB; Invitrogen) until day in vitro (DIV)9 for experimentation. Neurons represented >80% of the total cell population (Ramos et al., 2003; Pardo et al., 2006).
KD and βOHB treatments
For all the in vivo experiments, crosses between SVJ129 x C57BL/6 aralar+/− mice were set. Aralar+/− pregnant mothers were fed a standard diet (SD) or KD from the day they were crossed or 5 d after delivery. SD consisted of 4% fat, 60.5% carbohydrate, and 18% protein and offered a metabolizable energy of 16.95 MJ/kg (Safe-Diets, U8404610R); KD consisted of 79.2% fat, 6.3% carbohydrate, and 8% protein and offered a metabolizable energy of 31.6 MJ/kg (SSniff, E15149-30). For the experiments performed with βOHB, intraperitoneal injections of vehicle (i.e., 0.9% NaCl) or 290 mg/kg/d βOHB (DL-β-hydroxybutyric acid sodium salt, Sigma-Aldrich) were performed to WT and aralar-KO litters from PND12 to PND16. Gross abnormalities, body weight, and growth were checked for each pup. On PND17, pups were killed, and brains were extracted for dissection of cerebral regions and its preservation at −80°C.
Measurement of cellular oxygen consumption
Oxygen consumption rate (OCR) in intact neurons was measured using Seahorse XF24 Extracellular Flux Analyzer (Agilent) as previously described (Llorente-Folch et al., 2013a). Primary cortical neurons were maintained in NB (25 mm glucose) until DIV9–DIV10, or were preconditioned during 48 h in glucose-free NB A medium (NB-A; Invitrogen) supplemented with 5 mm glucose, B27, glutaMAX, and antibiotics. Cells were equilibrated with bicarbonate-free low-buffered DMEM (without pyruvate, L-lactate, glucose, glutamine, and calcium) supplemented with 2.5 mm glucose and 2 mm CaCl2 for 1 h before XF assay. Treatments with 5 mm βOHB, 5 mm acetoacetate (AcAc), 1 mm AR-C155858 (AR-C1) or 20 μm dimethylfumarate (DMF) were performed as detailed in figure legends. Sequential addition of medium or 50 μm glutamate (Glu50), 6 μm oligomycin (Oli), 0.5 mm 2,4-dinitrophenol (DNP), and 1 μm rotenone/1 μm antimycin (R/A) were performed when indicated. Basal O2 consumption, O2 consumption linked to ATP synthesis (ATP-linked), non-ATP linked O2 consumption (H+-leak), mitochondrial uncoupled respiration (MUR), and nonmitochondrial O2 consumption parameters were determined (Qian and Van Houten, 2010; Brand and Nicholls, 2011). Protein from each well was extracted with 0.1% NP-40 PBS solution and quantified with BCA protein assay kit (ThermoFisher), and data were normalized from protein concentration. Non-mitochondrial OCR was subtracted to OCR values and normalized from basal values.
Measurement of cytosolic pH
Cytosolic pH was measured in neurons with the fluorescent probe 2',7'-bis(2-carboxyethyl)−5(6)-carboxy-fluorescein (BCECF-AM; Invitrogen). At DIV9, neurons were incubated during 30 min at 37°C in HEPES-buffered control salted solution (HCSS; 137 mm NaCl, 1.25 mm MgSO4, 10 mm HEPES, 3 mm KCl, 2 mm NaHCO3, 2 mm CaCl2, and 1% BSA, pH 7.4), supplemented with 2.5 mm glucose, and containing 0.12 μm BCECF-AM and 0.025% pluronic F.127 (Invitrogen). After a 20-min wash in HCSS, BCECF fluorescence was imaged ratiometrically using alternate excitation at 450 and 490 nm, and a 530-nm emission filter with a Neofluar 40×/0.75 objective in an Axiovert 75 M microscope (Zeiss). When appropiate, neurons were pretreated with 1 mm AR-C1 for 20 min, and 5 mm βOHB was added during cytosolic pH imaging. Images were acquired with the Aquacosmos 2.5 software (Hamamatsu). The ratio of fluorescence intensity at 450 nm (F450) and 490 nm (F490), (F450/F490) was calculated for single cell analysis of cytosolic pH.
Cellular viability assay
Primary cortical neurons maintained in NB (25 mm glucose) were treated with 5 mm βOHB for 30 min (acute treatment) or once every 24 h for 96 h (chronic treatment). At DIV9, neurons were changed to Eagle's MEM and treated for 5 min with a glutamate concentration (10–25 μm) predetermined to give around 50% killing of neurons. Then cells were rinsed and maintained in fresh NB (25 mm glucose) for 24 h until calcein/propidium iodide (CA/PI) viability assay; 5 mm βOHB was present during glutamate stimulation and in the subsequent fresh media. Neurons were loaded with 1 μm calcein-AM (Invitrogen, wavelength excitation/emission 494/517 nm) and 2 μm PI (Sigma-Aldrich, wavelength excitation/emission 536/617 nm) for 5 min at 37°C, as previously described (Mattson et al., 1995). Images were captured with 40× objective with an inverted microscope AF6000 LX (Leica), and cells counts were determined in triplicate using ImageJ software.
3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide salt solution (MTT) reduction assay
Primary neuronal cultures maintained in NB (25 mm glucose) were supplemented with 5 mm βOHB once every 24 h from DIV5 to DIV9. Then MTT reduction capacity of neurons was assessed through their incubation with 0.5 mg/ml MTT (Sigma-Aldrich) during 1 h at 37°C. After a PBS rinse, reduced formazan products were solved in dimethylsulfoxide (DMSO), and absorbance at 540 nm was measured with FLUOstar optima microplate reader (BMG Labtech). Data are presented as percentage of basal condition in WT neurons.
Quantitative real-time PCR
Primary cortical neurons were supplemented with 5 mm βOHB once every 24 h from DIV5 to DIV9 and lysed in guanidinium thiocyanate (TRIzol; Sigma-Aldrich). After a 10-min incubation with added chloroform, samples were centrifuged 15 min at 13,000 rpm at 4°C. The RNA transparent phase was mixed with isopropanol and incubated for 2 h at −20°C. After centrifugation, pellets were washed in 80% ethanol, dried and solved in DNase and RNase free ultrapure water (Invitrogen). DNA residues were eliminated with DNase I recombinant RNase-free (Roche, 04-716-728-001) after a 30-min incubation at 37°C. Retrotranscription was performed with cDNA Reverse Transcription kit (Thermo Fisher Scientific). cDNA was amplified with Fast SYBR MasterMix probe (Thermo Fisher Scientific) in the ABI Prism 7900HT sequence detection system (Thermo Fisher Scientific) at the Genomics and Massive Sequencing Facility (CBMSO–UAM). The primers used for amplifying the target genes were: mouse PPARGC1A (5′-TGTCACCACCGAAATCCT-3′; 5′-CCTGGGGACCTTGATCTT-3′) and mouse β-actin (5′-CTAAGGCCAACCGTGAAAAG-3′; 5′-ACCAGAGGCATACAGGGACA-3′) as a housekeeping gene. Valid Prime expression (Tataa Biocenter, A106S25) was checked in all the samples and no significant gDNA expression was obtained. The relative expression of genes were calculated using the 2−ΔΔCt Livak comparative method (Schmittgen and Livak, 2008).
Amino acids and monoamines quantification
Mice on PND17 were killed by decapitation and brain regions were immediately dissected according to Carlsson and Lindqvist (1973) and Itier et al. (2003). Tissue samples were preserved at −80°C. Striatum samples were sonicated in 6 vol (weight/volume) of 0.4 n perchloric acid (PCA) for deproteinization and then centrifuged at 10,000 × g at 4°C for 20 min. Amino acids were determined by HPLC, as previously described (Perucho et al., 2015). Fluorescence detection was accomplished with Jasco detector (FP-2020) at 240 and 450 nm for excitation and emission wavelengths, respectively. Amino acids were identified by their retention times, and their concentrations were calculated by comparison to calibrated amino acid external standard solutions (1.5 μm). Dopamine (DA) and their metabolites were measured from supernatants by HPLC with an ESA Coulochem detector, according to Mena et al. (1984) with minor modifications. The chromatographic conditions were as follows: a column ACE 5 C18, 150 × 4.6 mm (UK); a citrate/acetate buffer 0.1 m, pH 3.9 with 10% methanol, 1 mm EDTA, and 1.2 mm heptane sulfonic acid, flow rate 1 ml/min. The detector voltage conditions were D1 (+0.05 V), D2 (−0.39 V), and the guard cell (+0.40 V). Monoamine levels were identified by their retention time and the amounts calculated against calibrated external standard solutions (0.6 μm).
Asp and N-acetyl-Asp (NAA) determination with HPLC-MS
In vivo-treated animals were killed at PND17, and their brains were halved for the determination of Asp and NAA. Primary cortical neurons treated with or without 5 mm βOHB for 96 h were rapidly rinsed in ultrapure milliQ water. Brain and neuronal samples were placed on dry ice and extraction buffer (i.e., methanol:chloroform:water, in a proportion of 700:200:50) was immediately added (1:9 volume/weight in brain tissue and 300 μl per million of neurons). Samples were homogenized, incubated 15 min on ice, centrifuged 10 min at 13,000 rpm, 4°C, and supernatants were preserved. Half-volume of extraction buffer was added to the pellets and the resultant supernatants after centrifugation were collected together with the first ones. Pellets were employed for protein quantification by BCA protein assay kit (ThermoFisher). HPLC-MS determinations were conducted in lyophilized supernatants by SIDI from Universidad Autónoma de Madrid in HPLC 1200 Series with Triple Quadrupolo 6410 mass detector. The chromatographic conditions were as follows: Ace 5AQ, C18, 5 µm, 250 × 4.6 mm ID column; two mobile phases (A: H2O milliQ + 0.1% formic acid or B: acetonitrile + 0.1% formic acid) injected at 0.4 ml/min at different times to create a gradient; ESI+/−; drying gas temperature: 325°C; drying gas flux: 12 min; nebulizer: 45 psi; capillary: +4500 V/−4500 V.
Western blotting
Cortical brain samples from in vivo-treated mice were sonicated in iced-cold lysis buffer (20 mm Tris-HCl, 10 mm AcK, 1 mm EDTA, and 0.25% NP-40; pH 7.4) freshly supplemented with 1 mm DTT, protease inhibitors [1 mm phenylmethylsulfonyl fluoride (PMSF), Fluka; and 1 mm iodoacetamide, Merck], and phosphatase inhibitors (phosSTOP, EDTA-free, Roche). Lysis buffer supplemented with 0.75% Na2CO3 was added to striatal pellets obtained after amino acid extraction, in a proportion of 1:9 versus initial tissue weight. Protein lysates (30 μg) were resolved by PAGE-SDS (10% polyacrylamide), and proteins were transferred to nitrocellulose membranes (GE HealthcareProtran 0.2, GE Healthcare Life Sciences). Blocking was performed in 5% (w/v) dry skimmed milk (Sveltesse, Nestle) in Tris-buffered saline [10 mm Tris-HCl (pH 7.5), 150 mm NaCl plus 0.05% (v/v) Tween 20] for 1 h at room temperature (RT). Membranes were incubated overnight at 4°C with primary antibodies against myelin-associated glycoprotein (MAG; monoclonal, 1:1000; Santa Cruz Biotechnology), myelin basic protein (MBP; monoclonal, 1:1000; Bio-Rad), vesicular monoamine transporter 2 (VMAT2; polyclonal, 1:500; Millipore), DA and cAMP-regulated phosphoprotein (32 kDa; DARPP-32; polyclonal, 1:5000; Millipore), and β-actin (1:5000; monoclonal; Sigma-Aldrich). Horseradish peroxidase (HRP)-conjugated secondary anti-rabbit (GARPO, 1:10,000; Bio-Rad) and anti-mouse (HAMPO, 1:5000; Vector Laboratories) were incubated for 1 h at RT. Signal detection was performed and enhanced with chemiluminescence substrate (Western lighting-ECL; PerkinElmer).
Statistical analysis
As a general rule, comparisons were planned between WT and aralar-KO groups, and between vehicle-treated and βOHB-treated aralar-KO groups. Vehicle-treated WT samples were considered as control group. In vitro experiments were conducted in primary neuronal cultures obtained from E15 to E16 embryos without gender consideration, and sample sizes contained a minimum of three embryos coming from at least two different primary cultures. Two-way ANOVA followed by post hoc Bonferroni-corrected t tests were performed except for AR-C1 seahorse XF assay and Asp and NAA determinations, in which one-way ANOVA followed by Newman–Keuls multiple comparison t test was made. In vivo experiments were performed in brain samples obtained from vehicle-injected and βOHB-injected WT and aralar-KO pups killed at PND17, without gender consideration. One-way ANOVA followed by post hoc Bonferroni-corrected t tests or Newman–Keuls multiple comparison t test was performed. Data are presented as mean ± SEM; the sample size for each experiment is indicated in the figure legends. Significance is presented as *p < 0.05, **p < 0.01, ***p < 0.001. All statistical analyses were performed using GraphPad Prism v.5.01 for Windows.
Results
Metabolic effects of βOHB in neurons derived from aralar-KO mice
KD was shown to ameliorate the Aralar/AGC1 deficiency-related phenotype in a human patient (Dahlin et al., 2015), the first child described with Aralar/AGC1 deficiency associated to global cerebral hypomyelination (Wibom et al., 2009). The diet was supplied to the affected patient at an advanced developmental stage when she was already six years old, and for at least 19 months. Also, KD reduces seizure frequency in a recently reported new human case of Aralar/AGC1 deficiency (Pfeiffer et al., 2020). Therefore, we assessed the therapeutic effectiveness of KD on the neurodevelopment of aralar-deficient pups (Fig. 1). To that end, KD was provided to the mothers of aralar-KO mice from one week before and during pregnancy and lactation (KD from gestation; Fig. 1B,C), or to aralar+/− females with pups of five postnatal days during lactation (KD at PND5; Fig. 1D–F). In both cases, KD had negative effects on females and offspring. As KD administration was not feasible for the aralar-KO mice, we turned to the metabolic product of KD, βOHB.
Figure 1.

Effect of KD on mice. A, Experimental design of treatments with SD and KD in mice. Experimental design 1: aralar+/− mice were fed a SD or a KD during one week before gestation and during both gestation and lactation of the offspring. However, KD had unwanted consequences in females' health, pregnancy, and number of offspring. B, Evolution of aralar+/− females body weight fed a SD or KD during the in vivo experiment. C, Percentage of survival, pregnancy and births of aralar+/− females fed a KD at experimental day 30. Experimental design 2: females were fed a KD when pups were at PND5. However, despite delaying KD administration, most of the offspring from females fed a KD were rickety; aralar−/− pups did not survive and a high percentage of aralar+/+ and aralar+/− siblings prematurely died. D, E, Evolution of individual offspring body weight under SD (D) and KD (E) from PND5 to PND28. F, Percentage of survival of aralar+/+, aralar+/−, and aralar−/− mice at PND20 SD, standard diet; KD, ketogenic diet.
The molecular mechanisms responsible for the beneficial effects of KD in each situation such as epilepsy, neurotrauma, genetic diseases as GLUT-1 deficiency or AGC1 deficiency (Klepper et al., 2007; Maalouf et al., 2009; for review, see Lutas and Yellen, 2013; Dahlin et al., 2015; Pfeiffer et al., 2020) need to be determined. KD is mainly composed of triglicerides, and of LCFA and MCFA which are metabolised mainly in liver giving rise to KBs: acetone, AcAc, and βOHB. Whereas LCFA do not cross the blood-brain barrier (BBB), and MCFA are only used by astrocytes but not by neurons (Thevenet et al., 2016), KBs can be efficiently oxidized by both neurons and astrocytes in brain (Edmond et al., 1987). For this reason, we focused on βOHB, the KB whose concentration maximally increases in blood during KD and is readily used by neurons, the main brain cell type expressing aralar (Ramos et al., 2003; Xu et al., 2007; Berkich et al., 2007; Pardo et al., 2011).
We started by investigating the effects of acetoacetate (AcAc) and βOHB on aralar-KO neurons as a first step toward a treatment of aralar-KO mice. βOHB in blood attains 1–2 mm or even higher concentrations (6–8 mm) during fasting or prolonged fasting, respectively, and reaches 5–7 mm in blood after KD in a patient with Aralar/AGC1 deficiency (Newman and Verdin, 2014; Dahlin et al., 2015). Therefore, primary neuronal cultures were treated with 5 mm βOHB to assess its neuroprotective capacity in aralar-deficient neurons.
Respiration of neurons in the presence of glucose in either basal non-stimulated conditions or in response to 50 μm glutamate (Glu) is significantly impaired by the lack of Aralar-MAS as determined by Seahorse OCR profiles (Llorente-Folch et al., 2013a, 2016; Fig. 2A–H). Besides, pyruvate supply is able to fully recover limited respiration in aralar-deficient neurons (Llorente-Folch et al., 2013a, 2016); showing that the lack of Aralar-MAS mainly prevents adequate glucose-derived pyruvate supply, producing a metabolic limitation to mitochondria. In this context, βOHB was provided as a likely energetic fuel to bypass Aralar/MAS deficiency in neurons, which is directly metabolized within the mitochondrial matrix. WT and aralar-KO neurons cultured in NB media supplemented with 25 mm (Fig. 2) or 5 mm (data not shown) glucose until DIV9 were subsequently switched to 2.5 mm glucose in the presence or absence of 5 mm βOHB for 30 min, and then tested for basal and glutamate-stimulated respiration (acute treatment). Figure 2A–D shows that acute βOHB treatment tends to increase basal respiration in both aralar-KO and WT neurons. We noted that acute βOHB markedly potentiated glutamate-induced stimulation of respiration in aralar-KO (Fig. 2C,D) but not in WT neurons (Fig. 2B,D). A chronic βOHB treatment (48 h) to neurons (5 mm βOHB in the presence of 5 mm glucose) was then applied, before the switch to 2.5 mm glucose used for OCR analysis. A chronic treatment with βOHB mimics in vivo conditions of KD. In this case, a marked increase of basal OCR (from 6.68 ± 0.42 nmol/min/mg to 10.54 ± 0.82 in the presence of βOHB) was observed in aralar-KO neurons along with a similar potentiation of glutamate-stimulated respiration, whereas no effect was reported in WT neurons (Fig. 2E–H). These results show a preferential effect of chronic βOHB on glutamate-stimulated respiration of aralar-KO as compared with control neurons, and even a specific effect of acute βOHB on aralar-KO neurons which become able to sustain 50 μm glutamate-stimulation of respiration to the same levels as control neurons. We have also tested the effect of the other major KB, AcAc, on Glu50-stimulated respiration of aralar-KO neurons. Acute treatment (30 min) with 5 mm AcAc elicited smaller stimulation than βOHB (Fig. 2I,J). For this reason, the study was focused on βOHB effects.
Figure 2.
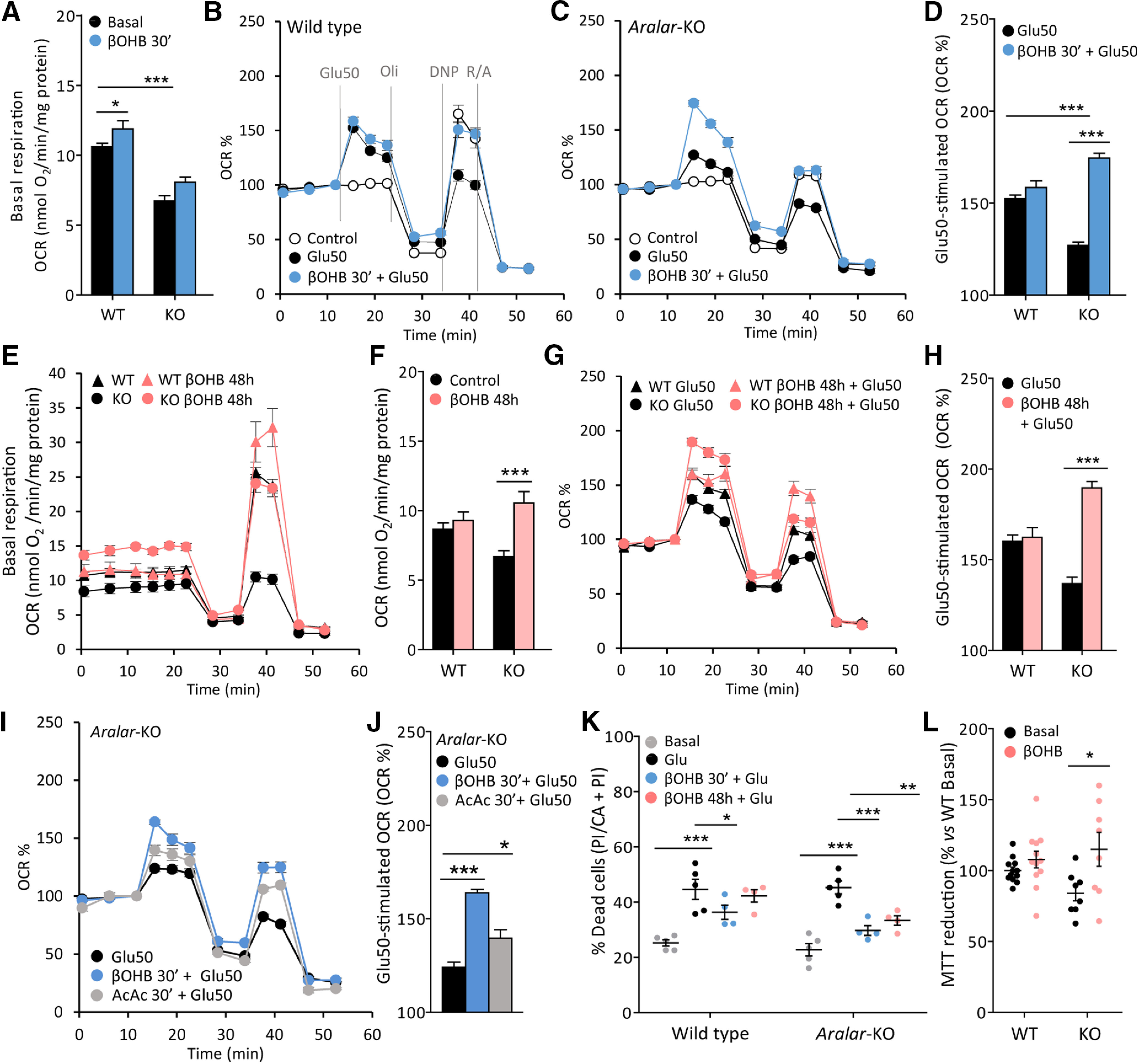
Effects of βOHB on mitochondrial respiration and neuroprotection of aralar-KO primary neuronal cultures. Effects of 30-min preincubation with 5 mm βOHB on WT or aralar-KO neurons maintained in 25 mm glucose NB until DIV9 (A–D), on basal respiration (A, OCR, nmol O2/min/mg protein) or on glutamate 50 μm (Glu50)-stimulated respiration (B–D, as % of basal values). E–H, Effects of 48-h βOHB treatment on WT and aralar-KO neurons maintained in 5 mm glucose NB-A from DIV7 to DIV9 on basal respiration (E, F) or Glu50-stimulated respiration (G, H). Mean ± SEM from three to six embryos per condition measured in three to nine replicates; ***p ≤ 0.001, *p ≤ 0.05 (two-way ANOVA followed by post hoc Bonferroni-corrected t test). I, J, Effects of 30-min treatment with 5 mm βOHB or 5 mm AcAc on glutamate 50 μm (Glu50)-stimulated respiration on aralar-KO neurons. Mean ± SEM (three to six wells/condition); ***p ≤ 0.001, *p ≤ 0.05 (one-way ANOVA followed by post hoc Bonferroni-corrected t test). Mitochondrial function was determined at DIV9 in 2.5 mm glucose and 2 mm Ca2+ DMEM. Sequential addition of glutamate (Glu50), 6 μm oligomycin (Oli), 0.5 mm dinitrophenol (DNP), and 1 μm rotenone/1 μm antimycin (R/A) was performed where indicated by the dashed lines in B. K, Percentage of dead cells maintained in 25 mm glucose NB in basal conditions of after treatment with glutamate (Glu), with or without 5 mm βOHB during 30 min or 96 h. Data calculated as an increase in % of dead cells versus basal. Mean ± SEM from four to six replicates coming from four to five different embryos per condition; ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05 (two-way ANOVA followed by post hoc Bonferroni-corrected t test). L, Percentage of MTT reduction in WT and aralar-KO neurons under basal conditions or preincubated 96 h with 5 mm βOHB. Data expressed as % of WT basal (mean ± SEM from 8 to 12 different embryos per condition measured in triplicate); *p ≤ 0.05 (two-way ANOVA followed by post hoc Bonferroni-corrected t test).
βOHB was also tested for neuroprotection against in vitro glutamate excitotoxicity in primary neuronal cultures from WT and aralar-KO embryos cultured in 25 mm glucose NB medium (Fig. 2K). Neurons in nutrient-restricted MEM were exposed to 10–25 μm Glu during 5 min after βOHB administration (acute, 30 min; or chronic, 96 h). βOHB was present during Glu exposure and 24 h after excitotoxic treatment until viability assay. Basal and Glu-induced neuronal death was similar in both WT and aralar-KO neurons (Fig. 2K), as previously reported (Llorente-Folch et al., 2016). βOHB administration protected from Glu-induced cell death in aralar-KO neurons in a 96.68% and 90.78% after acute and chronic treatments, respectively. In agreement with that observed on neuronal respiration (Fig. 2A–H), βOHB exerted a preferential effect on aralar-KO neurons.
One of the mechanisms through which βOHB could be exerting a neuroprotective effect is by allowing an improved redox status in mitochondria. While NADH/NAD+ ratio is low in aralar-KO brain mitochondria (Pardo et al., 2006) because of the lack of MAS and limited pyruvate supply (Llorente-Folch et al., 2013a), βOHB oxidation in mitochondria leads to the synthesis of NADH, which is necessary to maintain mitochondrial oxidative reactions and to produce mitochondrial NAD(P)H, through nicotinamide nucleotide transhydrogenase (NNT; Nesci et al., 2020). Accordingly, Figure 2L shows that βOHB increases MTT reduction specifically in aralar-KO neurons, an indicative of an increased level of NAD(P)H in these neurons which start from a more oxidized mitochondrial redox balance.
Molecular mechanisms involved in restoration of glutamate-stimulated respiration by βOHB in aralar-KO neurons
βOHB is considered to be more than a fuel, since it has a variety of signaling functions that might be involved in its overall metabolic effects (Fig. 3A). βOHB activates a Gi/o-protein-coupled receptor on the plasma membrane HCAR2/GPR109A, or may directly modulate gene expression by chromatin remodeling (Danial et al., 2013; Newman and Verdin, 2014; for review, see Grabacka et al., 2016). Therefore, it is relevant to find out the molecular mechanisms involved in βOHB neuroprotection of aralar-KO neurons.
Figure 3.
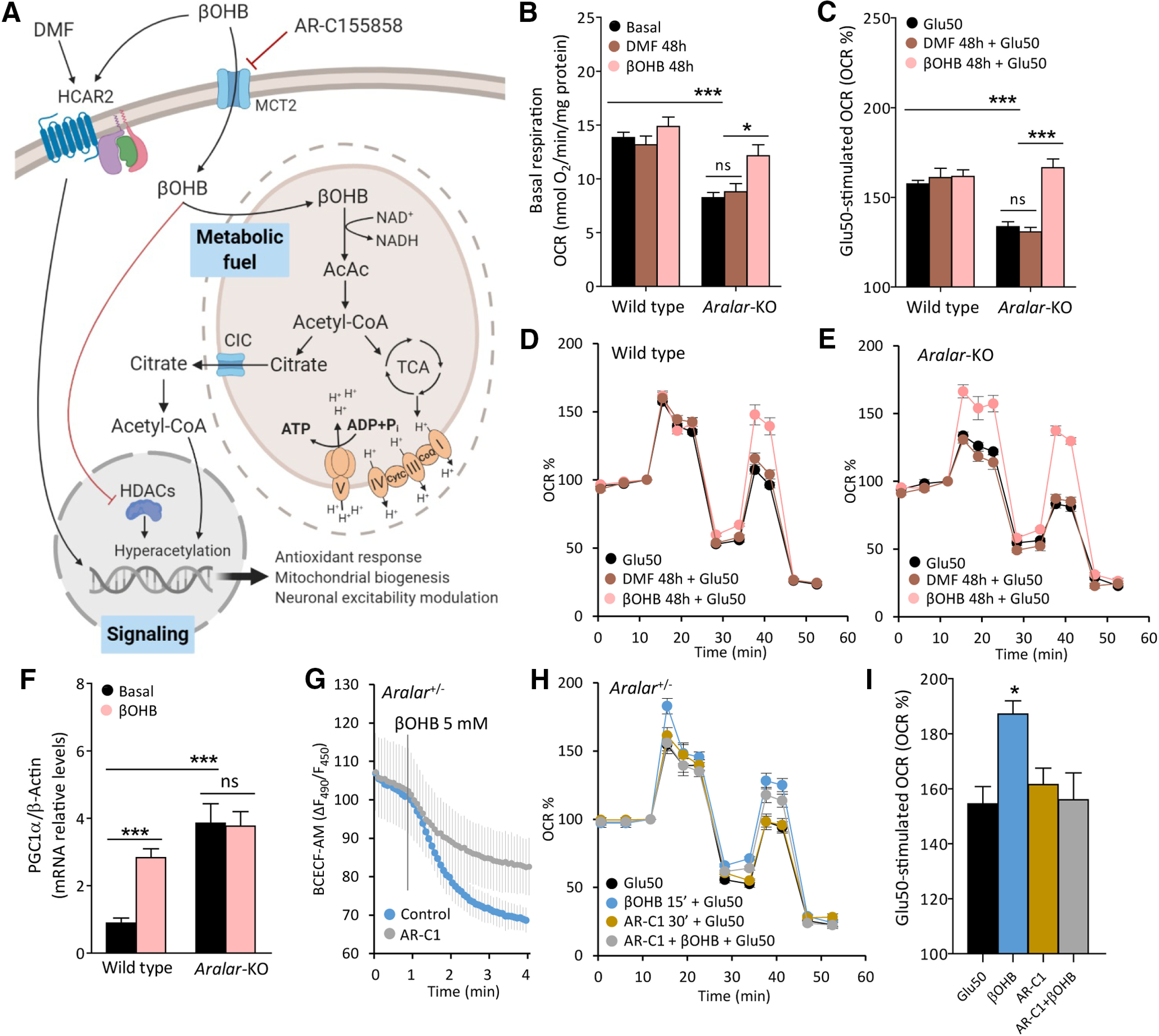
Molecular pathways involved in the effects of βOHB on neuronal respiration. A, Representative scheme of βOHB actions in neurons as a fuel and as a signaling molecule. Image created with BioRender.com. B, Effects of 20 μm DMF and 5 mm βOHB on basal respiration expressed as OCR (nmol O2/min/mg protein). C–E, Effects of DMF and βOHB on Glu50-stimulated respiration expressed as % of basal values (C), in WT (D), and aralar-KO (E) neurons. Mean ± SEM from three to seven embryos measured in triplicate; ***p ≤ 0.001, *p ≤ 0.05 (two-way ANOVA followed by post hoc Bonferroni-corrected t test). F, PGC1α mRNA levels in WT and aralar-KO neurons under basal conditions and after 96-h treatment with 5 mm βOHB. β-Actin used as a housekeeping gene. Mean ± SEM from 6–12 embryos measured in triplicate; ***p ≤ 0.001, versus WT basal (two-way ANOVA followed by post hoc Bonferroni-corrected t test). G, Intracellular pH variations of aralar+/− BCECF-AM loaded neurons expressed as variation in fluorescence ratio (ΔF490/F450), after 5 mm βOHB acute addition with or without a 30-min preincubation with 1 mm MCT2 inhibitor AR-C155858 (AR-C1). H, Effects of 1 mm AR-C1 30-min preincubation on Glu50-stimulated respiration, with or without a 5 mm βOHB 15-min preincubation in aralar+/− neurons. I, Glu50-stimulated respiration expressed as % of basal OCR. Mean ± SEM from one to two embryos in triplicates; *p ≤ 0.05 (one-way ANOVA followed by Newman–Keuls multiple comparison t test). Mitochondrial function determined in 2.5 mm glucose and 2 mm Ca2+ DMEM as indicated in Figure 1B. AcAc, acetoacetate; AcCoA, acetyl-CoA; CoQ, coenzyme Q; CytC, cytochrome C; DMF, dimethylfumarate; HCAR2, hydroxycarboxylic acid receptor 2; HDACs, histone deacetylases; MCT2, monocarboxylate transporter 2; TCA, trycarboxylic acid cycle; βOHB, β-hydroxybutyrate. ns, not significant.
HCAR2, a Gi/o-protein-coupled receptor of the hydroxy carboxylic acid receptor family, is activated by extracellular space metabolites resulting in reduced cAMP levels (Butcher et al., 1968; Tunaru et al., 2003). βOHB is the main endogenous ligand of this receptor (Offermanns et al., 2011). Human HCAR2 has an EC50 for its substrates of ∼700 μm, a concentration easily attainable during fasting. HCAR2 expression in brain is restricted to microglia and some sets of neurons, although more studies are needed to identify the neuronal cell populations expressing this receptor and its potential effects on neuronal metabolic modulation (Offermanns and Schwaninger, 2015; for review, see Katsu-Jiménez et al., 2017). To evaluate the role of HCAR2 activation in βOHB effects in aralar-KO neurons, we have used the synthetic HCAR2 ligand dimethylfumarate (DMF), with immunomodulatory and neuroprotective effects (Offermanns and Schwaninger, 2015; Graff et al., 2016; Peng et al., 2016). As shown in Figure 3B–E, 48-h treatment with DMF had no effect either in basal or in glutamate-stimulated respiration in WT and aralar-KO neuronal cortical cultures in vitro. This was also the case for 30-min acute treatment with DMF (data not shown). These results indicate that stimulation of respiration by βOHB is not because of effects through HCAR2, as DMF does not mimic the effects of βOHB.
KD increases mitochondrial mass in neuronal tissue (Lauritzen et al., 2016), presumably through up regulation of PGC1α, a master regulator of mitochondrial biogenesis. KD and βOHB treatments increase mitochondrial functional competence via PGC1α in hippocampal tissue in vivo and in cultures in vitro, respectively (Hasan-Olive et al., 2019). To analyze the effects of βOHB on PGC1-α levels, βOHB (5 mm) was administrated for 96 h to cultured neurons. This treatment was found to increase PGC1α mRNA levels in WT, but not in aralar-KO neurons, in which PGC1α was markedly increased compared with control ones (Fig. 3F). These results support the potential of βOHB to induce an increase of PGC1α expression in neurons and suggest that Aralar/AGC1 deficiency leads to PGC1α overexpression probably as a compensatory mechanism linked to the bioenergetic limitation of these neurons. Whether PGC1α is involved in the increase in glutamate-stimulated respiration in aralar-KO neurons caused by long-term treatment with βOHB remains to be established.
We have tested the role of βOHB as a fuel for neurons, after blocking βOHB transport into the cell with AR-C155858 (AR-C1), a potent inhibitor of monocarboxylate transporters (MCTs) MCT1 and MCT2 (Ovens et al., 2010). βOHB-treated aralar+/− neurons showed an increase in glutamate-stimulated respiration (Fig. 3H,I) although smaller than that showed for aralar-KO neurons in Figure 2C,D. The βOHB-dependent increase was abolished by incubation with 1 mm AR-C1 (30 min; Fig. 3H,I), a dose that clearly inhibits βOHB entry to the neuron. Indeed, monocarboxylate entry in cells is accompanied by a proton (Bröer et al., 1999), and the inhibition of βOHB entry was confirmed by the fact that βOHB-induced intracellular acidification was strongly decreased in the presence of AR-C1 (Fig. 3G). These results indicate that βOHB entry in the neuron is required for its effects in recovering neuronal respiration of aralar-KO neurons.
Therapeutic effects of intraperitoneal injections of βOHB in aralar-KO mice
We have studied the in vivo effects of βOHB based on its ability to recover glutamate-stimulated respiration and neuroprotect aralar-KO neurons in vitro, and also because it constitutes the main metabolic product of KD with wellknown beneficial effects in different paradigms (for review, see Maalouf et al., 2009). βOHB can cross the BBB through the MCTs (Halestrap and Meredith, 2004). WT and aralar-KO mice were injected intraperitoneally with βOHB (290 mg/kg/d) from PND12 to PND16 (Fig. 4A) to assess its effects on survival and recovery of brain deficits observed in aralar-deficient mice (Jalil et al., 2005; Ramos et al., 2011; Llorente-Folch et al., 2013b). Life expectancy of aralar-KO pups was not modified in βOHB-treated as compared with vehicle-treated mice. Indeed, 50% of aralar-KO mice died before finishing the experimental treatment, while all WT were alive until terminus (Fig. 4B). Also, the body weight was modified by genotype but not because of daily βOHB injections (Fig. 4C).
Figure 4.
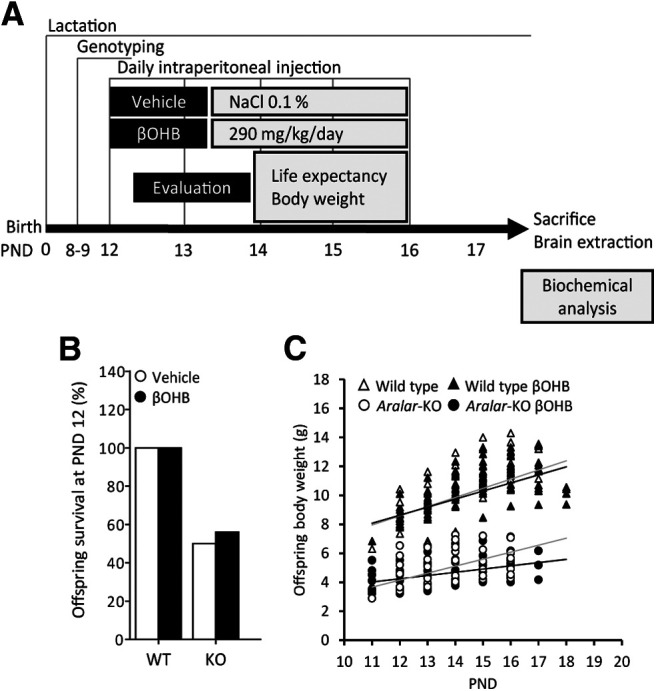
In vivo effects of βOHB on mice survival and body weight. A, Scheme of experimental design of βOHB in vivo treatments in WT and aralar-KO mice and the parameters studied. B, Percentage of litters' survival at PND12. C, Evolution of litters' body weight during βOHB in vivo treatments from PND12 to PND19.
Aralar-KO mice brain content of amino acids (i.e., Asp, glutamate, or glutamine) is drastically decreased in several brain regions (Jalil et al., 2005; Llorente-Folch et al., 2013b). In the present experiments, we found no recovery of amino acids levels by βOHB in the striatum of aralar-KO mice (Table 1). However, we have found a significant recovery in other traits of the aralar-KO mouse, particularly in the dopaminergic system (Llorente-Folch et al., 2013b) and in the postnatal myelination (Jalil et al., 2005; Ramos et al., 2011).
Table 1.
Striatal amino acid levels after intraperitoneal βOHB injections
| Amino acids (nmol/g tissue) | WT |
Aralar-KO |
||
|---|---|---|---|---|
| Mean ± SEM (% vs WT control) | Control | βOHB | Control | βOHB |
| Aspartate | 3113 ± 152 | 2656 ± 287 | 1061 ± 85*** | 918 ± 122*** |
| 100% | 85% | 34% | 29% | |
| Glutamate | 7869 ± 332 | 6683 ± 659 | 3592 ± 252*** | 3244 ± 437*** |
| 100% | 85% | 46% | 41% | |
| Serine | 1413 ± 156 | 1458 ± 295 | 436 ± 113** | 348 ± 47** |
| 100% | 103% | 31% | 25% | |
| Glutamine | 2739 ± 121 | 2550 ± 266 | 650 ± 63*** | 694 ± 106*** |
| 100% | 93% | 24% | 25% | |
| Histidine | 81 ± 8 | 87 ± 6 | 66 ± 3 | 59 ± 6* |
| 100% | 108% | 81% | 73% | |
| Glycine | 982 ± 60 | 1084 ± 163 | 984 ± 126 | 772 ± 124 |
| 100% | 110% | 100% | 79% | |
| Threonine | 299 ± 55 | 303 ± 66 | 121 ± 18* | 80 ± 14** |
| 100% | 101% | 41% | 27% | |
| Arginine | 158 ± 7 | 149 ± 12 | 112 ± 12* | 84 ± 10*** |
| 100% | 94% | 71% | 53% | |
| Taurine | 11668 ± 258 | 10090 ± 1174 | 9642 ± 1041 | 8736 ± 1000 |
| 100% | 86% | 83% | 75% | |
| Alanine | 1042 ± 96 | 908 ± 98 | 328 ± 31*** | 255 ± 24*** |
| 100% | 87% | 31% | 24% | |
| Tyrosine | 120 ± 33 | 143 ± 43 | 76 ± 28 | 32 ± 5 |
| 100% | 119% | 50% | 26% | |
| GABA | 1190 ± 38 | 1164 ± 121 | 1329 ± 168 | 984 ± 154 |
| 100% | 98% | 112% | 83% | |
Data are expressed as mean ± SEM and normalized to WT control (n = 6–8 mice per group);
***p ≤ 0.001,
**p ≤ 0.01,
*p ≤ 0.05 (one-way ANOVA followed by Student–Newman–Keuls t test).
Intraperitoneal βOHB recovers the striatal dopaminergic system of aralar-KO mice
Aralar/AGC1 deficiency resulted in changes in dopamine (DA) and its metabolites in the striatum, a brain region enriched in dopaminergic projections (Llorente-Folch et al., 2013b). In agreement with these findings, aralar-KO striatum from PND17 mice daily injected with vehicle showed a substantial reduction in DA (to 67% vs WT; Fig. 5A). Interestingly, 3,4-dihydroxyphenylacetic acid (DOPAC) content was not changed in aralar-KO striatum and the DOPAC/DA ratio was 1.6-fold larger than that of WT (Fig. 5A). An increment in DOPAC/DA is associated with increased MAO activity and related to oxidative stress in dopaminergic neurons (Spina and Cohen, 1989), as suggested to occur in aralar-KO striatum (Llorente-Folch et al., 2013b). However, βOHB treatment to aralar-KO mice caused a striking recovery of DA content and of the DOPAC/DA ratio which reached WT mice values (Fig. 5A).
Figure 5.
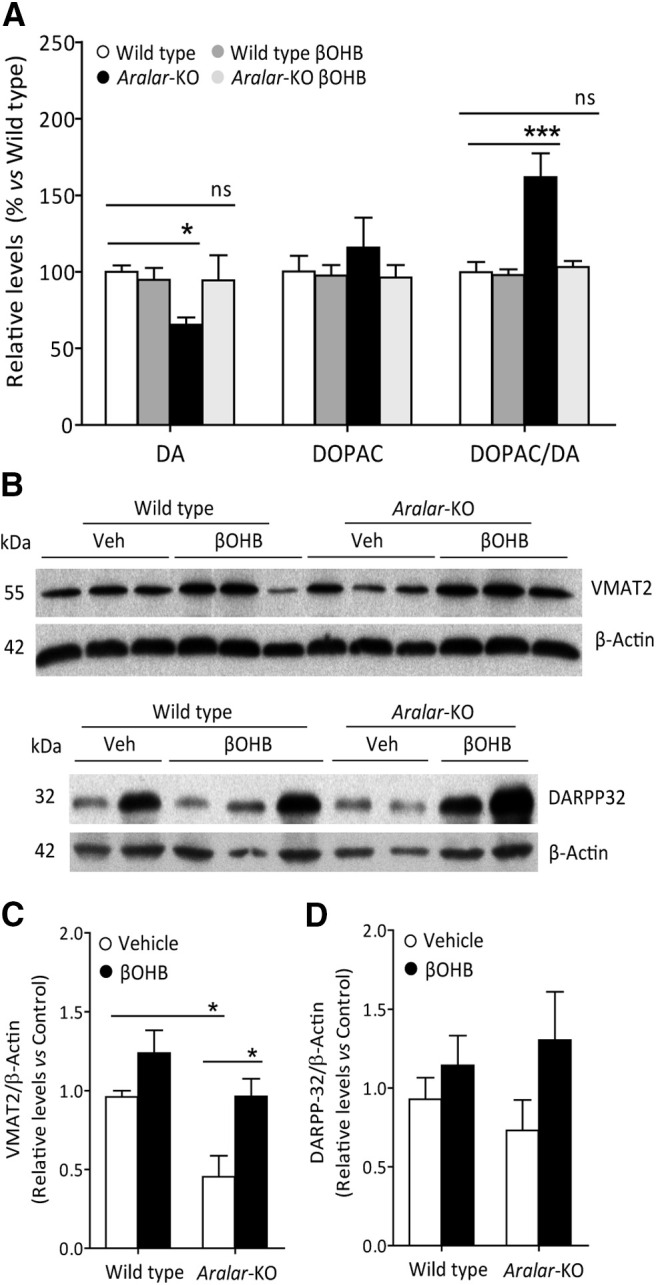
In vivo effects of βOHB on striatal dopaminergic system in aralar-KO mice. A, Striatum DA, DOPAC, and DOPAC/DA in WT and aralar-KO striatum after 290 mg/kg/d βOHB intraperitoneal injections. Data presented as % of WT. Mean ± SEM (n = 5–7 mice per group); ***p ≤ 0.001, *p ≤ 0.05 (one-way ANOVA followed by post hoc Bonferroni-corrected t test). B, Representative Western blotting images of VMAT2 and DARPP-32, with their respective densitometric histograms (C, D), in WT and aralar-KO striatum after βOHB intraperitoneal injections. β-Actin used as charge control. Mean ± SEM (n = 6–9 mice per group); *p ≤ 0.05 (one-way ANOVA followed by post hoc Bonferroni-corrected t test). ns, not significant.
We further investigated the content of specific dopaminergic markers as VMAT2, the presynaptic vesicular transporter of monoamines, and DARPP-32, found in postsynaptic medium spiny neurons in striatum (Fienberg et al., 1998). VMAT2 is decreased in aralar-KO striatum at PND17 (to 47% of control values), but not as much as at PND20 (Llorente-Folch et al., 2013b), whereas the decrease in DARPP-32 is not significant at this postnatal stage (Fig. 5B–D). Interestingly, βOHB supply in aralar-KO mice fully recovers VMAT2 protein level to control values and tends to increase DARPP-32 levels. Our previous results showed that striatum is particularly affected by Aralar/AGC1 deficiency (Llorente-Folch et al., 2013b), and a partial recovery of striatal dopaminergic system is revealed in the present study.
βOHB recovered Asp and NAA in neurons and myelin proteins in brain from aralar-KO mice
Aralar-KO mice show postnatal hypomyelination that is more marked in gray than in white matter regions in the brain (Ramos et al., 2011). A drastic decrease in Asp and NAA occurs in brain and neuronal cultures from aralar-KO cortices (Jalil et al., 2005). Quantification of specific myelin proteins is used as a reliable indicator for myelination. Indeed, we found a marked decrease in MBP and MAG in aralar-KO cortex (a gray matter-enriched region) at PND17 (to 7% and 18% vs WT, respectively; Fig. 6A–C), as described in aralar-KO brain at PND20 (Jalil et al., 2005; Ramos et al., 2011). Interestingly, βOHB-injected aralar-KO mice showed a recovery in the content of MAG (to 78% of controls; Fig. 6A,C), supporting an enhanced myelination in the aralar-KO mice after βOHB treatment. Although MBP values in vehicle-treated and βOHB-treated aralar-KO mice was not significantly different (Fig. 6A,B), βOHB caused a larger increase on MBP content in aralar-KO than in WT mice (243 ± 67% vs −16 ± 16%, respectively; p = 0.0038, Student's t test). This is reminiscent of the improvement in brain myelination observed in the Aralar/AGC1-deficient patient after KD (Dahlin et al., 2015).
Figure 6.

Effects of βOHB on cortical myelin protein levels and Asp/NAA brain and primary cortical neurons content. A, Representative Western Blotting images of MBP and MAG, with their respective densitometric histograms (B, C) in WT and aralar-KO brain cortex after 290 mg/kg/d βOHB intraperitoneal injections. β-Actin used as charge control. Mean ± SEM (n = 5–7 mice per group); ***p ≤ 0.001, **p ≤ 0.01 (one-way ANOVA followed by Newman–Keuls multiple comparisons t test). D, Asp and NAA levels after 290 mg/kg/d βOHB intraperitoneal injections in WT and aralar-KO brain. Mean ± SEM (n = 3 mice per group); **p ≤ 0.01, *p ≤ 0.05 (one-way ANOVA followed by Newman–Keuls multiple comparisons t test). E, F, Asp (E) and NAA (F) levels after 5 mm βOHB 96-h treatment in WT and aralar-KO primary neuronal cultures. Mean ± SEM from two to four embryos per condition measured in duplicates; ***p ≤ 0.001 (one-way ANOVA followed by Newman–Keuls multiple comparisons t test).
Impaired myelin synthesis in Aralar/AGC1 deficiency has been attributed to a lack of neuron-born NAA used as precursor of postnatal myelin lipid synthesis (Jalil et al., 2005; Satrústegui et al., 2007; Wibom et al., 2009; Ramos et al., 2011; Dahlin et al., 2015). However, myelin recovery obtained after intraperitoneal βOHB was not associated with an increase in Asp nor NAA in the brain of aralar-KO mice (Fig. 6D). Similarly, no increase in brain NAA was reported in the Aralar/AGC1-deficient patient with increased myelination resulting from KD (Dahlin et al., 2015). These observations appear to contradict the initial hypothesis regarding the role of NAA in postnatal myelination. However, we have also tested the effect of chronic treatment with 5 mm βOHB in vitro in neuronal cultures and found a significant increase in NAA (4-fold) and in Asp (3-fold) in cultures from aralar-KO cortex (Fig. 6E,F). The fact that neither Asp nor NAA levels increased in the brains of aralar-KO mice is probably because of their continuous use possibly by nearby glial cells.
Discussion
KD has been successfully used in two human patients with Aralar/AGC1 deficiency, observing resumed myelination (Dahlin et al., 2015) and preventing epilepsy (Dahlin et al., 2015; Pfeiffer et al., 2020), two of the main hallmarks of this neurodevelopmental rare disease (Wibom et al., 2009). Although the clinical benefits of long-term KD in the first Aralar/AGC1-deficient girl were markedly significant, the onset of the treatment was at an advanced neurodevelopmental stage (Dahlin et al., 2015). In the present study, to assess the therapeutic effectiveness of KD, the diet was administered earlier to aralar+/− mice females from pregnancy or during the postnatal life of the aralar-deficient pups. Unfortunately, testing the effects of KD on mice was unfeasible, since it affected maternal weight, fertility and increased mice mortality; as observed before in mice (Sussman et al., 2013a,b, 2015). The therapeutic potential of βOHB, the main KB produced during KD, was assessed in the aralar-KO mouse model. The lack of Aralar/MAS prevents adequate glucose-derived pyruvate supply to mitochondria, producing a metabolic limitation (Llorente-Folch et al., 2013a). Here, βOHB, was found to efficiently recover deficits in both basal and glutamate-stimulated respiration of aralar-deficient neurons. The effect of βOHB acting as an alternative fuel for aralar-deficient neurons was blunted in the presence of AR-C1, a selective MCT2 blocker that prevents βOHB uptake. On the other side, AcAc was far less effective than βOHB in increasing glutamate-stimulated respiration in aralar-KO neurons, possibly because beta-hydroxybutyrate dehydrogenase (BDH) is readily reversible (Williamson et al., 1967; Siess et al., 1976) and AcAc is expected to lower the mitochondrial NADH/NAD+ ratio, already low in aralar-KO neurons (Pardo et al., 2006; Fig. 7). Although βOHB induces neuroprotective effects (this work) and increases mitochondrial metabolism and glucose-sparing effects in WT neurons (Laird et al., 2013; Achanta and Rae, 2017), the βOHB-mediated neuroprotection, including that on glutamate excitotoxicity, is more pronounced on aralar-KO neurons. Thus, βOHB rather than AcAc constitutes an effective substrate able to bypass the energetic limitation imposed by aralar deficiency in neurons.
Figure 7.
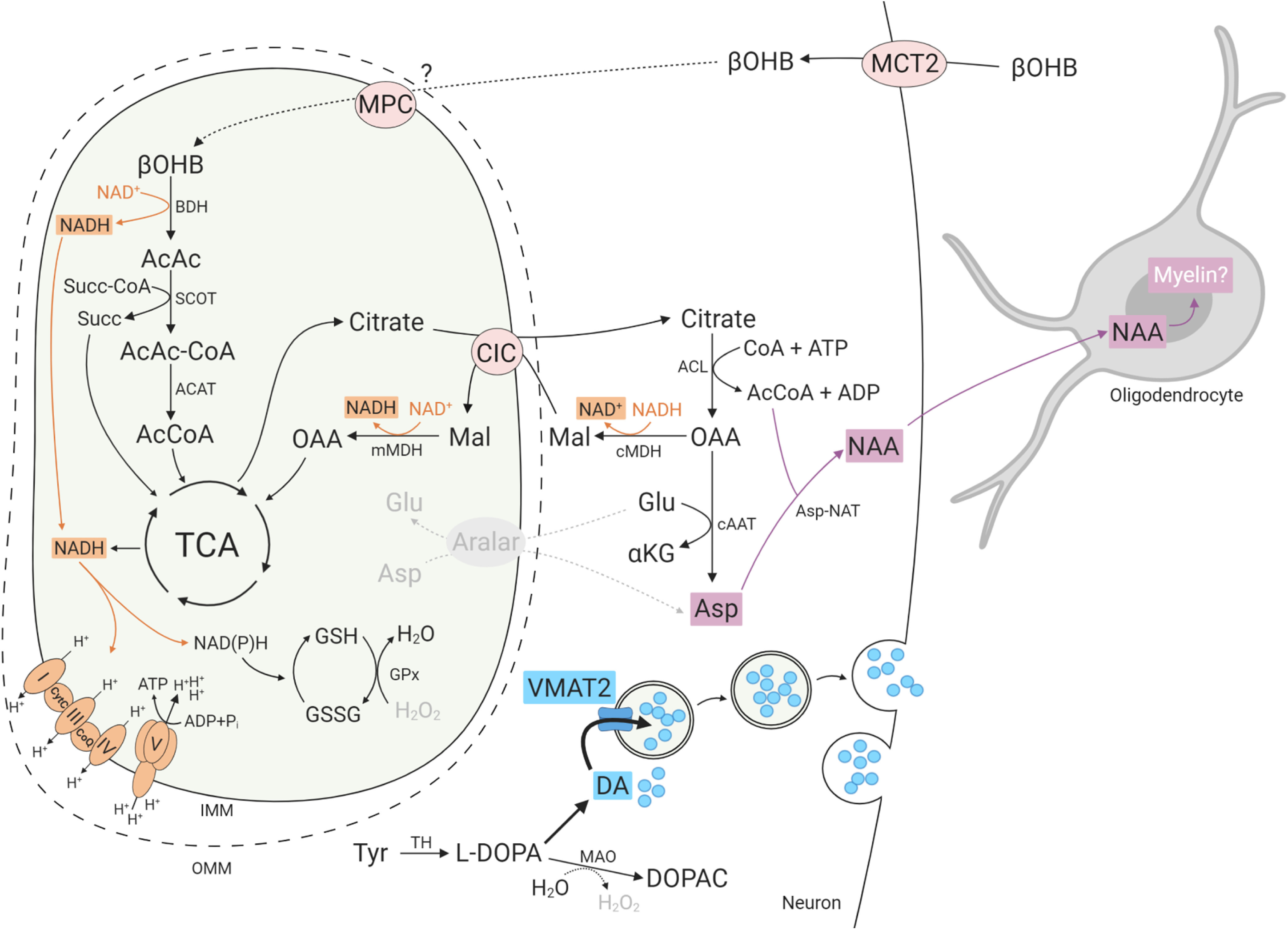
βOHB metabolism and effects in aralar-KO neurons and oligodendrocytes. βOHB metabolism leads to the synthesis of acetyl-CoA (AcCoA) and citrate which may be transported to the cytosol through the citrate carrier (CIC/Slc25a1). Citrate is converted to OAA through ACL reaction. Under conditions of high cytosolic NADH/NAD+ ratio as prevail in aralar-KO neurons (Pardo et al., 2006), a large fraction of OAA will be converted to malate (Mal) through malate DH reaction (cMDH) supplying the counter substrate for CIC to provide a citrate-malate redox shuttle (Palmieri, 2004; Pardo and Contreras, 2012). However, some of the OAA is also used in cytosolic Asp synthesis possibly through inverted Asp aminotransferase (cAAT) reaction, driven by the very low Asp levels (and also α-KG levels; Contreras et al., unpublished data) in aralar-KO neurons and retinas. The citrate-malate shuttle provides cytosolic AcCoA for NAA synthesis through Asp-NAT. This βOHB-induced NAA formed might serve as a precursor for myelin lipid synthesis in oligodendrocytes. Additionally, in the dopaminergic terminals, enhanced mitochondrial NADH production by βOHB would increase the GSH/GSSG ratio, reduce H2O2 levels and favor vesicular DA internalization through increased VMAT2 levels, avoiding its cytosolic oxidation occurring in aralar-KO mice (Llorente-Folch et al., 2013b). AcAc, acetoacetate; AcAc-CoA, acetoacetyl coenzyme A; ACAT, AcCoA C-acetyltransferase; Aralar, Asp-glutamate carrier isoform 1 (AGC1); BDH, b-hydroxybutyrate dehydrogenase; CoA, coenzyme A; CoQ, coenzyme Q; CytC, cytochrome C; DOPAC, 3,4-dihydroxyphenylacetic acid; Glu, glutamate; GPx, GSH peroxidase; GSH, glutathione; GSSG, GSH disulfure; MAO, monoamine oxidase; mMDH, mitochondrial malate dehydrogenase; MPC, mitochondrial pyruvate carrier; SCOT, succinyl-CoA-3-oxaloacid CoA transferase; Succ, succinate; Succ-CoA, succinil coenzyme A; TCA, tricarboxylic acid cycle; VMAT2, vesicular monoamine transporter 2; βOHB, β-hydroxybutyrate. Image created with BioRender.com.
A brief treatment of aralar-KO pups (from PND12 to PND16) with βOHB did not modify brain amino acid levels. However, it did elicited a marked positive effect on myelination and DA homeostasis, both of which are impaired in aralar-deficient mice (Jalil et al., 2005; Llorente-Folch et al., 2013b). Regarding the dopaminergic system, intraperitoneal βOHB recovers deficits in DA content and in VMAT2 protein level, and re-establishes the DOPAC/DA ratio in aralar-KO striatum. Curiously, short term treatment with βOHB recovers dopaminergic neurons from MPTP (Kashiwaya et al., 2000; Tieu et al., 2003) and rotenone (Imamura et al., 2006) neurotoxicity which, unlike Aralar/AGC1 deficiency, arises from their actions as Complex I inhibitors. Tieu et al. (2003) explained the protective effect of βOHB through the formation of succinate in the reactions leading to acetyl-CoA production in mitochondria (Fig. 7) and the ability of succinate Complex II respiration to increase ATP production. In the present case, aralar-KO mitochondria have no defects in Complex I but rather a depletion of the main respiratory substrate, pyruvate, and low mitochondrial NADH levels (Pardo et al., 2006; Llorente-Folch et al., 2013a). In aralar-KO brain, recovery of striatal dopaminergic neurons is explained most likely by mitochondrial consumption of βOHB enhancing mitochondrial NADH production, respiration and ATP synthesis as shown in PD mice models (Tieu et al., 2003). Aditionally, βOHB-induced recovery of mitochondrial NADH production may allow efficient NNT activity resulting in the formation of mitochondrial NADPH and glutathione (GSH), thus preventing mitochondrial ROS production and loss of cytosolic VMAT2. Presumably, VMAT2-mediated vesicular sequestration of cytosolic DA will allow to recover normal DA homeostasis in these terminals (Chen et al., 2008; Llorente-Folch et al., 2013b).
βOHB supply to aralar-KO mice resulted in a surprising improvement in myelination. Hypomyelination in Aralar/AGC1 deficiency was proposed to be related to the defective synthesis of Asp-derived NAA in aralar-KO brain (Satrústegui et al., 2007; Wibom et al., 2009). Asp and NAA production in brain takes place largely in neurons (Urenjak et al., 1992) in which these compounds attain higher concentrations than in whole brain. Resumed myelination by intraperitoneal βOHB in aralar-KO mice was not paralleled by a recovery in brain Asp or NAA content, a hallmark for Aralar/MAS deficiency in mice and humans (Jalil et al., 2005; Wibom et al., 2009). Moreover, no clear increase in brain NAA was found after a 19-month administration of KD in the patient with Aralar/AGC1 deficiency, except for a slight increase after six-month KD (Dahlin et al., 2015), although an augmented brain myelin was reported. This inability of βOHB to elevate brain Asp and NAA in vivo, despite resumed myelination, might be because of its high consumption precisely for myelin synthesis. Accordingly, Asp and NAA were both highly increased in aralar-KO neuronal cultures chronically treated with βOHB in which NAA is not used for myelination.
Citrate derived from βOHB metabolism (Fig. 7) may reach the cytosol through the citrate carrier (CIC/Slc25a1), leading to oxaloacetate (OAA) synthesis through ATP citrate lyase (ACL) reaction. Given the low Asp and α-ketoglutarate (α-KG; Contreras et al., unpublished data) levels in the cytosol of aralar-KO neurons, the Asp aminotransferase (cAAT) reaction may lead to cytosolic Asp synthesis. Asp together with acetyl-CoA provided through citrate-malate shuttle allow the synthesis of NAA through Asp N-acetyl transferase (Asp-NAT) enzyme, which is localized in microsomes and outer mitochondrial membrane (Madhavarao et al., 2003; Lu et al., 2004; Wiame et al., 2010). This proposed pathway is Aralar independent and would allow NAA formation available for transaxonal transport into olygodendrocytes for myelin lipid synthesis (Satrústegui et al., 2007; Pardo et al., 2011; Ramos et al., 2011; Fig. 7). βOHB-derived acetyl-CoA is important to maintain an activated CIC as this carrier increases its activity through acetylation (Palmieri and Monné, 2016), providing an activation loop of the citrate-malate shuttle. Interestingly, the βOHB-dependent recovery of Asp and NAA levels in aralar-KO neurons did not require lowering of carbohydrates, as it took place in neurons cultured in 25 mm glucose.
KD administration to the first Aralar-deficient patient was thought to be successful because limiting carbohydrates in the neuron would switch OAA away from malate and into Asp (Dahlin et al., 2015). The results from this study indicate that carbohydrate restriction is not strictly required for the improvement of aralar-KO mice phenotype by βOHB administration. Therefore, this treatment may provide an alternative therapy for Aralar/AGC1 deficiency devoid of the unwanted effects of the highly unpalatable, low carbohydrate, and low compliance KD. This study highlights positive effects of glucose unrestricted βOHB administration on Asp and NAA production, myelination, and in dopaminergic system function, all caused through βOHB actions in neurons. However, the possibility that βOHB is directly used by aralar-KO oligodendrocytes as a precursor for myelin lipid synthesis is not excluded. Brain cell-specific disruption of Aralar will be required to further clarify these issues.
Footnotes
This work was supported by Ministerio de Economía Grants SAF2014-56929R (to J.S. and B.P.) and SAF2017-82560R (AEI/FEDER, UE; to B.P.); the Centro de Investigación Biomédica en Red de Enfermedades Raras, an initiative of the Instituto de Salud Carlos III (ISCIII); a grant from the Fundación Ramon Areces (J.S.); the Irycis Chromatographic Services and Nervous System Markers Unit, UCS (2018/0135; to M.J.C.); and an institutional grant from the Fundación Ramon Areces to the Centro de Biología Molecular Severo Ochoa. I.P.-L. is the recipient of Contrato Predoctoral de Formación de Personal Investigador (FPI MINECO). We thank Dr. Antonio S. Herranz for his inputs as an expert in amino acid analysis by HPLC-UV, Dr. Araceli del Arco for critical reading of the manuscript, and Isabel Manso and Barbara Sesé for technical support. All experiments were conducted in compliance with the ARRIVE guidelines.
The authors declare no competing financial interests.
References
- Achanta LB, Rae CD (2017) β-Hydroxybutyrate in the brain: one molecule, multiple mechanisms. Neurochem Res 42:35–49. 10.1007/s11064-016-2099-2 [DOI] [PubMed] [Google Scholar]
- Alter AS, Engelstad K, Hinton VJ, Montes J, Pearson TS, Akman CI, De Vivo DC (2015) Long-term clinical course of Glut1 deficiency syndrome. J Child Neurol 30:160–169. 10.1177/0883073814531822 [DOI] [PubMed] [Google Scholar]
- Berkich DA, Ola MS, Cole J, Sweatt AJ, Hutson SM, LaNoue KF (2007) Mitochondrial transport proteins of the brain. J Neurosci Res 85:3367–3377. 10.1002/jnr.21500 [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 435:297–312. 10.1042/BJ20110162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröer S, Bröer A, Schneider HP, Stegen C, Halestrap AP, Deitmer JW (1999) Characterization of the high-affinity monocarboxylate transporter MCT2 in Xenopus laevis oocytes. Biochem. J 341:529–535. 10.1042/0264-6021:3410529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher RW, Baird CE, Sutherland EW (1968) Effects of lipolytic and antilipolytic substances on adenosine 3',5'-monophosphate levels in isolated fat cells. J Biol Chem 243:1705–1712. [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M (1973) Effect of ethanol on the hydroxylation of tyrosine and tryptophan in rat brain in vivo. J Pharm Pharmacol 25:437–440. 10.1111/j.2042-7158.1973.tb09129.x [DOI] [PubMed] [Google Scholar]
- Chen MK, Kuwabara H, Zhou Y, Adams RJ, Brasić JR, McGlothan JL, Verina T, Burton NC, Alexander M, Kumar A, Wong DF, Guilarte TR (2008) VMAT2 and dopamine neuron loss in a primate model of Parkinson's disease. J Neurochem 105:78–90. 10.1111/j.1471-4159.2007.05108.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin M, Martin DA, Hedlund Z, Jonsson M, Von Döbeln U, Wedell A (2015) The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 56:e176–e181. 10.1111/epi.13193 [DOI] [PubMed] [Google Scholar]
- Danial NN, Hartman AL, Stafstrom CE, Thio LL (2013) How does the ketogenic diet work? Four potential mechanisms. J Child Neurol 28:1027–1033. 10.1177/0883073813487598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J (1987) Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res 18:551–561. 10.1002/jnr.490180407 [DOI] [PubMed] [Google Scholar]
- Falk MJ, Li D, Gai X, McCormick E, Place E, Lasorsa FM, Otieno FG, Hou C, Kim CE, Abdel-Magid N, Vazquez L, Mentch FD, Chiavacci R, Liang J, Liu X, Jiang H, Giannuzzi G, Marsh ED, Yiran G, Tian L, et al. (2014) AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N-acetylaspartate. JIMD Rep 4:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fienberg AA, Hiroi N, Mermelstein PG, Song W, Snyder GL, Nishi A, Cheramy A, O'Callaghan JP, Miller DB, Cole DG, Corbett R, Haile CN, Cooper DC, Onn SP, Grace AA, Ouimet CC, White FJ, Hyman SE, Surmeier DJ, Girault J, et al. (1998) DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science 281:838–842. 10.1126/science.281.5378.838 [DOI] [PubMed] [Google Scholar]
- Gómez-Galán M, Makarova J, Llorente-Folch I, Saheki T, Pardo B, Satrústegui J, Herreras O (2012) Altered postnatal development of cortico-hippocampal neuronal electric activity in mice deficient for the mitochondrial aspartate-glutamate transporter. J Cereb Blood Flow Metab 32:306–317. 10.1038/jcbfm.2011.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabacka M, Pierzchalska M, Dean M, Reiss K (2016) Regulation of ketone body metabolism and the role of PPARα. Int J Mol Sci 17:2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff EC, Fang H, Wanders D, Judd RL (2016) Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism 65:102–113. 10.1016/j.metabol.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Meredith D (2004) The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch 447:619–628. 10.1007/s00424-003-1067-2 [DOI] [PubMed] [Google Scholar]
- Hasan-Olive MM, Lauritzen KH, Ali M, Rasmussen LJ, Storm-Mathisen J, Bergersen LH (2019) A ketogenic diet improves mitochondrial biogenesis and bioenergetics via the PGC1α-SIRT3-UCP2 axis. Neurochem Res 44:22–37. 10.1007/s11064-018-2588-6 [DOI] [PubMed] [Google Scholar]
- Imamura K, Takeshima T, Kashiwaya Y, Nakaso K, Nakashima K (2006) D-β-hydroxybutyrate protects dopaminergic SH-SY5Y cells in a rotenone model of Parkinson's disease. J Neurosci Res 84:1376–1384. 10.1002/jnr.21021 [DOI] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, et al. (2003) Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 12:2277–2291. 10.1093/hmg/ddg239 [DOI] [PubMed] [Google Scholar]
- Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, Li MX, Ramos M, Mármol P, Horiuchi M, Shimotsu K, Nakagawa S, Okubo A, Sameshima M, Isashiki Y, Del Arco A, Kobayashi K, Satrústegui J, Saheki T (2005) Reduced N -Acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem 280:31333–31339. 10.1074/jbc.M505286200 [DOI] [PubMed] [Google Scholar]
- Juaristi I, García-Martín ML, Rodrigues TB, Satrústegui J, Llorente-Folch I, Pardo B (2017) ARALAR/AGC1 deficiency, a neurodevelopmental disorder with severe impairment of neuronal mitochondrial respiration, does not produce a primary increase in brain lactate. J Neurochem 142:132–139. 10.1111/jnc.14047 [DOI] [PubMed] [Google Scholar]
- Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL (2000) D-beta -Hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci USA 97:5440–5444. 10.1073/pnas.97.10.5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsu-Jiménez Y, Alves RM, Giménez-Cassina A (2017) Food for thought: impact of metabolism on neuronal excitability. Exp Cell Res 360:41–46. 10.1016/j.yexcr.2017.03.002 [DOI] [PubMed] [Google Scholar]
- Kavanaugh BC, Warren EB, Baytas O, Schmidt M, Merck D, Buch K, Liu JS, Phornphutkul C, Caruso P, Morrow EM (2019) Longitudinal MRI findings in patient with SLC25A12 pathogenic variants inform disease progression and classification. Am J Med Genet A 179:2284–2291. 10.1002/ajmg.a.61322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler SK, Neal EG, Camfield CS, Kossoff EH (2011) Dietary therapies for epilepsy: future research. Epilepsy Behav 22:17–22. 10.1016/j.yebeh.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepper J, Engelbrecht V, Scheffer H, van der Knaap MS, Fiedler A (2007) GLUT1 deficiency with delayed myelination responding to ketogenic diet. Pediatr Neurol 37:130–133. 10.1016/j.pediatrneurol.2007.03.009 [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Zupec-Kania BA, Rho JM (2009) Ketogenic diets: an update for child neurologists. J Child Neurol 24:979–988. 10.1177/0883073809337162 [DOI] [PubMed] [Google Scholar]
- Laird MD, Clerc P, Polster BM, Fiskum G (2013) Augmentation of normal and glutamate-impaired neuronal respiratory capacity by exogenous alternative biofuels. Transl Stroke Res 4:643–651. 10.1007/s12975-013-0275-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen KH, Hasan-Olive MM, Regnell CE, Kleppa L, Scheibye-Knudsen M, Gjedde A, Gjedde A, Klungland A, Bohr VA, Storm-Mathisen J, Bergersen LH (2016) A ketogenic diet accelerates neurodegeneration in mice with induced mitochondrial DNA toxicity in the forebrain. Neurobiol Aging 48:34–47. 10.1016/j.neurobiolaging.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B, Satrústegui J (2013a) Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci 33:13957–13971. 10.1523/JNEUROSCI.0929-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Folch I, Sahún I, Contreras L, Casarejos MJ, Grau JM, Saheki T, Mena MA, Satrústegui J, Dierssen M, Pardo B (2013b) AGC1-malate aspartate shuttle activity is critical for dopamine handling in the nigrostriatal pathway. J Neurochem 124:347–362. 10.1111/jnc.12096 [DOI] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Pérez-Liébana I, Satrústegui J, Pardo B (2016) L-lactate-mediated neuroprotection against glutamate-induced excitotoxicity requires ARALAR/AGC1. J Neurosci 36:4443–4456. 10.1523/JNEUROSCI.3691-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZH, Chakraborty G, Ledeen RW, Yahya D, Wu G (2004) N-acetylaspartate synthase is bimodally expressed in microsomes and mitochondria of brain. Brain Res Mol Brain Res 122:71–78. 10.1016/j.molbrainres.2003.12.002 [DOI] [PubMed] [Google Scholar]
- Lutas A, Yellen G (2013) The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci 36:32–40. 10.1016/j.tins.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maalouf M, Rho JM, Mattson MP (2009) The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev 59:293–315. 10.1016/j.brainresrev.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Chinopoulos C, Chandrasekaran K, Namboodiri MA (2003) Characterization of the N-acetylaspartate biosynthetic enzyme from rat brain. J Neurochem 86:824–835. 10.1046/j.1471-4159.2003.01905.x [DOI] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Begley JG, Mark RJ (1995) Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol 46:187–216. [DOI] [PubMed] [Google Scholar]
- Mena MA, Aguado EG, Yebenes JG (1984) Monoamine metabolites in human cerebrospinal fluid. HPLC/ED method. Acta Neurol Scand 69:218–225. 10.1111/j.1600-0404.1984.tb07804.x [DOI] [PubMed] [Google Scholar]
- Nesci S, Trombetti F, Pagliarani A (2020) Nicotinamide nucleotide transhydrogenase as a sensor of mitochondrial biology. Trends Cell Biol 30:1–3. 10.1016/j.tcb.2019.11.001 [DOI] [PubMed] [Google Scholar]
- Newman JC, Verdin E (2014) β-Hydroxybutyrate: much more than a metabolite. Diabetes Res Clin Pract 106:173–181. 10.1016/j.diabres.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Schwaninger M (2015) Nutritional or pharmacological activation of HCA(2) ameliorates neuroinflammation. Trends Mol Med 21:245–255. 10.1016/j.molmed.2015.02.002 [DOI] [PubMed] [Google Scholar]
- Offermanns S, Colletti SL, Lovenberg TW, Semple G, Wise A, IJzerman AP (2011) Nomenclature and classification of hydroxy-carboxylic acid receptors. Pharmacol Rev 63:269–290. 10.1124/pr.110.003301 [DOI] [PubMed] [Google Scholar]
- Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP (2010) AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochem J 425:523–530. 10.1042/BJ20091515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F. (2004) The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 447:689–709. 10.1007/s00424-003-1099-7 [DOI] [PubMed] [Google Scholar]
- Palmieri F, Monné M (2016) Discoveries, metabolic roles and diseases of mitochondrial carriers: a review. Biochim Biophy Acta 1863:2362–2378. [DOI] [PubMed] [Google Scholar]
- Pardo B, Contreras L (2012) Redox shuttles in the brain. In: Advances in neurobiology. Neuronal metabolism in vivo (Choi IY, Gruetter R, eds), pp 841–883. Boston: Springer. [Google Scholar]
- Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, Saheki T, Satrústegui J (2006) Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem 281:1039–1047. 10.1074/jbc.M507270200 [DOI] [PubMed] [Google Scholar]
- Pardo B, Rodrigues TB, Contreras L, Garzón M, Llorente-Folch I, Kobayashi K, Saheki T, Cerdan S, Satrústegui J (2011) Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J Cereb Blood Flow Metab 31:90–101. 10.1038/jcbfm.2010.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Li H, Sheehy A, Cullen P, Allaire N, Scannevin RH (2016) Dimethyl fumarate alters microglia phenotype and protects neurons against proinflammatory toxic microenvironments. J Neuroimmunol 299:35–44. 10.1016/j.jneuroim.2016.08.006 [DOI] [PubMed] [Google Scholar]
- Perucho J, Gonzalo-Gobernado R, Bazan E, Casarejos MJ, Jiménez-Escrig A, Asensio MJ, Herranz AS (2015) Optimal excitation and emission wavelengths to analyze amino acids and optimize neurotransmitters quantification using precolumn OPA-derivatization by HPLC. Amino Acids 47:963–973. 10.1007/s00726-015-1925-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer B, Sen K, Kaur S, Pappas K (2020) Expanding phenotypic spectrum of cerebral aspartate-glutamate carrier isoform 1 (AGC1) deficiency. Neuropediatrics 51:160–163. 10.1055/s-0039-3400976 [DOI] [PubMed] [Google Scholar]
- Qian W, Van Houten B (2010) Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods 51:452–457. 10.1016/j.ymeth.2010.03.006 [DOI] [PubMed] [Google Scholar]
- Ramos M, Del Arco A, Pardo B, Martínez-Serrano A, Martínez-Morales JR, Kobayashi K, Yasuda T, Bogónez E, Bovolenta P, Saheki T, Satrústegui J (2003) Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res Dev Brain Res 143:33–46. 10.1016/S0165-3806(03)00097-X [DOI] [PubMed] [Google Scholar]
- Ramos M, Pardo B, Llorente-Folch I, Saheki T, del Arco A, Satrústegui J (2011) Deficiency of the mitochondrial transporter of aspartate/glutamate aralar/AGC1 causes hypomyelination and neuronal defects unrelated to myelin deficits in mouse brain. J Neurosci Res 89:2008–2017. 10.1002/jnr.22639 [DOI] [PubMed] [Google Scholar]
- Ruskin DN, Ross JL, Kawamura M, Ruiz TL, Geiger JD, Masino SA (2011) A ketogenic diet delays weight loss and does not impair working memory or motor function in the R6/2 1J mouse model of Huntington's disease. Physiol Behav 103:501–507. 10.1016/j.physbeh.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin DN, Svedova J, Cote JL, Sandau U, Rho JM, Kawamura M, Boison D, Masino SA (2013) Ketogenic diet improves core symptoms of autism in BTBR Mice. PLoS One 8:e65021 10.1371/journal.pone.0065021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satrústegui J, Pardo B, del Arco A (2007) Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev 87:29–67. 10.1152/physrev.00005.2006 [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Siess EA, Brocks DG, Wieland OH (1976) Subcellular distribution of key metabolites in isolated liver cells from fasted rats. FEBS Lett 69:265–271. 10.1016/0014-5793(76)80701-6 [DOI] [PubMed] [Google Scholar]
- Spina MB, Cohen G (1989) Dopamine turnover and glutathione oxidation: implications for Parkinson disease. Proc Natl Acad Sci USA 86:1398–1400. 10.1073/pnas.86.4.1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman D, Ellegood J, Henkelman M (2013a) A gestational ketogenic diet alters maternal metabolic status as well as offspring physiological growth and brain structure in the neonatal mouse. BMC Pregnancy Childbirth 13:198. 10.1186/1471-2393-13-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman D, van Eede M, Wong MD, Adamson SL, Henkelman M (2013b) Effects of a ketogenic diet during pregnancy on embryonic growth in the mouse. BMC Pregnancy Childbirth 13:109. 10.1186/1471-2393-13-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman D, Germann J, Henkelman M (2015) Gestational ketogenic diet programs brain structure and susceptibility to depression and anxiety in the adult mouse offspring. Brain Behav 5:e00300. 10.1002/brb3.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenet J, De Marchi U, Domingo JS, Christinat N, Bultot L, Lefebvre G, Sakamoto K, Descombes P, Masoodi M, Wiederkehr A (2016) Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J 30:1913–1926. 10.1096/fj.201500182 [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, Przedborski S (2003) D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest 112:892–901. 10.1172/JCI18797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S (2003) PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 9:352–355. 10.1038/nm824 [DOI] [PubMed] [Google Scholar]
- Urenjak J, Williams SR, Gadian DG, Noble M (1992) Specific expression of N-acetylaspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro. J Neurochem 59:55–61. 10.1111/j.1471-4159.1992.tb08875.x [DOI] [PubMed] [Google Scholar]
- Van Der Auwera I, Wera S, Van Leuven F, ST H (2005) A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer's disease. Nutr Metab 2:1–8. 10.1186/1743-7075-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderperre B, Herzig S, Krznar P, Hörl M, Ammar Z, Montessuit S, Pierredon S, Zamboni N, Martinou JC (2016) Embryonic lethality of mitochondrial pyruvate carrier 1 deficient mouse can be rescued by a ketogenic diet. PLoS Genet 12:e1006056. 10.1371/journal.pgen.1006056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanItallie TB, Nonas C, Di Rocco A, Boyar K, Hyams K, Heymsfield SB (2005) Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology 64:728–730. 10.1212/01.WNL.0000152046.11390.45 [DOI] [PubMed] [Google Scholar]
- Villeneuve N, Pinton F, Bahi-buisson N, Dulac O, Chiron C, Nabbout R (2009) The ketogenic diet improves recently worsened focal epilepsy. Dev Med Child Neurol 51:276–281. 10.1111/j.1469-8749.2008.03216.x [DOI] [PubMed] [Google Scholar]
- Wiame E, Tyteca D, Pierrot N, Collard F, Amyere M, Noel G, Desmedt J, Nassogne MC, Vikkula M, Octave JN, Vincent MF, Courtoy PJ, Boltshauser E, van Schaftingen E (2010) Molecular identification of aspartate N-acetyltransferase and its mutation in hypoacetylaspartia. Biochem. J 425:127–139. 10.1042/BJ20091024 10.1042/BJ20091024 [DOI] [PubMed] [Google Scholar]
- Wibom R, Lasorsa FM, Töhönen V, Barbaro M, Sterky FH, Kucinski T, Naess K, Jonsson M, Pierri CL, Palmieri F, Wedell A (2009) AGC1 deficiency associated with global cerebral hypomyelination. N Engl J Med 361:489–495. 10.1056/NEJMoa0900591 [DOI] [PubMed] [Google Scholar]
- Williamson JR, Olson MS, Browning ET (1967) Control of citrate formation in rat liver by the nicotinamide-adenine dinucleotide redox state. Biochem J 104:45P–46P. [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ola MS, Berkich DA, Gardner TW, Barber AJ, Palmieri F, Hutson SM, LaNoue KF (2007) Energy sources for glutamate neurotransmission in the retina: absence of the aspartate/glutamate carrier produces reliance on glycolysis in glia. J Neurochem 101:120–131. 10.1111/j.1471-4159.2006.04349.x [DOI] [PubMed] [Google Scholar]
- Zhao Z, Lange DJ, Voustianiouk A, MacGrogan D, Ho L, Suh J, Humala N, Thiyagarajan M, Wang J, Pasinetti GM (2006) A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci 7:29. 10.1186/1471-2202-7-29 [DOI] [PMC free article] [PubMed] [Google Scholar]