

Abstract
Ravulizumab, a novel long‐acting complement component 5 (C5) inhibitor administered every 8 weeks (q8w), was non‐inferior to eculizumab for all efficacy outcomes in two randomised, open‐label, phase 3 trials in C5 inhibitor‐naïve (Study 301) and eculizumab‐experienced (Study 302) adult patients with paroxysmal nocturnal haemoglobinuria (PNH). This pre‐specified analysis characterised ravulizumab pharmacokinetics (PK), pharmacodynamics (PD; free C5 levels), and PD differences between medications (Study 301, n = 246; Study 302, n = 195). Ravulizumab PK parameters were determined using non‐compartmental analysis. Serum free C5 was quantified with a Gyros‐based fluorescence assay (ravulizumab) and an electrochemiluminescence ligand‐binding assay (eculizumab). Ravulizumab PK parameters were numerically comparable in both studies; the median time to maximum concentrations ranged from 2·3 to 2·8 and 2·3 to 2·6 h in studies 301 and 302, respectively. Ravulizumab steady‐state serum concentrations were achieved immediately after the first dose and sustained throughout treatment. For ravulizumab, the mean (SD) post hoc terminal elimination half‐life was 49·7 (8·9) days. Serum free C5 concentrations <0·5 µg/ml were achieved after the first ravulizumab dose and sustained throughout treatment in both studies. In a minority of patients, free C5 concentrations <0·5 µg/ml were not consistently achieved with eculizumab in either study. Ravulizumab q8w was more consistent in providing immediate, complete, sustained C5 inhibition than eculizumab every‐2‐weeks in patients with PNH.
Keywords: complement C5, eculizumab, half‐life, L‐lactate dehydrogenase, ravulizumab
Ravulizumab (Ultomiris™; Alexion Pharmaceuticals, Inc., Boston, MA, USA), recently approved by the USA Food and Drug Administration (FDA), the European Medicines Agency, the Japanese Pharmaceutical and Medical Devices Agency, Health Canada, and the Brazilian Health Regulatory Agency, is a new long‐acting complement component 5 (C5) inhibitor developed from eculizumab (Soliris®; Alexion Pharmaceuticals, Inc.), which is the current standard of care for the treatment of patients with paroxysmal nocturnal haemoglobinuria (PNH). Although highly effective, eculizumab has a terminal half‐life that requires dosing every 2 weeks (q2w; Alexion Pharmaceuticals 2019). This is likely due to the eculizumab‐C5 complex not dissociating efficiently within the endosome, and therefore undergoing both lysosomal degradation and recycling. 1 In ravulizumab, the substitution of four amino acids results in enhanced endosomal dissociation of C5 and increased affinity for the neonatal FC receptor (FcRn), resulting in lysosomal degradation of free C5 and recycling of unbound ravulizumab back into the vascular compartment via the FcRn pathway. Consequently, ravulizumab has a mean terminal half‐life that is approximately fourfold longer than eculizumab, 2 , 3 which enables ravulizumab to be administered via an every‐8‐weeks (q8w) dosing interval. 4 , 5 , 6 , 7
In two phase 3 randomised, open‐label, multicentre clinical trials that compared q8w administration of ravulizumab with standard of care q2w administration of eculizumab in patients with PNH, ravulizumab was shown to be non‐inferior to eculizumab on all primary and key secondary outcomes assessed. 4 , 5 Study 301 (ClinicalTrials.gov identifier: NCT02946463) 5 was carried out in complement inhibitor‐naïve patients with lactate dehydrogenase (LDH) levels ≥1·5 times the upper limit of normal (ULN) and one or more PNH‐related signs or symptoms at screening. In that trial, ravulizumab was non‐inferior to eculizumab on the co‐primary endpoints of transfusion avoidance and LDH normalisation (LDH ULN = 246 u/l). 5 Ravulizumab was also non‐inferior to eculizumab on all four key secondary endpoints: percentage change from baseline in LDH levels, change from baseline in quality of life (QoL) assessed by the Functional Assessment of Chronic Illness Therapy (FACIT)‐Fatigue scale, proportion of patients with breakthrough haemolysis (BTH) and proportion of patients with stabilised haemoglobin levels. 5
Study 302 (ClinicalTrials.gov identifier: NCT03056040) 4 included complement inhibitor‐experienced patients who were stable for ≥6 months on eculizumab treatment, with LDH levels ≤1·5 × ULN at screening. In that trial, ravulizumab was shown to be non‐inferior to eculizumab on the primary endpoint of the percentage change from baseline in LDH levels. 4 Ravulizumab was also non‐inferior on all four key secondary endpoints: the proportion of patients with BTH, change from baseline in QoL assessed by the FACIT‐Fatigue scale, proportion of patients with transfusion avoidance and proportion of patients with stabilised haemoglobin levels. 4
The objectives of the present analysis were to characterise the relationship between plasma drug levels [pharmacokinetics (PK)] on corresponding plasma free and total C5 levels [pharmacodynamics (PD)] associated with ravulizumab q8w versus eculizumab q2w in adult patients with PNH in these clinical trials.
PATIENTS AND METHODS
Study designs
This was a pre‐specified analysis of PK and PD data from studies 301 and 302. 4 , 5 The protocols for studies 301 and 302 were approved by the Institutional Review Board or independent Ethics Committee at each participating centre, and the studies were conducted in accordance with the Declaration of Helsinki and Council for International Organization of Medical Sciences International Ethical Guidelines. Figure 1 presents an overview of the study designs. Briefly, in Study 301, patients with PNH who were complement inhibitor‐naïve and had LDH levels ≥1·5 × ULN were randomly assigned to ravulizumab or eculizumab. 4 , 5 In Study 302, patients with PNH on labelled‐dose (900 mg q2w) eculizumab for >6 months were randomly assigned 1:1 to switch to ravulizumab or continue eculizumab. 4 , 5 All patients gave written informed consent prior to study participation.
Fig 1.
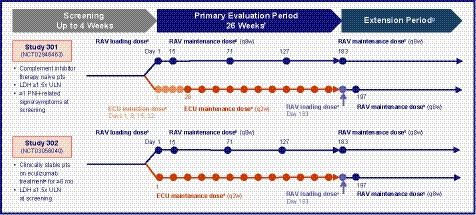
Study designs. aRavulizumab loading dose = 2400 mg for patients weighing ≥40 to <60 kg, 2700 mg for patients weighing ≥60 to <100 kg, and 3000 mg for patients weighing ≥100 kg. bRavulizumab maintenance dose = 3000 mg for patients weighing ≥40 to <60 kg, 3300 mg for patients weighing ≥60 to <100 kg, and 3600 mg for patients weighing ≥100 kg. cEculizumab induction dose = 600 mg (Study 301 only). dEculizumab maintenance dose = 900 mg. eApproved dose for PNH. fBlood was collected for serum drug assessment (ravulizumab and eculizumab) and free and total serum C5 quantitation on day 1 at the end of infusion, and anytime on days 8, 22, 29, 43, 57, 85, 99, 113, 141, 155, and 169, for the ravulizumab group and pre‐dose for the eculizumab group; at days 15, 71, and 127 data are from pre‐dose and end of infusion for both treatment groups; and at day 183 data are from end of the primary evaluation period (prior to dosing) for both treatment groups. gExtension period for Study 301 is planned to conclude at the end of 5 years; extension period for Study 302 is planned to conclude at the end of 3 years. C5, complement component 5; ECU, eculizumab; LDH, lactate dehydrogenase; PNH, paroxysmal nocturnal haemoglobinuria; q2w, every 2 weeks; q8w, every 8 weeks; RAV, ravulizumab; ULN, upper limit of normal (246 u/l).
Patients randomised to ravulizumab received weight‐based dosing: ≥40 to <60 kg received a loading dose of 2400 mg on day 1, then 3000 mg on day 15 and q8w thereafter; ≥60 to <100 kg received a loading dose of 2700 mg on day 1, then 3300 mg on day 15 and q8w thereafter; and ≥100 kg received a loading dose of 3000 mg on day 1, then 3600 mg on day 15 and q8w thereafter. 4 , 5 Patients randomised to eculizumab received 900 mg loading dose on day 1 and q2w thereafter. 4 , 5
Each study had a screening period of up to 4 weeks, a primary evaluation period of 26 weeks, and an extension period (concluding at the end of 5 years for Study 301 and at the end of 3 years for Study 302). Patients randomised to eculizumab were allowed to switch to an open‐label extension of ravulizumab after the 26‐week primary evaluation period. 4 , 5 Patients included in the present analysis had received at least one dose of ravulizumab or eculizumab and had evaluable PK data from the primary evaluation period of 26 weeks.
Details about the PK sampling methods for detecting free serum C5 levels have been published previously 4 , 5 and are summarised briefly in the Data S1. Detailed methods for detection of total serum C5 and for quantification of serum levels of eculizumab and ravulizumab are also presented in the Data S1.
Pharmacokinetic and pharmacodynamic investigations
PK parameters assessed included ravulizumab time to maximum observed serum concentration (tmax), maximum observed serum concentration (Cmax), and concentrations at the end of the dosing interval (Ctrough). PD assessments included change in serum free C5 levels over time and serum total C5 levels over time. Quantification of the key PD parameter, serum free C5, was carried out using validated bioanalytical assays that have a lower limit of quantification of <0·05 μg/ml (Data S1), which correlates with maximal intravascular haemolysis control (Alexion Pharmaceuticals 12/2018) and complete terminal complement inhibition (Alexion Pharmaceuticals 12/2018, Alexion Pharmaceuticals 2019).
Statistical analyses
Ravulizumab and eculizumab concentrations were summarised over time using descriptive statistics [number of patients, mean, standard deviation (SD), coefficient of variation (CV), median, minimum, maximum]. All evaluable PK data were used to derive PK parameters. Non‐compartmental PK parameters for ravulizumab were summarised by descriptive statistics (number of observations, mean, SD, CV, median, minimum, maximum, geometric mean, %CV). Summary statistics of the absolute values in total and free C5 serum concentrations were presented over time by treatment group using the full analysis set, which included all patients who received at least one dose of randomised study drug. All PK analyses were performed using Phoenix WinNonlin (version 7.1; Certara USA, Inc., Princeton, NJ, USA).
RESULTS
Patients
Baseline characteristics and disposition of the separate study populations were previously reported (Table S1). 4 , 5 In the total population of both studies, there was a slightly higher proportion of males [52·6% (232/441)] vs. females [47·4% (209/441)]. Study 301 had a higher proportion of Asian patients [52·4% (129/246)] than Study 302 [21·5% (42/195)]. In Study 301, 246 patients were included in the PK analysis population (ravulizumab, n = 125; eculizumab, n = 121). In Study 302, 195 patients were included in the PK analysis population (ravulizumab, n = 97; eculizumab, n = 98).
Ravulizumab pharmacokinetic profile
The mean steady‐state Cmax of ravulizumab was numerically comparable in both studies and when stratified by weight‐based dosing (Fig 2). There appeared to be a lower steady‐state Ctrough with increasing weight, but mean Ctrough levels remained above the PK threshold for maintaining complete terminal complement inhibition in all body weight groups. The median (minimum, maximum) tmax values (Study 301/Study 302) were 2·8 (2·4, 309·1)/2·6 (2·3, 3·5) h in the ≥40‐ to <60‐kg group, 2·3 (2·0, 335·8)/2·3 (2·0, 5·2) h in the ≥60‐ to <100‐kg group, and 2·6 (2·3, 2·9)/2·4 (2·3, 2·8) h in the ≥100‐kg group. The mean ravulizumab PK parameters were consistent across both studies. The mean (SD) ravulizumab and eculizumab serum concentration versus time profiles are shown in Fig 3. In both studies, steady‐state serum concentrations of ravulizumab were achieved immediately after the first dose and were sustained with q8w maintenance dosing throughout the entire 183‐day treatment period. In Study 301, the mean (SD) steady‐state Cmax was 1373·1 (286·3) µg/ml for ravulizumab and 450·2 (131·6) µg/ml for eculizumab (day 127, post‐dose) and the mean (SD) steady‐state Ctrough was 474·8 (159·1) µg/ml for ravulizumab and 212·6 (95·8) µg/ml for eculizumab (day 183, pre‐dose). In Study 302, the mean (SD) steady‐state Cmax was 1386·3 (268·4) µg/ml for ravulizumab and 492·7 (134·3) µg/ml for eculizumab (day 127, post‐dose) and the mean (SD) steady‐state Ctrough was 500·8 (143·2) µg/ml for ravulizumab and 263·7 (125·0) µg/ml for eculizumab (day 183, pre‐dose).
Fig 2.
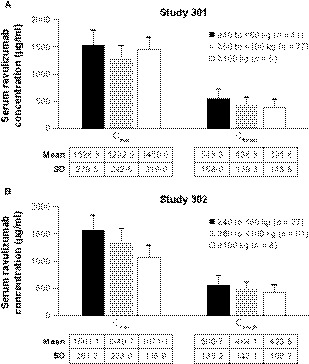
Mean (SD) maximum and trough serum drug concentrations for ravulizumab following the last maintenance dose, stratified by dose group (PK analysis population). (A) Study 301. (B) Study 302. Cmax, maximum observed serum concentration; Ctrough, concentration at the end of the dosing interval; PK, pharmacokinetic; SD, standard deviation.
Fig 3.
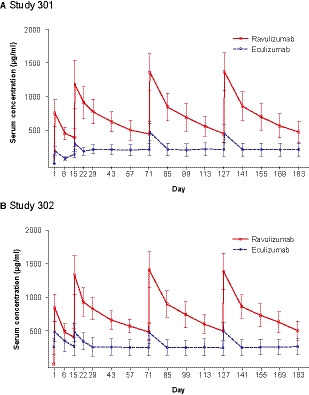
Mean (SD) serum drug concentrations over time (PK analysis population). (A) Study 301. (B) Study 302. Blood was collected for serum drug assessment (ravulizumab and eculizumab) and free and total serum C5 quantitation on day 1 at the end of infusion, and anytime on days 8, 22, 29, 43, 57, 85, 99, 113, 141, 155, and 169, for the ravulizumab group and pre‐dose for the eculizumab group; at days 15, 71, and 127 data are from pre‐dose and end of infusion for both treatment groups; and at day 183 data are from end of the primary evaluation period (prior to dosing) for both treatment groups. Lower limit of quantitation was 1·00 µg/ml for ravulizumab and 5·00 µg/ml for eculizumab. PK, pharmacokinetic; SD, standard deviation. [Colour figure can be viewed at wileyonlinelibrary.com]
Pharmacodynamics
Serum free C5 levels
The mean (SD) baseline serum free C5 levels were 104·1 (27·9) µg/ml and 144·4 (33·2) µg/ml in the ravulizumab and eculizumab groups, respectively, in Study 301 and 0·02 (0·02) µg/ml and 0·05 (0·03) µg/ml, respectively, in Study 302. Immediate, complete, and sustained inhibition of serum free C5 (<0·5 μg/ml) was achieved by the end of the first ravulizumab infusion and maintained throughout the entire 26‐week treatment period in both studies (Fig 4A, B). In both studies, no patient who received ravulizumab was observed to have a post‐baseline serum free C5 level of ≥0·5 µg/ml, whereas among patients who received eculizumab, 15 of the 121 patients (12·4%) in Study 301 and seven of the 98 patients (7·1%) in Study 302 had one or more individual post‐baseline serum free C5 levels ≥0·5 µg/ml over the 26‐week treatment period (treatment difference Study 301: −12·4%, Fisher’s exact P < 0·0001, treatment difference Study 302: −7·1%, Fisher’s exact P = 0·0140). Statistical analyses indicated that the treatment differences in both studies were significant; however, this result should be interpreted with caution as it was not a pre‐specified analysis and the method used to evaluate free C5 levels differed between the treatment groups. Of the 15 eculizumab‐treated patients with a post‐baseline serum free C5 level of ≥0·5 µg/ml in Study 301, nine patients exceeded this level at only one time point, and the other six patients at multiple time points [two time points (n = 1), three time points (n = 1), four time points (n = 1), five time points (n = 2) and nine time points (n = 1)]. Of the seven eculizumab‐treated patients with a post‐baseline serum free C5 level of ≥0·5 µg/ml in Study 302, three patients exceeded this level at one time point, and the other four patients at multiple time points [two time points (n = 2), six time points (n = 1), 10 time points (n = 1)].
Fig 4.
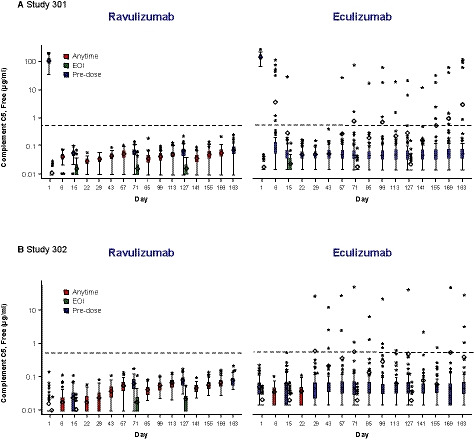
Serum free C5 concentrations over time. (A) Study 301. (B) Study 302. Dashed horizontal lines indicate serum free C5 concentration of 0·5 µg/ml. Horizontal line in the middle of each box is the median; the top and the bottom mark the 75th and 25th percentiles, respectively. The diamond indicates the mean and whiskers represent the 1·5 interquartile range of the lower quartile and upper quartile. Asterisks represent outliers. Y‐axis is a log scale. For the ravulizumab group, for free C5 values that were BLOQ, LLOQ/2 = 0·00915 μg/ml was utilised. For the eculizumab group, for free C5 values that were BLOQ, LLOQ/2 = 0·0137 μg/ml was utilised. For Study 301, the following free C5 samples were excluded as they were considered biologically implausible: ravulizumab, n = 3 day 1 free C5 samples at EOI had values similar to pre‐treatment values; eculizumab, n = 3 Day 1 free C5 samples were BLOQ at pre‐treatment, n = 5 Day 1 free C5 samples at EOI had values similar to pre‐treatment values. For Study 302, day 1 baseline free C5 sample from each treatment group was excluded as the data were considered biologically implausible. The exclusions were corroborated with the paired PK data, as the PK and free C5 samples were collected from the same blood draw. BLOQ, below limit of quantitation; C5, complement component 5; EOI, end of infusion; LLOQ, lower limit of quantitation
Serum total C5 levels
The mean (SD) baseline serum total C5 levels were 104·0 (24·4) µg/ml and 106·5 (26·3) µg/ml in the ravulizumab and eculizumab groups, respectively, in Study 301 and 202·8 (29·5) µg/ml and 206·7 (37·3) µg/ml, respectively, in Study 302. In Study 301, the total mean (SD) C5 levels increased in the ravulizumab and eculizumab groups from 104·0 (24·4) µg/ml and 106·5 (26·3) μg/ml, respectively, at baseline to 183·3 (36·5) µg/ml and 196·1 (38·5) μg/ml, respectively, at day 183. In Study 302, the total mean (SD) C5 levels were elevated in the ravulizumab and eculizumab groups at baseline [202·8 (29·5) µg/ml and 206·7 (37·3) μg/ml, respectively] and remained elevated at day 183 [196·6 (29·3) µg/ml and 226·5 (40·2) μg/ml, respectively]. The rate and magnitude of change in mean serum total C5 over time were numerically comparable between the treatment groups throughout the 26‐week treatment period in both studies, as indicated by the overlapping confidence intervals (Fig 5).
Fig 5.
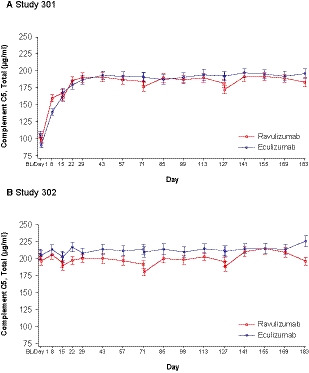
Serum total C5 concentrations over time [mean (95% CI)]. (A) Study 301. (B) Study 302. Lower limit of quantitation was 2·50 µg/ml. Baseline was defined as the last non‐missing assessment value prior to the first dose of study drug. Two patients treated with eculizumab in Study 301 had missing baseline values and were omitted from this analysis. BL, baseline; C5, complement component 5; CI, confidence interval. [Colour figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
In the two largest phase 3 randomised clinical trials conducted to date in patients with PNH, ravulizumab, a novel long‐acting C5 inhibitor derived from targeted modification of eculizumab, was dosed based on body weight and administered q8w. Ravulizumab was shown to achieve better free C5 control compared with eculizumab, the first approved C5 inhibitor. Steady‐state therapeutic serum concentrations of ravulizumab were achieved after the first ravulizumab dose, with a greater than four‐times longer mean (SD) terminal half‐life observed in ravulizumab‐treated patients across both studies [49·7 (8·9) days] (Alexion Pharmaceuticals 12/2018) compared with the previously reported terminal half‐life of eculizumab [11·3 (3·4) days] (Alexion Pharmaceuticals 2019). 8 Immediate and complete terminal complement inhibition was attained by the end of the first ravulizumab infusion and sustained throughout the entire 26‐week treatment period. In contrast, suppression of serum free C5 levels with the approved dosing regimen of eculizumab (900 mg q2w) was inconsistent.
Prior to this analysis, little was known about the PK/PD of ravulizumab. Preclinical analysis described the significant increase in terminal half‐life due to the four amino acid substitutions that differentiate ravulizumab from eculizumab 1 and an analysis of PK/PD in a phase 1 study of single subtherapeutic doses of ravulizumab demonstrated that the mean Cmax increased in a dose‐proportional manner and that the mean terminal half‐life was approximately 32 days. 9 In contrast, there is significantly more known about eculizumab. An overview of PK/PD data compiled from previous eculizumab studies has recently been reported. 10 An analysis of data from patients in the TRIUMPH 11 (ClinicalTrials.gov Identifier: NCT00122330) and SHEPHERD 12 (ClinicalTrials.gov Identifier: NCT00130000) studies to assess the relationship between eculizumab plasma levels and haemolytic activity showed that 36% of eculizumab‐treated patients (49/135 patients with available data) were observed to have trough concentrations below the threshold needed for adequate complement inhibition (<35 µg/ml), and 74% of these patients (36/49) exhibited intravascular haemolysis. 13 Another study in paediatric patients with PNH (n = 7) reported by Reiss et al. 14 showed that eculizumab concentrations ≥124 µg/ml corresponded with complete complement inhibition. Finally, the French group 15 analysed the association between eculizumab concentrations and haemolytic complement activity (reported as CH50) in eculizumab‐treated patients with PNH (n = 22). In that study, none of the patients who had eculizumab serum levels >150 µg/ml exhibited BTH during the study period. However, suboptimal complement inhibition, defined as CH50 values >10%, was observed in 49% (180/364) of the patient samples obtained.
One factor with the potential to have affected the PK/PD characteristics of eculizumab in these studies could be body weight, as eculizumab was given at a fixed dose every 14 days. As with many therapeutic monoclonal antibodies, 16 , 17 , 18 , 19 body weight is a covariate that affects the PK of ravulizumab, with higher body weight patients experiencing lower exposures to ravulizumab than those with lower body weights. Thus, a weight‐based dosing regimen for ravulizumab was developed and evaluated in these phase 3 studies in order to limit inter‐patient variability in exposures across a wide range of patient weights.
In clinical practice, patients treated with eculizumab who had experienced BTH due to suboptimal C5‐inhibition (ranging from 11% to 27%) have been successfully managed by adjusting either the dosage or frequency of maintenance doses to achieve and maintain efficacy of treatment, adjustments that may differ from the approved regimen. 20 , 21 , 22 , 23 , 24 As shown in these clinical studies, the overall incidence of BTH (defined as one or more new or worsening signs or symptoms of intravascular haemolysis in the presence of LDH ≥2 × ULN after prior reduction of LDH to <1·5 × ULN on treatment) 5 observed with ravulizumab in Study 301 [4·0% of ravulizumab patients vs. 10·7% of eculizumab patients, between‐group difference [95% confidence interval (CI), −6·7% (−14·21, 0·180; P inf <0·0001], 5 and Study 302 [0% of ravulizumab patients vs. 5·1% of eculizumab patients, between‐group difference (95% CI) 5·1% (−8·89, 18·99); P inf <0·0004] 4 showed non‐inferiority between treatment groups. However, no BTH events in patients receiving ravulizumab in either study were associated with elevations in free C5. 25 In contrast, patients who received eculizumab experienced BTH events associated with elevations in free C5, suggesting these patients had suboptimal C5 control. 25 A preliminary analysis of the correlation between free C5 levels and BTH episodes was presented previously 25 and a more detailed analysis will be presented separately (manuscript in preparation). Inconsistency and variability in inhibition of free C5 with eculizumab could possibly be associated with manifestations of free C5‐related BTH, thereby driving the numerical difference in that clinical endpoint. However, elevated C5 levels, for example due to infection, may also be able to cause BTH by overwhelming C5 inhibition by eculizumab or ravulizumab. 25
Treatment with eculizumab and ravulizumab was associated with increased total C5 levels. In eculizumab‐naïve patients, a two‐fold increase in total C5 over baseline was observed, and comparable results were seen in patients who were stable on prior eculizumab treatment before randomisation to either ravulizumab or eculizumab. The increase in total C5 levels requires additional analysis and further study.
There are several limitations to the present study. Direct (1:1) correlations between free C5 levels and efficacy as reflected by LDH levels were not assessed. Also, the clinical utility of free C5 levels requires further study as free C5 assays are not often used in clinical practice. In addition, eculizumab dosing in the 301 and 302 studies was per the approved labelled dosage versus a weight‐based dosing regimen for ravulizumab.
Conclusion
Ravulizumab represents a major advance in C5‐inhibitor therapy. Ravulizumab, derived from targeted modification of eculizumab and dosed based on body weight, achieves steady‐state therapeutic serum concentrations immediately after the first dose that are sustained throughout the entire treatment period, and has a mean half‐life of approximately 50 days. In addition, ravulizumab q8w achieved immediate, complete, and sustained terminal complement inhibition throughout the entire 26‐week primary treatment period in two large phase 3 studies. These results provide a mechanistic basis for the overall efficacy of ravulizumab and have implications with respect to free C5‐related BTH.
Conflict of interest
Régis Peffault de Latour has received honoraria, consulting fees, and research support from Alexion Pharmaceuticals, Inc., Pfizer, and Novartis, and has received research support from Amgen. Robert A. Brodsky is a member of the scientific advisory board for and receives grant funding from Alexion Pharmaceuticals, Inc. Stephan Ortiz is an employee and stockholder of Alexion Pharmaceuticals, Inc. Antonio M. Risitano has received research support, honoraria, and consulting fees from Alexion Pharmaceuticals, Inc., Novartis, Alnylam, and Ra Pharma, lecture fees from Alexion Pharmaceuticals, Inc., Novartis, Pfizer, and Jazz, and served as an advisory board member for Alexion Pharmaceuticals, Inc., Novartis, Pfizer, and Jazz, as well as a consultant for Amyndas. Jun H. Jang has no conflicts to declare. Peter Hillmen has received honoraria from and has been a consultant for Alexion Pharmaceuticals, Inc. Alexander D. Kulagin has received research support, honoraria, and consulting fees from Alexion Pharmaceuticals, Inc., Novartis, and JSC GENERIUM. Austin G. Kulasekararaj has received honoraria, travel support, and consulting fees from Alexion Pharmaceuticals, Inc. Scott T. Rottinghaus is an employee and stockholder of Alexion Pharmaceuticals, Inc. Rasha Aguzzi is an employee and stockholder of Alexion Pharmaceuticals, Inc. Xiang Gao is an employee and stockholder of Alexion Pharmaceuticals, Inc. Richard A. Wells has received honoraria, research support, and travel support from Alexion Pharmaceuticals, Inc. Jeff Szer has received research support (to Royal Melbourne Hospital), honoraria, consulting fees, and travel support from Alexion Pharmaceuticals, Inc.
Funding information
The clinical studies were sponsored by Alexion Pharmaceuticals, Inc. Alexion Pharmaceuticals, Inc. also reviewed the manuscript for scientific accuracy of the data. Manuscript revision was made by the authors to address reviewer comments on the basis of scientific and editorial merit. All authors had full access to all the data in the study. The interpretation of study results and the final decision to submit the manuscript for publication was made independently by the authors with no involvement from the sponsor of the study.
Author contributions
Régis Peffault de Latour, Antonio M. Risitano, Peter Hillmen, Austin G. Kulasekararaj and Scott T. Rottinghaus designed the study. Régis Peffault de Latour, Robert A. Brodsky, Antonio M. Risitano, Jun H. Jang, Peter Hillmen, Alexander D. Kulagin, Austin G. Kulasekararaj, Richard A. Wells and Jeff Szer served as study investigators. Régis Peffault de Latour, Robert A. Brodsky, Antonio M. Risitano, Jun H. Jang, Peter Hillmen, Alexander D. Kulagin, Austin G. Kulasekararaj, Richard A. Wells and Jeff Szer enrolled patients. All authors collected data; Stephan Ortiz, Antonio M. Risitano and Scott T. Rottinghaus analysed data. All authors interpreted the data, critically reviewed and revised, and gave final approval of the manuscript.
Data sharing
All relevant data are within the manuscript and its supporting information files. Qualified researchers may request participant‐level, de‐identified clinical data and supporting documents (statistical analysis plan and protocol) pertaining to this study at the following link: https://alexion.com/en/contact‐alexion/medical‐information. Further details regarding data availability, instructions for requesting information, and our data disclosure policy is available on the alexion.com website: http://alexion.com/research‐development.
Supporting information
Data S1. Supplementary material.
Table S1. Demographics and baseline characteristics from the 301 and 302 studies 4 , 5 .
Acknowledgments
The sponsor and investigators thank the patients and their families for their participation in and support for this clinical study. The authors thank Rodrigo Pavani, MD, PhD, and Masayo Ogawa, MD, of Alexion Pharmaceuticals, Inc., for their contribution to the implementation of the 301 and 302 studies. Statistical analysis, PK/PD assessments, and editorial review were provided by Andrew I. Damokosh, PhD, and Rajendra Pradhan, PhD, of Alexion Pharmaceuticals, Inc. Editorial review for scientific accuracy was also provided by Shweta Rane, PhD, CMPP, and Kenneth Pomerantz, PhD, of Alexion Pharmaceuticals, Inc. Medical writing and editorial support were provided by Jennifer M. Kulak, PhD (ApotheCom) and Michael D. Morren, RPh, MBA (Peloton Advantage, LLC, an OPEN Health Company), with funding from Alexion Pharmaceuticals, Inc.
Statement of prior presentation: Portions of this work were presented at the American Society of Hematology 60th Annual Meeting, 1–4 December 2018, San Diego, CA, USA.
References
- 1. Sheridan D, Yu ZX, Zhang Y, Patel R, Sun F, Lasaro MA, et al Design and preclinical characterization of ALXN1210: A novel anti‐C5 antibody with extended duration of action. PLoS One. 2018;13:e0195909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soliris [prescribing information]. New Haven, CT: Alexion Pharmaceuticals, Inc.; 2019. [Google Scholar]
- 3. Peffault de Latour R, Brodsky RA, Ortiz S, Risitano AM, Jang JH, Hillmen P, et al Ravulizumab (ALXN1210) versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria: pharmacokinetics and pharmacodynamics observed in two phase 3 randomized, multicenter studies. Blood. 2018;132(Suppl. 1):626. [Google Scholar]
- 4. Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez‐Fernandez FA, et al Ravulizumab (ALXN1210) vs eculizumab in C5‐inhibitor‐experienced adult patients with PNH: the 302 study. Blood. 2019;133:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, Pessoa V, Gualandro S, Füreder W, et al Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133:530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roth A, Rottinghaus ST, Hill A, Bachman ES, Kim JS, Schrezenmeier H, et al Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of 2 phase 1b/2 studies. Blood Adv. 2018;2:2176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ultomiris [prescribing information]. Boston, MA: Alexion Pharmaceuticals, Inc.; 2018. [Google Scholar]
- 8. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nature Biotechnol. 2007;25:1256–64. [DOI] [PubMed] [Google Scholar]
- 9. Sahelijo L, Mujeebuddin A, Mitchell D, Larouche R, Yu ZX, Zhang Y, et al First in human single‐ascending dose study: safety, biomarker, pharmacokinetics and exposure‐response relationships of ALXN1210, a humanized monoclonal antibody to C5, with marked half‐life extension and potential for significantly longer dosing intervals. Blood. 2015;126:4777. [Google Scholar]
- 10. Wijnsma KL, Ter Heine R, Moes D, Langemeijer S, Schols SE, Volokhina EB, et al Pharmacology, pharmacokinetics and pharmacodynamics of eculizumab, and possibilities for an individualized approach to eculizumab. Clin Pharmacokinet. 2019;58:859–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233–43. [DOI] [PubMed] [Google Scholar]
- 12. Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111:1840–7. [DOI] [PubMed] [Google Scholar]
- 13. Hillmen P, Muus P, Roth A, Elebute MO, Risitano AM, Schrezenmeier H, et al Long‐term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162:62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reiss UM, Schwartz J, Sakamoto KM, Puthenveetil G, Ogawa M, Bedrosian CL, et al Efficacy and safety of eculizumab in children and adolescents with paroxysmal nocturnal hemoglobinuria. Pediatr Blood Cancer. 2014;61:1544–50. [DOI] [PubMed] [Google Scholar]
- 15. Peffault de Latour R, Fremeaux‐Bacchi V, Porcher R, Xhaard A, Rosain J, Castaneda DC, et al Assessing complement blockade in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Blood. 2015;125:775–83. [DOI] [PubMed] [Google Scholar]
- 16. Bai S, Jorga K, Xin Y, Jin D, Zheng Y, Damico‐Beyer LA, et al A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet. 2012;51:119–35. [DOI] [PubMed] [Google Scholar]
- 17. Freshwater T, Kondic A, Ahamadi M, Li CH, de Greef R, de Alwis D, et al Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hendrikx JJ, Haanen JB, Voest EE, Schellens JH, Huitema AD, Beijnen JH. Fixed dosing of monoclonal antibodies in oncology. Oncologist. 2017;22:1212–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang DD, Zhang S, Zhao H, Men AY, Parivar K. Fixed dosing versus body size‐based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol. 2009;49:1012–4. [DOI] [PubMed] [Google Scholar]
- 20. Almomen A, Al Bakistani A, Alsaeed A, Al Olama A, Hejazi A, Awarji C, et al Paroxysmal nocturnal hemoglobinuria: diagnosis and management protocol. J Appl Hematol. 2014;5:37–44. [Google Scholar]
- 21. Brodsky RA. Eculizumab: another breakthrough. Blood. 2017;129:922–3. [DOI] [PubMed] [Google Scholar]
- 22. Harder MJ, Kuhn N, Schrezenmeier H, Hochsmann B, von Zabern I, Weinstock C, et al Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129:970–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelly R, Arnold L, Richards S, Hill A, vanBijnen S, Muus P, et al Modification of the eculizumab dose to successfully manage intravascular breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;112:3441. [Google Scholar]
- 24. Nakayama H, Usuki K, Echizen H, Ogawa R, Orii T. Eculizumab dosing intervals longer than 17 days may be associated with greater risk of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Biol Pharm Bull. 2016;39:285–8. [DOI] [PubMed] [Google Scholar]
- 25. Brodsky RA, Peffault De Latour R, Rottinghaus ST, Röth A, Risitano AM, Weitz IC, et al A prospective analysis of breakthrough hemolysis in 2 phase 3 randomized studies of ravulizumab (ALXN1210) versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Blood. 2018;132(Suppl. 1):2330. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary material.
Table S1. Demographics and baseline characteristics from the 301 and 302 studies 4 , 5 .