

Abstract
Background
Conventional type 1 dendritic cells (cDC1s) control anti‐viral and anti‐tumor immunity by inducing antigen‐specific cytotoxic CD8+ T‐cell responses. Controversy exists whether cDC1s also control CD4+ T helper 2 (Th2) cell responses, since suppressive and activating roles have been reported. DC activation status, controlled by the transcription factor NF‐κB, might determine the precise outcome of Th‐cell differentiation upon encounter with cDC1s. To investigate the role of activated cDC1s in Th2‐driven immune responses, pulmonary cDC1s were activated by targeted deletion of A20/Tnfaip3, a negative regulator of NF‐κB signaling.
Methods
To target pulmonary cDC1s, Cd207 (Langerin)‐mediated excision of A20/Tnfaip3 was used, generating Tnfaip3 fl/flxCd207 +/cre (Tnfaip3 Lg‐KO) mice. Mice were exposed to house dust mite (HDM) to provoke Th2‐mediated immune responses.
Results
Mice harboring Tnfaip3‐deficient cDC1s did not develop Th2‐driven eosinophilic airway inflammation upon HDM exposure, but rather showed elevated numbers of IFNγ‐expressing CD8+ T cells. In addition, Tnfaip3 Lg‐KO mice harbored increased numbers of IL‐12–expressing cDC1s and elevated PD‐L1 expression in all pulmonary DC subsets. Blocking either IL‐12 or IFNγ in Tnfaip3 Lg‐KO mice restored Th2 responses, whereas administration of recombinant IFNγ during HDM sensitization in C57Bl/6 mice blocked Th2 development.
Conclusions
These findings indicate that the activation status of cDC1s, shown by their specific expression of co‐inhibitory molecules and cytokines, critically contributes to the development of Th2 cell–mediated disorders, most likely by influencing IFNγ production in CD8+ T cells.
Keywords: A20/Tnfaip3, CD8+ T cells, interferon gamma, Th2‐driven airway inflammation, Type 1 conventional dendritic cells
Mice harboring Tnfaip3‐deficient cDC1s (Tnfaip3 Lg‐KO mice) do not develop Th2‐driven eosinophilic airway inflammation upon house dust mite exposure but have elevated numbers of pulmonary IFNγ‐expressing CD8+ T‐cells. In the lungs of Tnfaip3 Lg‐KO mice, cDC1s have increased IL‐12 and PD‐L1 expression. Both IL‐12 and IFNγ are involved in the reduced Th2‐response observed in Tnfaip3 Lg‐KO mice.
Abbreviations: cDC, conventional dendritic cell; PD‐L1, programmed death ligand 1; Tnfaip3, TNF alpha‐induced protein 3.
Abbreviations
- BAL
bronchoalveolar lavage
- cDC
conventional dendritic cell
- DC
dendritic cell
- HDM
house dust mite
- Lg
Langerin
- MLN
mediastinal lymph node
- moDC
monocyte‐derived dendritic cell
- NF‐κB
nuclear factor‐κB
- PBS
phosphate buffered saline
- pDC
plasmacytoid dendritic cell
- PRR
pattern recognition receptors
- Th
T helper
- TNFAIP3
TNF‐α‐induced protein 3
- TNIP
TNFAIP3‐interacting protein
- Treg
regulatory T cell
- WT
wild‐type
- YFP
yellow fluorescent protein
1. INTRODUCTION
T helper (Th) 2‐mediated diseases such as allergic asthma affect people all over the world. Th2 cytokines facilitate the classical allergic response, such as IgE class switching by B cells (IL‐4), eosinophilic inflammation (IL‐5), and goblet cell hyperplasia (IL‐13). 1 Dendritic cells (DCs) are potent antigen‐presenting cells that induce the differentiation of naïve Th cells into Th2 cells or various other T helper cell subsets. Based on surface markers and transcription factor expression, two main conventional DC (cDC) subsets can be identified: type 1 cDCs (cDC1s) and cDC2s. 2 During inflammation, a third population of monocyte‐derived DCs (moDCs) arises. 3 IRF‐4–dependent cDC2s are considered to be efficient at priming CD4+ T cells through MHC class II‐restricted antigen (Ag) presentation. 4 , 5 , 6 Upon allergen inhalation, cDC2s drive Th2 differentiation, whereas moDCs maintain Th2 inflammation through local secretion of chemokines. 7 cDC1s have superior Ag‐cross‐presenting capabilities and play pivotal roles in antiviral and antitumor immunity through the induction of antigen‐specific cytotoxic CD8+ T‐cell responses. 8 , 9 Conflicting data exist concerning cDC1 function in Th2‐mediated diseases, whereby cDC1s were reported to either suppress allergic airway inflammation, 10 , 11 , 12 be redundant, 7 , 11 or essential for Th2 immune responses. 13 , 14 , 15 During experimental helminth infection, cDC1s limit the amplitude of Th2 immune responses through their IL‐12 production. 16
DC activation is needed for proper Th‐cell differentiation into Th1, Th2, and Th17cells. 17 Although there is evidence that pattern recognition receptors (PRRs) on lung epithelial cells contribute to allergic airway inflammation, 18 activation of DCs in Th2‐mediated diseases also often occurs through allergen‐mediated triggering of PRRs on DCs. 19 PRR triggering on DCs activates the transcription factor NF‐κB, which initiates transcription of pro‐inflammatory cytokines. NF‐κB activation is negatively regulated by TNFAIP3 (TNFα‐induced protein 3, also known as A20), a ubiquitin‐modifying enzyme that can (de)ubiquitinate several NF‐κB signaling molecules to terminate NF‐κB activation. 20 , 21 Several models of DC‐specific deletion of TNFAIP3 in CD11chi cells have shown that this molecule limits DC activation, and in the absence of TNFAIP3, DCs produce higher levels of cytokines, express higher co‐stimulatory molecules, resist to cell death, and activate auto‐reactive T‐ and B‐cell responses. 22 , 23 TNFAIP3 is also implicated in allergic disorders in humans, as genetic polymorphisms in the TNFAIP3 and TNFAIP3 interacting protein (TNIP) loci have been associated with asthma and allergies. 24 , 25 , 26 Also, reduced TNFAIP3 expression in peripheral blood mononuclear cells was observed in asthmatic children compared healthy controls. 27 Recently, we found that increasing the activation status of DCs by ablation of the Tnfaip3 gene in myeloid cells induced a neutrophilic inflammation in a house dust mite (HDM)‐mediated murine asthma model, which was accompanied by elevated numbers of Th17‐cells. 23
However, whether TNFAIP3 levels in cDC1s affect the development of asthma is currently unknown. Therefore, in this study, we investigated whether TNFAIP3 depletion in cDC1s would affect their activation status and thereby affect asthma development. To this end, we crossed Tnfaip3floxed mice 22 with a transgenic mouse line that expresses Cre under the control of the Langerin (Cd207) promotor, which targets Langerin‐expressing cDC1s, 28 and performed HDM‐driven allergic airway inflammation experiments. Our data indicate that TNFAIP3 depletion in Langerin‐expressing cDC1s inhibited Th2‐mediated immune responses. This was associated with an increase in the production of the Th2‐suppressive cytokine IL‐12 by cDC1s, leading to increased IFNγ production by CD8+ T cells. Increased production of IFNγ subsequently increased the expression of the co‐inhibitory molecule PD‐L1 on all pulmonary DC subsets. Strikingly, administration of IFNγ only during HDM sensitization was already sufficient to abrogate Th2 development and eosinophilic inflammation.
2. MATERIALS AND METHODS
2.1. Mice
Tnfaip3 fl/fl mice 22 were crossed to Cd207 CRE/+ (Langerin‐CRE) mice 28 to generate Tnfaip3 Lg mice. Tnfaip3 Lg mice were crossed to Rosa26‐stop‐EYFP reporter (ROSA26flEYFP) mice. Mice were backcrossed to the C57BL/6 genetic background for at least six generations. Mice were housed and bred under SPF conditions at the Erasmus MC and analyzed at 6‐12 weeks of age. All experiments were performed with approval by the animal ethics committee of the Erasmus MC.
2.2. HDM‐induced allergic airway inflammation
During HDM exposures, mice were anesthetized using isoflurane. Mice were sensitized intranasally (i.n.) with 1 μg/40 μL HDM (Greer) or 40 μL PBS as a control on day 1 (GIBCO Life Technologies), and challenged i.n. on days 7‐11 with 10 μg/40 μL HDM. 7 Mice were killed on day 15. Bronchoalveolar lavage (BAL) was obtained by flushing the lungs three times with 1 mL PBS containing 0.5 mM EDTA (Sigma‐Aldrich). Lungs were either inflated with PBS/OCT (1:1) solution and placed in 4% PFA and embedded in paraffin or single‐cell suspensions were prepared.
2.3. HDM‐induced acute immune responses
For the induction of an HDM‐mediated acute innate response, mice were treated with 100 µg/80 µL of HDM extract intra‐tracheally (i.t). Twenty‐four hours later, single‐cell suspensions were obtained from lungs by digesting the lungs using DNAse (Sigma) and Liberase TM (Roche) for 30 minutes at 37°C. 7 After digestion, the lungs were homogenized through a 100 μm cell strainer (BD Biosciences). Red blood cells were lysed using osmotic lysis buffer (8.3% NH4Cl, 1% KHCO3, and 0.04% NA2EDTA in Milli‐Q). Cell suspensions were prepared and used for flow cytometry procedures.
2.4. Antibody treatment of mice during HDM‐induced allergic airway inflammation
To study the effect of IL‐12, IFNγ, and PD‐L1 on development of HDM‐induced allergic airway inflammation in Tnfaip3 Lg mice, mice were treated i.p. with 500 μg anti–IL‐12p40 (clone C17/8), 500 μg anti‐IFNy (clone XMG1.2) antibodies, 250 μg anti–PD‐L1 (clone MIH5) antibodies or with monoclonal antibody β‐galactosidase (GL113) as isotype control, 11, 7, 4, and day prior to 1 µg/40 µL HDM sensitization on day 0, and at days 0, 3, 7, and 10 during the HDM‐induced allergic airway inflammation model. Fifty ng recombinant IFNγ (R&D systems) was administered together with 1 µg/40 µL HDM sensitization on day 0. Mice were killed on day 15 after HDM sensitization. For the HDM‐induced acute immune response, mice were treated with monoclonal antibodies or isotype control 10, 7, 4, and 1 day prior to 100 µg/80 µL HDM administration on day 0. Mice were killed on day 1.
2.5. Flow cytometry
Single‐cell suspensions were prepared from bronchoalveolar lavage (BAL) and MLN using standard procedures. MLNs were homogenized through a 100 μm cell strainer (BD Biosciences). Flow cytometry surface and intracellular staining procedures have been described previously. 23 Monoclonal antibodies used for mouse flow cytometric analyses are listed in Table S1. In all experiments, dead cells were excluded using Fixable viability dye (eBioscience). To measure cytokine production by T cells, cells were stimulated at 37°C using 10 ng/mL PMA (Sigma‐Aldrich), 250 ng/mL ionomycin (Sigma‐Aldrich) and GolgiStop (BD Bioscience), for 4 hours. To measure cytokine production by DCs, cells were incubated at 37°C in the presence of GolgiPlug (BD Biosciences) for 4 hours. Data were acquired using an LSR II flow cytometer (Beckton Dickinson) and FACS software (Beckton Dickinson) and analyzed by FlowJo version 9 (Tree Star Inc software).
2.6. Lung histology
Five‐um‐thick paraffin‐embedded lung sections were stained with periodic acid and Schiffs reagents to visualize mucus‐producing cells.
2.7. ELISA
Total IgE and HDM‐specific IgG1 were measured in serum (Opteia, BD Biosciences).
2.8. Statistical analysis
Mann‐Whitney U tests were used for comparison between two relevant groups, which either differ only in genotype of the mice, or in the protocol/treatment received, and a P‐value of <.05 was considered statistically significant. Analysis was determined using Prism (GraphPad Software).
3. RESULTS
3.1. Depletion of TNFAIP3 in Langerin+ cDC1s increases PD‐L1 expressing lung DCs
Langerin (CD207) is expressed in Langerhans cells in the skin and in a proportion of pulmonary cDC1s. Consistent with other publications, we also found that Langerin was expressed by 15% of cDC1s in the lung, whereas in other pulmonary DC subsets, Langerin expression was not detected (Figure 1A,B). 28 , 29 To determine whether Langerin‐Cre‐mediated targeting of cDC1s reflects CD207 expression, we crossed Langerin‐Cre mice 28 to Rosa26‐stopflEYFP mice (Langerin‐cre x Rosa26‐stopflEYFP mice). DC subsets were examined according to the gating strategy as shown in Figure S1. In the lungs and lung‐draining mediastinal lymph node (MLN) of naive mice at the age of 6‐8 weeks, approximately 10% of the cDC1s expressed EYFP, whereas in the bronchoalveolar lavage (BAL), 35% of cDC1s expressed EYFP (Figure 1C,D). Expression of EYFP in other DC subsets in BAL, lung, or MLN was below 3%. TNFAIP3 deletion did not affect EYFP expression as the proportions of EYFP‐expressing pulmonary DCs were not different between Tnfaip3 Lg‐WT and Tnfaip3 Lg‐KO Rosa26‐stopflEYFP mice (Figure 1E). Since TNFAIP3 deletion affects the activation status of DCs, 22 , 23 we determined the expression of MHCII, CD86, and PD‐L1 on cDC1s. MHCII expression was not altered, whereas CD86 expression was significantly decreased, and PD‐L1 expression was increased on pulmonary cDC1s of Tnfaip3 Lg‐KO mice compared with Tnfaip3 Lg‐WT mice (Figure 1F). As the Cd207 promotor targets only a fraction of pulmonary cDC1s, we compared the expression of PD‐L1 in EYFP+ and EYFP‐ cDC1s of Tnfaip3 Lg x Rosa26‐stopflEYFP mice. PD‐L1 expression was especially increased in EYFP+ cDC1s compared to EYFP‐ cDC1s in Tnfaip3 Lg‐KO x Rosa26‐stopflEYFP mice, suggesting that the increased expression of PD‐L1 was caused by a cell intrinsic defect caused by loss of Tnfaip3 in cells expressing langerin (Figure S2).
FIGURE 1.
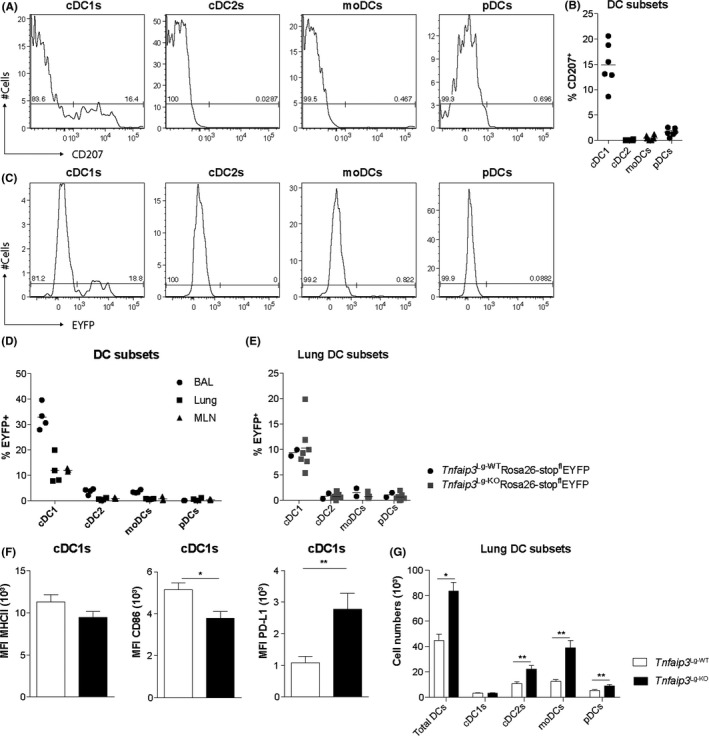
Depletion of TNFAIP3 in Langerin+ cDC1s increased PD‐L1 expression and pulmonary DC numbers. (A) Flow cytometry histograms of lung DC subsets in C57BL/6 mice showing CD207 expression in cDC1s, cDC2s, moDCs, and pDCs. (B) Quantification of the percentage CD207 expression in different DC subsets. (C) Flow cytometric histograms of EYFP expression in lung DC subsets of Langerin‐cre x Rosa26‐stopflEYFP mice. (D) Quantification of the percentage EYFP expression in different DC subsets by flow cytometry in BAL, lung, and MLN. (E) EYFP expression in different DC subsets in the lung of Tnfaip3 Lg‐WTRosa26‐stopflEYFP and Tnfaip3 Lg‐KORosa26‐stopflEYFP mice. (F) Quantification of mean fluorescent intensity of MHCII, CD86, and PD‐L1 in lung cDC1s of Tnfaip3 Lg‐WT mice (n = 6) and cDC1s of Tnfaip3 Lg‐KO (n = 4) by flow cytometry. (G) Quantification of PD‐L1 in EYFP‐ and EYFP+ cDC1s in the lung of Tnfaip3 Lg‐WT x Rosa26‐stopflEYFP (n = 6) and Tnfaip3 Lg‐KO x Rosa26‐stopflEYFP mice (n = 4). Results are presented as mean ± SEM of n = 2‐7 per group and representative of two independent experiments
We next investigated whether the Langerin‐Cre‐mediated TNFAIP3 deletion in a fraction of pulmonary cDC1s affected the numbers of pulmonary DC subsets. Total DCs and specifically cDC2s, moDCs, and plasmacytoid (pDCs) were increased in lungs of Tnfaip3 Lg‐KO mice as compared to controls. The number of cDC1s was unaffected in Tnfaip3 Lg‐KO mice (Figure 1G). As Langerin is mainly expressed by Langerhans cells in the skin, we also evaluated the frequency and activation status of Langerhans cells in the epidermis, and found no differences in their frequencies between Tnfaip3 Lg‐KO and Tnfaip3 Lg‐WT mice. In contrast to Tnfaip3‐deficient cDC1s in the lungs, expression of the co‐stimulatory molecule CD86 was increased in Langerhans cells of Tnfaip3 Lg‐KO mice as compared to Tnfaip3 Lg‐WT mice (Figure S3).
As DC activation can affect T‐cell homeostasis 30 and TNFAIP3 deletion in DCs has been shown to induce autoimmune disease, we evaluated Tcells in the lung, MLN, and spleen. Both CD4+ and CD8+ T‐cell numbers were similar between lung, MLN, and spleen of Tnfaip3 Lg‐KO and Tnfaip3 Lg‐WT mice (Figure S4), indicating that TNFAIP3 deletion selectively in Langerin‐expressing DC subsets does not induce systemic inflammation or signs of autoimmunity.
In conclusion, these data show that Langerin‐expressing cDC1s comprise a small proportion of lung cDC1s. TNFAIP3 deletion in cDC1s altered their phenotype by decreasing CD86 expression, while increasing the expression of the co‐inhibitory molecule PD‐L1. Ablation of TNFAIP3 in Langerin+ cDC1s augmented the numbers of pulmonary cDC2s, moDCs, and pDCs, whereas it had no effect on T‐cell numbers locally nor systemically.
3.2. Th2‐mediated HDM‐induced airway inflammation is reduced in Tnfaip3 Lg‐KO mice
To investigate the effects of cDC1‐specific TNFAIP3 deficiency on Th2‐cell differentiation, we exposed Tnfaip3 Lg‐KO mice to inhaled HDM to induce allergic airway inflammation (Figure 2A). As previously reported, 31 HDM sensitization followed by repetitive HDM challenge increased eosinophils, B cells, and Tcells in the BAL as compared with PBS sensitization in WT mice (Figure 2B,C). Strikingly, HDM‐sensitized and HDM‐challenged Tnfaip3 Lg‐KO had reduced eosinophil and T‐cell numbers in the BAL compared to Tnfaip3 Lg‐WT mice, whereas the numbers of neutrophils, macrophages, and B cells were unchanged (Figure 2B,C). Th2 cytokine‐secreting CD4+ T‐cells were increased in HDM‐sensitized Tnfaip3 Lg‐WT mice, whereas IL‐5+ and IL‐13+ Thcells in HDM‐sensitized Tnfaip3 Lg‐KO were not elevated as compared with PBS‐sensitized controls. In this model, IL‐17+ was not induced in Th cells (Figure 2E). Serum IgE increased upon HDM sensitization and challenge in Tnfaip3 Lg‐WT mice, but not in Tnfaip3 Lg‐KO mice, compared to PBS‐sensitized control mice (Figure 2F). Accordingly, upon immunohistochemistry analysis of the lungs, HDM‐sensitized Tnfaip3 Lg‐KO mice showed no lung inflammation and mucus production, which were readily observed in Tnfaip3 Lg‐WT mice (Figure 2G). As Th2‐mediated airway inflammation was hampered in HDM‐sensitized Tnfaip3 Lg‐KO mice, we wondered whether anti‐inflammatory cells would be increased in HDM‐treated Tnfaip3 Lg‐KO mice. MLNs of both PBS‐ and HDM‐sensitized Tnfaip3 Lg‐KO mice had increased proportions of Foxp3+CD25+ Tregs compared to PBS‐ and HDM‐sensitized Tnfaip3 Lg‐WT mice, respectively (Figure S5). However, this increase in Tregs in Tnfaip3 Lg‐KO mice was not responsible for the reduced Th2‐driven immune responses, as Tnfaip3 Lg‐KO mice depleted of Tregs by an anti‐CD25–depleting antibody (PC61), still failed to develop Th2‐driven inflammation (Figure S6).
FIGURE 2.
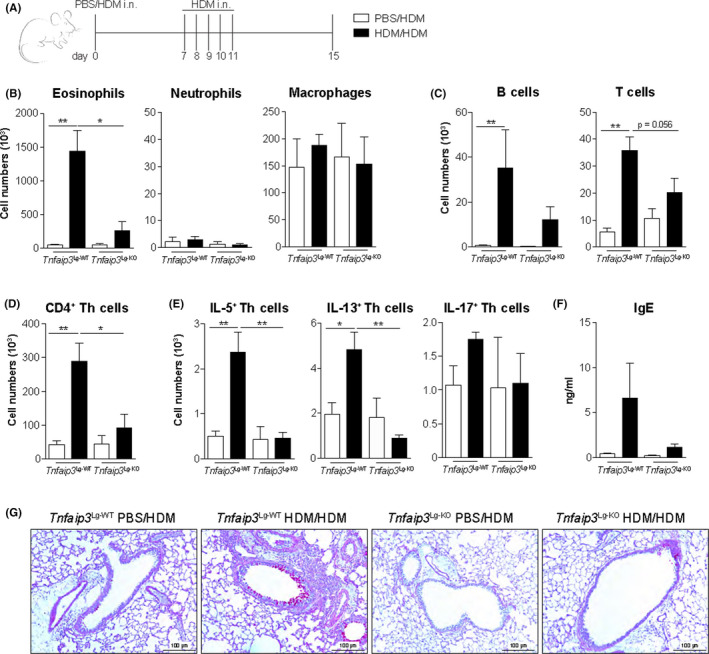
Tnfaip3 Lg‐KO mice develop reduced Th2‐mediated inflammation. (A) Tnfaip3 Lg mice were sensitized i.n. with PBS or 1 μg HDM on day 0 and challenged i.n. with 10 μg HDM daily between days 7 and 11 to induce allergic airway inflammation. Analysis was performed at day 15. (B‐C) Numbers of eosinophils, neutrophils, macrophages, B cells, and Tcells determined in BAL by flow cytometric analysis. (D) Number of CD4+Tcells in BAL by flow cytometric analysis. (E) Number of IL‐5+, IL‐13+, and IL‐17+ CD4 Tcells in the BAL by flow cytometric analysis. (F) Histological analysis of mucus‐producing goblet cells in the airways by periodic acid staining. (G) Serum total IgE levels determined by ELISA. Results are presented as mean ± SEM of 6 PBS or HDM‐sensitized Tnfaip3 Lg‐WT mice, 2 PBS‐sensitized Tnfaip3 Lg‐KO mice, and 5 HDM‐sensitized Tnfaip3 Lg‐KO mice. Results depicted are representative of two or more independent experiments. *P < .05, **P < .01
Taken together, these findings indicate that TNFAIP3 deletion in cDC1s hampers the induction of eosinophilia, Th2 cytokine‐producing Tcells, and increased serum IgE upon HDM exposure. Furthermore, Tnfaip3 Lg‐KO mice had higher numbers of anti‐inflammatory Tregs following HDM exposure. Depleting Tregs in Tnfaip3 Lg‐KO mice did not restore Th2‐mediated inflammation, indicating that Tregs are not essential for the suppression of Th2‐mediated inflammation by TNFAIP3‐deficient cDC1s.
3.3. TNFAIP3 deletion in cDC1s increases their PD‐L1 and IL‐12 expression
Tnfaip3 Lg‐KO mice developed reduced eosinophilic inflammation upon HDM sensitization and challenge exposure, and this effect could be due to effects during priming or allergen re‐challenge. To study effect of TNFAIP3 loss on DCs during priming, we examined DC subset frequencies, migratory capacity, and co‐stimulatory marker expression following a single intra‐tracheal high dose of HDM. After HDM administration, total DC numbers and moDCs in the lung increased in Tnfaip3 Lg‐WT mice, but not in Tnfaip3 Lg‐KO mice. cDC1s, cDC2s, and pDC numbers were not altered upon HDM exposure in lungs of Tnfaip3 Lg‐WT mice (Figure S7A). Pulmonary cDC1s and cDC2s decreased upon HDM exposure in Tnfaip3 Lg‐KO mice. Since activated pulmonary DCs migrate toward the MLN, we also evaluated DC numbers in the MLN. Only cDC1s were increased in PBS‐treated Tnfaip3 Lg‐KO mice compared to PBS‐treated Tnfaip3 Lg‐WT mice. HDM exposure increased the numbers of total DCs, cDC1s, and cDC2s similarly in both Tnfaip3 Lg‐KO and Tnfaip3 Lg‐WT mice (Figure S7B), indicating that DC migration to the MLN is not affected by Tnfaip3 deletion in langerin‐expressing cDC1s.
As co‐inhibitory marker expression and cytokine production by DCs are implicated in immune tolerance during a first antigen exposure, 32 we examined DC subsets for the expression of the co‐inhibitory molecules PD‐L1, PD‐L2, and ICOSL, and the regulatory cytokines TGF‐β and IL‐10 after a single HDM administration. No differences were observed in PD‐L2, ICOSL, TGF‐β, or IL‐10 expression between pulmonary DCs of Tnfaip3 Lg‐WT and Tnfaip3 Lg‐KO mice (data not shown). PD‐L1 was upregulated on all DCs of Tnfaip3 Lg‐WT mice after HDM exposure yet was already elevated in all DCs of Tnfaip3 Lg‐KO mice in steady state (PBS) and further increased by HDM in cDC1s of Tnfaip3 Lg‐KO mice. However, after HDM provocation, PD‐L1 levels are similar between DC subsets of Tnfaip3 Lg‐WT and Tnfaip3 Lg‐KO mice (Figure 3A,B and Figure S8A). Surprisingly, cDC2s and moDCs in Tnfaip3 Lg‐KO mice fail to upregulate MHCII in response to HDM as compared to Tnfaip3 Lg‐WT mice (Figure 3C,D and Figure S8B).
FIGURE 3.
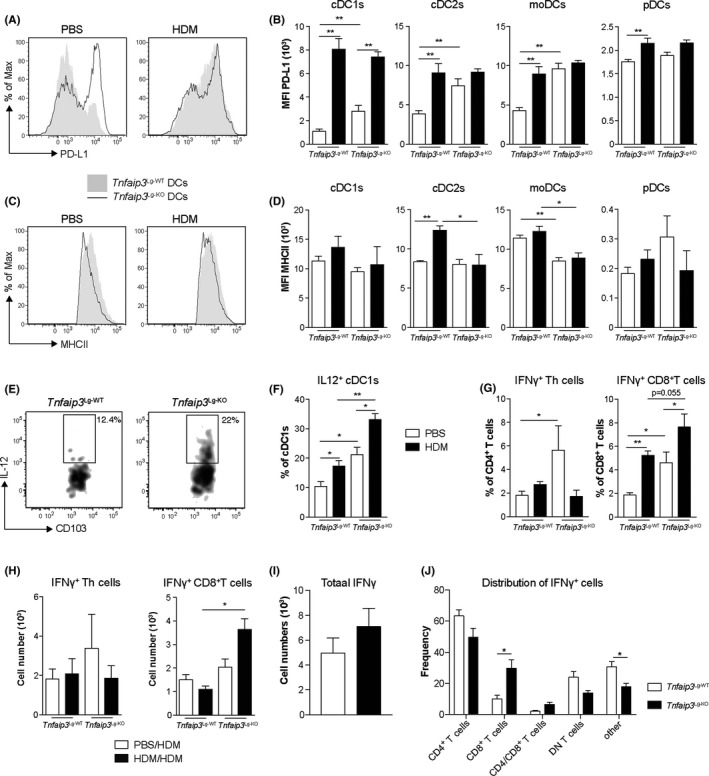
TNFAIP3 deletion in cDC1s increases PD‐L1 and IL‐12 expression in all pulmonary DC subsets, together with increased IFNγ expression by CD8+ T cells. (A) Flow cytometric histogram showing PD‐L1 expression in pulmonary DCs in Tnfaip3 Lg mice upon a single provocation of PBS or 100 μg HDM. (B) Quantification of MFI of PD‐L1 in cDC1s, cDC2s, moDCs, and pDCs of PBS or HDM‐treated Tnfaip3 Lg‐WT (n = 6) or Tnfaip3 Lg‐KO (n = 4) mice by flow cytometry. (C) Flow cytometric histogram of MHCII expression in pulmonary DCs upon PBS or HDM treatment. (D) Quantification of MFI of MHCII in cDC1s, cDC2s, moDCs, and pDCs of PBS or HDM‐treated Tnfaip3 Lg‐WT (n = 6) or Tnfaip3 Lg‐KO (n = 4) mice by flow cytometry. (E) Flow cytometric gating of pulmonary IL‐12‐secreting cDC1s. (F) Percentage of IL‐12 secreting cDC1s in lungs of PBS or HDM‐treated Tnfaip3 Lg‐WT (n = 6) or Tnfaip3 Lg‐KO (n = 4) mice by flow cytometry. (G) Proportion of IFNγ + CD4+ and CD8+ T cells in lungs of Tnfaip3 Lg mice 24 h after single PBS or HDM provocation by flow cytometry. (H) Quantification of the number of IFNγ+ CD4+ and CD8+ T cells in BAL of Tnfaip3 Lg‐WT mice (n = 6) and Tnfaip3 Lg‐KO mice (n = 4) after HDM sensitization and challenge. (I) Quantification of the total numbers of IFNγ‐producing cells in BAL of HDM‐sensitized and HDM‐challenged Tnfaip3 Lg‐WT mice (n = 6) and Tnfaip3 Lg‐KO mice (n = 6) (J) Distribution of IFNγ‐expressing cells in the BAL of HDM‐sensitized and HDM‐challenged Tnfaip3 Lg‐WT mice (n = 6) and Tnfaip3 Lg‐KO mice (n = 6). Results are presented as mean ± SEM of n = 2‐6 mice per group and representative of two or more independent experiments. *P < .05, **P < .01
Everts et al 16 previously demonstrated that IL‐12 secretion by cDC1s is essential for inhibiting Th2 immune responses against helminths. Alongside the Th2 suppressive properties of IL‐12, it is also essential for IFNγ production by CD8+ T cells. 33 Similarly, we found enhanced production of IL‐12, most predominantly in lung cDC1s but not in other DC subsets of PBS‐treated Tnfaip3 Lg‐KO mice (Figure 3E,F, Figure S8C and data not shown).
These data imply that TNFAIP3 deletion in cDC1s specifically elevated the expression of IL‐12 only in Tnfaip3‐deficient cDC1s. Furthermore, the co‐inhibitory molecule PD‐L1 was increased on Tnfaip3‐deficient cDC1s, and unexpectedly also in all other lung DC subsets that did not intrinsically lacked TNFAIP3.
3.4. IFNγ expression is increased by T cells of Tnfaip3 Lg‐KO mice
As IL‐12 expression by cDC1s and PD‐L1 in all DC subsets was increased in PBS‐treated and HDM‐treated Tnfaip3 Lg‐KO mice, we wondered whether this was accompanied by induction of IFNγ expression in T cells upon HDM exposure, as PD‐L1 is a type I interferon‐inducible protein. 34 We first investigated whether IFNγ expression was already induced upon a single HDM exposure. Both the proportion of IFNγ‐producing CD4+ and CD8+ T cells were already augmented in lungs of PBS‐treated Tnfaip3 Lg‐KO mice as compared to control mice (Figure 3G). HDM exposure specifically induced IFNγ‐producing CD8+ T cells in both Tnfaip3 Lg‐WT and Tnfaip3 Lg‐KO mice (Figure 3G). In contrast, in the acute HDM‐driven allergic inflammation model IFNγ expression in CD4+ T cells was not induced upon HDM sensitization, whereas the number of IFNγ+ CD8+ T cells was specifically increased in BAL of HDM‐sensitized Tnfaip3 Lg‐KO mice, whereas this was not observed in Tnfaip3 Lg‐WT mice (Figure 3G,H).
Next, we evaluated whether the increase in IFNγ expression was specific for CD8+ T cells, and not by other cells, by gating first all IFNγ expressing cells (Figure S9). The total amount of IFNγ expressing cells in the BAL was not different between HDM‐sensitized Tnfaip3 Lg‐WT and Tnfaip3 Lg‐KO mice (Figure 3I). Also, of all IFNγ expressing cells, the majority is expressed by CD4+ T cells, but only the expression of IFNγ in CD8+ T cells was increased in HDM‐sensitized and HDM‐challenged Tnfaip3 Lg‐KO mice (Figure 3J). Also, IFNγ expression by other cells, that are not T cells, which could include ILC1s or macrophages was decreased in BAL of HDM‐sensitized and HDM‐challenged Tnfaip3 Lg‐KO mice (Figure 3J).
In conclusion, these data indicate that mice harboring TNFAIP3‐deficient Langerin+ cDC1s especially displayed increased numbers of IFNγ‐producing CD8+ T cells upon either a single HDM exposure or repetitive exposure with HDM.
3.5. Blockade of IFNγ restores eosinophilic airway inflammation in Tnfaip3 Lg‐KO mice
Both the numbers of IL‐12–expressing cDC1s and IFNγ‐producing CD8+ T cells were enhanced in Tnfaip3 Lg‐KO mice. Previous work demonstrated that both of these populations can efficiently suppress Th2 immune responses. 16 , 35 , 36 , 37 Accordingly, we found that when we exposed mice to recombinant IFNγ during HDM sensitization, eosinophilia was significantly reduced (data not shown). To determine whether IL‐12 or IFNγ were crucial in the suppression of Th2‐mediated eosinophilic airway inflammation in Tnfaip3 Lg‐KO mice, we used antibodies to block IL‐12p40 or IFNγ starting 10 days before antigen sensitization in the HDM‐induced allergic airway inflammation model (Figure 4A). Blocking IFNγ completely restored eosinophilic, neutrophil, macrophage, B‐cell, and T‐cell infiltration in BAL of HDM‐treated Tnfaip3 Lg‐KO mice to levels found in HDM‐treated Tnfaip3 Lg‐WT mice (Figure 4B,C). Blockade of IL‐12 partially (eosinophils, neutrophils) or completely (macrophages, B cells, and T cells) restored infiltration in BAL of HDM‐exposed Tnfaip3 Lg‐KO mice, as compared to HDM‐treated Tnfaip3 Lg‐WT mice (Figure 4B,C). Blocking either IL‐12 or IFNγ did not alter Th or CD8+ T‐cell numbers, but increased IL‐5 expression specifically in CD8+ T cells as compared to isotype‐treated Tnfaip3 Lg‐KO mice (Figure 4D,E). Furthermore, anti‐IFNγ antibody treatment strongly reduced the number of IFNγ+ Th cells and CD8+ T cells and IL‐10+ Th cells and CD8+ T cells in Tnfaip3 Lg‐KO mice as compared to isotype‐treated Tnfaip3 Lg‐KO mice to similar numbers as observed in Tnfaip3 Lg‐WT mice (Figure 4F,G).
FIGURE 4.
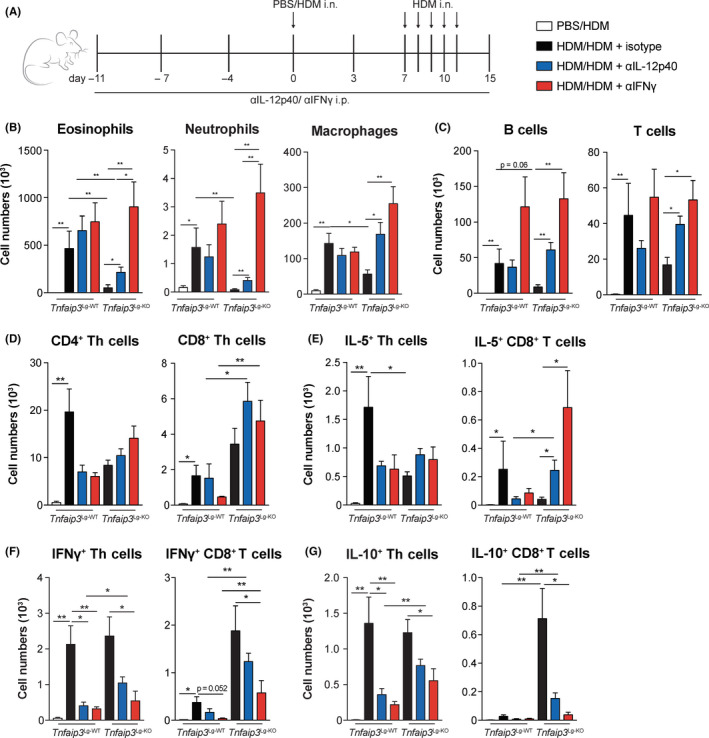
Blockage of IFNγ restores eosinophilic airway inflammation in Tnfaip3 Lg‐KO mice. (A) IL‐12p40 and IFNγ were neutralized in Tnfaip3 Lg mice by i.p. injections with anti–IL‐12p40 and anti‐IFNγ on days −11, −7, −4, and −1. Tnfaip3 Lg mice were sensitized i.n. with PBS or 1 μg HDM on day 0 and challenged i.n. with 10 μg HDM daily between days 7 and 11 to induce allergic asthma. Analysis was performed at day 15. (B‐C) Numbers of eosinophils, neutrophils, macrophages, B cells, and T cells determined in BAL by flow cytometric analysis. (D) Enumeration of CD4+ and CD8+ T cells in BAL by flow cytometric analysis. (E‐G) Quantification of the number of IL‐5+, IFNγ+, and IL‐10+ CD4+ and CD8+ T cells in BAL by flow cytometry. Results are presented as mean ± SEM of n = 4 PBS‐sensitized Tnfaip3 Lg‐WT mice, n = 6 HD‐sensitized Tnfaip3 Lg‐WT mice, n = 5 HDM‐sensitized Tnfaip3 Lg‐WT mice that were treated with either anti–IL‐12p40 and anti‐IFNγ, n = 6 HD‐sensitized Tnfaip3 Lg‐KO mice, n = 5 HD‐sensitized Tnfaip3 Lg‐KO mice that were treated with anti–IL‐12p40, and n = 6 HD‐sensitized Tnfaip3 Lg‐KO mice that were treated with anti‐IFNγ. Results are representative of two independent experiments. *P < .05, **P < .01
These data imply that blocking IFNγ restores development of Th2‐mediated allergic airway inflammation in Tnfaip3 Lg‐KO mice and IL‐12 is partially or indirectly involved.
3.6. IL‐12 and IFNγ are essential for the enhanced PD‐L1 expression on DC subsets in Tnfaip3 Lg‐KO mice
IL‐12 is essential for the induction of IFNγ secretion by CD8+ T cells, 33 and IFNγ can subsequently induce PD‐L1 expression. 38 To determine whether IL‐12, IFNγ, or both are responsible for the increased PD‐L1 expression as observed in pulmonary DCs of Tnfaip3 Lg‐KO mice, we treated mice with anti–IL‐12p40 or anti‐IFNγ antibodies for 10 days prior to a single HDM exposure (Figure 5A). Strikingly, blocking IL‐12 completely prevented PD‐L1 upregulation after HDM exposure in all pulmonary DC subsets of Tnfaip3 Lg‐KO mice. Anti‐IFNγ antibodies also inhibited the upregulation of PD‐L1 in DCs of Tnfaip3 Lg‐KO mice, although less vigorously than anti–IL‐12 treatment (Figure 5B).
FIGURE 5.
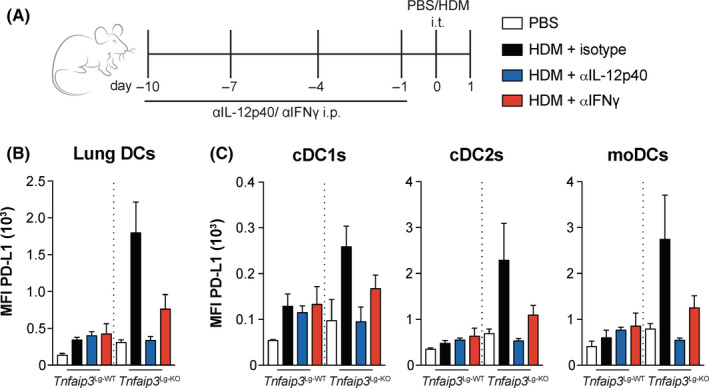
IL‐12 and IFNγ are essential for PD‐L1 expression on DC subsets in Tnfaip3 Lg‐KO mice. (A) IL‐12p40 and IFNγ were neutralized in Tnfaip3 Lg mice by i.p. injections with anti–IL‐12p40 and anti‐IFNγ on days −10, −7, −4, and −1. Tnfaip3 Lg mice were treated i.t. with PBS or 100 μg HDM on day 0. Analysis was performed on day 1. (B‐C) Quantification of MFI of PD‐L1 expression in pulmonary total DCs (B) and DC subsets (C) by flow cytometry. Results are presented as mean ± SEM of n = 3‐4 mice per group and representative of two independent experiments. *P < .05, **P < .01
We therefore conclude that IL‐12p40 or IFNγ blockade can separately reduce the increased expression of PD‐L1 on DC subsets in Tnfaip3 Lg‐KO mice, and could therefore be essential for the increase in PD‐L1 expression on all pulmonary DC subsets after HDM exposure in Tnfaip3 Lg‐KO mice.
4. DISCUSSION
DCs are critically involved in the pathogenesis of Th2‐mediated disorders; however, the exact function of cDC1s in allergic asthma is still debated, as it has been shown that cDC1s can either suppress or induce Th2‐mediated inflammation. 11 , 13 , 14 , 16 Our results indicate that TNFAIP3 deletion in Langerin+ cDC1s elevated IL‐12 expression specifically in cDC1s. Increased IL‐12 expression by Tnfaip3‐deficient cDC1s could provoke an augmented IFNγ production by CD8+ T cells. In turn, IFNγ was responsible for the enhanced PD‐L1 expression on all pulmonary DC subsets. IFNγ during sensitization controlled the development of Th2‐mediated allergic inflammation upon HDM.
Our observation that mice harboring TNFAIP3‐deficient Langerin+ cDC1s do not develop Th2‐mediated inflammation upon exposure to HDM agrees with other reports that cDC1s suppress Th2‐mediated pulmonary inflammation upon helminth infection, 16 ovalbumin sensitization, 10 , 39 and chronic HDM exposure. 11 Additionally, Helicobacter pylori infection suppresses allergic airway inflammation through activation of cDC1s. 40 In contrast, others have reported that cDC1s are essential for the induction of Th2‐mediated differentiation in response to HDM 13 , 15 and that depleting cDC1s attenuates Th2‐mediated eosinophilic inflammation. 13 , 14 , 41 These conflicting findings may be due to the type of allergen used, timing of administration, or the use of different mouse models, such as Basic Leucine Zipper ATF‐Like Transcription Factor 3 (BATF3)KO mice, 10 , 11 , 16 BHX2 mice 13 that harbor a spontaneous point mutation (R294C) of IRF8, Langerin‐DTR mice, 7 CD103KO mice, 39 , 41 or XCR1KO mice. 14 Furthermore, the lung is unique, as pulmonary cDC1s contain a mixture of Langerin+ and Langerin‐ cDC1s. 28 , 42 Differences in ontogeny and function between Langerin+ and Langerin‐ cDC1s are currently unknown. Specific deletion of lung Langerin+ cDC1s using Langerin‐DTR mice did not affect eosinophilic inflammation, 7 which could indicate that activation of these Langerin+ cDC1s provides an additional trigger enhancing their Th2‐suppressive character.
Various reports have established cDC1s as major IL‐12 producers that drive protective Th1 immunity against several pathogens. 43 IL‐12 production by cDC1s is also known to inhibit Th2 immune responses during chronic Schistosoma mansoni infection. 16 Additionally, IL‐12–producing DCs, established through retroviral overexpression, are also unable to prime mice for pulmonary Th2‐mediated eosinophilic inflammation. 44 There are case reports describing that blocking IL‐12 with ustekinumab (monoclonal antibody to the p40 subunit of IL‐12 and IL‐23) in psoriasis patients exacerbated their atopic dermatitis, 45 indicating a possible role for IL‐12 in regulating ongoing Th2 responses, next to controlling the induction of Th2 immunity. Somewhat unexpected considering the increased IL‐12 expression, we did not observe enhanced Th1‐cell differentiation, but rather an increase in IFNγ‐production by CD8+ T cells, which is also dependent on IL‐12. 33 The specific increase in IFNγ‐producing CD8+ T cells and not IFNγ‐producing Th cells may be related to the unique cross‐presenting capacities of cDC1s, which allow cDC1s to load exogenous peptides on MHCI molecules, making them superior inducers of CD8+ T‐cell activation. 8 , 9 Increased numbers of IFNγ‐expressing T cells were already found in the lungs of naïve Tnfaip3 Lg‐KO mice. These are likely induced by TNFAIP3‐deficient cDC1s in the lungs; however, we cannot exclude that the IFNγ‐expressing T cells in the lung were partly induced by TNFAIP3‐deficient Langerhans cells in the skin, because skin DCs have been reported to mediate inflammation in the airways through the skin‐lung axis. 5
Furthermore, the presence of CD8+ T cells, and specifically allergen‐specific CD8+ T cells, hampers the development of allergic diseases. 37 , 46 , 47 The inhibitory effect of CD8+ T cells is likely caused by increased IFNγ production and was most pronounced during the sensitization phase, as blocking IFNγ during the challenge phase had no effect on Th2‐mediated inflammation. 46 This was confirmed by our findings that administration of IFNγ during sensitization reduced Th2‐mediated HDM‐triggered inflammation. Strikingly, CD8+ T cells also contribute to Th2‐mediated inflammation by secretion of type‐2 cytokines. 46 We also found that blocking IL‐12 or IFNγ promoted IL‐5 expression and reduced IFNγ expression in CD8+ T cells, which is in line with previous reports that IL‐12 is implicated in the induction of IFNγ. 48 In conclusion, this indicates that the environment is a crucial determinant of cytokine expression by CD8+ T cells.
In our experiments, the IL‐12p40 subunit was blocked, which is used by both IL‐12 and IL‐23. However, previous reports showed that Tnfaip3‐deficient cDC1s do not express IL‐23, 23 and identified cDC1s as key producers of IL‐12, 16 , 43 , 49 making it most likely that the observed effects were mediated by blocking IL‐12.
In conclusion, our data establish that mice harboring Tnfaip3‐deficient Langerin+ cDC1s develop a strongly reduced Th2‐mediated inflammation in response to HDM which is accompanied by the induction of IFNγ‐producing CD8+ T cells and increased PD‐L1 expression on pulmonary DCs. This indicates that the activation status of pulmonary cDC1s critically controls development of Th2‐mediated allergic disorders.
CONFLICT OF INTEREST
Dr Vroman reports grants from Dutch Pulmonary Foundation, during the conduct of the study. Dr van Uden, Dr Bergen, Dr van Hulst, Dr Lukkes, Dr van Loo, Dr Clausen, Dr Boon, Dr Lambrecht, Dr Hammad, and Dr Hendriks have nothing to disclose. Dr Kool reports grants from the Netherlands Lung Foundation, grants from European Framework program 7, and grants from NWO‐Veni, during the conduct of the study.
AUTHOR CONTRIBUTIONS
HV, BNL, HH, RWH, and MK designed the experiments. HV, IB, JvH, ML, DvU, and IT performed experiments and analyzed data. BNL, GvL, and BEC provided transgenic mouse strains used for the experiments. LB provided therapeutic blocking antibodies. HV, RWH, and MK wrote the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig S1
{kind=link}
Fig S2
{kind=link}
Fig S3
{kind=link}
Fig S4
{kind=link}
Fig S5
{kind=link}
Fig S6
Fig S7
{kind=link}
Fig S8
{kind=link}
Fig S9
{kind=link}
Table S1
Supplementary Material
ACKNOWLEDGMENTS
These studies were partly supported by NWO‐VENI (916.11.067), European Framework program 7 (FP7‐MC‐CIG grant 304221), and the Netherlands Lung Foundation (3.2.12.087, 4.2.13.054JO, and 9.2.15.065FE). We would like to thank Martijn Schuijs (VIB, Ghent, Belgium), Boudewijn van der Wel, Lisette Krassenburg (Erasmus MC), and the Erasmus MC Animal Facility (EDC) staff for their assistance during the project.
Vroman H, van Uden D, Bergen IM, et al. Tnfaip3 expression in pulmonary conventional type 1 Langerin‐expressing dendritic cells regulates T helper 2‐mediated airway inflammation in mice. Allergy. 2020;75:2587–2598. 10.1111/all.14334
REFERENCES
- 1. Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18(5):673‐683. [DOI] [PubMed] [Google Scholar]
- 2. Guilliams M, Dutertre CA, Scott CL, et al. Unsupervised high‐dimensional analysis aligns dendritic cells across tissues and species. Immunity. 2016;45:669‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012;18(5):684‐692. [DOI] [PubMed] [Google Scholar]
- 4. Dudziak D, Kamphorst AO, Heidkamp GF, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315(5808):107‐111. [DOI] [PubMed] [Google Scholar]
- 5. Deckers J, Sichien D, Plantinga M, et al. Epicutaneous sensitization to house dust mite allergen requires interferon regulatory factor 4–dependent dermal dendritic cells. J Allergy Clin Immunol. 2017;140(5):1364‐1377.e2. [DOI] [PubMed] [Google Scholar]
- 6. Williams JW, Tjota MY, Clay BS, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun. 2013;4:2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plantinga M, Guilliams M, Vanheerswynghels M. Conventional and monocyte‐derived CD11b+ dendritic cells initiate and maintain T Helper 2 cell‐mediated immunity to house dust mite allergen. Immunity. 2013;38:322‐335. [DOI] [PubMed] [Google Scholar]
- 8. Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med. 2011;208(10):2005‐2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khare A, Krishnamoorthy N, Oriss TB, Fei M, Ray P, Ray A. Cutting edge: inhaled antigen upregulates retinaldehyde dehydrogenase in lung CD103+ but not plasmacytoid dendritic cells to induce Foxp3 de novo in CD4+ T cells and promote airway tolerance. J Immunol. 2013;191:25‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conejero L, Khouili SC, Martínez‐Cano S, Izquierdo HM, Brandi P, Sancho D. Lung CD103+ dendritic cells restrain allergic airway inflammation through IL‐12 production. JCI Insight. 2017;2(10):90420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Semmrich M, Plantinga M, Svensson‐Frej M, et al. Directed antigen targeting in vivo identifies a role for CD103 + dendritic cells in both tolerogenic and immunogenic T‐cell responses. Mucosal Immunol. 2012;5:150‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakano H, Free ME, Whitehead GS, et al. Pulmonary CD103 + dendritic cells prime Th2 responses to inhaled allergens. Mucosal Immunol. 2012;5(1):53‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woo YD, Koh J, Kang H‐R, Kim HY, Chung DH. The invariant natural killer T cell–mediated chemokine X‐C motif chemokine ligand 1–X‐C motif chemokine receptor 1 axis promotes allergic airway hyperresponsiveness by recruiting CD103+dendritic cells. J Allergy Clin Immunol. 2018;142(6):1781‐1792.e12. [DOI] [PubMed] [Google Scholar]
- 15. Ortiz‐Stern A, Kanda A, Mionnet C, et al. Langerin dendritic cells are responsible for LPS‐induced reactivation of allergen‐specific Th2 responses in postasthmatic mice. Mucosal Immunol. 2011;4(3):343‐353. [DOI] [PubMed] [Google Scholar]
- 16. Everts B, Tussiwand R, Dreesen L, et al. Migratory CD103 + dendritic cells suppress helminth‐driven type 2 immunity through constitutive expression of IL‐12. J Exp Med. 2016;213(1):35‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vroman H, van den Blink B, Kool M. Mode of dendritic cell activation: The decisive hand in Th2/Th17 cell differentiation. Implications in asthma severity? Immunobiology. 2015;220(2):254‐261. [DOI] [PubMed] [Google Scholar]
- 18. Hammad H, Chieppa M, Perros F, et al. House dust mite allergen induces asthma via Toll‐like receptor 4 triggering of airway structural cells. Nat Med. 2009;15(4):410‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J‐Y. The innate immune response in house dust mite‐induced allergic inflammation. Allergy Asthma Immunol Res. 2013;5(2):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma A, Malynn BA. A20: Linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. 2012;12(11):774‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol. 2014;35(1):22‐31. [DOI] [PubMed] [Google Scholar]
- 22. Kool M, van Loo G, Waelput W, et al. The ubiquitin‐editing protein a20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. 2011;35(1):82‐96. [DOI] [PubMed] [Google Scholar]
- 23. Vroman H, Bergen IM, van Hulst JAC, et al. TNF‐α–induced protein 3 levels in lung dendritic cells instruct TH2 or TH17 cell differentiation in eosinophilic or neutrophilic asthma. J Allergy Clin Immunol. 2018;141:1620–1633. [DOI] [PubMed] [Google Scholar]
- 24. Schuijs M, Willart M, Vergote K. Farm dust and endotoxin protect against allergy. Science. 2015;349:1106‐1110. [DOI] [PubMed] [Google Scholar]
- 25. Li X, Ampleford EJ, Howard TD, et al. Genome‐wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J Allergy Clin Immunol. 2012;130(4):861‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stein MM, Hrusch CL, Gozdz J, et al. innate immunity and asthma risk in Amish and Hutterite Farm Children. N Engl J Med. 2016;375(5):411‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krusche J, Twardziok M, Rehbach K, et al. TNFAIP3 is a key player in childhood asthma development and environment‐mediated protection. J Allergy Clin Immunol. 2019;144:1684‐1686. [DOI] [PubMed] [Google Scholar]
- 28. Zahner SP, Kel JM, Martina CAE, et al. Conditional deletion of TGF‐βR1 using Langerin‐Cre mice results in Langerhans cell deficiency and reduced contact hypersensitivity. J Immunol. 2011;187(10):5069‐5076. [DOI] [PubMed] [Google Scholar]
- 29. GeurtsvanKessel CH, Willart MAM, van Rijt LS, et al. Clearance of influenza virus from the lung depends on migratory langerin + CD11b − but not plasmacytoid dendritic cells. J Exp Med. 2008;205(7):1621‐1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kool M, Hammad H, Lambrecht B. Cellular networks controlling Th2 polarization in allergy and immunity. F1000 Biol Rep. 2012;4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vroman H, Bergen IM, Li BWS, et al. Development of eosinophilic inflammation is independent of B‐T cell interaction in a chronic house dust mite‐driven asthma model. Clin Exp Allergy. 2017;47:551‐564. [DOI] [PubMed] [Google Scholar]
- 32. Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The importance of dendritic cells in maintaining immune tolerance. J Immunol. 2017;198(6):2223‐2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rutishauser RL, Kaech SM. Generating diversity: transcriptional regulation of effector and memory CD8+ T‐cell differentiation. Immunol Rev. 2010;235:219‐233. [DOI] [PubMed] [Google Scholar]
- 34. Lee SJ, Jang BC, Lee SW, et al. Interferon regulatory factor‐1 is prerequisite to the constitutive expression and IFN‐γ‐induced upregulation of B7–H1 (CD274). FEBS Lett. 2006;580(3):755‐762. [DOI] [PubMed] [Google Scholar]
- 35. Oriss TB, McCarthy SA, Morel BF, Campana MA, Morel PA. Crossregulation between T helper cell (Th)1 and Th2: inhibition of Th2 proliferation by IFN‐γ involves interference with IL‐1. J Immunol. 1997;158(8):3666‐3672. [PubMed] [Google Scholar]
- 36. Gouveia ACC, Braga FG, Mota M, et al. Enhanced expression of PD‐L1 and IFN‐γ on dendritic cells is associated with BCG‐induced Th2 inhibition. Cytokine. 2017;99:163‐172. [DOI] [PubMed] [Google Scholar]
- 37. Suzuki M, Maghni K, Molet S, Shimbara A, Hamid QA, Martin JG. IFN‐γ secretion by CD8 + T cells inhibits allergen‐induced airway eosinophilia but not late airway responses. J Allergy Clin Immunol. 2002;109:803‐809. [DOI] [PubMed] [Google Scholar]
- 38. Abiko K, Matsumura N, Hamanishi J, et al. IFN‐γ from lymphocytes induces PD‐L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112:1501‐1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bernatchez E, Gold MJ, Langlois A, et al. Pulmonary CD103 expression regulates airway inflammation in asthma. Am J Physiol Lung Cell Mol Physiol. 2015;308:L816‐L826. [DOI] [PubMed] [Google Scholar]
- 40. Engler DB, Reuter S, van Wijck Y, et al. Effective treatment of allergic airway inflammation with Helicobacter pylori immunomodulators requires BATF3‐dependent dendritic cells and IL‐10. Proc Natl Acad Sci USA. 2014;111(32):11810‐11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fear VS, Lai SP, Zosky GR, et al. A pathogenic role for the integrin CD103 in experimental allergic airways disease. Physiol Rep. 2016;4:13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sung S‐SJ, Fu SM, Rose CE, Gaskin F, Ju ST, Beaty SR. A major lung CD103 ( E)‐ 7 integrin‐positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176:2161‐2172. [DOI] [PubMed] [Google Scholar]
- 43. Martínez‐López M, Iborra S, Conde‐Garrosa R, Sancho D. Batf3‐dependent CD103+ dendritic cells are major producers of IL‐12 that drive local Th1 immunity against Leishmania major infection in mice. Eur J Immunol. 2015;45:119‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuipers H, Heirman C, Hijdra D, et al. Dendritic cells retrovirally overexpressing IL‐12 induce strong Th1 responses to inhaled antigen in the lung but fail to revert established Th2 sensitization. J Leukoc Biol. 2004;76:1028‐1038. [DOI] [PubMed] [Google Scholar]
- 45. Deleanu D, Nedelea I. Biological therapies for atopic dermatitis: An update (Review). Exp Ther Med. 2018;4:1061‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stock P, Kallinich T, Akbari O, et al. CD8(+) T cells regulate immune responses in a murine model of allergen‐induced sensitization and airway inflammation. Eur J Immunol. 2004;34(7):1817‐1827. [DOI] [PubMed] [Google Scholar]
- 47. Wells JW, Cowled CJ, Giorgini A, Kemeny DM, Noble A. Regulation of allergic airway inflammation by class I‐restricted allergen presentation and CD8 T‐cell infiltration. J Allergy Clin Immunol. 2007;119:226‐234. [DOI] [PubMed] [Google Scholar]
- 48. Meyaard L, Hovenkamp E, Otto SA, Miedema F. IL‐12‐induced IL‐10 production by human T cells as a negative feedback for IL‐12‐induced immune responses. J Immunol. 1996;156:2776‐2782. [PubMed] [Google Scholar]
- 49. Mittal D, Vijayan D, Putz EM, et al. Interleukin‐12 from CD103+ Batf3‐dependent dendritic cells required for NK‐cell suppression of metastasis. Cancer Immunol Res. 2017;5:1098‐1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Table S1
Supplementary Material