

Summary
We studied the efficacy and safety of humanized CAR‐T therapy following intensive chemotherapy for refractory/relapsed (R/R) acute lymphoblastic leukaemia (B‐ALL). Twenty‐three patients with R/R B‐ALL were pretreated with intensive chemotherapy (fludarabine combined with medium‐dose cytarabine) 12 days before CAR‐T therapy. Adverse events (AEs), curative effects, infection indicators and cytokine release syndrome (CRS) were monitored. Each of the 23 patients received a dose of 1·0 × 106 cells/kg CAR‐T cell infusion on day 0. After 14 days, 19 patients (82·61%) achieved complete response (CR) or CR with incomplete count recovery. No survival benefit was achieved with consolidative haematopoietic stem‐cell transplantation (HSCT), with a median follow‐up of 14·0 months (range, 1·5–21·0 months). The notable AEs were grade 1–2 CRS in 18 patients, while the other five patients were grade 3 CRS. No patients died of CRS. Only one patient died of respiratory failure due to cytomegalovirus infection 24 days after infusion. The proportion of leukaemic cells in bone marrow on infusion day and the peaks of IL‐6, TNF‐α and IL‐8 levels were correlated with CRS levels. A lower disease burden was achieved by intensive lymphodepleting chemotherapy, and the subsequent CAR‐T therapy had a high response and manageable toxicity.
Trial registration: The patients were enrolled in a clinical trial of ChiCTR‐ONN‐16009862, and ChiCTR1800019622.
Keywords: chimaeric antigen receptor, acute lymphoblastic leukaemia, refractory, lymphodepleting chemotherapy, cytokine release syndrome
Refractory/relapsed (R/R) B acute lymphoblastic leukaemia (B‐ALL) patients have a poor prognosis, with a complete response (CR) less than 45%, and even allogeneic haematopoietic stem‐cell transplantation (allo‐HSCT) can hardly improve the outcome. 1 , 2 Novel therapeutic strategies to effectively treat these R/R B‐ALL patients are urgently needed.
The second‐generation anti‐CD19‐CAR‐T cells expressing anti‐CD19 scFv and 4‐1BB‐CD3ζ costimulatory activation domains have been shown to be effective in the activation and expansion of CAR‐T cells to eliminate malignant B cells. 3 In recent years, anti‐CD19‐CAR‐T‐cell therapy has been one of the most promising therapeutic approaches for R/R B‐ALL patients. 4 Previous clinical trials showed that anti‐CD19‐CAR‐T therapy is associated with a high remission rate in 83% in patients with R/R B‐ALL. 2 , 5 , 6 , 7 , 8 To increase the potency of anti‐CD19‐CAR‐T cells, patients receive lymphodepleting chemotherapy regimens before the anti‐CD19‐CAR‐T‐cell therapy. The lymphodepleting chemotherapy includes cyclophosphamide alone, 2 fludarabine combined with cyclophosphamide (FC) 6 , 9 and bendamustine‐based regimens. 10 The primary aim of lymphodepleting chemotherapy is to reduce host T cells, and this procedure has been shown to increase the expansion and persistence of anti‐CD19‐CAR‐T cells as well as the event‐free survival (EFS) time of R/R B‐ALL patients. 11 However, none of the lymphodepleting chemotherapy regimens are superior in efficacy or toxicity as compared to other regimens in the clinical trials with anti‐CD19‐CAR T cells.
Although anti‐CD19‐CAR‐T‐cell therapy has been effective in the treatment of R/R B‐ALL, it often causes severe toxicities, including cytokine release syndrome (CRS), neurological toxicities and B‐cell aplasia. CRS with significantly elevated serum levels of inflammatory cytokines is the most common fatal toxicity associated with anti‐CD19‐CAR‐T‐cell therapy. 4 , 12 , 13 , 14 , 15 , 16 Higher anti‐CD19‐CAR‐T‐cell doses (3 × 106/kg, 2 × 107/kg) 7 and lymphodepleting chemotherapy, including fludarabine to enhance the activity of CAR‐T cells, 9 , 17 , 18 , 19 have been associated with severe CRS and neurological toxicities. Another critical factor that influenced the toxicities of this therapy is tumour burden. 20 A high tumour burden was shown to indicate poor prognosis in R/R B‐ALL patients. 20 This observation suggested that we should reduce the tumour burden before introducing the anti‐CD19‐CAR‐T cells to reduce side effects. Lymphodepleting chemotherapy usually includes cyclophosphamide, fludarabine, and bendamustine. A combination regimen containing cytarabine is also a common regimen for R/R B‐ALL. 21 The synergistic effect of fludarabine and cytarabine (FA) has been demonstrated. 22 Therefore, the combination regimen of FA could reduce the tumour burden and increase the expansion and persistence of anti‐CD19‐CAR‐T.
In our study, 23 patients with R/R B‐ALL were enrolled in an open‐label, single‐centre and single‐arm pilot study of intensive chemotherapy, which was utilized to reduce tumour load, followed by humanized anti‐CD19‐CAR‐T therapy. We analyzed the efficacy and safety of this therapeutic strategy in the R/R B‐ALL patients.
Patients and methods
Participants in the clinical trial
In the study, 23 patients with R/R B‐ALL were admitted to the Department of Hematology in Tianjin First Center Hospital (Tianjin, China) between April 2018 and April 2019. All of these B‐ALL patients met the diagnostic criteria according to the WHO classification. They all had high CD19 expression on malignant B cells analyzed by flow cytometry (FCM). These R/R B‐ALL patients were enrolled in a clinical trial of anti‐CD19‐CAR‐T cells expressing humanized anti‐CD19 scFv and 4‐1BB‐CD3ζ costimulatory activation domains (ChiCTR‐ONN‐16009862 and ChiCTR1800019622). None of the patients had been exposed to blinatumomab before. They or their representatives provided informed consent before enrolment. The cut‐off date for data collection was January 31, 2020.
Generation and detection of anti‐CD19‐CAR‐T cells
Peripheral blood mononuclear cells (PBMCs) were collected from all the R/R B‐ALL patients. Cell production and quality‐control assays were conducted according to procedures described in the literature. 23 The lenti‐CD19‐2rd‐CAR was provided by Shanghai Genbase Biotechnology Co., Ltd. Shanghai, China. The culture time of anti‐CD19‐CAR‐T cells in vitro was approximately 12 days.
Intensive chemotherapy in advance and anti‐CD19‐CAR‐T‐cell infusion
All patients enrolled in this clinical trial received intensive chemotherapy (FA chemotherapy regimens) first, i.e. fludarabine (30 mg/m2/day) followed by cytarabine (1,000 mg/m2/day) for five days, after the PBMCs were collected (Fig 1). Leucocytes were assessed by FCM before the FA regimen and on day 0 (i.e. when patients received the CAR‐T‐cell infusion). Some patients were given the FA chemotherapy regimen only for 3–4 days because the proportion of leukaemic cells in the bone marrow (BM) was less than 20% before the FA regimen, or severe granulocytosis occurred quickly during the treatment. Three patients with central nervous system leukaemia (CNSL) received FA lymphodepleting chemotherapy combined with intrathecal chemotherapy before CAR‐T cell infusion. Humanized anti‐CD19‐CAR‐T cells were infused on day 0 at a dose of 1 × 106/kg. 7 , 24 , 25
Fig 1.
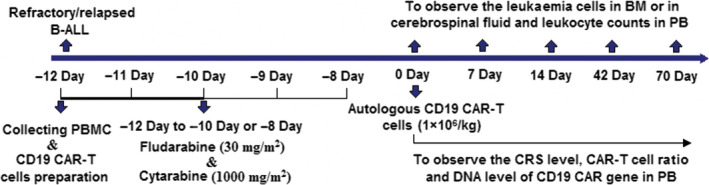
Procedures for the treatment and observation of side effects.
Adverse events observed after infusion of anti‐CD19‐CAR‐T cells
The secretion levels of cytokines, including interleukin‐6 (IL‐6), IL‐2R, tumour necrosis factor‐α (TNF‐α), and IL‐8, were measured on days 0, 4, 7, 14, 21 and 28 by enzyme‐linked immunosorbent assay with the double antibody one‐step sandwich method. The level of these cytokines in the cerebrospinal fluid (CSF) was also measured in patients who suffered from CNSL. Adverse events (AEs) were monitored. CRS was graded according to the adopted CRS scoring system and the National Cancer Institute Common Terminology Criteria for AE v4.03 after CAR‐T‐cell infusion. 14 Treatment‐related mortalities were calculated from the date of CAR‐T‐cell infusion to the date when the AE of this therapy disappeared.
Supportive care and monitoring of infectious disease after infusion of anti‐CD19‐CAR‐T cells
Granulocyte colony‐stimulating factor at 5 μg/kg/day was administered subcutaneously after intensive chemotherapy when the absolute neutrophil count was below 0·5 × 109/l. From the day of CAR‐T‐cell infusion to 28 days after infusion, the level of procalcitonin was observed by quantitative chemiluminescence to evaluate bacterial infections. A (1,3)‐beta‐D‐glucan assay was performed by dynamic colour rendering, and a galactomannan ELISA (GM) test was done by enzyme immunoassay to evaluate fungal infections. A quantitative polymerase chain reaction (PCR) was carried out to test for Epstein–Barr virus and cytomegalovirus in the plasma. Infectious diseases were recorded when there were microbiologic diagnosis or clinical symptoms of infections.
The expansion and persistence of anti‐CD19‐CAR‐T cells
The expansions of anti‐CD19‐CAR‐T cells in peripheral blood and in CSF were examined by FCM on days 0, 4, 7, 14, 21 and 28. At the same time, the DNA level of the anti‐CD19‐CAR gene was detected by PCR. Similarly, these assays were also carried out on the CSF from patients with CNSL.
Clinical response criteria
From the date of anti‐CD19‐CAR‐T‐cell infusion, follow‐up was done until the cut‐off date or until the patients died. Therapy responses were assessed and the minimal residual disease (MRD) analyzed on day 14 after infusion. Disease status was defined as complete response (CR), CR with incomplete count recovery (Cri), or no remission (NR). Leukaemia‐free survival (LFS) was calculated from the date of CR to date of relapse.
Statistical analysis
Data were expressed as mean + SE, along with the number of repeated experiments. Non‐normal distribution data are expressed as their median and interquartile range (IQR) unless otherwise indicated. The Wilcoxon paired test was used to compare leukaemia cells in BM and peripheral blood before and after intensive chemotherapy. CRS was grouped and compared by Mann–Whitney rank and inspection. The Pearson correlation coefficient was used for evaluating the correlation between different factors. Linear regression was used for correlation analysis. The probabilities of overall survival (OS) and LFS were estimated using the Kaplan–Meier method and were compared using the log‐rank test. The median follow‐up was estimated using the reverse Kaplan–Meier method. All statistical analyses were performed with Graphpad Prism 8 (GraphPad Software Inc., La Jolla, CA, USA) and SPSS 17.0 (SPSS Inc., Chicago, IL, USA) software. P‐values of less than 0·05 were considered significant.
Results
Patient characteristics
The medical history and the tumour burden of the R/R B‐ALL patients at the time of enrolment, as well as the parameters of the anti‐CD19‐CAR‐T‐cell infusion, are listed in Table I. Patients with a median age of 42 years (range 10–67 years) were enrolled in this trial. All 25 patients recruited for this trial had CD19 expression on the malignant B cells analyzed by FCM. Two patients withdrew from our clinical trial four and two days before the infusion because of cerebral haemorrhage and severe infection respectively. The eligible patients included three patients with CNSL.
Table I.
Patients' baseline and therapy‐related characteristics.
| Subject | Sex (M/F) | Age (yrs) | WBC (109/l) | Blasts in BM by FCM (%) | Immunophenotyping | High‐risk cytogenetics | Previous therapies | CNSL | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before leukapheresis | On the day of CD19‐CAR‐T‐cell infusion | Before leukapheresis | On the day of CD19‐CAR‐T‐cell infusion | Early pre‐B‐cell | pre‐B‐cell | mature B‐cell ALL | Lines of therapy | Relapse after autologous stem‐cell transplantation | |||||||
| P1# | M | 66 | 6·41 | 8·64 | 26·2 | 3·2 | Y | t(9;22)(q34;q11) | 2 | Y | |||||
| P2# | M | 67 | 24·43 | 0·14 | 60·5 | 10·5 | Y | t(9;22)(q34;q11) | 2 | Y | |||||
| P3# | F | 54 | 16·05 | 0·01 | 54·2 | 12·3 | Y | t(9;22)(q34;q11) | 2 | ||||||
| P4# | F | 54 | 56·5 | 0·11 | 87·5 | 70·2 | Y | t(9;22)(q34;q11) | 2 | ||||||
| P5# | M | 29 | 5·07 | 2·66 | 23·4 | 3·2 | Y | 2 | |||||||
| P6# | M | 30 | 16·07 | 0·27 | 48·6 | 40·6 | Y | 2 | |||||||
| P7# | M | 46 | 4·44 | 6·53 | 37·26 | 30·5 | Y | 2 | |||||||
| P8# | F | 63 | 1·93 | 0·06 | 70·5 | 4·6 | Y | 3 | Y | ||||||
| P9# | M | 42 | 4·39 | 0·08 | 70·71 | 18·5 | Y | t(9;22)(q34;q11) | 3 | Y | |||||
| P10# | F | 56 | 36·79 | 0·93 | 25·2 | 2·6 | Y | 2 | Y | ||||||
| P11# | M | 10 | 42·03 | 2·49 | 19·8 | 4·4 | Y | 43,XY, −7, −8, −9,add(12)(p11), −17,−22,+mar1 | 3 | Y | |||||
| P12# | F | 31 | 1·15 | 0·62 | 82·6 | 13·6 | Y | t[4;11] | 2 | ||||||
| P13# | F | 15 | 8·94 | 0·24 | 80·86 | 66·8 | Y | 2 | |||||||
| P14# | F | 51 | 0·74 | 4·22 | 12·5 | 2·2 | Y | 2 | |||||||
| P15# | M | 18 | 5·14 | 0·57 | 60·62 | 12·4 | Y | 2 | |||||||
| P16# | F | 16 | 2·6 | 1·81 | 12·5 | 1·8 | Y | 2 | |||||||
| P17# | F | 14 | 3·43 | 2·16 | 8·2 | 4·6 | Y | 2 | |||||||
| P18# | M | 62 | 2·98 | 3·3 | 12·5 | 6·8 | Y | 2 | |||||||
| P19# | M | 25 | 4·67 | 0·22 | 40·4 | 28·5 | Y | 3 | Y | ||||||
| P20# | M | 16 | 10·3 | 0·31 | 72·5 | 20·4 | Y | t[4;11] | 2 | ||||||
| P21# | M | 61 | 22·5 | 0·56 | 12·6 | 4 | Y | t(9;22)(q34;q11) | 3 | Y | |||||
| P22# | M | 13 | 80·3 | 0·45 | 80·2 | 32·5 | Y | 2 | |||||||
| P23# | M | 66 | 20·1 | 0·94 | 20·6 | 2·4 | Y | t(9;22)(q34;q11) | 2 | ||||||
Abbreviations: B‐ALL, B acute lymphoblastic leukaemia; BM, bone marrow; CNSL, central nervous system leukaemia; FCM, flow cytometry; WBC, white blood cell count.
BM condition before the anti‐CD19‐CAR‐T‐cell infusion
Twenty‐three patients received intensive chemotherapy. The median proportion of leukaemia cells in BM was 40·4% (IQR 19·8–70·7) before the FA regimen. After the FA regimen, the proportion was reduced to 10·5% (IQR 3·2–28·5) (P < 0·0001) on the infusion day. The proportion of leukaemia cells in the BM on the day of infusion was less than 20% in 16 of the 23 patients. The other seven patients had leukaemia cell proportions ranging from 20·4% to 70·2%. The median leukocyte count in peripheral blood after intensive chemotherapy was 0·6 × 109/l (IQR 0·2–2·5), which is lower than that before the chemotherapy 6·4 × 109/l (IQR 3·4–22·5) (P = 0·0002) (Fig 2).
Fig 2.
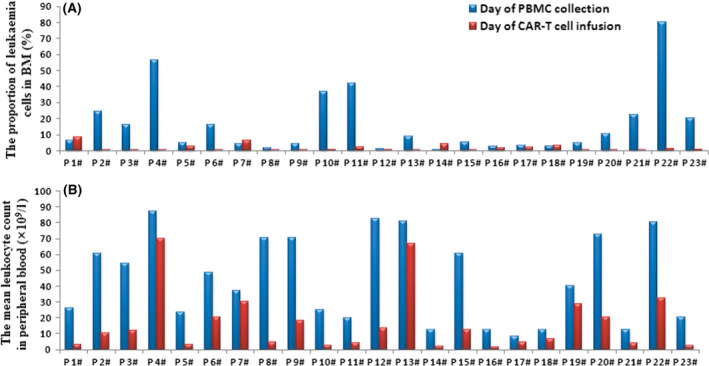
Proportional changes of leukaemia cells in BM and leukocyte counts in peripheral blood before and after the intensive chemotherapy. (A) The proportion of leukaemia cells in the bone marrow detected by flow cytometry (FCM) before intensive chemotherapy and on the day of infusion. (B) The leukocyte count detected by FCM in peripheral blood before intensive chemotherapy and on the infusion day.
The three patients with CNSL received FA chemotherapy combined with six, four, or two treatments of intrathecal chemotherapy, depending on the response. The leukaemic cell proportions in CSF decreased from 35·6%, 98·1% and 12·5% on the day of enrolment to 9. 7%, 13·2% and 1·0% on the day of CAR‐T‐cell infusion respectively.
Transduction efficiency, amplification and infusion of the anti‐CD19‐CAR‐T cell
The mean CD19‐CAR transduction efficiency in the final products of the 23 patients was 55·3 ± 12·7%. Except for one case in which the T cells were transduced with CD19‐CAR lentivirus on day six in vitro due to leukaemia cells detected on day four (1·1%), T cells from the other 22 patients were transduced on day four in vitro. When the cells were harvested, the mean anti‐CD19‐CAR‐T‐cell numbers of all patients (23) were 6·2 ± 2·2 × 106 cells/kg.
All 23 patients received an infusion of 1·0 × 106 cells/kg humanized anti‐CD19‐CAR‐T cells on day 0. Notably, bacteria, mycoplasma, fungus and endotoxin were not detected in the final products before infusion.
Clinical responses
After 14 days of CAR‐T‐cell infusion, 19 patients (19 /23, 82·6%) achieved CR/Cri, 12 patients (12/23, 52·2%) achieved CR and 18 patients (18/23, 78·3%) were MRD‐negative. The other four patients were evaluated as NR. One of the NR patients achieved CR after two months of infusion with anti‐CD22‐CAR‐T cells. If these patients were younger than 60 years old and had a suitable donor, they would receive allo‐HSCT therapy. Of the 20 patients that achieved CR/Cri, five received allo‐HSCT after 28–84 days of CAR‐T‐cell therapy. Seven patients were diagnosed as suffering from a recurrent disease from the anti‐CD19‐CAR‐T‐cell therapy from 14 days to 19 months after CR. The other eight patients remained CR/Cri up to the writing of the manuscript (for 4–15 months). By the cut‐off date, patients in LFS and OS received allo‐HSCT and non‐allo‐HSCT. CD19 expression in the first recurrence after infusion is shown in Fig 3. Figure 3 also shows the time of loss of CD19 CAR‐T cells in all the patients except for the nine patients who received allo‐HSCT, or were evaluated as NR. The three patients with CNSL achieving CR were MRD‐negative in their BM and CSF 14 days after infusion.
Fig 3.
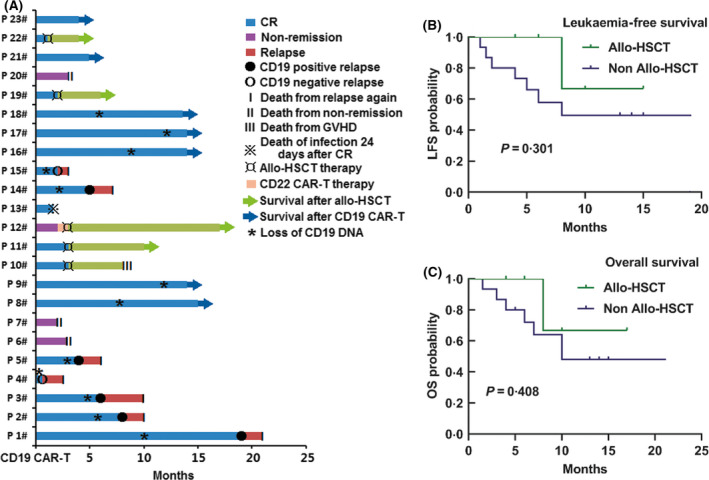
Outcomes in patients with B acute lymphoblastic leukaemia (B‐ALL) receiving anti‐CD19‐CAR‐T therapy. (A) The response in the 23 B‐ALL patients, the time of loss of CD19 CAR‐T cells, and the CD19 expression at the first recurrence. (B, C) The leukaemia‐free survival (LFS) and overall survival (OS) in patients who received allo‐HSCT and no allo‐HSCT. Of the patients who received no allo‐HSCT, the median LFS was 8·0 months (95% confidence interval 1·9–14·1; P = 0·301) (Fig 3B), and the median OS was 10·0 months (95% confidence interval 4·1–15·9; P = 0·408) (Fig 3C). The median LFS and OS in the allogeneic haematopoietic stem‐cell transplantation (allo‐HSCT) group were not achieved, with a median follow‐up of 14·0 months (range, 1·5–21·0 months). Patients who did not have a relapse at the last follow‐up were censored. CR, complete response; GVHD, graft versus host disease.
Expansion of the anti‐CD19‐CAR‐T cells
The proportions of CAR‐T cells and the DNA levels of anti‐CD19‐CAR gene in peripheral blood were determined throughout the anti‐CD19‐CAR‐T‐cell therapy. The median expansion peak of the anti‐CD19‐CAR‐T cells was 11·6% (range, 0·4–70·9) on day 11 (range, 7–14) after infusion. DNA level of the anti‐CD19‐CAR gene showed the same trend. The median number of copies of the anti‐CD19‐CAR gene was 12 650 copies/μg (range, 187–44 509). The two levels began to decline 21 days after the infusion (Fig 4).
Fig 4.
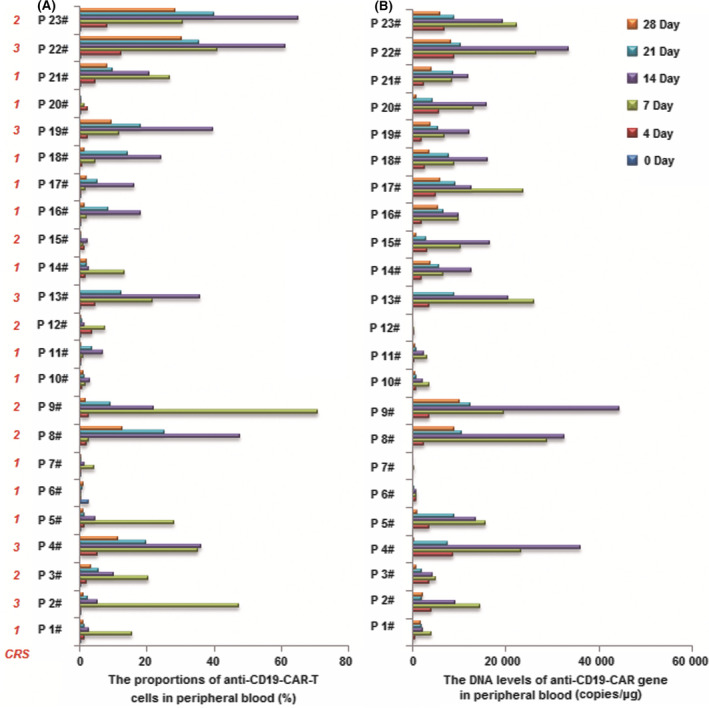
Cellular kinetics of the anti‐CD19‐CAR‐T cells. (A) Expansion of the anti‐CD19‐CAR‐T cells in peripheral blood. (B) DNA level of the anti‐CD19‐CAR gene in peripheral blood.
Adverse effects
Patients receiving CAR‐T‐cell infusion showed pyrexia with chills, accompanied with fatigue, nausea, decreased appetite, hypoalbuminaemia and tachycardia (Table II). Haematological toxicity was monitored. The notable AEs graded 1–2 CRS in 18 patients (18/23, 78·3%) and grade 3 CRS in five patients (5/23, 21·7%).
Table II.
Patients' baseline and therapy‐related characteristics.
| Any | Worst grade 1 | Worst grade 2 | Worst grade 3 | Worst grade 4 | Worst grade 5 | |
|---|---|---|---|---|---|---|
| Any | 23 (100%) | 0 | 13 (57%) | 7 (30%) | 3 (13%) | 0 |
| Pyrexia | 22 (96%) | 5 (22%) | 8 (35%) | 9 (39%) | 0 | 0 |
| Chills | 22 (96%) | 10 (43%) | 12 (52%) | 0 | 0 | 0 |
| Peripheral oedema | 3 (13%) | 2 (9%) | 1 (4%) | 0 | 0 | 0 |
| Hypoalbuminaemia | 21 (91%) | 21 (91%) | 0 | 0 | 0 | 0 |
| Hyponatraemia | 5 (22%) | 5 (22%) | 0 | 0 | 0 | 0 |
| Hypokalaemia | 10 (43%) | 8 (35%) | 2 (9%) | 0 | 0 | 0 |
| Hypocalcaemia | 14 (61%) | 13 (56%) | 1 (4%) | 0 | 0 | 0 |
| Hypophosphataemia | 4 (17%) | 2 (9%) | 1 (4%) | 1 (4%) | 0 | 0 |
| Hypomagnesaemia | 4 (17%) | 3 (13%) | 1 (4%) | 0 | 0 | 0 |
| Hyperglycaemia | 2 (9%) | 1 (4%) | 1 (4%) | 0 | 0 | 0 |
| Increased alanine aminotransferase | 13 (57%) | 4 (17%) | 7 (30%) | 0 | 2 (9%) | 0 |
| Increased aspartate aminotransferase | 9 (39%) | 4 (17%) | 4 (17%) | 0 | 1 (4%) | 0 |
| Hypoxia | 5 (22%) | 0 | 1 (4%) | 3 (13%) | 1 (4%) | 0 |
| Dyspnoea | 4 (17%) | 3 (13%) | 0 | 1 (4%) | 0 | 0 |
| Cough | 1 (4%) | 1 (4%) | 0 | 0 | 0 | 0 |
| Pleural effusion | 8 (35%) | 8 (35%) | 0 | 0 | 0 | 0 |
| Encephalopathy | 1 (4%) | 1 (4%) | 0 | 0 | 0 | 0 |
| Headache | 5 (22%) | 4 (17%) | 1 (4%) | 0 | 0 | 0 |
| Tremor | 1 (4%) | 1 (4%) | 0 | 0 | 0 | 0 |
| Confusion | 3 (13%) | 1 (4%) | 2 (9%) | 0 | 0 | 0 |
| Dizziness | 3 (13%) | 2 (9%) | 1 (4%) | 0 | 0 | 0 |
| Tachycardia | 3 (13%) | 0 | 2 (9%) | 1 (4%) | 0 | 0 |
| Sinus tachycardia | 23 (100%) | 23 (100%) | 0 | 0 | 0 | 0 |
| Hypertension | 4 (17%) | 4 (17%) | 0 | 0 | 0 | 0 |
| Hypotension | 2 (9%) | 2 (9%) | 0 | 0 | 0 | 0 |
| Nausea | 19 (83%) | 19 (83%) | 0 | 0 | 0 | 0 |
| Vomiting | 3 (13%) | 3 (13%) | 0 | 0 | 0 | 0 |
| Fatigue | 23 (100%) | 13 (57%) | 10 (43%) | 0 | 0 | 0 |
| Decreased appetite | 23 (100%) | 23 (100%) | 0 | 0 | 0 | 0 |
| Constipation | 10 (43%) | 10 (43%) | 0 | 0 | 0 | 0 |
| Diarrhoea | 2 (9%) | 2 (9%) | 0 | 0 | 0 | 0 |
| Weight loss | 0 | 0 | 0 | 0 | 0 | 0 |
Data are n (%). All 23 patients who received treatment are shown. The AEs are according to the National Cancer Institute's Common Terminology Criteria for Adverse Events (version 4.03).
Grade 3–4 leucopenia and neutropenia were observed in 17 patients (17/23, 74·9%), anaemia was measured in seven patients (7/23, 30·4%) and thrombocytopenia in nine patients (9/23, 39·1%). Grade 3–4 haematological toxicity occurred five days after strong pretreatment and recovery occurred 14–18 days after the CAR‐T‐cell infusion. The three patients with CNSL were diagnosed as grade 1, grade 3, and grade 1 CAR‐T‐cell‐related encephalopathy syndrome (CRES). 16 , 26 None of the patients experienced neurologic toxicity associated with CAR‐T cells, except for the above three. All the patients were given antipyretic drugs and methylprednisolone to overcome the AEs. No patients received tocilizumab during therapy, because of their rapid recovery from the AEs. The AEs related to the anti‐CD19‐CAR‐T‐cell therapy in all patients were relieved 12–14 days after the CAR‐T‐cell infusion. No patients died of CRS or CRES after the anti‐CD19‐CAR‐T‐cell infusion.
In the anti‐CD19‐CAR‐T‐cell therapy, the serum levels of IL‐6, IL‐2R, TNF‐α, and IL‐8 peaked on days four to seven after infusion of anti‐CD19‐CAR‐T cells and then declined after 14 days of infusion (Fig 5).
Fig 5.
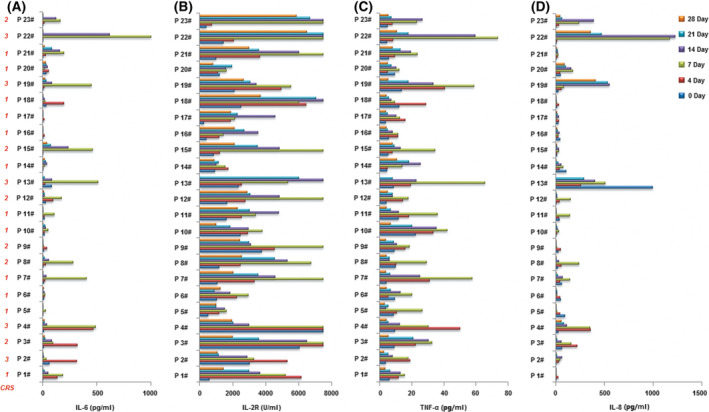
The serum levels of IL‐6, IL‐2R, TNF‐α, and IL‐8 after infusion of anti‐CD19‐CAR‐T‐cells. The levels of IL‐6 (A), IL‐2R (B), TNF‐α (C) and IL‐8 (D) in serum after 0,4,7,14,21 and 28 days of anti‐CD19‐CAR‐T cell infusion.
The 23 patients were divided into a grade 1–2 CRS group and a grade 3 or greater CRS group. On the day of anti‐CD19‐CAR‐T‐cell infusion, the proportion of leukaemic cells in BM was lower in the grade 1–2 CRS group than in the grade 3 or greater CRS group (Fig 6A). However, the leukocyte count in peripheral blood on the day of the anti‐CD19‐CAR‐T‐cell infusion was uncorrelated with CRS levels (Fig 6B). The peaks of IL‐6, TNF‐α and IL‐8 were lower in the grade 1–2 CRS group than in the grade 3 or greater CRS group (Fig 6C–E). However, the peak of the IL‐2R level was not correlated with CRS levels (Fig 6F). The peak proportion of the anti‐CD19‐CAR‐T cells and the peak of the anti‐CD19‐CAR gene level were not correlated with CRS levels (Fig 6G, H). Could this observation suggest that the peak proportion of the CAR‐T cells is independent of tumour burden?
Fig 6.
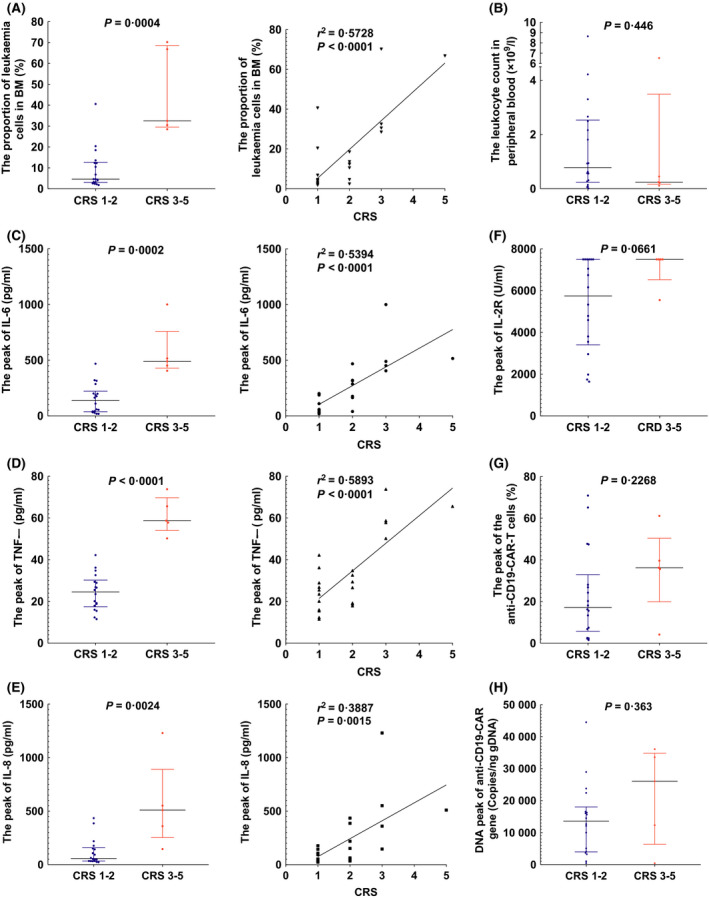
Factors in anti‐CD19‐CAR‐T‐cell therapy related to cytokine release syndrome (CRS) levels. (A) The proportion of leukaemic cells in bone marrow (BM) correlated with CRS levels [4·6% (IQR 3·1–12·7) to 32·5% (IQR 29·5–68·5)]. (B) The leukocyte count in peripheral was not correlated with CRS levels [0·8 × 109/l (IQR 0·3–2·5) to 0·2 × 109/l (IQR 0·2–3·5)]. (C) The peak of IL‐6 level correlated with CRS levels [137·0 pg/ml (IQR 37·9–222·3) to 489·5 pg/ml (IQR 428·5–758·0)]. (D) The peak of TNF‐α level correlated with CRS levels [24·5 pg/ml (IQR 17·4–30·2) to 58·7 pg/ml (IQR 54·0–69·7)]. (E) The peak of IL‐8 level correlated with CRS levels [57·6 pg/ml (IQR 34·1–158·3) to 510·0 pg/mL (IQR 253·0–890·5)]. (F) The peak of IL‐2R level was not correlated with CRS levels [5,747 U/ml (IQR 3,404–7,500) to 7,500 U/ml (IQR 6,525–7,500)]. (G) The peak proportion of the anti‐CD19‐CAR‐T cells was not correlated with CRS levels [17·2% (IQR 5·7–32· 9) to 36·2% (IQR 19. 9–50·4)]. (H) The peak of anti‐CD19‐CAR gene level was not correlated with CRS levels [13,578 Copies/ng gDNA (IQR 3,949–18,009) to 26,045 Copies/ng gDNA (IQR 6,363–34,819)].
Incidence and prognosis of the infections after anti‐CD19‐CAR‐T‐cell infusion
According to a microbiological examination for infectious diseases, six patients developed infectious diseases within 28 days after the CAR‐T‐cell infusion. Bacterial infections were detected in five patients (5/23, 21·7%), among which three were gram‐negative and two gram‐positive. All patients with bacterial infectious diseases were cured by antibiotics and supportive treatment. The other patient was diagnosed with cytomegalovirus infection and died of respiratory failure 24 days after CR from anti‐CD19‐CAR‐T‐cell therapy. Patients were diagnosed with bacterial infections 4–24 days after the CAR‐T‐cell infusion (the absolute neutrophil count was 0·0–0·5 × 109/l). No patients were diagnosed with invasive fungal disease.
Discussion
In recent years, clinical trials have shown that anti‐CD19‐CAR‐T‐cell therapy is effective in R/R B‐ALL patients, with a high response rate in many studies. 7 , 8 , 27 CRS and neurologic toxicity in the course of anti‐CD19‐CAR‐T cell therapy have been the main obstacles for this treatment. 20 The tumour burden and design of the lymphodepleting chemotherapy are all critical factors that might influence the toxicity. In our study, after the initial intensive lymphodepleting chemotherapy, R/R B‐ALL patients achieved 82·6% CR/Cri and suffered relatively mild side effects (78·3% in grade 1–2 CRS). With a median follow‐up of 14·0 months (range, 1·5–21·0 months), no survival benefit was achieved with consolidative HSCT.
Fludarabine induces cellular cytotoxicity by inhibition of DNA synthesis. The cellular cytotoxicity and lymphodepleting property of fludarabine depend on the activity of deoxycytidine kinase in lymphocytes. 28 FC, as a lymphodepletion drug, has been the most common regimen applied to anti‐CD19‐CAR‐T‐cell therapy. 29 , 30 FC enhanced the persistence of anti‐CD19‐CAR‐T cells by 2·9‐fold and improved disease‐free survival compared to cyclophosphamide alone in CAR‐T therapy. 9 , 11
It is worth noting that neurotoxicity of fludarabine might occur in therapy with anti‐CD19‐CAR‐T cells. Two patients died of cerebral oedema, leading to the suspension of a clinical trial. 31 Thus, fludarabine was subsequently removed from the lymphodepleting chemotherapy in this clinical trial. However, two other patients died of cerebral oedema shortly after the trial restarted, suggesting that fludarabine was not the cause of the fatal cerebral oedema. In a subsequent study, 17 the fatal neurotoxicity was attributed to the cytokines produced during CRS that were transported into the central nervous system.
In this clinical trial, a combination of fludarabine and cytarabine was chosen instead of FC regimens for synergistic effect. The purpose of the FA programme in our clinical trial was to reduce tumour load before the anti‐CD19‐CAR‐T‐cell therapy. The action of cytarabine depends on the phosphorylation of Ara‐C to Ara‐C‐triphosphate (Ara‐CTP). The rate‐limiting enzyme in this phosphorylation is the cytoplasmatic deoxycytidine kinase. The saturation concentration of cytarabine to the rate‐limiting enzyme is 500–1,000 mg/m2. 32 This was the basis on which we designed our clinical trial for the cytarabine dose. Fludarabine increases the concentration of Ara‐CTP by 2·2‐ to 2·8‐fold in vitro. 33 The synergistic effect of cytarabine and fludarabine had been demonstrated, as fludarabine increased the concentration of Ara‐CTP in leukaemia cells. 22 This was the basis for the use of FA as the lymphodepleting chemotherapy in our clinical trial. We are the first to use this combination to treat R/R B‐ALL or to prepare R/R B‐ALL patients for anti‐CD19‐CAR‐T‐cell therapy. The synergistic effect of cytarabine and fludarabine was utilized to reduce tumour load before anti‐CD19‐CAR‐T‐cell therapy. In this study, most of the R/R B‐ALL patients in this group had a decrease in the percentage of leukaemic cells in BM before the anti‐CD19‐CAR‐T‐cell infusion. The low proportion of leukaemic cells in the BM was correlated with 1–2 grade CRS levels. Furthermore, the peaks in the IL‐6, TNF‐α and IL‐8 levels were correlated with CRS levels.
In our study, a lower disease burden was achieved by intensive lymphodepleting chemotherapy, and the subsequent CAR‐T therapy had a high response and manageable toxicity. We will conduct a randomized controlled study to identify whether enhanced chemotherapy could ultimately benefit survival.
Author contributions
Concept and design: DQ. Drafted or revised the manuscript: WJ. Acquisition of data: JYY, LQ, MN, WJ, YZX. Analysis and interpretation of data: MJX, LXX, WJ. Writing, review and/or revision of manuscript: all authors. Study supervision: DQ.
Conflicts of interest
No conflict of interest exits in the submission of this manuscript, and the manuscript is approved by all authors for publication.
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of the Department of Hematology, Tianjin First Center Hospital (Tianjin, China). (Approval No. of Ethic Committee: 2015002X and 2018N105KY). Patients gave their written informed consent in accordance with the Declaration of Helsinki. This Clinical trial is registered at http://www.chictr.org.cn/index.aspx as ChiCTR‐ONN‐16009862, and ChiCTR1800019622. The patients agreed to the use of their specimens and data for our study.
Acknowledgements
We thank the patients for their participation in our experimental studies and clinical trials. We thank the Shanghai Genbase Biotechnology Co., Ltd. for providing us with anti‐CD19‐CAR‐T‐cells and technical support.
References
- 1. Gokbuget N, Kneba M, Raff T, Trautmann H, Bartram CR, Arnold R, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120:1868–76. [DOI] [PubMed] [Google Scholar]
- 2. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long‐term follow‐up of CD19 CAR therapy in acute lymphoblastic leukemia. New England Journal of Medicine. 2018;378:449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campana D, Schwarz H, Imai C. 4–1BB chimeric antigen receptors. Cancer Journal. 2014;20:134–40. [DOI] [PubMed] [Google Scholar]
- 4. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine. 2014b;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science Translational Medicine. 2014;6:224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent‐to‐treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee DW, Kochenderfer JN, Stetler‐Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. The Lancet. 2015;385:517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. New England Journal of Medicine. 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science Translational Medicine. 2015;7:303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19‐targeted T cells in patients with relapsed or chemotherapy refractory B‐cell leukemias. Blood. 2011;118:4817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clinical Cancer Research. 2007;13:5426–35. [DOI] [PubMed] [Google Scholar]
- 13. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B‐cell depletion and remissions of malignancy along with cytokine‐associated toxicity in a clinical trial of anti‐CD19 chimeric‐antigen‐receptor‐transduced T cells. Blood. 2012;119:2709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maude SL, Pulsipher MA, Boyer MW, Grupp SA, Davies SM, Phillips CL, et al. Efficacy and safety of CTL019 in the first US phase II multicenter trial in pediatric relapsed/refractory acute lymphoblastic leukemia: results of an interim analysis. Blood. 2016;128:2801–2801. [Google Scholar]
- 16. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T‐cell therapy ‐ assessment and management of toxicities. Nature Reviews Clinical Oncology. 2018;15:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez‐Cuyar LF, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov. 2017;7:1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor‐modified T‐cell therapy. Blood. 2017;130:2295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T‐cell therapy in patients with B‐cell acute lymphoblastic leukemia. Cancer Discov. 2018;8:958–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brudno JN, Kochenderfer JN. Recent advances in CAR T‐cell toxicity: mechanisms, manifestations and management. Blood Rev. 2019;34:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiss MA, Aliff TB, Tallman MS, Frankel SR, Kalaycio ME, Maslak PG, et al. A single, high dose of idarubicin combined with cytarabine as induction therapy for adult patients with recurrent or refractory acute lymphoblastic leukemia. Cancer. 2002;95:581–7. [DOI] [PubMed] [Google Scholar]
- 22. Clavio M, Carrara P, Miglino M, Pierri I, Canepa L, Balleari E, et al. High efficacy of fludarabine‐containing therapy (FLAG‐FLANG) in poor risk acute myeloid leukemia. Haematologica. 1996;81:513–20. [PubMed] [Google Scholar]
- 23. Wang J, Deng Q, Jiang YY, Zhang R, Zhu HB, Meng JX, et al. CAR‐T 19 combined with reduced‐dose PD‐1 blockade therapy for treatment of refractory follicular lymphoma: a case report. Oncol Lett. 2019;18:4415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jaspers JE, Brentjens RJ. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacology & Therapeutics. 2017;178:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T‐cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33:2854–66. [DOI] [PubMed] [Google Scholar]
- 26. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell‐engaging therapies. Cancer Journal. 2014a;20:119–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kenderian SS, Porter DL, Gill S. Chimeric Antigen Receptor T Cells and Hematopoietic Cell Transplantation: How Not to Put the CART Before the Horse. Biology of Blood and Marrow Transplantation. 2017;23:235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clinical Pharmacokinetics. 2002;41:93–103. [DOI] [PubMed] [Google Scholar]
- 29. Rapoport AP, Stadtmauer EA, Binder‐Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY‐ESO‐1‐specific TCR‐engineered T cells mediate sustained antigen‐specific antitumor effects in myeloma. Nature Medicine. 2015;21:914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riaz IB, Zahid U, Kamal MU, Husnain M, McBride A, Hua A, et al. Anti‐CD 19 and anti‐CD 20 CAR‐modified T cells for B‐cell malignancies: a systematic review and meta‐analysis. Immunotherapy. 2017;9:979–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DeAngelo DJ, Ghobadi A, Park JH, Dinner SN, Mannis GN, Lunning MA, et al. Clinical outcomes for the phase 2, single‐arm, multicenter trial of JCAR015 in adult B‐ALL (ROCKET Study). J Immunother Cancer. 2017;5:86. https:// doi.org/10.1186/s40425‐017‐0289‐3 [Google Scholar]
- 32. Kell J. Treatment of relapsed acute myeloid leukaemia. Rev Recent Clin Trials. 2006;1:103–11. [DOI] [PubMed] [Google Scholar]
- 33. Seymour JF, Huang P, Plunkett W, Gandhi V. Influence of fludarabine on pharmacokinetics and pharmacodynamics of cytarabine: implications for a continuous infusion schedule. Clinical Cancer Research. 1996;2:653–8. [PubMed] [Google Scholar]