

Abstract
Aims
The effects of vericiguat vs. placebo on high‐sensitivity C‐reactive protein (hsCRP) and serum uric acid (SUA) were assessed in patients with heart failure with reduced ejection fraction (HFrEF) in the Phase 2 SOCRATES‐REDUCED study (NCT01951625).
Methods and results
Changes from baseline hsCRP and SUA values at 12 weeks with placebo and vericiguat (1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg, respectively) were assessed. The probability of achieving an hsCRP value of ≤3.0 mg/L or SUA value of <7.0 mg/dL at week 12 was tested. Median baseline hsCRP and SUA levels were 3.68 mg/L [interquartile range (IQR) 1.41–8.41; n = 335] and 7.80 mg/dL (IQR 6.40–9.33; n = 348), respectively. Baseline‐adjusted mean percentage changes in hsCRP were 0.2%, −19.5%, −24.3%, −25.7% and −31.9% in the placebo and vericiguat 1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg groups, respectively; significance vs. placebo was observed in the vericiguat 10.0 mg group (P = 0.035). Baseline‐adjusted mean percentage changes in SUA were 5.0%, −1.3%, −1.1%, −3.5% and −5.3% in the placebo, and vericiguat 1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg groups, respectively; significance vs. placebo was observed in the 5.0 mg and 10.0 mg groups (P = 0.0202 and P = 0.004, respectively). Estimated probability for an end‐of‐treatment hsCRP value of ≤3.0 mg/L and SUA value of <7.0 mg/dL was higher with vericiguat compared with placebo. The effect was dose‐dependent, with the greatest effect observed in the 10.0 mg group.
Conclusions
Vericiguat treatment for 12 weeks was associated with reductions in hsCRP and SUA, and a higher likelihood of achieving an hsCRP value of ≤3.0 mg/L and SUA value of <7.0 mg/dL.
Keywords: Heart failure, Ventricular ejection fraction, Biomarker, C‐reactive protein, Uric acid, Vericiguat
Introduction
Inflammation and oxidative stress play key roles in the development and progression of heart failure (HF). 1 , 2 High‐sensitivity C‐reactive protein (hsCRP) is an established marker of systemic inflammation and is associated with HF severity and outcomes. 1 , 3 , 4 , 5 , 6 , 7 Uric acid, the end product of purine metabolism, circulates at increased serum levels in conditions of high oxidative stress and is frequently elevated in patients with HF, in which it correlates with HF severity and outcomes. 6 , 8 , 9 , 10 Under conditions of oxidative stress, endothelial dysfunction creates a deficiency in nitric oxide (NO) and leads to impaired NO‐soluble guanylate cyclase‐cyclic guanosine monophosphate (NO‐sGC‐cGMP) signalling, and contributes, amongst other pathways, to the development of HF. 11 , 12
Vericiguat is a stimulator of sGC under investigation as a first‐in‐class therapy for worsening chronic HF with reduced ejection fraction (HFrEF). 13 Although vericiguat did not meet the primary endpoint [change from baseline in log‐transformed N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP) compared with placebo at week 12] in the Phase 2 dose‐finding SOCRATES‐REDUCED study (NCT01951625) in patients with HFrEF, exploratory analyses suggested a dose–response association between higher vericiguat dose and greater reductions in NT‐proBNP compared with placebo. 14 As vericiguat may address inflammation and oxidative stress via its action on the NO‐sGC‐cGMP pathway, we aimed to evaluate the effects of vericiguat on hsCRP and serum uric acid (SUA) (markers of inflammation and oxidative stress) in patients with HFrEF using data from the Phase 2 SOCRATES‐REDUCED study.
Methods
Study design, treatment and assessments
SOCRATES‐REDUCED (n = 456; protocol BAY 1021189/15371) was a randomized, placebo‐controlled, double‐blind, dose‐finding, Phase 2 study of vericiguat in patients with HFrEF. 14 In brief, the study population comprised patients who were post‐hospitalization for HF or had received outpatient treatment with i.v. diuretics for HF. Patients were eligible for inclusion in the SOCRATES‐REDUCED study if they had a left ventricular ejection fraction (LVEF) of <45% within 4 weeks of a symptomatic HF event. Patients were randomized to 12 weeks of treatment with vericiguat (1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg once daily) or placebo after clinical stabilization or within 4 weeks after discharge. Dose up‐titration (dose doubling) or sham titration occurred at week 2 (visit 2) and week 4 (visit 3) after randomization, with dose titration predicated on safety assessments and systolic blood pressure criteria. 15 The study design was published 15 and is summarized in supplementary material online Figure S1 . The study conformed with the principles outlined in the Declaration of Helsinki. 16 Institutional review board or ethics committee approval was obtained at each study site. All patients provided written informed consent.
Blood samples were collected at all visits and centrifuged at 4°C. Serum and ethylenediaminetetraacetic acid plasma were aliquoted and stored immediately at −20°C or lower at study sites until they were transported on dry ice to the central laboratory (Covance Central Laboratory Services, Geneva, Switzerland), where samples were stored at −70°C or lower until analysis.
High‐sensitivity CRP was measured using an immunoturbidimetry assay (Roche Diagnostics, Indianapolis, IN, USA). Assays for SUA (Roche Diagnostics) were performed with the Roche Modular and Cobas Analyzer. Baseline data were collected at visit 1, before randomization and before the first intake of study treatment.
Other biomarkers investigated included bone alkaline phosphatase (bAP), cGMP, C‐terminal telopeptide of type 1 collagen (CTX), galectin‐3 (Gal‐3), growth differentiation factor 15 (GDF‐15), high‐sensitivity troponin T (hsTnT), osteopontin (OPN), pro‐collagen III peptide (PIIINP), soluble suppression of tumorigenicity 2 (sST2) and tissue metallopeptidase inhibitor 4 (TIMP‐4).
Statistical analysis and model generation
The impacts of treatment in terms of biomarker changes from baseline to end of treatment were evaluated with a linear mixed model in order to account for intra‐patient variability and differences in biomarker baseline levels. All analyses were based on log‐transformed biomarker levels and the per‐protocol population. Baseline‐adjusted mean hsCRP and SUA changes in each vericiguat treatment arm were statistically compared with those in the placebo arm using two‐sided t‐tests on the model contrasts. Dose dependency was assessed by a linear trend test. The analyses for hsCRP and SUA were performed with and without additional clinical covariates (age group, sex, New York Heart Association functional class, diabetes and atrial fibrillation).
Building on this analysis, risk cut‐offs of hsCRP and SUA established from prior studies were examined: hsCRP concentrations of <1.0 mg/L, 1.0–3.0 mg/L and >3.0 mg/L, respectively, are associated with low, moderate and high categories of relative risk for cardiovascular disease. 17 Based on the linear mixed model, the expected end of treatment value for a given baseline value and treatment was calculated. The estimated probabilities of biomarker decreases to levels below the risk cut‐off (hsCRP ≤3.0 mg/L, SUA <7.0 mg/dL) during the course of the study were calculated for all treatment groups and selected baseline values within the observed range. These calculations were performed with consideration of the observed inter‐patient variability and on the assumption of a log‐normal distribution of the biomarkers.
The associations of hsCRP and SUA changes from baseline to end of treatment with clinical outcomes (cardiovascular death, cardiovascular hospitalization and emergency presentation caused by worsening chronic HF) were assessed. The relative odds (odds ratio) of a clinical outcome in each group (segmented by the direction of biomarker change from baseline to end of treatment) were calculated and Fisher's exact test was conducted.
Results
Patients
The SOCRATES‐REDUCED study was conducted across Europe, North America and Asia between November 2013 and January 2015, with follow‐up ending in June 2015. Randomized patients with no major protocol deviations and biomarker values at baseline and week 12 were included in the analysis (n = 328 and n = 345 in the hsCRP and SUA analyses, respectively) (supplementary material online Figure S2 ). Baseline characteristics for the full analysis set are presented in supplementary material online Table S1 .
Biomarker evaluation at baseline
Overall median hsCRP was 3.68 mg/L [interquartile range (IQR) 1.41–8.41 mg/L] at baseline. Within each treatment arm, baseline median hsCRP values were 3.98 mg/L (IQR 1.54–8.46 mg/L), 5.45 mg/L (IQR 1.78–10.75 mg/L), 3.62 mg/L (IQR 1.29–7.58 mg/L), 3.88 mg/L (IQR 1.29–8.60 mg/L) and 2.81 mg/L (IQR 1.37–6.62 mg/L) in the placebo, and vericiguat 1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg groups, respectively.
Median SUA was 7.80 mg/dL (IQR 6.40–9.33 mg/dL) at baseline. Within each treatment arm, baseline median SUA values were 7.50 mg/dL (IQR 6.30–9.90 mg/dL), 7.40 mg/dL (IQR 6.10–8.70 mg/dL), 7.90 mg/dL (IQR 6.88–9.13 mg/dL), 8.25 mg/dL (IQR 6.63–9.80 mg/dL) and 7.95 mg/dL (IQR 6.55–9.38 mg/dL) in the placebo, and vericiguat 1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg groups, respectively.
Patient demographics by baseline hsCRP and SUA subgroups are shown in Tables 1 and 2, respectively. There were several differences between patients with lower and higher hsCRP and SUA values at baseline: patients with higher hsCRP and SUA values at baseline were of a slightly younger age and had a higher body mass index; patients with higher SUA at baseline had a lower LVEF, systolic blood pressure, serum sodium and estimated glomerular filtration rate (eGFR), and a higher serum creatinine level.
Table 1.
Baseline characteristics by high‐sensitivity C‐reactive protein subgroup
| Baseline characteristic | Total | hsCRP <1.0 mg/L n = 53 | hsCRP 1.0–3.0 mg/L n = 92 | hsCRP >3.0 mg/L n = 183 | P‐valuea |
|---|---|---|---|---|---|
| N = 328 | (16.2%) | (28.0%) | (55.8%) | ||
| Age, years, median (IQR) | 68.0 (58.0–76.0) | 69.0 (60.0–78.0) | 70.0 (60.8–79.2) | 66.0 (57.0–74.5) | 0.035 |
| BMI, kg/m2, median (IQR) | 27.6 (24.0–31.4) | 26.5 (23.7–29.6) | 27.1 (24.0–30.9) | 28.2 (24.5–31.9) | 0.041 |
| Female sex, n | 69 (21.0%) | 15 (28.3%) | 21 (22.8%) | 33 (18.0%) | 0.231 |
| Non‐White race, n | 78 (23.8%) | 15 (28.3%) | 21 (22.8%) | 42 (23.0%) | 0.716 |
| NYHA class III/IV, n | 139 (42.4%) | 16 (30.2%) | 40 (43.5%) | 83 (45.4%) | 0.140 |
| Atrial fibrillation, n | 157 (47.9%) | 29 (54.7%) | 46 (50.0%) | 82 (44.8%) | 0.402 |
| Arterial hypertension, n | 254 (77.4%) | 44 (83.0%) | 65 (70.7%) | 145 (79.2%) | 0.163 |
| Coronary artery disease, n | 167 (50.9%) | 25 (47.2%) | 48 (52.2%) | 94 (51.4%) | 0.844 |
| Chronic kidney disease, n | 121 (36.9%) | 20 (37.7%) | 25 (27.2%) | 76 (41.5%) | 0.063 |
| Diabetes mellitus, n | 155 (47.3%) | 21 (39.6%) | 43 (46.7%) | 91 (49.7%) | 0.421 |
| DBP, mmHg, median (IQR) | 75.2 (69.6–81.7) | 75.3 (69.0–79.7) | 75.3 (68.3–82.0) | 75.0 (70.0–83.0) | 0.814 |
| SBP, mmHg, median (IQR) | 121.7 (115.6–134.4) | 120.0 (114.7–128.7) | 125.3 (117.0–135.5) | 121.0 (115.0–136.5) | 0.104 |
| Heart rate, b.p.m., median (IQR) | 70.5 (62.0–81.0) | 69.3 (61.0–78.7) | 71.0 (60.7–81.5) | 70.7 (63.1–81.2) | 0.655 |
| LVEF, %, median (IQR) | 29.2 (23.3–36.0) | 29.8 (24.5–33.9) | 31.2 (24.9–36.8) | 28.5 (23.0–37.1) | 0.350 |
| Serum sodium, mmol/L, median (IQR) | 139.0 (137.0–141.0) | 139.0 (137.0–141.0) | 139.5 (137.0–141.0) | 139.0 (136.0–140.0) | 0.194 |
| Serum creatinine, mg/dL, median (IQR) | 1.2 (1.0–1.4) | 1.1 (1.0–1.3) | 1.2 (1.0–1.4) | 1.2 (1.0–1.5) | 0.397 |
| eGFR, mL/min/1.73 m2, median (IQR) | 56.3 (44.4–72.6) | 55.3 (47.9–64.6) | 56.3 (41.3–75.0) | 56.3 (44.5–73.8) | 0.689 |
BMI, body mass index; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; hsCRP, high‐sensitivity C‐reactive protein; IQR, interquartile range; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association; SBP, systolic blood pressure.
P‐values are based on Fisher's exact tests for sex, race, NYHA class, atrial fibrillation, arterial hypertension, coronary artery disease, chronic kidney disease and diabetes mellitus, and on Kruskal–Wallis tests for other baseline characteristics.
Table 2.
Baseline characteristics by serum uric acid subgroup
| Baseline characteristic | Total | SUA <7.0 mg/dL n = 116 | SUA 7.0–10.0 mg/dL n = 159 | SUA >10.0 mg/dL n = 70 | P‐valuea |
|---|---|---|---|---|---|
| N = 345 | (33.6%) | (46.1%) | (20.3%) | ||
| Age, years, median (IQR) | 68.0 (59.0–77.0) | 71.0 (62.0–79.0) | 67.0 (59.0–75.0) | 63.0 (54.0–73.0) | 0.001 |
| BMI, kg/m2, median (IQR) | 27.7 (24.3–31.5) | 26.8 (23.2–30.2) | 27.6 (24.8–31.4) | 29.1 (26.2–33.6) | 0.004 |
| Female sex, n | 76 (22.0%) | 29 (25.0%) | 32 (20.1%) | 15 (21.4%) | 0.637 |
| Non‐White race, n | 76 (22.0%) | 24 (20.7%) | 32 (20.1%) | 20 (28.6%) | 0.344 |
| NYHA class III/IV, n | 152 (44.1%) | 52 (44.8%) | 67 (42.1%) | 33 (47.1%) | 0.761 |
| Atrial fibrillation, n | 167 (48.4%) | 56 (48.3%) | 78 (49.1%) | 33 (47.1%) | 0.970 |
| Arterial hypertension, n | 268 (77.7%) | 89 (76.7%) | 124 (78.0%) | 55 (78.6%) | 0.956 |
| Coronary artery disease, n | 174 (50.4%) | 57 (49.1%) | 81 (50.9%) | 36 (51.4%) | 0.950 |
| Chronic kidney disease, n | 128 (37.1%) | 49 (42.2%) | 52 (32.7%) | 27 (38.6%) | 0.267 |
| Diabetes mellitus, n | 168 (48.7%) | 55 (47.4%) | 80 (50.3%) | 33 (47.1%) | 0.874 |
| DBP, mmHg, median (IQR) | 75.0 (69.7–81.3) | 74.8 (69.7–79.2) | 75.3 (69.0–81.5) | 76.3 (70.2–83.2) | 0.480 |
| SBP, mmHg, median (IQR) | 121.3 (115.0–133.7) | 125.3 (116.7–140.7) | 120.7 (115.0–131.7) | 119.3 (113.9–127.8) | 0.010 |
| Heart rate, b.p.m., median (IQR) | 70.7 (62.3–81.0) | 70.5 (61.7–76.8) | 70.0 (62.5–81.0) | 74.5 (64.7–85.3) | 0.144 |
| LVEF, %, median (IQR) | 29.0 (23.2–35.8) | 30.2 (25.6–37.6) | 29.6 (23.0–36.0) | 25.8 (21.8–33.1) | 0.005 |
| Serum sodium, mmol/L, median (IQR) | 139.0 (137.0–141.0) | 140.0 (138.0–141.0) | 139.0 (136.0–140.5) | 138.0 (136.0–140.0) | 0.006 |
| Serum creatinine, mg/dL, median (IQR) | 1.2 (1.0–1.4) | 1.1 (0.9–1.4) | 1.2 (1.0–1.4) | 1.4 (1.1–1.7) | <0.001 |
| eGFR, mL/min/1.73 m2, median (IQR) | 56.3 (44.9–72.7) | 60.2 (45.8–80.8) | 59.2 (48.2–70.9) | 47.2 (38.2–59.1) | <0.001 |
BMI, body mass index; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; IQR, interquartile range; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association; SBP, systolic blood pressure; SUA, serum uric acid.
P‐values are based on Fisher's exact tests for sex, race, NYHA class, atrial fibrillation, arterial hypertension, coronary artery disease, chronic kidney disease and diabetes mellitus, and on Kruskal–Wallis tests for other baseline characteristics.
Biomarker evaluation following treatment
Baseline‐adjusted mean hsCRP and SUA changes (%) from baseline to week 12 across the placebo and vericiguat dose groups are shown in Figure 1. Following 12 weeks of treatment, baseline‐adjusted mean percentage changes from baseline in hsCRP were 0.2% [95% confidence interval (CI)] −22.4 to 29.3], −19.5% (95% CI −37.8 to 4.1), −24.3% (95% CI −41.1 to −2.7), −25.7% (95% CI −42.6 to −3.7) and −31.9% (95% CI −47.2 to −12.3) in the placebo, and vericiguat 1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg groups, respectively (Figure 1 A). Dose‐dependent reductions in hsCRP were observed; the analysis indicated a significantly greater reduction in hsCRP with the vericiguat target dose of 10.0 mg relative to placebo (−31.9% vs. 0.2%; P = 0.035) and a significant linear trend in the reduction of hsCRP from baseline to end of treatment from placebo up to the highest vericiguat dose of 10.0 mg (P = 0.039) (Table 3).
Figure 1.
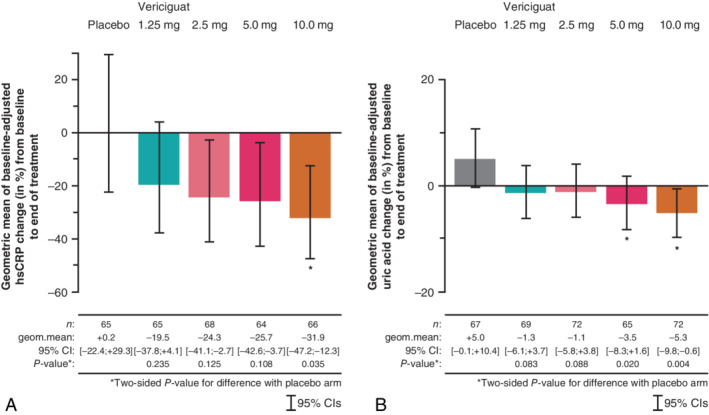
Baseline‐adjusted mean changes (%) in (A) high‐sensitivity C‐reactive protein (hsCRP) and (B) serum uric acid (SUA) from baseline to end of treatment. CI, confidence interval; geom.mean, geometric mean. *Statistical significance relative to placebo.
Table 3.
Analysis of changes in log‐transformed high‐sensitivity C‐reactive protein and serum uric acid levels from baseline to end of treatment with and without additional clinical covariates
| hsCRP changes | SUA changes | |||||||
|---|---|---|---|---|---|---|---|---|
| Adjusted for hsCRP baseline level (log‐transformed) | Adjusted for hsCRP baseline level (log‐transformed) and clinical covariates | Adjusted for SUA baseline level (log‐transformed) | Adjusted for SUA baseline level (log‐transformed) and clinical covariates | |||||
| Effect ratioa (95% CI) | P‐value | Effect ratioa (95% CI) | P‐value | Effect ratioa (95% CI) | P‐value | Effect ratioa (95% CI) | P‐value | |
| Treatment groups | ||||||||
| Vericiguat 1.25 mg vs. placebo | 0.80 (0.56–1.15) | 0.235 | 0.84 (0.59–1.22) | 0.367 | 0.94 (0.88–1.01) | 0.083 | 0.94 (0.88–1.01) | 0.081 |
| Vericiguat 2.5 mg vs. placebo | 0.76 (0.53–1.08) | 0.125 | 0.71 (0.50–1.03) | 0.069 | 0.94 (0.88–1.01) | 0.088 | 0.95 (0.88–1.01) | 0.114 |
| Vericiguat 5.0 mg vs. placebo | 0.74 (0.52–1.07) | 0.108 | 0.69 (0.48–1.00) | 0.049 | 0.92 (0.86–0.99) | 0.020 | 0.91 (0.85–0.98) | 0.010 |
| Vericiguat 10.0 mg vs. placebo | 0.68 (0.47–0.97) | 0.035 | 0.66 (0.46–0.96) | 0.024 | 0.90 (0.84–0.97) | 0.004 | 0.90 (0.84–0.97) | 0.004 |
| Linear trend test | 0.039 | 0.013 | 0.004 | 0.003 | ||||
CI, confidence interval; hsCRP, high‐sensitivity C‐reactive protein: SUA, serum uric acid.
As biomarker levels were log‐transformed for the analyses, statistical testing is based on the effect ratio instead of the effect difference. The effect ratio of group 1 vs. group 2 is the ratio of the baseline‐adjusted biomarker levels at end of treatment in groups 1 and 2.
After 12 weeks of treatment, baseline‐adjusted mean percentage changes from baseline in SUA were 5.0% (95% CI −0.1 to 10.4), −1.3% (95% CI −6.1 to 3.7), −1.1% (95% CI −5.8 to 3.8), −3.5% (95% CI −8.3 to 1.6) and −5.3% (95% CI −9.8 to −0.6) in the placebo, and vericiguat 1.25 mg, 2.5 mg, 5.0 mg and 10.0 mg groups, respectively (Figure 1 B). Significant reductions from baseline in SUA were observed in the vericiguat 5.0 mg and 10 mg groups relative to placebo [−3.5% vs. 5.0% (P = 0.02) and −5.3% vs. 5.0% (P = 0.004), respectively] and a significant linear trend in the reduction of SUA from baseline to end of treatment from placebo up to the highest vericiguat dose of 10.0 mg (P = 0.004) was apparent (Table 3), indicating a dose‐dependent effect.
Additional consideration of clinical covariates in the analysis did not lead to noteworthy changes in the treatment effects (e.g. effect ratios of 0.66 vs. 0.68 for hsCRP and 0.90 vs. 0.90 for SUA in comparisons of 10.0 mg vericiguat and placebo in the model with and without clinical covariates).
Estimated probability of achieving biomarker thresholds at end of treatment
The estimated probability for an end‐of‐treatment (week 12) hsCRP value of ≤3.0 mg/L and SUA value of <7.0 mg/dL was higher in all vericiguat treatment groups compared with placebo (Figure 2). The estimated probability for an end‐of‐treatment hsCRP value of ≤3.0 mg/L and SUA value of <7.0 mg/dL varied by treatment and baseline biomarker level. The estimated probability differences between all vericiguat treatment groups and placebo for an end‐of‐treatment hsCRP value of ≤3.0 mg/L were comparable across baseline hsCRP values (Figure 2 A). The estimated probability increase (shift on y‐axis) from placebo to vericiguat for an end‐of‐treatment hsCRP value of ≤3.0 mg/L ranged from 8.3 to 14.6 percentage points in patients with a baseline hsCRP value of 4 mg/L (left dashed vertical line) and from 6.5 to 12.1 percentage points in patients with a baseline hsCRP value of 10 mg/L (right dashed vertical line) in the vericiguat 1.25 mg and 10.0 mg groups, respectively.
Figure 2.
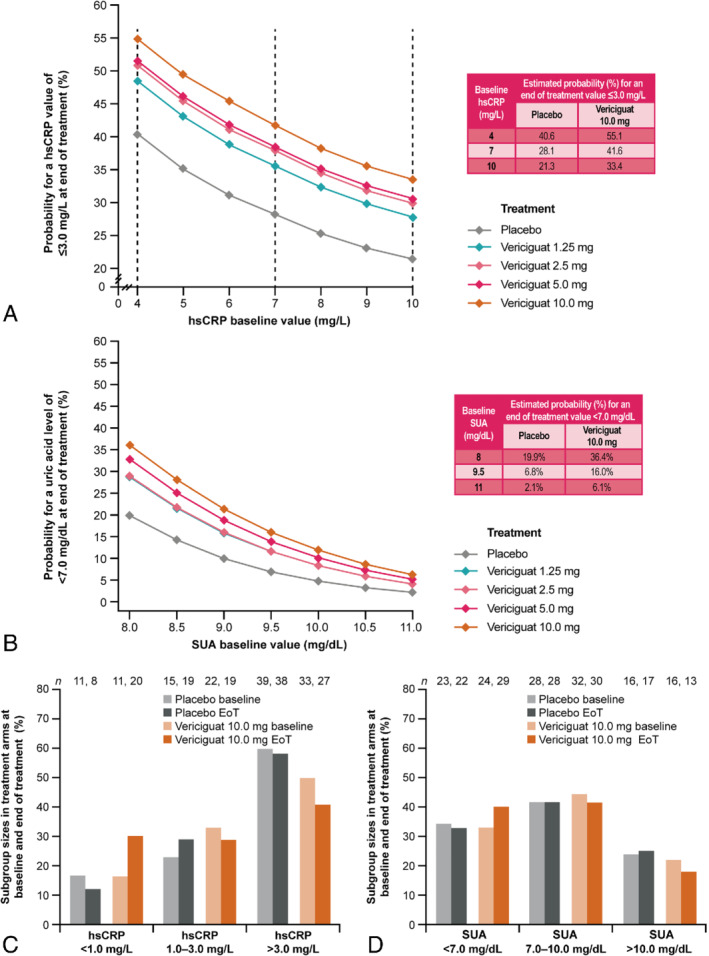
Estimated probabilities for (A) high‐sensitivity C‐reactive protein (hsCRP) and (B) serum uric acid (SUA) reduction to below risk level and (C, D) subgroup sizes per treatment arm at baseline and end of treatment (EoT). In (B), 1.25 mg and 2.5 mg vericiguat trend lines overlap SUA data. Numbers within the bar charts represent the numbers of patients with specified (C) hsCRP or (D) SUA values at baseline or EoT. The total numbers of patients per treatment arm for placebo and vericiguat 10.0 mg were 65 and 66, respectively, for hsCRP data, and 67 and 72, respectively, for SUA data.
The SUA model indicated that the estimated probability difference between all vericiguat treatment groups and placebo for an end‐of‐treatment SUA value of <7.0 mg/dL was decreased in patients with higher baseline SUA levels (Figure 2 B). The estimated probability increase from placebo to vericiguat for an end‐of‐treatment SUA value of <7.0 mg/L ranged from 9.1 to 16.5 percentage points in patients with a baseline SUA value of 8.0 mg/dL and from 1.9 to 4.0 percentage points in patients with a baseline SUA value of 11.0 mg/dL across the vericiguat treatment groups.
The distributions of the hsCRP and SUA subgroups in the placebo and 2.5–10.0 mg vericiguat treatment arms at baseline and end of treatment are shown in Figure 2 C and 2 D. At baseline, 11, 22 and 33 patients in the 10.0 mg vericiguat treatment arm had hsCRP values of <1.0 mg/L, 1.0–3.0 mg/L and >3.0 mg/L, respectively (Figure 2C). Following 12 weeks of treatment with vericiguat, 20, 19 and 27 patients had hsCRP values of <1.0 mg/L, 1.0–3.0 mg/L and >3.0 mg/L, respectively. Changes in the distribution of the hsCRP subgroups were also observed in the placebo treatment group, with fewer patients having an hsCRP value of <1.0 mg/L at week 12 than at baseline. These changes reflect a near doubling of patients (from 11 to 20) with hsCRP values of <1.0 mg/L by week 12 in the 10.0 mg vericiguat treatment group.
At baseline, 24, 32 and 16 patients in the 10.0 mg vericiguat treatment arm had SUA values of <7.0 mg/dL, 7.0–10.0 mg/dL and >10.0 mg/dL, respectively (Figure 2 D). At the end of vericiguat treatment, 29, 30 and 13 patients had SUA values of <7.0 mg/dL, 7.0–10.0 mg/dL and >10.0 mg/dL, respectively. The distribution of SUA subgroups in the placebo treatment arm was largely unchanged throughout the study.
Association of biomarker changes from baseline with clinical outcomes
The proportion of clinical outcome events (cardiovascular death, cardiovascular hospitalization and emergency presentation caused by worsening chronic HF) was numerically smaller in patients with decreases in hsCRP and SUA than in those with increases from baseline (Table 4). Clinical outcome events occurred in 14.0% of patients who had a decrease in hsCRP compared with 21.8% who had an increase in hsCRP, relative to baseline, at the end of treatment (P = 0.08). Similarly, clinical outcome events occurred in 14.8% of patients who had a decrease in SUA compared with 19.9% who had an increase in SUA, relative to baseline, at the end of treatment (P = 0.26).
Table 4.
Association of changes in high‐sensitivity C‐reactive protein and serum uric acid with clinical outcomes
| Direction of biomarker change from baseline to end of treatment | n | Patients with an event, n | Event proportion, % | Odds ratio | 95% CI | P‐value |
|---|---|---|---|---|---|---|
| hsCRP | ||||||
| Increase | 142 | 31 | 21.8% | 0.58 | 0.31–1.08 | 0.08 |
| Decrease | 186 | 26 | 14.0% | |||
| SUA | ||||||
| Increase | 176 | 35 | 19.9% | 0.70 | 0.38–1.27 | 0.26 |
| Decrease | 169 | 25 | 14.8% | |||
CI, confidence interval; hsCRP, high‐sensitivity C‐reactive protein; SUA, serum uric acid.
Other exploratory biomarkers
Geometric means at baseline and summary statistics of percentage changes from baseline in other exploratory biomarkers (bAP, cGMP, CTX, Gal‐3, GDF‐15, hsTnT, OPN, PIIINP, sST2 and TIMP‐4) are presented in supplementary material online Table S2 . There was no effect of treatment with vericiguat compared with placebo on these biomarkers at week 12 (P‐value of linear trend test >0.05).
Discussion
Vericiguat treatment for 12 weeks was associated with a decrease in hsCRP (significant in the vericiguat 10.0 mg group and a significant dose‐dependent trend) and SUA concentrations (significant in the vericiguat 5.0 mg and 10.0 mg groups and a significant dose‐dependent trend) from baseline in patients with HFrEF. Statistically significant dose‐dependent reductions in hsCRP and SUA were observed in vericiguat‐treated patients with HFrEF when data were adjusted for baseline levels and clinical covariates.
Vericiguat treatment was also associated with a higher probability of an hsCRP value of ≤3.0 mg/L at the end of the study than placebo. The distribution of hsCRP subgroups in the 10.0 mg vericiguat treatment arm revealed that the increase in patients with an end‐of‐treatment hsCRP value of <1.0 mg/L corresponded with fewer patients having an hsCRP value of ≥1.0 mg/L at week 12. These novel results point to a potential anti‐inflammatory effect of sGC stimulation in patients with HFrEF after a recent worsening HF event who are post‐hospitalization for HF or have required outpatient treatment with i.v. diuretics.
The mechanism underlying the lowering of hsCRP by vericiguat in patients with HFrEF is not directly addressed by these data. The anti‐inflammatory capacity of sGC stimulation has been shown in a murine model of interleukin‐1β‐induced leucocyte rolling and adhesion, in which sGC stimulation with BAY 41–2272 (a predecessor to the sGC activator) down‐regulated P‐selectin expression and inhibited leucocyte recruitment. 18 However, studies using tumour necrosis factor inhibitors demonstrated no improvement in HF‐related outcomes. 4 Similarly, although statin treatment in the CORONA study was associated with a reduction in CRP, the study did not meet its primary endpoint. 4 It remains to be determined whether hsCRP is a true prognostic biomarker in relation to clinical outcomes.
Systemic inflammation in HF has previously been postulated to result from an impaired intestinal mucosal barrier function secondary to mesenteric venous congestion in right ventricular (RV) HF. The intestinal damage leads to translocation of lipopolysaccharides from the intestinal lumen into the circulation, and endotoxaemia subsequently causes production of proinflammatory cytokines. 19 , 20 , 21 , 22 , 23 Thus, a decrease in systemic inflammatory markers could be hypothesized to reflect decreased peripheral venous congestion as a potential consequence of improved RV function and decreased right heart filling pressures. Reduced congestion may also result in less hepatic congestion, 24 which may, in turn, result in lower levels of inflammation and oxidative stress. 25 However, observed correlations between changes in hsCRP and SUA and changes in echocardiographic parameters following 12 weeks of treatment were only very small and not supportive of an imaging correlate reflecting lower RV filling pressures as an underlying mechanism (data not shown). In patients who experienced a decrease from baseline in hsCRP or SUA at week 12, the relative odds of a clinical outcome were non‐significantly reduced compared with patients who had an increase from baseline in hsCRP or SUA at end of treatment. The collection of biomarkers from the Phase 3 vericiguat study VICTORIA (NCT02861534) in patients with HFrEF will provide an opportunity to assess clinical laboratory markers of haemodynamic right heart unloading in parallel with echocardiography in an imaging ancillary study as potential determinants of reduced systemic inflammation in response to treatment with vericiguat. 13 In addition, the longer duration and increased number of clinical events in the VICTORIA study will enable a more robust assessment of the associations between inflammatory biomarkers and clinical outcomes.
Treatment with vericiguat in patients with HFrEF was associated with a higher likelihood of SUA levels falling below the threshold of hyperuricaemia (SUA ≥7.0 mg/dL) at the end of the study. The magnitude of SUA reduction observed with vericiguat treatment in this analysis is comparable with that achieved by sacubitril/valsartan treatment in the PARADIGM‐HF study. 10 Hyperuricaemic levels are associated with increased risk for cardiovascular death, hospitalization for HF and all‐cause mortality. 26 , 27 , 28 The observed reduction in uric acid levels following treatment with vericiguat may also represent a potential protective mechanism to prevent the deterioration of kidney function in HF and ameliorate cardiorenal syndrome. Although they are not strongly correlated with changes in eGFR (Spearman correlation −0.363 (95% CI −0.452 to −0.267) (supplementary material online Table S3 ), the reductions from baseline in SUA reported here (geometric mean: <0.5 mg/dL) are smaller than those reported in two dedicated SUA‐lowering trials in patients with HF (reductions of 2 mg/dL and 4.2 mg/dL), which found no improvements in survival, hospitalization for HF or LVEF. 29 , 30 Thus, although effectively reducing SUA, the inconsistent clinical efficacy of xanthine oxidase inhibitors in these studies called into question the promise of SUA reduction to improve clinical outcomes. This contrasts with the broad evidence base in support of the pathophysiological relevance of an increase in SUA as a risk factor for adverse outcomes in both HF and chronic kidney disease. The SUA‐reducing effect of vericiguat may support the hypothesis that mechanisms other than the inhibition of SUA generation via xanthine oxidase may have promise for tackling this risk factor.
The analysis of baseline characteristics by biomarker subgroup revealed that the baseline demographic characteristics of younger age and a higher body mass index were associated with higher hsCRP and SUA values. Higher SUA but not higher hsCRP were found at baseline in patients with worse renal function, more severely reduced LVEF and lower serum sodium. With the exception of serum sodium, these findings are consistent with those reported for SUA in the PARADIGM‐HF study 10 and suggest that the clinical phenotype associated with higher hsCRP and SUA is consistent with more advanced HF.
There was no effect of treatment with vericiguat compared with placebo on other exploratory biomarkers associated with HF (bAP, cGMP, CTX, Gal‐3, GDF‐15, hsTnT, OPN, PIIINP, sST2, TIMP‐4) at week 12; these results are consistent with those reported in the full analysis set. 14
One limitation of this analysis is that the mechanisms behind the effects of vericiguat on hsCRP and SUA are unknown and further studies are warranted to validate the prognostic value of reducing SUA as a treatment target in therapeutic approaches in HFrEF. Given the exploratory post hoc nature of the analysis, these results should be considered as hypothesis‐generating. It should also be considered that elevated hsCRP levels at baseline may reflect the presence of aetiologies other than HF, such as metabolic conditions including insulin resistance. 7 The analysis of SUA changes from baseline to week 12 did not take into consideration the concomitant use of SUA‐altering therapies such as diuretics. Additionally, this study was not powered for outcomes and the reported associations between biomarkers and adverse clinical outcomes should be interpreted with caution.
In conclusion, dose‐dependent decreases in hsCRP and SUA concentrations from baseline in patients with HFrEF were observed following 12 weeks of treatment with vericiguat. Whether the reductions in hsCRP and SUA with vericiguat are sustained with longer periods of treatment and are associated with improved clinical outcomes remains to be demonstrated in the Phase 3 VICTORIA study (NCT02861534). 13
Supporting information
Table S1. Baseline characteristics of patients in the full analysis set and biomarker analysis sets.
Table S2. Baseline levels and changes from baseline to end of treatment of additional biomarkers (per‐protocol population).
Table S3. Correlation analysis of changes in estimated glomerular filtration rate and body mass index with changes in log‐transformed high‐sensitivity C‐reactive protein and serum uric acid levels from baseline to end of treatment.
Figure S1. SOCRATES‐REDUCED study design.
Figure S2. Patient disposition in the SOCRATES‐REDUCED study.
Acknowledgements
Part of this analysis was presented at the 2018 European Society of Cardiology Heart Failure Congress. Medical writing and editorial assistance were provided by Laila Guzadhur, PhD, Moamen Hammad, PhD and Annabel Ola, MSc, all of Scion (London, UK). This assistance was funded by Bayer AG (Berlin, Germany) and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. (Kenilworth, NJ, USA).
Funding
Funding for this research was provided by Bayer AG (Berlin, Germany) and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. (Kenilworth, NJ, USA).
Conflict of interest: F.K. and L.R. are employees of Bayer AG. S.V. is an employee of Chrestos Concept, a contract partner of Bayer. B.‐W.I. was an employee of Bayer during the course of the SOCRATES‐REDUCED study and is currently employed by Boehringer Ingelheim. J.B. has received research support from the US National Institutes of Health, the European Union, and the Patient‐Centered Outcomes Research Institute, and consults for Abbott, Adrenomed, Amgen, Array, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Cardiocell, Corvidia, CVRx, G3 Pharmaceutical, Innolife, Janssen, LivaNova, Luitpold, Medtronic, Merck Sharp & Dohme, Novartis, NovoNordisk, Relypsa, Roche, Sanofi, V‐Wave and Vifor. C.S.P.L. has received research support from Boston Scientific, Bayer, Roche Diagnostics, AstraZeneca, Medtronic and Vifor Pharma, and has served as a consultant or on the advisory board/steering committee/executive committee for Boston Scientific, Bayer, Roche Diagnostics, AstraZeneca, Medtronic, Vifor Pharma, Novartis, Amgen, Merck Sharp & Dohme, Janssen Research & Development, Menarini, Boehringer Ingelheim, NovoNordisk, Abbott Diagnostics, Corvia, Stealth BioTherapeutics, JanaCare, Biofourmis, Darma, Applied Therapeutics, MyoKardia, WebMD Global, Radcliffe Group and Corpus. A.P.M. has served as a committee member on clinical studies sponsored by Bayer and Novartis. S.J.S. reports the receipt of research grants from the National Institutes of Health (R01 HL107577, R01 HL127028, R01 HL140731, R01 HL149423), Actelion, AstraZeneca, Corvia and Novartis, and has served as a consultant/advisory board member for Abbott, Actelion, AstraZeneca, Amgen, Bayer, Boehringer‐Ingelheim, Cardiora, Coridea, CVRx, Eisai, Ionis, Ironwood, Merck Sharp & Dohme, MyoKardia, Novartis, Pfizer, Sanofi, Tenax and United Therapeutics. B.P. is a steering committee member of the SOCRATES‐REDUCED study and a consultant/steering committee member for Bayer, Merck Sharp & Dohme, Novartis, Stealth Peptides, Daiichi‐Sankyo, AstraZeneca, BMS and Servier.
References
- 1. Williams ES, Shah SJ, Ali S, Ya Na B, Schiller NB, Whooley MA. C‐reactive protein, diastolic dysfunction, and risk of heart failure in patients with coronary disease: heart and soul study. Eur J Heart Fail 2008;10:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ayoub KF, Pothineni NVK, Rutland J, Ding Z, Mehta JL. Immunity, inflammation, and oxidative stress in heart failure: emerging molecular targets. Cardiovasc Drugs Ther 2017;31:593–608. [DOI] [PubMed] [Google Scholar]
- 3. Shah SJ, Marcus GM, Gerber IL, Mckeown BH, Vessey JC, Jordan MV, Huddleston M, Foster E, Chatterjee K, Michaels AD. High‐sensitivity C‐reactive protein and parameters of left ventricular dysfunction. J Card Fail 2006;12:61–65. [DOI] [PubMed] [Google Scholar]
- 4. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 2015;116:1254–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McMurray JJ, Kjekshus J, Gullestad L, Dunselman P, Hjalmarson A, Wedel H, Lindberg M, Waagstein F, Grande P, Hradec J, Kamensky G, Korewicki J, Kuusi T, Mach F, Ranjith N, Wikstrand J; CORONA Study Group . Effects of statin therapy according to plasma high‐sensitivity C‐reactive protein concentration in the controlled rosuvastatin multinational trial in heart failure (CORONA): a retrospective analysis. Circulation 2009;120:2188–2196. [DOI] [PubMed] [Google Scholar]
- 6. Anand IS, Latini R, Florea VG, Kuskowski MA, Rector T, Masson S, Signorini S, Mocarelli P, Hester A, Glazer R, Cohn JN; Val‐HeFT Investigators. C‐reactive protein in heart failure prognostic value and the effect of valsartan. Circulation 2005;112:1428–1434. [DOI] [PubMed] [Google Scholar]
- 7. Castro AR, Silva SO, Soares SC. The use of high sensitivity C‐reactive protein in cardiovascular disease detection. J Pharm Pharm Sci 2018;21:496–503. [DOI] [PubMed] [Google Scholar]
- 8. Anker SD, Doehner W, Rauchhaus M, Sharma R, Francis D, Knosalla C, Davos CH, Cicoira M, Shamim W, Kemp M, Segal R, Osterziel KJ, Leyva F, Hetzer R, Ponikowski P, Coats AJ. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation 2003;107:1991–1997. [DOI] [PubMed] [Google Scholar]
- 9. Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med 2008;359:1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mogensen UM, Kober L, Jhund PS, Desai AS, Senni M, Kristensen SL, Dukat A, Chen CH, Ramires F, Lefkowitz MP, Prescott MF, Shi VC, Rouleau JL, Solomon SD, Swedberg K, Packer M, McMurray JJV. Sacubitril/valsartan reduces serum uric acid concentration, an independent predictor of adverse outcomes in PARADIGM‐HF. Eur J Heart Fail 2018;20:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J 2009;73:411–418. [DOI] [PubMed] [Google Scholar]
- 12. Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO‐independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov 2006;5:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Armstrong PW, Roessig L, Patel MJ, Anstrom KJ, Butler J, Voors AA, Lam CSP, Ponikowski P, Temple T, Pieske B, Ezekowitz J, Hernandez AF, Koglin J, O'Connor CM. A multicenter, randomized, double‐blind, placebo‐controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator: the VICTORIA trial. JACC Heart Fail 2018;6:96–104. [DOI] [PubMed] [Google Scholar]
- 14. Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CS, Maggioni AP, Ponikowski P, Shah SJ, Solomon SD, Kraigher‐Krainer E, Samano ET, Muller K, Roessig L, Pieske B; SOCRATES‐REDUCED Investigators and Coordinators. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: the SOCRATES‐REDUCED randomized trial. JAMA 2015;314:2251–2262. [DOI] [PubMed] [Google Scholar]
- 15. Pieske B, Butler J, Filippatos G, Lam C, Maggioni PA, Ponikowski P, Shah S, Solomon S, Kraigher‐Krainer E, Samano ET, Scalise AV, Müller K, Roessig L, Gheorghiade M; SOCRATES Investigators and Coordinators . Rationale and design of the SOluble guanylate cyclase stimulatoR in heArT failurE studies (SOCRATES). Eur J Heart Fail 2014;16:1026–1038. [DOI] [PubMed] [Google Scholar]
- 16. Rickham PP. Human experimentation. Code of ethics of the World Medical Association. Declaration of Helsinki. Br Med J 1964;2:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO III, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, Rifai N, Smith SC, Taubert K, Tracy RP, Vinicor F. Markers of inflammation and cardiovascular disease: application to clinical and public health practice. Circulation 2003;107:499–511. [DOI] [PubMed] [Google Scholar]
- 18. Ahluwalia A, Foster P, Scotland RS, McLean PG, Mathur A, Perretti M, Moncada S, Hobbs AJ. Antiinflammatory activity of soluble guanylate cyclase: cGMP‐dependent down‐regulation of P‐selectin expression and leukocyte recruitment. Proc Natl Acad Sci U S A 2004;101:1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bahls M, Felix SB. Cachexia and right ventricular dysfunction in chronic heart failure: what is the chicken and what the egg? Eur Heart J 2016;37:1692–1694. [DOI] [PubMed] [Google Scholar]
- 20. Valentova M, von Haehling S, Bauditz J, Doehner W, Ebner N, Bekfani T, Elsner S, Sliziuk V, Scherbakov N, Murin J, Anker SD, Sandek A. Intestinal congestion and right ventricular dysfunction: a link with appetite loss, inflammation, and cachexia in chronic heart failure. Eur Heart J 2016;37:1684–1691. [DOI] [PubMed] [Google Scholar]
- 21. Pasini E, Aquilani R, Testa C, Baiardi P, Angioletti S, Boschi F, Verri M, Dioguardi F. Pathogenic gut flora in patients with chronic heart failure. JACC Heart Fail 2016;4:220–227. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail 2017;19:1379–1389. [DOI] [PubMed] [Google Scholar]
- 23. Laurent S, Bruno RM. Gut microbiome composition, a third player in the inflammation–arterial stiffness relationship. Eur Heart J 2018;39:2398–2400. [DOI] [PubMed] [Google Scholar]
- 24. Alvarez AM, Mukherjee D. Liver abnormalities in cardiac diseases and heart failure. Int J Angiol 2011;20:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maleki M, Vakilian F, Amin A. Liver diseases in heart failure. Heart Asia 2011;3:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Amin A, Chitsazan M, Shiukhi Ahmad Abad F, Taghavi S, Naderi N. On admission serum sodium and uric acid levels predict 30 day rehospitalization or death in patients with acute decompensated heart failure. ESC Heart Fail 2017;4:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palazzuoli A, Ruocco G, Pellegrini M, Beltrami M, Giordano N, Nuti R, McCullough PA. Prognostic significance of hyperuricemia in patients with acute heart failure. Am J Cardiol 2016;117:1616–1621. [DOI] [PubMed] [Google Scholar]
- 28. Okazaki H, Shirakabe A, Kobayashi N, Hata N, Shinada T, Matsushita M, Yamamoto Y, Shibuya J, Shiomura R, Nishigoori S, Asai K, Shimizu W. The prognostic impact of uric acid in patients with severely decompensated acute heart failure. J Cardiol 2016;68:384–391. [DOI] [PubMed] [Google Scholar]
- 29. Givertz MM, Anstrom KJ, Redfield MM, Deswal A, Haddad H, Butler J, Tang WH, Dunlap ME, LeWinter MM, Mann DL, Felker GM, O'Connor CM, Goldsmith SR, Ofili EO, Saltzberg MT, Margulies KB, Cappola TP, Konstam MA, Semigran MJ, McNulty SE, Lee KL, Shah MR, Hernandez AF. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT‐HF) study. Circulation 2015;131:1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hare JM, Mangal B, Brown J, Fisher C Jr, Freudenberger R, Colucci WS, Mann DL, Liu P, Givertz MM, Schwarz RP. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT‐CHF study. J Am Coll Cardiol 2008;51:2301–2309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Baseline characteristics of patients in the full analysis set and biomarker analysis sets.
Table S2. Baseline levels and changes from baseline to end of treatment of additional biomarkers (per‐protocol population).
Table S3. Correlation analysis of changes in estimated glomerular filtration rate and body mass index with changes in log‐transformed high‐sensitivity C‐reactive protein and serum uric acid levels from baseline to end of treatment.
Figure S1. SOCRATES‐REDUCED study design.
Figure S2. Patient disposition in the SOCRATES‐REDUCED study.