

Abstract
Roxadustat is a hypoxia‐inducible factor prolyl hydroxylase inhibitor approved in China for anemia of dialysis‐dependent chronic kidney disease (CKD). Japanese hemodialysis patients with anemia of CKD previously naïve to, or converted from, erythropoiesis‐stimulating agents (ESAs) were enrolled in two open‐label, noncomparative studies of titrated oral roxadustat administered three times weekly. ESA‐naïve patients (n = 75) were randomized to roxadustat (initial dose, 50 or 70 mg) for 24 weeks; ESA‐converted patients (n = 164) were assigned to roxadustat (initial dose, 70 or 100 mg based on prior ESA dose) for 52 weeks. Efficacy outcomes included average hemoglobin (Hb, weeks 18‐24 or 46‐52), change of Hb from baseline to weeks 18 to 24 (ΔHb18‐24) or weeks 46 to 52 (ΔHb46‐52), and maintenance rate (proportion of patients who achieved average Hb of 10.0‐12.0 g/dL for weeks 18‐24 or weeks 46‐52). Treatment‐emergent adverse events (TEAEs) were monitored. Mean (SD) Hb was 10.93 (0.79) g/dL (weeks 18‐24) (ESA‐Naïve Study), and 10.93 (0.69; weeks 18‐24) g/dL and 11.11 (0.67; weeks 46‐52) g/dL (ESA‐Converted Study). Mean (SD) ΔHb18‐24 was 2.26 (1.02) g/dL (ESA‐Naïve Study) and −0.03 (0.90) g/dL (ESA‐Converted Study); mean (SD) ΔHb46‐52 was 0.12 (0.83) g/dL (ESA‐Converted Study). The overall maintenance rate was 73.0% (54/74) (ESA‐Naïve Study) (weeks 18‐24), and 79.1% (129/163; weeks 18‐24) and 71.2% (116/163; weeks 46‐52) (ESA‐Converted Study). Nasopharyngitis was the most common TEAE. Two deaths, considered unrelated to roxadustat, occurred in the ESA‐Converted Study. Roxadustat effectively corrected and maintained Hb, regardless of previous ESA treatment, in Japanese anemic CKD patients on hemodialysis.
Keywords: anemia, chronic kidney disease, erythropoiesis‐stimulating agent, hemodialysis, roxadustat
1. INTRODUCTION
Renal anemia is a common complication of chronic kidney disease (CKD) that is associated with decreased erythropoiesis by the malfunctioning kidneys and is commonly treated with erythropoiesis‐stimulating agents (ESAs). 1 Safety concerns, particularly in patients with cancer, diabetes, and cardiovascular comorbidities, have resulted in a reduction in the use and dosage of ESAs worldwide in accordance with current clinical guidelines. 2 , 3 Furthermore, a significant proportion of CKD patients with anemia do not adequately respond to ESA treatment, 4 , 5 and ESA hyporesponsiveness has been reported in patients with inflammation, which is prevalent in 30% to 50% of CKD patients. 6
Hypoxia‐inducible factor prolyl hydroxylase inhibitors (HIF‐PHIs) represent an alternative class of agents for the treatment of anemia of CKD. Hypoxia‐inducible factor is a transcription factor that responds to decreased cellular oxygen levels by activating the transcription of several genes, including erythropoietin. At normal oxygen levels, the hypoxia‐inducible factor prolyl hydroxylases catalyze the degradation of HIFα subunits. In the presence of low oxygen, the enzyme activity is suppressed and HIF‐α dimerizes with HIF‐β and accumulates in the nucleus, leading to increased erythropoiesis, transferrin receptor expression, and iron absorption. The transient inhibition of hypoxia‐inducible factor prolyl hydroxylases by HIF‐PHIs mimics the body's natural response to hypoxia in the presence of normal oxygen levels. 7
Roxadustat (ASP1517, FG‐4592, AZD9941) is an oral, first‐in‐class HIF‐PHI that has demonstrated efficacy and tolerability in phase 2 studies in patients with dialysis‐dependent 8 , 9 , 10 and non‐dialysis‐dependent (NDD) 9 , 11 , 12 , 13 anemia of CKD. Phase 3 studies conducted in China and Japan have reported the efficacy of roxadustat in CKD patients on peritoneal dialysis (PD) 14 , 15 and hemodialysis (HD), 15 and also in NDD 16 patients.
Herein, we report the results of two related phase 3 studies that evaluated the efficacy and safety of treatment with roxadustat for up to 1 year in Japanese patients with anemia of CKD on HD who were either ESA naïve (ESA‐Naïve Study) or who were converted from ESA to roxadustat (ESA‐Converted Study).
2. PATIENTS AND METHODS
2.1. Study design
Both studies were multicenter (ESA‐Naïve Study, 47 sites; ESA‐Converted Study, 25 sites), open‐label, noncomparative, phase 3 studies (ClinicalTrials.gov Identifier: NCT02780141 and NCT02779764) conducted in Japan. Prescreening was conducted on the day of dialysis after the longest dialysis interval to determine if patients satisfy inclusion and exclusion criteria. Screening to confirm patients′ eligibility occurred 1 to 10 weeks after prescreening. In the ESA‐Naïve Study, patients were randomized to oral roxadustat at an initial dose of either 50 or 70 mg three times weekly (TIW) for a maximum of 24 weeks; in the ESA‐Converted Study, patients were assigned to one of two initial conversion doses of roxadustat (70 or 100 mg), determined by the prior ESA dose, and roxadustat was administered TIW for a maximum of 52 weeks. Starting at week 4, roxadustat dose was titrated throughout the study to maintain hemoglobin (Hb) levels between 10.0 and 12.0 g/dL. Both studies were conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice, the International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines, and applicable laws and regulations. The clinical study protocol was reviewed and approved by each Institutional Review Board and all subjects signed a written informed consent form.
2.2. Study population
Patients in both studies were aged ≥20 years with anemia of CKD. Major inclusion criteria for patients in the ESA‐Naïve Study were: (a) must have been receiving HD once weekly or more at prescreening; (b) had not received ESA after the start of dialysis; (c) had a mean of the two most recent Hb levels ≤10.0 g/dL with the absolute difference between the two measurements ≤1.0 g/dL (Hb measurements were conducted at least 1 week apart during the screening period); and (d) transferrin saturation (TSAT) ≥5% or serum ferritin ≥30 ng/mL during the screening period. Major inclusion criteria for patients in the ESA‐Converted Study were: (a) must have been receiving stable maintenance HD TIW for ≥12 weeks before prescreening; (b) must have been receiving intravenous (IV) ESA for >8 weeks before prescreening; (c) had a mean of the two most recent Hb levels between 10.0 and 12.0 g/dL (Hb measurements were conducted at least 1 week apart during the screening period); and (d) TSAT ≥20% or serum ferritin ≥100 ng/mL during the screening period. A full list of eligibility criteria is reported in Table S1.
2.3. Study drug administration
In both studies, roxadustat was supplied as 20‐, 50‐, and 100‐mg tablets and was self‐administered TIW at 2‐ or 3‐day intervals (eg, Monday‐Wednesday‐Friday or Tuesday‐Thursday‐Saturday). These doses and regimens of administration were based on previous studies. Roxadustat had to be taken at least 1 hour before or after any concomitant phosphate binders. Oral iron supplementation was allowed throughout both studies based on individual patient needs.
For patients in the ESA‐Naïve Study, randomization to an initial dose of roxadustat 50 or 70 mg was based on dynamic allocation conducted using a biased‐coin minimization approach with study site, body weight, and the average of the two most recent Hb measurements taken before registration as assignment factors. Concomitant IV iron was allowed only to maintain TSAT ≥5% and/or serum ferritin ≥30 ng/mL when TSAT was <5% or serum ferritin was <30 ng/mL.
For patients in the ESA‐Converted Study, the initial roxadustat dose was based on the average weekly dose of ESA received during the 4 weeks prior to registration in patients receiving recombinant human erythropoietin or darbepoetin alfa, or the average 4‐week dose during the 8 weeks prior to registration in patients receiving epoetin beta pegol in accordance with the dose conversion criteria (Table S2). Concomitant IV iron was allowed only to maintain TSAT ≥20% and/or serum ferritin ≥100 ng/mL when TSAT was <20% or serum ferritin was <100 ng/mL.
In both studies, roxadustat dose was titrated at every even‐week visit starting at week 4 in accordance with the adjustment rules (Tables S3 and S4) to maintain Hb levels between 10.0 and 12.0 g/dL. As a general rule, any dose adjustment had to be maintained for ≥4 weeks before any further adjustment, and the maximum dose of roxadustat was not to exceed 3 mg/kg. If Hb level increased to >12.5 g/dL, dosing had to be stopped and could be resumed at a one‐step reduction after the Hb level had decreased to <11.0 g/dL. In both studies, the use of ESAs, protein anabolic hormone, testosterone enanthate, and mepitiostane was prohibited, and the use of statins was recommended at doses not exceeding the recommended maximum dose.
2.4. Study outcomes
Efficacy outcomes common to both studies included average Hb levels of weeks 18 to 24; change of average Hb levels from baseline to weeks 18 to 24 (ΔHb18‐24); maintenance rate of target Hb level (proportion of patients in the full analysis set [FAS] who achieved an average Hb level of 10.0‐12.0 g/dL) for weeks 18 to 24; and levels of iron, ferritin, transferrin, total iron binding capacity (TIBC), soluble transferrin receptors (sTfR), TSAT, and reticulocyte Hb content (CHr). All efficacy outcomes common to both studies were also assessed at weeks 46 to 52 in the ESA‐Converted Study.
Efficacy outcomes specific for the ESA‐Naïve Study included response rate (Hb ≥10.0 g/dL and Hb increase ≥1.0 g/dL from baseline) from baseline to end of treatment (EoT); proportion of patients who achieved, and time to achieve, the lower limit of the target Hb level (10.0 g/dL); and rate of rise (g/dL/week) in Hb levels from week 0 to week 4, time of discontinuation, or time of dose adjustment. An efficacy outcome specific for the ESA‐Converted Study was the proportion of Hb measurements that met the target level (10.0‐12.0 g/dL) between weeks 18‐24 and weeks 46‐52. Serum hepcidin levels were measured as an exploratory outcome at weeks 0, 4, 12, and 24, or at discontinuation in both studies and at weeks 36 and 52 in the ESA‐Converted Study.
In both studies, patient compliance to study treatment was confirmed by monitoring patient diaries, the number of prescribed, returned, or lost roxadustat tablets, and by patient comments. Safety assessments in both studies included the occurrence of treatment‐emergent adverse events (TEAEs), laboratory tests, vital signs, and 12‐lead electrocardiograms. Schedules of assessments for both studies are reported in Tables S5 and S6.
2.5. Statistical methods
A sample size of 70 patients (n = 35 per dose group) was planned for the ESA‐Naïve Study based on feasibility considerations, and a sample size of 160 patients was planned for the ESA‐Converted Study to ensure that ≥100 patients would be available to provide safety and efficacy data after 1 year of treatment, taking possible dropouts into account. Analyses of efficacy endpoints were performed on the FAS, comprising all patients who received ≥1 dose of roxadustat and who had data for ≥1 efficacy measurement after the start of the study treatment. For the assessment of maintenance rate, any patient who did not have at least one Hb value during weeks 18 to 24 was treated as a nonresponder. A separate analysis of maintenance rate was conducted including only those patients who had at least one Hb value during weeks 18 to 24. Analysis of safety was conducted on the safety analysis set, comprising all patients who received ≥1 dose of roxadustat. Descriptive statistics were used to summarize demographics and baseline characteristics, and efficacy and exploratory endpoints. No formal statistical comparisons were made. To investigate the effect of roxadustat in the presence of inflammation, a subgroup analysis of the mean changes of Hb levels from baseline was conducted using high‐sensitivity C‐reactive protein (hs‐CRP; ie, <28.57 and ≥28.57 nmol/L) as a factor.
3. RESULTS
3.1. Patient disposition
3.1.1. ESA‐Naïve Study
Of 94 patients who provided informed consent, 19 discontinued before randomization and 75 were randomized to receive titrated roxadustat at initial doses of 50 mg (N = 37) or 70 mg (N = 38). A total of 65 (86.7%) patients (roxadustat 50 mg, N = 33; roxadustat 70 mg, N = 32) completed the study and 10 (13.3%) patients (roxadustat 50 mg, N = 4; roxadustat 70 mg, N = 6) discontinued. Reasons for discontinuation were TEAEs (N = 3, 4.0%; roxadustat 50 mg, N = 1; roxadustat 70 mg, N = 2), lack of efficacy (N = 1, 1.3%; roxadustat 50 mg, N = 1), and other (N = 6, 8.0%; roxadustat 50 mg, N = 2; roxadustat 70 mg, N = 4) (Figure 01A). Patient demographics and baseline efficacy variables are reported in Table 1.
FIGURE 1.
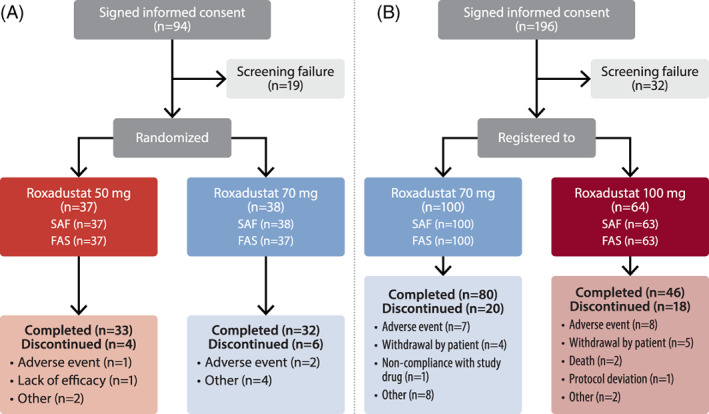
Flow diagram of the ESA‐Naïve (A) and ESA‐Converted (B) Studies. ESA, erythropoiesis‐stimulating agent; FAS, full analysis set; SAF, safety analysis set
TABLE 1.
Patient demographics and baseline efficacy variables (full analysis set)
| Parameter | ESA‐Naïve Study | ESA‐Converted Study | ||
|---|---|---|---|---|
| Roxadustat 50 mga | Roxadustat 70 mga | All ESA‐Naïve | All ESA‐Converted | |
| N = 37 | N = 37 | N = 74 | N = 163 | |
| Demographics | ||||
| Sex, n (%) | ||||
| Male | 28 (75.7) | 27 (73.0) | 55 (74.3) | 98 (60.1) |
| Female | 9 (24.3) | 10 (27.0) | 19 (25.7) | 65 (39.9) |
| Age (years) | ||||
| Mean (SD) | 65.1 (12.1) | 67.2 (12.2) | 66.2 (12.1) | 62.8 (11.8) |
| Median | 67.0 | 72.0 | 69.0 | 63.0 |
| Range | 38‐83 | 42‐86 | 38‐86 | 28‐91 |
| Age (years), n (%) | ||||
| <65 | 15 (40.5) | 14 (37.8) | 29 (39.2) | 86 (52.8) |
| ≥65 | 22 (59.5) | 23 (62.2) | 45 (60.8) | 77 (47.2) |
| Weight (after hemodialysis) at week 0 (kg) | ||||
| Mean (SD) | 60.60 (12.57) | 62.06 (14.70) | 61.33 (13.60) | 58.35 (12.79) |
| Median | 58.20 | 58.90 | 58.55 | 57.70 |
| Range | 41.9‐95.2 | 39.6‐120.5 | 39.6‐120.5 | 35.3‐109.2 |
| Height (cm) | ||||
| Mean (SD) | 163.23 (10.41) | 160.89 (8.33) | 162.06 (9.44) | 161.30 (9.04) |
| Median | 165.00 | 161.00 | 162.75 | 162.30 |
| Range | 145.3‐185.3 | 144.4‐183.0 | 144.4‐185.3 | 141.0‐181.0 |
| BMI (kg/m2) | ||||
| Mean (SD) | 22.69 (2.65) | 24.04 (4.35) | 23.36 (3.64) | 22.26 (3.69) |
| Median | 22.26 | 22.63 | 22.42 | 21.59 |
| Range | 18.3‐28.6 | 17.6‐36.8 | 17.6‐36.8 | 14.1‐37.4 |
| Duration of anemia of CKDb (months) | ||||
| n | 34 | 33 | 67 | 115 |
| Mean (SD) | 27.07 (24.06) | 24.92 (25.15) | 26.01 (24.44) | 98.40 (69.47) |
| Median | 18.35 | 15.40 | 15.70 | 83.60 |
| Range | 2.0‐84.1 | 0.8‐114.4 | 0.8‐114.4 | 7.6‐366.3 |
| Primary disease of CKD, n (%) | ||||
| Chronic glomerular nephritis | 7 (18.9) | 8 (21.6) | 15 (20.3) | 64 (39.3) |
| Diabetic nephropathy | 14 (37.8) | 17 (45.9) | 31 (41.9) | 54 (33.1) |
| Chronic pyelonephritis | 0 | 0 | 0 | 1 (0.6) |
| Polycystic kidney | 5 (13.5) | 2 (5.4) | 7 (9.5) | 6 (3.7) |
| Nephrosclerosis | 8 (21.6) | 6 (16.2) | 14 (18.9) | 18 (11.0) |
| Other | 3 (8.1) | 4 (10.8) | 7 (9.5) | 20 (12.3) |
| Hemodialysis vintage (days) | ||||
| Mean (SD) | 17.1 (16.6) | 21.8 (20.2) | 19.5 (18.5) | NA |
| Median | 11.0 | 14.0 | 12.0 | |
| Range | 3‐77 | 3‐83 | 3‐83 | |
| Hemodialysis vintage (months) | ||||
| Mean (SD) | NA | NA | NA | 89.69 (78.83) |
| Median | 66.90 | |||
| Range | 3.8‐362.4 | |||
| hs‐CRP (nmol/L) | ||||
| Mean (SD) | 16.964 (26.446) | 37.662 (102.387) | 27.313 (74.988) | 13.038 (22.043) |
| Median | 7.730 | 10.000 | 8.435 | 5.040 |
| Range | 1.01‐143.81 | 0.48‐560.96 | 0.48‐560.96 | 0.48‐163.81 |
| hs‐CRP group (nmol/L), n (%) | ||||
| <28.57 | 32 (86.5) | 28 (75.7) | 60 (81.1) | 144 (88.3) |
| ≥28.57 | 5 (13.5) | 9 (24.3) | 14 (18.9) | 19 (11.7) |
| Baseline efficacy variables | ||||
| Hemoglobin (g/dL)c | ||||
| Mean (SD) | 8.63 (0.77) | 8.67 (0.79) | 8.65 (0.78) | 10.96 (0.57) |
| Median | 8.50 | 8.70 | 8.55 | 11.00 |
| Range | 7.0‐10.0 | 7.0‐10.0 | 7.0‐10.0 | 9.7‐12.3 |
| Iron (μmol/L) | ||||
| Mean (SD) | 12.6 (4.7) | 11.3 (3.6) | 11.9 (4.2) | 12.7 (4.3) |
| Median | 11.0 | 11.0 | 11.0 | 12.0 |
| Range | 6‐25 | 4‐19 | 4‐25 | 5‐27 |
| Ferritin (ng/mL) | ||||
| Mean (SD) | 125.26 (80.37) | 129.02 (89.87) | 127.14 (84.69) | 108.30 (93.18) |
| Median | 98.70 | 110.00 | 106.50 | 87.40 |
| Range | 25.9‐284.0 | 22.1‐422.0 | 22.1‐422.0 | 11.1‐572.0 |
| TSAT (%) | ||||
| Mean (SD) | 30.22 (11.66) | 26.79 (8.51) | 28.51 (10.29) | 29.10 (10.42) |
| Median | 26.50 | 26.30 | 26.40 | 26.60 |
| Range | 13.9‐70.7 | 11.1‐48.3 | 11.1‐70.7 | 8.1‐71.2 |
| CHr (pg) | ||||
| Mean (SD) | 34.43 (2.06) | 33.79 (2.03) | 34.11 (2.05) | 35.03 (2.25) |
| Median | 34.40 | 33.80 | 33.95 | 35.10 |
| Range | 30.3‐39.0 | 26.7‐37.5 | 26.7‐39.0 | 22.5‐39.7 |
| Transferrin (g/L) | ||||
| Mean (SD) | 1.785 (0.286) | 1.785 (0.317) | 1.785 (0.300) | 1.869 (0.333) |
| Median | 1.710 | 1.800 | 1.765 | 1.860 |
| Range | 1.32‐2.68 | 1.05‐2.83 | 1.05‐2.83 | 1.16‐2.96 |
Abbreviations: BMI, body mass index; CHr, reticulocyte hemoglobin; CKD, chronic kidney disease; ESA, erythropoiesis‐stimulating agent; hs‐CRP, high‐sensitivity C‐reactive protein; NA, not applicable; TSAT, transferrin saturation.
The initial dose only.
The diagnosis and time of onset of CKD anemia were confirmed by a physician.
Defined as the mean of three Hb values: two latest Hb values prior to registration and one Hb value at week 0.
3.1.2. ESA‐Converted Study
Of 196 patients who provided informed consent, 32 discontinued before registration and 164 were registered to receive titrated roxadustat at initial doses of 70 mg (N = 100) or 100 mg (N = 64) TIW. One patient registered in the roxadustat 100‐mg group did not receive roxadustat. A total of 126 (76.8%) patients completed the study and 38 (23.2%) discontinued. Reasons for discontinuation were TEAEs (N = 15, 9.1%), withdrawal by patient (N = 9, 5.5%), death (N = 2, 1.2%), protocol deviation (N = 1, 0.6%), noncompliance with study drug (N = 1, 0.6%), and other (N = 10, 6.1%) (Figure 1B). Patient demographics and baseline efficacy variables are reported in Table 1. The use of prior and concomitant iron in both studies is reported in Table 2.
TABLE 2.
Prior and concomitant iron use (safety analysis set)
| Parameter | ESA‐Naïve Studya | ESA‐Converted Studyb | ||
|---|---|---|---|---|
| Roxadustat 50 mg | Roxadustat 70 mg | All ESA‐Naïve | All ESA‐Converted | |
| N = 37 | N = 38 | N = 75 | N = 163 | |
| Prior oral iron use | 4 (10.8) | 12 (31.6) | 16 (21.3) | 21 (12.9) |
| Prior IV iron use | 5 (13.5) | 4 (10.5) | 9 (12.0) | 65 (39.9) |
| Concomitant oral iron use | 10 (27.0) | 12 (31.6) | 22 (29.3) | 35 (21.5) |
| Concomitant IV iron use | 6 (16.2) | 5 (13.2) | 11 (14.7) | 71 (43.6) |
Note: Data are presented as n (%).
Abbreviations: ESA, erythropoiesis‐stimulating agent; IV, intravenous.
The study duration was 24 weeks.
The study duration was 52 weeks.
3.2. Treatment compliance and exposure
The mean (SD) treatment compliance was 99.18% (2.15) in the ESA‐Naïve Study and 98.36% (4.43) in the ESA‐Converted Study. The mean (SD) allocated dose of titrated roxadustat per intake at week 22 was 68.4 (41.3) mg (1.17 [SD, 0.70] mg/kg) and 86.4 (39.9) mg (1.45 [SD, 0.72] mg/kg) in the ESA‐Naïve Study at the initial doses of 50 and 70 mg, respectively, and 73.8 (43.0) mg (1.29 [SD, 0.74] mg/kg) and 74.1 (43.6) mg (1.30 [SD, 0.75] mg/kg) in the ESA‐Converted Study at week 22 and at week 50, respectively. Over the course of the study, the mean (SD) number of changes (increases and/or decreases) per patient in roxadustat dosing was 3.2 (1.4) times and 3.1 (1.4) times for the roxadustat 50‐ and 70‐mg groups, respectively, in the ESA‐Naïve Study, and 5.5 (2.8) times in the ESA‐Converted Study.
3.3. Efficacy outcomes
3.3.1. Changes in Hb levels
In the ESA‐Naïve Study, the mean levels of Hb increased from week 0 to week 16 and then remained stable through the EoT with both doses of titrated roxadustat (Figure 2A), whereas in the ESA‐Converted Study, no remarkable changes in Hb levels were observed from week 0 to the EoT (Figure 2B).
FIGURE 2.
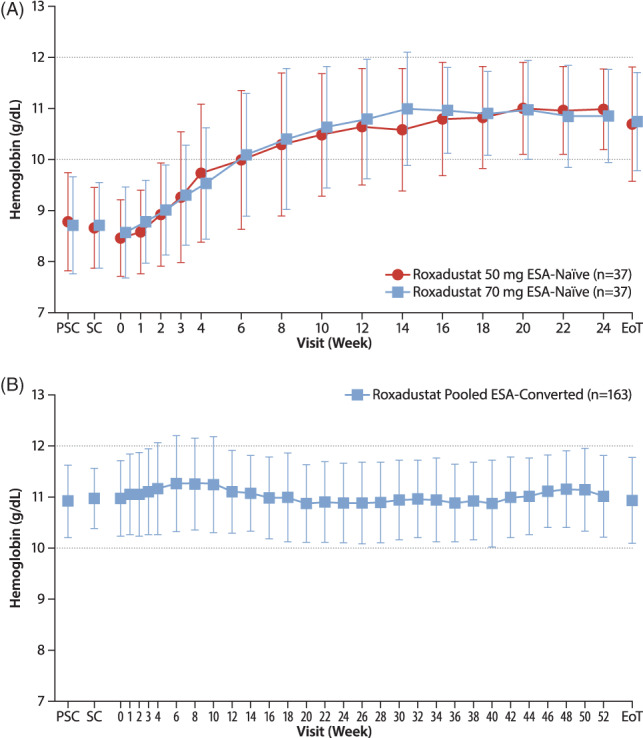
Mean (SD) hemoglobin levels in the ESA‐Naïve Study (A) and ESA‐Converted Study (B) (full analysis set). EoT, end of treatment; ESA, erythropoiesis‐stimulating agent; PSC, prescreening; SC, screening; w, week [Correction added on 29 June 2020, after first online publication: figure 2a has been amended to correct the data points.]
The mean (SD) of average Hb levels was 10.93 (0.79) g/dL (10.96 [0.78] g/dL, roxadustat 50 mg; 10.90 [0.81] g/dL, roxadustat 70 mg) in the ESA‐Naïve Study during weeks 18 to 24, and 10.93 (0.69) g/dL and 11.11 (0.67) g/dL in the ESA‐Converted Study during weeks 18 to 24 and weeks 46 to 52, respectively. The mean (SD) of ΔHb18‐24 was 2.26 (1.02) g/dL in the ESA‐Naïve Study (2.29 [1.05] g/dL, roxadustat 50 mg; 2.24 [1.01] g/dL, roxadustat 70 mg) and −0.03 (0.90) g/dL in the ESA‐Converted Study, whereas the mean (SD) of ΔHb46‐52 was 0.12 (0.83) g/dL in the ESA‐Converted Study (Table 3). In the ESA‐Naïve Study, the mean (SD) change of Hb levels from week 0 to EoT was similar among patients with hs‐CRP ≥28.57 nmol/L (2.22 [1.21] g/dL) and those with CRP <28.57 nmol/L (2.19 [1.25] g/dL), whereas in the ESA‐Converted Study, the mean (SD) of ΔHb18‐24 and of ΔHb46‐52 was higher among patients with hs‐CRP ≥28.57 nmol/L (0.44 [0.75] g/dL and 0.46 [0.82] g/dL, respectively) than among those with hs‐CRP <28.57 nmol/L (−0.09 [0.90] g/dL and 0.08 [0.82] g/dL, respectively). The mean (SD) Hb level at week 0 was 10.81 (0.80) g/dL among patients with hs‐CRP ≥28.57 nmol/L and 11.00 (0.73) g/dL among those with CRP <28.57 nmol/L.
TABLE 3.
Changes of average Hb (g/dL) levels at weeks 18 to 24 and weeks 46 to 52 from baseline (full analysis set)
| Parameter | ESA‐Naïve Studya | ESA‐Converted Studyb | ||
|---|---|---|---|---|
| Roxadustat 50 mg | Roxadustat 70 mg | All ESA‐Naïve | All ESA‐Converted | |
| N = 37 | N = 37 | N = 74 | N = 163 | |
| Weeks 18‐24 | ||||
| n | 33 | 32 | 65 | 148 |
| Mean (SD) | 2.29 (1.05) | 2.24 (1.01) | 2.26 (1.02) | −0.03 (0.90) |
| Median | 2.40 | 2.25 | 2.30 | 0.00 |
| Range | 0.2‐4.2 | 0.4‐4.5 | 0.2‐4.5 | −3.2 to 1.9 |
| Weeks 46‐52 | ||||
| n | — | — | — | 128 |
| Mean (SD) | — | — | — | 0.12 (0.83) |
| Median | — | — | — | 0.10 |
| Range | — | — | — | −2.6 to 2.0 |
Abbreviations: ESA, erythropoiesis‐stimulating agent; Hb, hemoglobin.
The study duration was 24 weeks.
The study duration was 52 weeks.
In the ESA‐Naïve Study, the mean (SD) rate of rise in Hb levels was 0.268 (0.352) g/dL/week (0.297 [0.337] g/dL/week, roxadustat 50 mg; 0.238 [0.368] g/dL/week, roxadustat 70 mg).
3.3.2. Maintenance of target Hb levels and response rate
Maintenance rates of target Hb levels (proportions of patients who achieved an average Hb level of 10.0‐12.0 g/dL) in the FAS were ≥70% in all treatment groups and were comparable among the two doses of titrated roxadustat in the ESA‐Naïve Study and between ESA‐Naïve and ESA‐Converted Studies. Among all patients in the ESA‐Naïve Study, the overall maintenance rate (95% CI [confidence interval]) of the target Hb level in the FAS during weeks 18 to 24 was 73.0% (54/74; 61.4, 82.6) (75.7% [58.8, 88.2], roxadustat 50 mg; 70.3% [53.0, 84.1], roxadustat 70 mg; Table 4). Among those patients who had at least one Hb value during weeks 18 to 24, the maintenance rate (95% CI) was 83.1% (54/65; 71.7, 91.2) (84.8% [68.1, 94.9], roxadustat 50 mg; 81.3% [63.6, 92.8], roxadustat 70 mg). In the ESA‐Converted Study, the overall maintenance rate (95% CI) was 79.1% (129/163; 72.1, 85.1) at weeks 18 to 24 and 71.2% (116/163; 63.6, 78.0) at weeks 46 to 52 (Table 4). Among patients with at least one Hb value at the respective time points, the maintenance rate (95% CI) was 87.2% (129/148; 80.7, 92.1) during weeks 18 to 24 and 90.6% (116/128; 84.2, 95.1) during weeks 46 to 52.
TABLE 4.
Maintenance rate of target Hb level (full analysis set)
| ESA‐Naïve StudyaWeeks 18‐24 | ESA‐Converted Studyb All ESA‐Converted | ||||
|---|---|---|---|---|---|
| Roxadustat 50 mg | Roxadustat 70 mg | All ESA‐Naïve | N = 163 | ||
| Parameter | N = 37 | N = 37 | N = 74 | Weeks 18‐24 | Weeks 46‐52 |
| All patients | |||||
| Maintenance rate, n (%) | 28/37 (75.7) | 26/37 (70.3) | 54/74 (73.0) | 129/163 (79.1) | 116/163 (71.2) |
| 95% CI, % | 58.8, 88.2 | 53.0, 84.1 | 61.4, 82.6 | 72.1, 85.1 | 63.6, 78.0 |
| Patients with at least one Hb value | |||||
| Maintenance rate, n (%) | 28/33 (84.8) | 26/32 (81.3) | 54/65 (83.1) | 129/148 (87.2) | 116/128 (90.6) |
| 95% CI, % | 68.1, 94.9 | 63.6, 92.8 | 71.7, 91.2 | 80.7, 92.1 | 84.2, 95.1 |
Note: Target Hb level, mean Hb 10.0 to 12.0 g/dL.
Abbreviations: CI, confidence interval; ESA, erythropoiesis‐stimulating agent; Hb, hemoglobin.
The study duration was 24 weeks.
The study duration was 52 weeks.
In the ESA‐Naïve Study, the mean (95% CI) response rate (Hb ≥10.0 g/dL and Hb increase ≥1.0 g/dL from baseline) from baseline to EoT was 87.8% (65/74; 78.2, 94.3) (86.5% [71.2, 95.5], roxadustat 50 mg; 89.2% [74.6, 97.0], roxadustat 70 mg), and the proportion of patients who achieved an Hb level of 10.0 g/dL increased from 6.8% at week 1 to 87.7% at week 16 and then remained stable through EoT. In the ESA‐Converted Study, the overall mean proportion of Hb measurements that met the target Hb level (10.0‐12.0 g/dL) for all patients was 82.09% (95% CI: 77.57, 86.61) between weeks 18 and 24, and 85.35% (95% CI: 80.89, 89.82) between weeks 46 and 52.
3.3.3. Iron parameters
The inclusion criteria of TSAT ≥5% and ferritin ≥30 ng/mL for the ESA‐Naïve Study and TSAT ≥20% and ferritin ≥100 ng/mL for the ESA‐Converted Study were selected in order to allow the investigation of the efficacy of titrated roxadustat in a patient population with low iron stores. In the ESA‐Naïve Study, the course of iron parameters was reflective of robust erythropoiesis with increase in mean levels of transferrin, TIBC, and sTfR, whereas mean ferritin and TSAT values showed a decrease in the first 8 weeks of treatment followed by stabilization. Serum iron and reticulocyte Hb remained stable with 14.7% of patients receiving concomitant IV iron. No apparent difference in any iron parameters was observed between treatment groups (Table 5 and Figures [Link], [Link]).
TABLE 5.
Iron parameters (full analysis set)
| Parameter | Baseline | EoT | Change From Baseline to EoT |
|---|---|---|---|
| ESA‐Naïve Studya; roxadustat 50 mg, N = 37 | |||
| Iron (μmol/L) | 12.6 (4.7) | 12.8 (6.2) | 0.2 (7.1) |
| Ferritin (ng/mL) | 125.26 (80.37) | 50.58 (51.41) | −74.68 (69.55) |
| Transferrin (g/L) | 1.785 (0.286) | 2.485 (0.626) | 0.700 (0.592) |
| TIBC (μmol/L) | 42.3 (5.9) | 56.9 (12.7) | 14.6 (11.8) |
| TSAT (%) | 30.22 (11.66) | 24.41 (13.52) | −5.81 (16.14) |
| Reticulocyte Hb (pg) | 34.43 (2.06) | 34.08 (4.02) | −0.35 (4.01) |
| Soluble transferrin receptor (nmol/L) | 16.82 (7.54) | 26.71 (14.92) | 9.89 (13.43) |
| ESA‐Naïve Studya; roxadustat 70 mg, N = 37 | |||
| Iron (μmol/L) | 11.3 (3.6) | 11.9 (5.8) | 0.6 (6.2) |
| Ferritin (ng/mL) | 129.02 (89.87) | 55.01 (49.10) | −74.01 (67.94) |
| Transferrin (g/L) | 1.785 (0.317) | 2.380 (0.453) | 0.595 (0.499) |
| TIBC (μmol/L) | 42.4 (6.8) | 54.7 (9.4) | 12.3 (9.9) |
| TSAT (%) | 26.79 (8.51) | 22.56 (11.11) | −4.23 (13.59) |
| Reticulocyte Hb (pg) | 33.79 (2.03) | 32.44 (4.61) | −1.35 (4.88) |
| Soluble transferrin receptor (nmol/L) | 20.27 (10.43) | 31.18 (21.80) | 10.91 (20.70) |
| All ESA‐Naïve Studya; N = 74 | |||
| Iron (μmol/L) | 11.9 (4.2) | 12.4 (6.0) | 0.4 (6.6) |
| Ferritin (ng/mL) | 127.14 (84.69) | 52.79 (49.97) | −74.34 (68.28) |
| Transferrin (g/L) | 1.785 (0.300) | 2.432 (0.545) | 0.648 (0.546) |
| TIBC (μmol/L) | 42.3 (6.3) | 55.8 (11.2) | 13.5 (10.9) |
| TSAT (%) | 28.51 (10.29) | 23.49 (12.32) | −5.02 (14.84) |
| Reticulocyte Hb (pg) | 34.11 (2.05) | 33.26 (4.37) | −0.85 (4.46) |
| Soluble transferrin receptor (nmol/L) | 18.55 (9.20) | 28.95 (18.69) | 10.40 (17.34) |
| All ESA‐Converted Studyb; N = 163 | |||
| Iron (μmol/L) | 12.7 (4.3) | 13.1 (6.8) | 0.4 (7.4) |
| Ferritin (ng/mL) | 108.30 (93.18) | 84.31 (96.25) | −23.99 (76.33) |
| Transferrin (g/L) | 1.869 (0.333) | 2.363 (0.557) | 0.495 (0.467) |
| TIBC (μmol/L) | 44.5 (7.1) | 54.5 (11.4) | 10.0 (9.6) |
| TSAT (%) | 29.10 (10.42) | 25.17 (14.03) | −3.93 (15.42) |
| Reticulocyte Hb (pg) | 35.03 (2.25) | 34.14 (3.74) | −0.89 (3.84) |
| Soluble transferrin receptor (nmol/L) | 23.36 (10.99) | 27.08 (21.14) | 3.72 (19.78) |
Note: Data are presented as mean (SD).
Abbreviations: EoT, end of treatment; ESA, erythropoiesis‐stimulating agent; Hb, hemoglobin; TIBC, total iron binding capacity; TSAT, transferrin saturation.
The study duration was 24 weeks.
The study duration was 52 weeks.
In the ESA‐Converted Study, the mean levels of ferritin decreased slightly during the first 2 weeks of treatment with subsequent stabilization until EoT. The mean values of transferrin and TIBC increased slightly throughout the first 4 weeks and then remained stable through EoT. No remarkable changes were observed in the mean values of iron, TSAT, CHr, and sTfR at each visit and EoT (Table 5 and Figures [Link], [Link]).
3.4. Exploratory outcomes
In the ESA‐Naïve Study, the mean (SD) hepcidin values decreased from week 0 (41.673 [25.296] ng/mL, roxadustat 50 mg; 38.015 [22.001] ng/mL, roxadustat 70 mg) through week 4 and then remained stable through EoT (15.494 [17.315] ng/mL, roxadustat 50 mg; 17.797 [17.981] ng/mL, roxadustat 70 mg). A small decrease in the overall mean (SD) hepcidin levels was also observed in the ESA‐Converted Study (28.777 [25.142] ng/mL, week 0; 22.618 [26.449] ng/mL, EoT; Table 6).
TABLE 6.
Hepcidin (ng/mL) levels (full analysis set)
| ESA‐Naïve Studya | ESA‐Converted Studyb | |||
|---|---|---|---|---|
| Roxadustat 50 mg (N = 37) | Roxadustat 70 mg (N = 37) | All ESA‐Naïve (N = 74) | All ESA‐Converted (N = 163) | |
| Baseline | 41.673 (25.296) | 38.015 (22.001) | 39.844 (23.615) | 28.777 (25.142) |
| Week 4 | 13.241 (17.391) | 14.919 (19.206) | 14.080 (18.208) | 19.547 (22.194) |
| EoT | 15.494 (17.315) | 17.797 (17.981) | 16.645 (17.568) | 22.618 (26.449) |
| Change from baseline to EoT | −26.179 (23.383) | −20.218 (22.183) | −23.199 (22.832) | −6.159 (29.026) |
Note: Data are reported as mean (SD).
Abbreviations: EoT, end of treatment; ESA, erythropoiesis‐stimulating agent.
The study duration was 24 weeks.
The study duration was 52 weeks.
3.5. Safety findings
3.5.1. ESA‐Naïve Study
The incidence of TEAEs during the 24‐week ESA‐Naïve Study was 90.7% (86.5% [N = 32/37], roxadustat 50 mg; 94.7% [N = 36/38], roxadustat 70 mg). Serious TEAEs were reported in 29.3% (N = 22/75) of patients overall. Treatment‐emergent adverse events leading to withdrawal of treatment were 4.0% (N = 3/75) overall. No deaths occurred throughout the study. Common (≥5%) TEAEs included nasopharyngitis (20.0%); contact dermatitis (13.3%); shunt occlusion (9.3%); constipation, shunt stenosis, and hyperphosphatemia (6.7% each); and diarrhea, vomiting, eczema, contusion, back pain, and insomnia (5.3% each) (Table 7).
TABLE 7.
Overview of treatment‐emergent adverse events in the ESA‐Naïve Study (safety analysis set)
| Roxadustat 50 mg (N = 37) | Roxadustat 70 mg (N = 38) | All ESA‐Naïve (N = 75) | |
|---|---|---|---|
| Any TEAEs | 32 (86.5) | 36 (94.7) | 68 (90.7) |
| Serious TEAEs | 9 (24.3) | 13 (34.2) | 22 (29.3) |
| TEAEs leading to withdrawal of treatment | 1 (2.7) | 2 (5.3) | 3 (4.0) |
| TEAEs occurring in ≥5% of patients by MedDRA version 19.0 | |||
| System organ class and preferred term, n (%) | |||
| Gastrointestinal disorders | |||
| Constipation | 1 (2.7) | 4 (10.5) | 5 (6.7) |
| Diarrhea | 1 (2.7) | 3 (7.9) | 4 (5.3) |
| Vomiting | 3 (8.1) | 1 (2.6) | 4 (5.3) |
| Infections and infestations | |||
| Nasopharyngitis | 3 (8.1) | 12 (31.6) | 15 (20.0) |
| Musculoskeletal and connective tissue disorders | |||
| Back pain | 3 (8.1) | 1 (2.6) | 4 (5.3) |
| Skin and subcutaneous tissue disorders | |||
| Contact dermatitis | 4 (10.8) | 6 (15.8) | 10 (13.3) |
| Eczema | 2 (5.4) | 2 (5.3) | 4 (5.3) |
| Injury, poisoning, and procedural complications | |||
| Shunt occlusion | 3 (8.1) | 4 (10.5) | 7 (9.3) |
| Shunt stenosis | 2 (5.4) | 3 (7.9) | 5 (6.7) |
| Contusion | 2 (5.4) | 2 (5.3) | 4 (5.3) |
| Metabolism and nutrition disorders | |||
| Hyperphosphatemia | 2 (5.4) | 3 (7.9) | 5 (6.7) |
| Psychiatric disorders | |||
| Insomnia | 2 (5.4) | 2 (5.3) | 4 (5.3) |
Note: Data are presented as n (%).
Abbreviations: ESA, erythropoiesis‐stimulating agent; TEAEs, treatment‐emergent adverse events.
3.5.2. ESA‐Converted Study
The overall incidence of TEAEs during the 52‐week ESA‐Converted Study was 95.7% (N = 156/163). Serious TEAEs were reported in 28.2% (N = 46/163) of patients overall. The overall incidence of TEAEs leading to withdrawal of treatment was 10.4% (N = 17/163). Two deaths (1.2%) occurred during the study, both in the 100‐mg titrated roxadustat treatment group. One of the deaths occurred in a 79‐year‐old man with a history of chronic cardiac failure, myocardial infarction, and chronic obstructive pulmonary disease. Treatment with titrated roxadustat was discontinued in this patient on day 218 of the study following the diagnosis of pancreatic carcinoma, and the patient died on day 255. Based on the duration of treatment and the timing of the confirmation of multiple metastases to the liver, it was deemed unlikely that the pancreatic cancer had developed after initiation of the study drug; therefore this event was not considered related to the study drug. The second death event occurred in a 67‐year‐old man with a history of type 2 diabetes mellitus, hypertension, arteriosclerosis, and cerebral infarction. On day 250 of the study, the patient died from hemorrhagic shock due to bleeding from multiple gastric and duodenal ulcers, and from sepsis. The event was not considered related to the study drug. Common (≥5%) TEAEs included nasopharyngitis (52.8%); diarrhea (11.0%); vomiting (10.4%); contusion (9.8%); shunt stenosis and back pain (7.4% each); constipation and shunt occlusion (6.1% each); and dental caries and headache (5.5% each) (Table 8).
TABLE 8.
Overview of treatment‐emergent adverse events and death in the ESA‐Converted Study (safety analysis set)
| ESA‐Converted Study (N = 163) | |
|---|---|
| Any TEAEs | 156 (95.7) |
| Serious TEAEs | 46 (28.2) |
| TEAEs leading to withdrawal of treatment | 17 (10.4) |
| Death | 2 (1.2) |
| TEAEs occurring in ≥5% of patients by MedDRA version 19.0 | |
| System organ class and preferred term, n (%) | |
| Gastrointestinal disorders | |
| Diarrhea | 18 (11.0) |
| Vomiting | 17 (10.4) |
| Constipation | 10 (6.1) |
| Dental caries | 9 (5.5) |
| Infections and infestations | |
| Nasopharyngitis | 86 (52.8) |
| Musculoskeletal and connective tissue disorders | |
| Back pain | 12 (7.4) |
| Injury, poisoning, and procedural complications | |
| Contusion | 16 (9.8) |
| Shunt stenosis | 12 (7.4) |
| Shunt occlusion | 10 (6.1) |
| Nervous system disorders | |
| Headache | 9 (5.5%) |
Note: Data are presented as n (%).
Abbreviations: ESA, erythropoiesis‐stimulating agent; TEAEs, treatment‐emergent adverse events.
4. DISCUSSION
The efficacy and safety of titrated roxadustat for correcting and maintaining target levels of Hb in patients with anemia of CKD were prospectively investigated in two separate studies of Japanese HD patient populations with different histories of anemia treatment. In patients who were ESA‐naïve, a 24‐week treatment with titrated roxadustat increased the mean Hb levels above 10.0 g/dL and maintained them within the target range of 10.0 to 12.0 g/dL. In patients who were previously treated with ESA, a 52‐week treatment with titrated roxadustat maintained Hb levels within the target range for the full course of the study. Among patients who had at least one Hb value during each evaluation period, the proportions (95% CI) of those who achieved an average Hb level of 10.0 to 12.0 g/dL were 84.8% (68.1, 94.9; roxadustat 50 mg) and 81.3% (63.6, 92.8; roxadustat 70 mg) during weeks 18 to 24 in the ESA‐Naïve Study, and 87.2% (80.7, 92.1) and 90.6% (84.2, 95.1) during weeks 18 to 24 and weeks 46 to 52, respectively, in the ESA‐Converted Study. Furthermore, a subgroup analysis showed that the mean changes of Hb levels from week 0 to EoT were comparable (ESA‐Naïve Study) and the mean of ΔHb18‐24 and of ΔHb46‐52 were higher (ESA‐Converted Study) among patients with hs‐CRP ≥28.57 nmol/L vs those with hs‐CRP <28.57 nmol/L, suggesting that roxadustat is effective regardless of inflammation status.
Most iron parameters were clinically stable in the presence of robust erythropoiesis without increased administration of IV iron, which suggests improved iron metabolism due to increased iron absorption by roxadustat. An increase in the mean levels of sTfR, which is in line with roxadustat's mechanism of action, was observed —particularly in the ESA‐Naïve Study. Inhibition of HIF‐PH activity by roxadustat results in increased gene transcription and upregulation of the expression of genes involved in iron transport, such as the transferrin receptor, which leads to improved iron transport to tissues and to developing erythrocytes. Interestingly, a reduction from baseline in the mean (SD) levels of ferritin was observed with both doses of titrated roxadustat (−74.68 [69.55] ng/mL, roxadustat 50 mg; −74.01 [67.94] ng/mL, roxadustat 70 mg) at EoT in the ESA‐Naïve Study. This finding is in contrast with the results from the ESA‐Converted Study, and from previous Japanese studies of roxadustat in HD (manuscript submitted) or PD 14 patients with anemia that showed that the levels of ferritin remained clinically stable throughout the treatment period. This difference is thought to be due to the higher amount of iron required when correcting low Hb in ESA‐naïve patients to support the higher degree of erythropoiesis necessary to increase the levels of Hb to the target range and then maintain them within that range. Less supplemental iron was required in ESA‐converted patients whose Hb levels were already within the target range at the beginning of the study. Furthermore, a decrease in the levels of hepcidin, which was observed in both studies and was most pronounced in patients who were ESA‐naïve, may be associated with an improvement in iron mobilization. As reported in previous studies, roxadustat was generally safe and well tolerated in patients with CKD and anemia on HD. Overall, these studies demonstrate that titrated roxadustat successfully treated anemia in both ESA‐naïve and ESA‐converted Japanese patients on HD.
The findings of these studies are consistent with those from previous studies. The ESA‐Naïve Study is the longest study submitted for publication to date that examines the efficacy of roxadustat in HD patients who were not previously treated with ESA. In a previous study conducted in the United States, Russia, and Hong Kong, a 12‐week treatment with roxadustat in ESA‐naïve HD or PD patients resulted in similar increases in Hb levels. 8 In another study conducted in China, an 8‐week treatment with doses of roxadustat in the same dose range as those used in this study in patients who were not previously treated with ESA and not dialysis‐dependent resulted in a trend in Hb levels comparable with the one observed in the ESA‐Naïve Study, with the target level reached after 6 weeks of treatment. 9 Moreover, in HD patients who were previously treated with ESA, a 6‐week treatment with roxadustat doses similar to those used in this study resulted in a similar trend in Hb levels. 9 Similar findings were also reported in two recent phase 3 studies. A Japanese study showed that in CKD patients on PD who were ESA‐naïve, roxadustat increased and maintained Hb in the target range; however, the time to reach the target was shorter (2‐4 weeks) than that observed in this study (6 weeks), possibly due to higher Hb levels at baseline. 14 Comparable results were also reported in a Chinese study in CKD patients on HD or PD. 15 Results from the ESA‐Converted Study reported herein provide data on the longest duration of roxadustat efficacy and safety assessment in Japanese patients to date and showed that titrated roxadustat is effective in maintaining Hb levels within the target range for up to 52 weeks.
The patient populations in both studies were exclusively Japanese with a mean age of ≥60 years, which limits the generalizability of these results to other ethnicities and age groups. Another limitation of both studies is their open‐label design, which may increase the risk of bias in the results since awareness of treatment assignments by investigators and patients may affect the study outcomes. Furthermore, this study was noncomparative, a design that may reduce the interpretability of the results due to the lack of a control group.
5. CONCLUSIONS
Overall, these two studies showed that titrated roxadustat was effective in increasing and maintaining Hb levels within the target range of 10.0 to 12.0 g/dL for up to 24 weeks in Japanese patients with anemia of CKD who were ESA‐naïve or ESA‐converted. Titrated roxadustat also maintained Hb levels for up to 52 weeks in those who were switched from ESA to roxadustat.
CONFLICT OF INTEREST
Tadao Akizawa reports personal fees from Astellas during the conduct of the study, and personal fees from: Bayer Yakuhin, Ltd.; Japan Tobacco, Inc.; GlaxoSmithKline; Kissei Pharmaceutical Co., Ltd.; Chugai Pharmaceutical Co. Ltd; Ono Pharmaceutical Co. Ltd.; Fuso Pharmaceutical Industries, Ltd.; Nipro Corporation; Kyowa Kirin; Otsuka; Sanwa; and Torii Pharmaceutical Co., Ltd. outside of the submitted work. Michael Reusch is an employee of Astellas Pharma Europe B.V. Mai Ueno and Takanori Shiga own stock in Astellas Pharma, Inc. and are employees of Astellas Pharma, Inc.
Supporting information
FIGURE S1 Mean (SD) concentrations of iron in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S2 Mean (SD) Concentrations of ferritin in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S3 Mean (SD) of TSAT in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S4 Mean (SD) of reticulocyte Hb content in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S5 Mean (SD) Concentrations of transferrin in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S6 Mean (SD) concentrations of total iron binding capacity in ESA‐Naïve, A, and ESA‐Converted B, patients (full analysis set)
FIGURE S7 Mean (SD) concentrations of soluble transferrin receptor in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
TABLE S1 Inclusion and exclusion criteria
TABLE S2. Dose conversion between average doses of ESA before study registration and roxadustat
TABLE S3. Dose‐adjusting criteria
TABLE S4. Dose‐adjustment steps for roxadustat
TABLE S5. Schedule of assessments from week 0 to week 24 or discontinuation for both studies
TABLE S6. Schedule of assessments from week 26 to week 52 or discontinuation for the ESA‐Converted Study
ACKNOWLEDGMENTS
This study was funded by Astellas Pharma, Inc. Medical writing and editorial support were provided by Mike Zbreski, PharmD, Rosalba Satta, PhD, and Elizabeth Hermans, PhD, of OPEN Health Medical Communications, and were funded by Astellas Pharma, Inc. Roxadustat is being developed by FibroGen, AstraZeneca, and Astellas. The authors would like to thank all principal investigators and patients for participating in the study: Shigeru Miyazaki (Shinrakuen Hospital), Zenzo Fujii (St. Hill Hospital), Tomohiko Naruse (Kasugai Municipal Hospital), Naoaki Hayama (Keijukai Healthcare Corporation, Saitama Tsukinomori Clinic), Akihisa Nakaoka (Sanin Rosai Hospital), Yoshitaka Maeda (JA Toride Medical Center), Yutaka Kanno (Kanno Dialysis & Vascular Access Clinic), Atsushi Kyan (Shirakawa Kosei General Hospital), Kazue Ueki (Sanshikai Toho Hospital), Hidetoshi Yoshinaga (Social Medical Corporation the Chiyukai Foundation Fukuoka Wajiro Hospital), Shinji Ako (Matsumoto City Hospital), Daisuke Yoshida (Oji General Hospital), Tadashi Iitsuka (Ibaraki Seinan Medical Center Hospital), Masaki Hashimoto (Chiba Tokushukai Hospital), Takashi Sekikawa (Matsuyama Shimin Hospital), Kazuya Takasawa (Public Central Hospital of Matto Ishikawa), Nobuo Hashimoto (Medical Corporation H.N. Medic), Teruo Kowatari (Nanbu Tokushukai Hospital), Tetsuya Shigehara (Jyoumou Ohashi Clinic), Shunichi Umeda (Iida Hospital), Sumiko Homma (Japanese Red Cross Koga Hospital), Yoichi Iwafuchi (Sanjo General Hospital), Takayuki Toda (Tsuchiura Kyodo General Hospital), Taku Miyoshi (Kumamoto General Hospital), Toshiro Shibata (Takayama Red Cross Hospital), Kazushige Ejiri (Koujyukai Clinic), Kenji Yaginuma (Japan Community Health Care Organization Nihonmatsu Hospital), Shigeichi Shoji (Shirasagi Hospital), Shoji Fujisawa (Tamana Urological Clinic), Hironori Kobayashi (Japanese Red Cross Asahikawa Hospital), Akihiko Nakamura (Osafune Clinic), Hideya Niimura (Ueyama Hospital), Masayoshi Yamaha (Daiyukai Health System Daiyukai Dai‐ichi Hospital), Sekiya Shibazaki (National Hospital Organization Hokkaido Medical Center), Yasufumi Takahashi (National Hospital Organization Shinshu Ueda Medical Center), Katsuhiko Tamura (Shinonoi General Hospital), Shintaro Yano (Maebashi Hirosegawa Clinic), Kuniko Takayama (Takayama Hospital), Akira Ohishi (Association of Healthcare Corporation Meiko‐kai Ohishi Naika Clinic), Hisanori Azekura (Yuujinkai Sanaru Sun Clinic), Motoko Tanaka (Akebono Clinic), Naoya Kodama (Kodama Hospital), Tomio Suzuki (Medical Corporation Suzuki Hinyoukika), Hideto Emoto (Shuyukai Corporate Medical Association, Tokai Hospital), Morihiro Kondo (Rakuwakai Otowa Kinen Hospital; Present affiliation: Rakuwakai Tojiminami Hospital), Yoshinari Tsuruta (Meiyo Clinic Hemodialysis Center), Masanori Matsukawa (Takikawa Municipal Hospital).
Akizawa T, Ueno M, Shiga T, Reusch M. Oral roxadustat three times weekly in ESA‐naïve and ESA‐converted patients with anemia of chronic kidney disease on hemodialysis: Results from two phase 3 studies. Ther Apher Dial. 2020;24:628–641. 10.1111/1744-9987.13468
Funding information Astellas Pharma, Inc.
DATA AVAILABILITY STATEMENT
Access to anonymized individual participant level data collected during the study, in addition to supporting clinical documentation, is planned for studies conducted with approved product indications and formulations, as well as compounds terminated during development. Studies conducted with product indications or formulations that remain active in development are assessed after study completion to determine if individual participant data can be shared. Conditions and exceptions are described under the sponsor‐specific details for Astellas on www.clinicalstudydatarequest.com. Study‐related supporting documentation is redacted and provided if available, such as the protocol and amendments, statistical analysis plan, and clinical study report. Access to participant level data is offered to researchers after publication of the primary manuscript (if applicable) and is available as long as Astellas has legal authority to provide the data. Researchers must submit a proposal to conduct a scientifically relevant analysis of the study data. The research proposal is reviewed by an Independent Research Panel. If the proposal is approved, access to the study data is provided in a secure data sharing environment after receipt of a signed Data Sharing Agreement.
REFERENCES
- 1. Locatelli F, Fishbane S, Block GA, Macdougall IC. Targeting hypoxia‐inducible factors for the treatment of anemia in chronic kidney disease patients. Am J Nephrol. 2017;45(3):187–199. [DOI] [PubMed] [Google Scholar]
- 2. Del Vecchio L, Locatelli F. An overview on safety issues related to erythropoiesis‐stimulating agents for the treatment of anaemia in patients with chronic kidney disease. Expert Opin Drug Saf. 2016;15(8):1021–1030. [DOI] [PubMed] [Google Scholar]
- 3. KDIGO . KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl. 2012;2(4):279–335. [Google Scholar]
- 4. Sui Z, Wang M, Zuo L. Statin therapy and erythropoiesis‐stimulating agent hyporesponsiveness in patients with nondialysis chronic kidney disease: A retrospective study in Beijing, China. Medicine (Baltimore). 2019;98(2):e13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akizawa T, Okumura H, Alexandre AF, Fukushima A, Kiyabu G, Dorey J. Burden of anemia in chronic kidney disease patients in Japan: A literature review. Ther Apheresis Dial. 2018;22(5):444–456. [DOI] [PubMed] [Google Scholar]
- 6. Johnson DW, Pollock CA, Macdougall IC. Erythropoiesis‐stimulating agent hyporesponsiveness. Nephrology (Carlton). 2007;12(4):321–330. [DOI] [PubMed] [Google Scholar]
- 7. Gupta N, Wish JB. Hypoxia‐inducible factor prolyl hydroxylase inhibitors: A potential new treatment for anemia in patients with CKD. Am J Kidney Dis. 2017;69(6):815–826. [DOI] [PubMed] [Google Scholar]
- 8. Besarab A, Chernyavskaya E, Motylev I, et al. Roxadustat (FG‐4592): Correction of anemia in incident dialysis patients. J Am Soc Nephrol. 2016;27(4):1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen N, Qian J, Chen J, et al. Phase 2 studies of oral hypoxia‐inducible factor prolyl hydroxylase inhibitor FG‐4592 for treatment of anemia in China. Nephrol Dial Transplant. 2017;32(8):1373–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Provenzano R, Besarab A, Wright S, et al. Roxadustat (FG‐4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: A phase 2, randomized, 6‐ to 19‐week, open‐label, active‐comparator, dose‐ranging, safety and exploratory efficacy study. Am J Kidney Dis. 2016;67(6):912–924. [DOI] [PubMed] [Google Scholar]
- 11. Besarab A, Provenzano R, Hertel J, et al. Randomized placebo‐controlled dose‐ranging and pharmacodynamics study of roxadustat (FG‐4592) to treat anemia in nondialysis‐dependent chronic kidney disease (NDD‐CKD) patients. Nephrol Dial Transplant. 2015;30(10):1665–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akizawa T, Iwasaki M, Otsuka T, Reusch M, Misumi T. Roxadustat treatment of chronic kidney disease‐associated anemia in Japanese patients not on dialysis: A phase 2, randomized, double‐blind, placebo‐controlled trial. Adv Ther. 2019;36(6):1438–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Provenzano R, Besarab A, Sun CH, et al. Oral hypoxia‐inducible factor prolyl hydroxylase inhibitor roxadustat (FG‐4592) for the treatment of anemia in patients with CKD. Clin J Am Soc Nephrol. 2016;11(6):982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akizawa T, Otsuka T, Reusch M, Ueno M. Intermittent oral dosing of roxadustat in peritoneal dialysis chronic kidney disease patients with anemia: A randomized, phase 3, multicenter, open‐label study. Ther Apheresis Dial. 2019. 10.1111/1744-9987.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen N, Hao C, Liu BC, et al. Roxadustat treatment for anemia in patients undergoing long‐term dialysis. N Engl J Med. 2019;381:1011–1022. 10.1056/NEJMoa1901713. [DOI] [PubMed] [Google Scholar]
- 16. Chen N, Hao C, Peng X, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med. 2019;381:1001–1010. 10.1056/NEJMoa1813599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Mean (SD) concentrations of iron in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S2 Mean (SD) Concentrations of ferritin in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S3 Mean (SD) of TSAT in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S4 Mean (SD) of reticulocyte Hb content in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S5 Mean (SD) Concentrations of transferrin in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
FIGURE S6 Mean (SD) concentrations of total iron binding capacity in ESA‐Naïve, A, and ESA‐Converted B, patients (full analysis set)
FIGURE S7 Mean (SD) concentrations of soluble transferrin receptor in ESA‐Naïve, A, and ESA‐Converted, B, patients (full analysis set)
TABLE S1 Inclusion and exclusion criteria
TABLE S2. Dose conversion between average doses of ESA before study registration and roxadustat
TABLE S3. Dose‐adjusting criteria
TABLE S4. Dose‐adjustment steps for roxadustat
TABLE S5. Schedule of assessments from week 0 to week 24 or discontinuation for both studies
TABLE S6. Schedule of assessments from week 26 to week 52 or discontinuation for the ESA‐Converted Study
Data Availability Statement
Access to anonymized individual participant level data collected during the study, in addition to supporting clinical documentation, is planned for studies conducted with approved product indications and formulations, as well as compounds terminated during development. Studies conducted with product indications or formulations that remain active in development are assessed after study completion to determine if individual participant data can be shared. Conditions and exceptions are described under the sponsor‐specific details for Astellas on www.clinicalstudydatarequest.com. Study‐related supporting documentation is redacted and provided if available, such as the protocol and amendments, statistical analysis plan, and clinical study report. Access to participant level data is offered to researchers after publication of the primary manuscript (if applicable) and is available as long as Astellas has legal authority to provide the data. Researchers must submit a proposal to conduct a scientifically relevant analysis of the study data. The research proposal is reviewed by an Independent Research Panel. If the proposal is approved, access to the study data is provided in a secure data sharing environment after receipt of a signed Data Sharing Agreement.