

Abstract
The prototype of transmissible neurodegenerative proteinopathies is prion diseases, characterized by aggregation of abnormally folded conformers of the native prion protein. A wealth of mechanisms has been proposed to explain the conformational conversion from physiological protein into misfolded, pathological form, mode of toxicity, propagation from cell‐to‐cell and regional spread. There is increasing evidence that other neurodegenerative diseases, most notably Alzheimer’s disease (Aβ and tau), Parkinson’s disease (α‐synuclein), frontotemporal dementia (TDP43, tau or FUS) and motor neurone disease (TDP43), exhibit at least some of the misfolded prion protein properties. In this review, we will discuss to what extent each of the properties of misfolded prion protein is known to occur for Aβ, tau, α‐synuclein and TDP43, with particular focus on self‐propagation through seeding, conformational strains, selective cellular and regional vulnerability, stability and resistance to inactivation, oligomers, toxicity and summarize the most recent literature on transmissibility of neurodegenerative disorders.
Keywords: amyloid‐β, neurodegeneration, prion‐like, Tau, TDP43, α‐synuclein
Introduction
Protein aggregation diseases refer to a variety of disorders which develop as a result of the deposition of misfolded peptides or proteins in various organs, causing their dysfunction. The majority of adult neurodegenerative disorders is characterized by intra‐ or extracellular aggregation of misfolded proteins, leading to progressive neuronal and glial cell dysfunction and relentless progression of clinical symptoms. Over the last two centuries, there has been a continued growth of knowledge of the different phenotypes, pathologies and causes of neurodegenerative diseases (Figure 1).
Figure 1.

Timeline of landmark discoveries in neurodegenerative diseases. Graphical representation of the history of the discoveries and descriptions of diseases or symptoms, hallmark pathology, proteins, underlying genetic causes and first reported transmissions, as indicated in the left column. The timescale is represented as a Renard series to visualise recent discoveries in higher temporal resolution. Various discoveries related to different misfolded protein pathologies are represented colour‐coded, as indicated at the bottom of the figure. Row 1, first descriptions of the disease or symptom complexes, with separate representation of the time of the initial report and time of the introduction of the eponym, shown below in a corresponding lighter shade. Row 2 shows the timeline of the description of hallmark neuropathology for each disease. Row 3, the first discovery of the protein (for tau, α‐synuclein and TDP43 this precedes association of the misfolded protein with a specific disease). Row 4, the discovery of pathogenic mutations in various genes causing neurodegenerative diseases. The establishment of the journal Neuropathology Applied Neurobiology is displayed here to provide the context to other discoveries. Rows 5 and 6 display experimental transmission (from humans to rodents and primates, respectively) and row 7 indicates the first descriptions of human transmission. References: A 154; B 155, 156; C 156, 157; D 158; E 159, 160; F 161; G 162; H 163; I 164; J 165, 166; K 167; L 168; M 169; N 170; O 171, 172; P 173, 174; Q 175, 176; R 177, 178; S 179; T 170, 180, 181; U 174, 182; V 156, 183; W 184; X 185, 186; Y 185, 186; Z 187; AA 188; AB 189, 190, 191; AC 9, 10; AD 192; AE 193; AF 10; AG 11; AH 11; AI 175, 176; AJ 6; AK 194; AL 7, 195; AM 14; AN 13; AO 12; AP 196; AQ 10; AR 197; AS 11; AT 16; AU 198, 201; AV 202, 203; AW 204, 205; AX 187; AY 206; AZ 207; BA 208; BB 209; BC 210; BD 211; BE 212; BF 213; BG 214; BH 215, 216, 217, 218; BI 219; BJ 220, 221, 222; BK 223; BL 224, 225; BM 226, 227; BN 228; BO 229, 230, 231; BP 232, 233; BQ 224, 225, 226; BR 237; BS 238, 239; BT 240; BU 241, 242; BV 243; BW 244; BX 144; BY 59; BZ 117; CA 79; CB 245; CC 130; CD 126; CE 128; CF 41; CG 246; CH 137.
Seminal studies on prion disease, undertaken mostly during the second half of the 20th century, have shown that the normal host prion protein (cellular PrP, PrPC) can undergo conformational change, and when misfolded (then referred to as PrPSc) not only causes prion disease in its host but can also under certain circumstances be transmitted to the same or to other species. An essential prerequisite for such transmission and toxicity, discussed later in this review, is the presence of PrPC in the recipient organism 1, 2. Over the last two decades, research into neurodegeneration has widened the scope, with a significant focus on investigating if and to what extent tau, α‐synuclein, Aβ peptide and more recently TDP43 exhibit similar properties. Stanley Prusiner, who received the Nobel Prize in 1997 for his work on prion disease, has always been a strong proponent that misfolded proteins associated with other neurodegenerative diseases may have similar properties. More than 30 years ago he hypothesized that experimental propagation of these proteins would require identification of a permissive host, appropriate conditions for replication and that transmission may manifest after very long incubation time 3. PrPSc displays characteristic properties, including (i) the formation of β‐sheet structures with a striking ability to form aggregates, often with protease resistance and with resistance to inactivation with conventional decontamination methods, (ii) seed formation with self‐propagation (i.e. an ability to induce abnormal conformation in a protein of the same kind, initiating a self‐amplifying cascade – termed template‐directed misfolding), (iii) the ability to form distinct conformations or strains, (iv) to propagate along neural pathways in the presence of host PrPC 4, (v) exerting neurotoxicity and (vi) showing a selective regional vulnerability, which is modulated by strains and genetic polymorphisms, (vii) transmission within a species and across species, usually with a species barrier, and adaptation by serial passaging.
The term ‘prion’ was coined by Prusiner, with a loose reference to ‘proteinaceous infectious particle’ 5, describing an agent of transmissible spongiform encephalopathy with unconventional properties. This was intended to be an operational term that does not have structural implications other than that a protein is an essential component.
Subsequently, and particularly over the last decade, the term ‘prion‐like’ or ‘prionoid’ has been introduced into the literature to describe a misfolding and disease‐initiating mechanisms similar to, yet distinct from, prion disease. Numerous studies show that the proteins associated with neurodegenerative disorders such as Alzheimer’s disease (Aβ 6 and tau 7, 8), Parkinson’s and multiple system atrophy (α‐synuclein 9, 10), frontotemporal dementia (TDP43 11, tau 12, 13, 14 or FUS 15) and motor neurone disease (TDP43 11), exhibit at least some of the properties of abnormal prion protein 16 (the first reports of the misfolded protein presence in respective diseases are referenced here and also shown in Figure 1). However, the term ‘prion‐like’ is arguably one of the most overused terms in original studies and reviews published in the last 5 years and is applied with reference to different properties of misfolded prion protein or prion disease.
In an attempt to unriddle the controversies around the semantics, it has recently been proposed to define misfolded proteins which only partially display the properties of bona fide prions as ‘proteinaceous nucleating particles’ as opposed to the original definition of ‘prion’ 17. This is to emphasize that neurodegenerative disorders other than prion diseases may lack the same efficacy of transmission.
In this review, we will discuss to what extent each of the properties of prions is known to occur for Aβ, tau, α‐synuclein and TDP43.
Amyloid structure
Amyloids are proteins with a propensity to aggregate into fibrils composed of cross‐β structures, which can be visualized with amyloid‐binding dyes, such as thioflavin S, thioflavin T or Congo red. Aβ, tau, α‐synuclein and, as shown also TDP43 18, birefringe under polarized light after Congo red staining, or fluoresce when stained with thioflavin S or thioflavin T. Instead, only FUS and SOD1 do not stain with amyloid dyes and therefore probably are not rich in β‐pleated sheets 18. Oligomers, the smaller assemblies of the protein fibrils, do not or only weakly react with amyloid dyes.
Self‐propagation through seeding
The hypothesis of self‐replication of a protein in the absence of nucleic acids was first proposed in 1967 by the British scientists John Stanley Griffith, Tikvar Alper, Ian H Pattisson and Katharine M Jones, at the time working for the British government 19, 20. This hypothesis was later, in early 1980s, refined by the American neurologist Stanley Prusiner, who proposed that a protease‐resistant protein is a structural component of the scrapie agent 5.
The ability of misfolded proteins to seed, that is, to recruit physiological proteins of the same kind and to induce their conversion into a pathological form, and propagate from cell‐to‐cell with the continuous conversion of normal protein into misfolded form is the tenet of the protein‐only hypothesis. The ability of self‐propagation through seeding is the feature most frequently referred to when describing ‘prion‐like’ properties of other misfolded peptides and proteins. Growing experimental in vitro and in vivo evidence, indeed, indicates that templated corruption of like proteins is characteristic not only of PrPSc but also Aβ, tau, α‐synuclein (reviewed in 21) and TDP43 22. Furthermore, the ability of Aβ to enhance tau pathology, similar to what is speculated to occur in AD, has been demonstrated in several in vitro experiments, using tau seeding assays, and in vivo experiments, using transgenic and wild‐type mice 23, 24, 25, 26. Whether in AD Aβ, indeed, enhances tau pathology through mechanisms, such as neurotoxicity or proteostasis and clearance failure, or induces tau aggregation by direct cross‐seeding (or heterologous seeding), needs clarification through further research. Of note, in iatrogenic Creutzfeldt–Jakob disease (CJD) with concomitant Aβ pathology, it has been speculated that Aβ may have been induced by misfolded prion protein through cross‐seeding rather than iatrogenically transmitted. However, a recent experimental study does not support the cross‐seeding hypothesis 27.
Strains (conformations) and seeding unit
The first description of strains (which derives from the early assumption of the causative agent of prion disease to be a virus) came from the observations in the early 1960s of distinct clinical phenotypes in goats, following experimental infection with scrapie brain extracts 28. These experiments also showed that the distinct clinical phenotypes were present even after several passages to other sheep.
There are at least four different human prion strains which are determined by distinct glycosylation characteristics of PrPSc and lead to a variation in the anatomical distribution of prion disease pathology, clinical symptoms, transmission properties, seeding efficiencies and in vitro amplification characteristics. It is now fairly established that the strain‐specific properties are encoded in the structure of the aggregated proteins and are retained when serially passaged in vitro or in vivo 29.
It has long been speculated that distinct structural conformations (strains) could also be a feature of other misfolded proteins and may, at least in part, explain distinct pathological and clinical phenotypes, and experimental transmission properties underlying AD, tauopathies, α‐synucleinopathies and TDP43 proteinopathies. Novel in vitro and in vivo models are constantly being developed to examine conformation, aggregation and propagation of misfolded proteins. These models are developed to specifically determine the minimal seeding unit required for initiating an efficient cascade of the seed amplification, propagation from cell‐to‐cell and spread from region‐to‐region. Recent reviews are available on the molecular basis and strain heterogeneity in neurodegeneration 30, the utility of bioassays to determine transmission 31 and cell free‐based methods for improved amplification and selective detection of PrPSc and other proteopathic seeds in biological materials, such as various tissues and fluids 32.
Aβ strains and Alzheimer’s disease (AD)
Memory impairment is the most classic presentation of AD. However other initial symptoms, such as visual impairment characteristic of posterior cortical atrophy (PCA) clinical syndrome or behavioural changes and language impairment in the spectrum of frontotemporal dementia, are well known. It is tempting to speculate that this phenotypic diversity could be due to structurally distinct Aβ strains 33. Indeed, the existence of at least two distinct Aβ strains in AD has been shown in transmission studies in susceptible transgenic mice using sporadic AD and familial AD (FAD) brain homogenates 34. Another study, using solid‐state nuclear magnetic resonance, showed structural differences of Aβ species in patients with rapidly progressive, pathologically confirmed AD and those with PCA clinical phenotype 35. Conformational heterogeneity of Aβ42 in rapidly progressive AD has also been demonstrated using conformation‐dependent immunoassay and conformational stability assay 36. A further study, using luminescent conjugated oligothiophene binding to amyloid, showed differences in Aβ spectral properties in patients with sporadic AD and FAD 37.
The concept of the seeding unit is supported by in vitro data showing that intracellular oligomers can constitute such units and that seeded nucleation of Aβ can be intracellularly induced in an APP‐producing cell line when exposed to FAD brain extract 38. Using two transgenic mouse models (APP23 and APPPS1), it has been demonstrated that Aβ seeding potency is greatest during the earliest stages of Aβ deposition and coincides with a transient increase in the Aβ42/Aβ40 ratio; Aβ seeding potency decreases with increasing Aβ accumulation in the brain 39. This study highlights the potential importance of an early intervention with compounds that can interfere with Aβ seed formation well before amyloid deposits have aggregated to an extent to be detectable on imaging or post mortem. In vivo, Aβ oligomers in the CSF can be successfully detected with protein misfolding cyclic amplification assay (PMCA) 40.
Human transmission of Aβ pathology via medical procedures, such as contaminated cadaver‐derived pituitary hormone extracts, dura mater transplants and surgical procedures have been described by us and others 41, 42, 43, 44, 45, 46, 47, 48, 49. Transmitted Aβ pathologies cannot currently be distinguished histopathologically from sporadic or familial forms, unlike prion diseases, where, in some instances, iatrogenic transmission can be suspected on histological and biochemical grounds. This is possible in patients who are homozygous for methionine at codon 129 of the PRNP gene (129MM), but show a molecular prion strain type and kuru‐plaque pathology akin to prion disease patients with valine at codon 129 (i.e. 129MV or 129VV), suggestive of the pathogen originating from a patient with at least one PRNP 129V allele 50, 51.
Tau
Tau deposition can be seen as primary pathology or as an accompanying secondary process in multiple neurodegenerative diseases. It is characterized by the predominance of either 3‐repeat (3R) tau, 4‐repeat (4R) tau or a mixture of 3R and 4R tau isoforms, generated by alternative splicing of exon 10 of the tau (MAPT) gene. The investigation, whether different tauopathies may be due to distinct tau conformers or strains, is an active area of research. Cellular and structural heterogeneity of tau conformations, as well as tau seeding, is demonstrated in several models in vitro and in vivo using nontransgenic and transgenic (P301S) mice and fluorescence resonance energy transfer‐based flow cytometry biosensor assays or Real‐Time Quaking Induced Conversion assay (RT‐QuIC) for tau seed detection 52, 53, 54, 55, 56. Tau substrates used in these experiments are of wide range including recombinant tau and preparations from human brains or CSF from patients with AD, chronic traumatic encephalopathy or Pick’s disease. Some research suggests that tau aggregation is induced by phosphorylated, high‐molecular weight tau fractions and not monomers 52, with the former also present in the CSF of an AD mouse model or AD patients 56. Recently, however, self‐assembly and aggregation properties have also been demonstrated in vitro for seed‐competent monomers extracted from AD brain tissue 57.
There is compelling experimental evidence that tau, similar to prions, stably propagates multiple conformations from different sporadic tauopathies in vitro 58 and induces distinct pathologies in vivo in mouse models reflecting the diversity of tauopathies in humans 55, 59, 60. Importantly, these tau conformations are transmissible and can be serially passaged in vivo and in vitro 58. However, there is also a key difference to prion diseases: mice expressing human P301L tau in the entorhinal cortex, crossed onto mice with tau‐null background show tau propagation and accumulation both in the presence and absence of endogenous tau, although lack of endogenous tau greatly reduces toxicity and brain atrophy 61. This finding indicates that, in contrast to prion protein, for tau transfer between neurones endogenous tau may not be obligatory. Further research is warranted to determine if distinct clinical presentations, disease progression dynamics and neuropathologies within a single disease entity [e.g. progressive supranuclear palsy (PSP)] can be explained by finer variations of the main tau strain or are determined by other factors, such as genetic, epigenetic or environmental influences.
Seeding activity in vivo has been established with insoluble tau aggregates in the form of short fibrils, but not small (>6mer) oligomeric tau assemblies 62. While in an in vitro study tau trimer is reported to be the smallest assembly that promotes dimerization of tau and could seed intracellular tau aggregation 63. This latter study, however, did not investigate tau trimer seeding and propagation ability in vivo, hence the smallest tau assembly capable to seed in vivo remains to be determined. The ability to seed tau from both soluble oligomers and insoluble tau fibrils suggests the existence of different tau seeds or conformers which may influence the speed of pathology as well as cellular response. Furthermore, the finding of widespread tau seeding activity in tissues containing both soluble and insoluble tau aggregates from brain regions preceding predicted Braak and Braak stages supports the hypothesis of tau spread along anatomically connected regions 64. Most seeding studies so far have used fresh or frozen tissue derived from human or experimental animal brains, but recently a protocol has been developed to extract and measure tau seeding activity from small volumes (0.04 mm3) of formaldehyde‐fixed tissue, which may enable successful studies of tau seeding activity in diverse tauopathies using archival formalin‐fixed brain tissues 65.
α‐synuclein
There are three diseases characterized by pathological α‐synuclein deposits, affecting neurones and glial cells. Dementia with Lewy bodies (DLB) and Parkinson’s disease (PD) show predominantly neuronal Lewy body pathology, and for the third, multiple system atrophy (MSA), hallmark pathologies are glial cytoplasmic, and to a lesser extent, neuronal inclusions. Both PD and DLB show similar pathology, with differences in clinical phenotypes. MSA pathology can present with several distinct clinical phenotypes with variations of the pathology burden in affected anatomical regions.
Both in vivo and in vitro experiments indicate that brain homogenates from patients with MSA, but not with PD, induce neurodegeneration in transgenic mice (TgM83+/‐) and in cell models (α‐syn*A53T‐YFP) 66. Furthermore, an experiment involving serial propagation of MSA α‐synuclein between two different mouse models showed retained specificity of pathological conformers 67.
Recently, further differences in seed characteristics and seeding activity have been demonstrated between soluble and insoluble fractions of PD and MSA extracts, which persist in second‐generation biosensor cells 68. In this study, MSA extracts showed seeding activity for soluble and insoluble fractions, while for PD extracts it was only the insoluble fraction which displayed seeding activity 68. Findings from a study specifically looking into the seeding activity of pathological α‐synuclein extracted from DLB patients, measured with RT‐QuIC, suggests that in extracts from DLB patient brains prefibrillar soluble α‐synuclein oligomers, and not insoluble forms, exert seeding activity 69. Structural differences in the seeding kinetics using RT‐QuIC have been also observed when comparing DLB and PD cases 70. Experiments like these, support at least indirectly the notion that MSA, PD and DLB, are caused by different α‐synuclein strains. RT‐QuIC is a sensitive and specific method for misfolded prion protein detection 71 and it can also detect with high sensitivity and specificity pathological α‐synuclein in both, brain homogenates and, more importantly, CSF from patients with PD and DLB 72, 73. α‐synuclein seeding activity, using specially optimized PMCA protocol, has been demonstrated in formalin‐fixed MSA brain tissue 74.
TDP43
The majority of motor neurone disease (MND) and all FTLD‐TDP43 cases are caused by aggregated phosphorylated TDP43. There are five subtypes of FTLD‐TDP43, designated types A‐E, which differ in terms of the shapes of pathological aggregates, affected brain regions and clinical phenotypes. TDP43 pathology is also seen in elderly patients in the context of certain other neurodegenerative diseases (e.g. PSP and AD), or as part of the ageing process. As with other neurodegenerative diseases, it has been hypothesized that the distinct FTLD‐TDP43 types may be due to different TDP43 conformations, but this is currently not yet underpinned by strong experimental evidence. However, biochemical differences between phosphorylated TDP43 (pTDP43) extracted from distinct FTLD‐TDP43 types and differences in their abilities to interfere with neurite growth have been recently shown 75. Formation of oligomers during the early stages of TDP43 misfolding 76, 77 and differences in staining properties with TDP43 oligomer‐specific antibodies has also recently been demonstrated for distinct FTLD‐TDP43 types and in patients with hippocampal sclerosis with TDP43 pathology 78. In a cell‐based seeding assay, using inducible GFP‐tagged cytoplasmic TDP43, extracts derived from brains with sporadic and familial FTLD‐TDP43, indeed, show more seeding than control brain extracts, with the highest seeding activity seen in samples with GRN mutation, while C9ORF72 mutant and sporadic cases show no difference 79. All these experiments can be regarded as circumstantial evidence of distinct TDP43 strains.
TDP43 consists of three domains, an N‐terminal Dix‐like domain mediating self‐assembly, two RNA recognition motifs, and an intrinsically disordered low complexity domain (LCD) at the C terminus. Majority of TDP43 inclusions contain C‐terminal fragments of LCD, which have a tendency to form amyloid fibrils 80. TDP43 aggregation, at least in part, is governed by a process called liquid–liquid phase separation (LLPS), which is a reversible process, characterized by fluid de‐mixing into distinct liquid phases. LLPS is increasingly recognized as a possible source of protein self‐assembly into larger aggregates. In neurodegeneration, this has been most studied concerning RNA‐binding proteins TDP43 and FUS, although LLPS also occurs in tau 81 and prion protein 82. Recent studies show that for TDP43 to undergo LLPS as few as three tryptophan residues are required 83, however, minimum seeding unit required for efficient induction of TDP43 pathology is not yet ascertained.
Selective regional vulnerability
A characteristic spatiotemporal distribution of Lewy bodies in PD, Aβ and tau in AD and grain pathology in argyrophilic grain disease is typically seen during disease progression. This progression extends across neuro‐anatomically connected regions (Lewy body pathology and neurofibrillary tangle pathology) or to some degree in relation to spatial proximity (Aβ 84). These observations have formed the basis of various pathological staging schemes 85, 86, 87. Similar patterns of spread may also exist for other α‐synucleinopathies, tauopathies and TDP43 proteinopathies, and for some (notably secondary TDP43 in AD), such staging schemes are suggested 88, 89.
Prion diseases are a prime example where selective regional vulnerability is associated with clinical symptoms and syndromes, and genetic modifiers of the pathology and phenotypes have been well characterized. For example, inherited prion disease due to D178N mutation in the PRNP gene can cause two distinct clinical phenotypes and pathologies. These phenotypes are determined by the methionine (M) and valine (V) polymorphism on the codon 129 of the PRNP gene. Coupling of the D178N mutated codon with codon 129M on the same allele results in fatal familial insomnia with the main pathology in the thalamus, while CJD phenotype with dominant cortical pathology requires coupling of the mutated codon with codon 129V.
Such striking correlate has not yet been established for any other neurodegenerative disease, although selective regional vulnerability is a feature of most, leading to differences in clinical phenotypes. For instance, the same mutation in the MAPT gene can show marked phenotypic and pathological diversity, but factors contributing to this heterogeneity remain to be determined 90. In fact, the MAPT A152T mutation may confer risk to completely different diseases, such as AD, corticobasal degeneration (CBD), PSP and DLB.
Of note, phenotypic heterogeneity in inherited prion disease due to P102L mutation in the PRNP gene, in part, is explained by variable involvement of wild‐type PrPSc 91. By drawing further parallels with inherited prion disease, one explanation for phenotypic variability in inherited tauopathies, TDP43 proteinopathies and AD, could be the extent of non‐mutant protein misfolding and propagation.
Tauopathies are among the most striking examples of heterogeneity of affected cell types and anatomical regions, resulting in distinct clinical phenotypes (Figure 2). Even within 4R tauopathies, there is a great variation in tau pathology, atrophy and clinical presentation. For example, there are eight known clinical phenotypes of PSP, each with overlapping but also distinct anatomical involvement. Current experimental research suggests the existence of different pathological tau strains in AD, PSP and CBD, which maintain cell‐type specificity in nontransgenic mice 55. Hence, selective cellular vulnerability in different tauopathies may, at least in part, be strain‐related. However, the same study, by injecting comparable concentrations of tau purified from AD, CBD and PSP brains, into the brains of nontransgenic mice, showed similar neuroanatomical distribution of tau pathology, albeit with differences in density between different diseases or the same disease, but with distinct phenotypes. Similar neuroanatomical distribution of transmitted tau pathology from different tauopathies suggests that selective regional anatomical vulnerability is not directly strain related. However, the observed differences of tau pathology burden using different human brain samples with the same disease, indicates the existence of variably potent tau strains, resulting in variably severe regional burden of tau pathology and diverse clinical phenotypes of the same disease (Figure 2).
Figure 2.
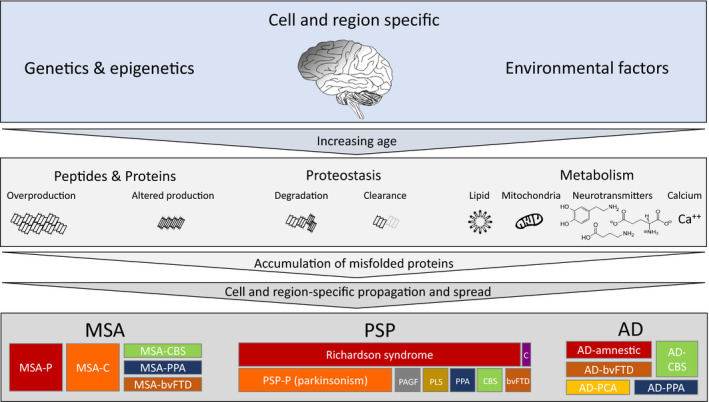
Determinants of phenotypic diversity of neurodegenerative diseases. Cell‐ and region‐specific genetic, epigenetic and environmental factors with increasing age play an essential role in neurodegeneration development, highlighted in the top row. The central panel illustrates the most significantly involved or best characterised subcellular processes, which lead to the initiation of pathological protein aggregation. These include overproduction or abnormal production of specific peptides and proteins, disturbance in the proteostasis with impaired degradation of misfolded proteins or reduced clearance through various fluids and compromised metabolism (mitochondria, lipid metabolism, neurotransmitter, such as glutamate, dopamine or GABA, and calcium homeostasis). Impairment of one or a combination of the above, leads to initiation of misfolded protein aggregation and self‐amplification, with a cell‐specific propagation and region‐specific spread, resulting in distinct clinical phenotypes, represented in the bottom panel. Multiple system atrophy (MSA), α‐synucleinopathy with its hallmark glial cytoplasmic inclusion pathology, shown on the left side, typically presents with parkinsonism (MSA‐P) or cerebellar signs (MSA‐C). However rare clinical presentations, such as corticobasal syndrome (MSA‐CBS), primary progressive aphasia (MSA‐PPA), and behavioural variant frontotemporal dementia (MSA‐bvFTD) are well known. Progressive supranuclear palsy (PSP), a primary tauopathy with characteristic tufted astrocyte pathology, shown in the centre of the panel. In addition to the most widely known Richardson syndrome (PSP‐RS), PSP can have other clinical presentations, such as PSP‐P which closely resembles parkinsonism; pure akinesia with gait freezing (PSP‐PAGF), primary lateral sclerosis dominated by corticospinal tract degeneration (PSP‐PLS), primary progressive aphasia (PSP‐PPA), corticobasal syndrome (PSP‐CBS), behavioural variant frontotemporal dementia (PSP‐bvFTD) or the rarest presentation dominated by cerebellar signs (PSP‐C). Also for Alzheimer’s disease, in addition to the most common amnestic presentation, other clinical phenotypes include behavioural variant frontotemporal dementia (bvFTD), posterior cortical atrophy (AD‐PCA), corticobasal syndrome (AD‐CBS) and primary progressive aphasia (AD‐PPA).
Large gaps remain in the understanding of the pathobiology of selective regional vulnerability in neurodegeneration. Increasingly it is recognized that complex and diverse genome‐wide alterations, gene expression patterns and epigenome‐wide DNA methylation profiles play a role in cellular and anatomical vulnerability in neurodegeneration. They modify neuronal, glial, and microglial cell function, clearance and degradation of misfolded proteins, factors influencing spread, such as exocytosis and cellular uptake, or cause alterations in signalling pathways. Furthermore, genetic and epigenetic factors also modulate resistance to harmful proteopathic seeds and eventually much of the phenotypic and pathological variability is probably caused by a combination of the above (Figure 2). A recent systematic genome‐wide review discusses common molecular pathways involved in neurodegeneration and highlights the complexity of involved pathways spanning from protein degradation, mitochondrial function, inflammatory response, metabolism of lipids and cholesterol 92.
Of note, recent research suggests that at least for PD, the genetic risk loci do not lie in specific brain regions or restricted (e.g. solely neuronal or glial) cell types, but, instead, they are present across a range of cell types with involvement of a number of global pathways 93. These observations highlight the need for further research to better understand the interplay between cell and region specific (epi)‐genetic alterations and selective cellular and regional vulnerability in various neurodegenerative diseases.
Stability and resistance to inactivation
One of the defining features of PrPSc is the relative resistance to proteolysis by proteases. This was discovered in the 1930s in the context of vaccine development against louping‐ill virus, a devastating endemic tick‐borne illness in sheep. The virus was isolated from spleens and brains of affected sheep, and inactivated with formalin. However, scrapie agent, unknowingly present in the starting material, was transmitted into vaccinated sheep who succumbed to typical scrapie 94. More recently, it has been shown that also Aβ from AD brains and α‐synuclein from MSA brains is not inactivated by formalin; and formalin‐fixed human brain tissue extracts from patients with AD and MSA or transgenic mice with respective pathologies have been transmitted to susceptible transgenic mice where they elicit corresponding pathology 95, 96, 97. Such stability is also relevant for the decontamination of surgical instruments. PrPSc, Aβ and α‐synuclein adhere to steel and show remarkable resistance to ‘inactivation’ with standard decontamination procedures and elicit prion disease, Aβ pathology 98 and MSA 96 pathology respectively. Instead, the transmission of tau, TDP43 or Lewy body pathologies from formalin‐fixed tissues or through steel wires has not been reported to date. It is also worthwhile to note, that studies showing transmission of tau, TDP43 and Lewy body pathology after intracerebral injections in various experimental models have utilized tissue enriched in pathology with excessive quantities of the pathological conformers. Furthermore, it is curious, that differences in proteinase K proteolytic properties are documented in DLB and PD patient brain extracts subjected to RT‐QuIC analysis, with amplified α‐synuclein from DLB, but not PD or control cases, showing proteinase K resistant species 70.
Oligomers and toxicity
The term ‘toxicity’ may imply a number of different effects at biochemical and molecular level involving cells and their networks. This also means different measurements of the toxicity, which may vary, for example, from the assessment of cognition with an array of behaviour tests, or assessment of the synaptic or mitochondrial function in tissues using neurophysiological and biochemical assays 99. Seeded templating and neurotoxicity are two of the most critical properties attributed to oligomers. Oligomeric states have been documented for misfolded proteins involved in neurodegeneration, that is, prion protein, Aβ, tau, α‐synuclein, huntingtin, TDP43, mutant nuclear (but not cytoplasmic) FUS 100 and mutant SOD1 in MND 101.
A growing body of in vivo data suggests that soluble oligomers of PrPSc 102, Aβ 103, tau 99, α‐synuclein 104 and TDP43 77 are among the key components to induce seeding and cause neurotoxicity with neurodegeneration. It has been proposed that the efficiency of self‐amplification occurs at oligomer concentrations that are different from those causing toxicity, as it has been demonstrated for α‐synuclein 105. For Aβ, specifically the longer forms (1–42 and 1–46) show greatest synaptotoxicity, and synaptic function can be restored upon prevention of soluble Aβ formation and/or treatment with antibodies targeting specifically N‐terminal residues 103.
Differences in neurotoxicity and seeding activity have recently also been established in FTLD‐TDP43 subtypes. pTDP43 extracts from FTLD‐TDP43 type A show the most severe toxicity with a reduction in neurite length 75 and similar findings are described for in vitro generated α‐synuclein oligomers 106. Yet, it is not established if (or how) oligomers initiate neurodegeneration or if neurodegeneration could be merely the consequence of misfolded protein aggregation intra‐ and extracellularly, separate from propagation and toxicity.
A number of key questions related to oligomers, toxicity and propagation remain to be answered: (i) are oligomers responsible for cell‐to‐cell propagation across specified networks or defined anatomical pathways; (ii) to what extent aggregation and propagation of oligomers, protofibrils and larger fibrils corresponds to toxicity, and if therapeutic attempts to inhibit oligomer formation or fibril aggregation and propagation can eliminate neurotoxicity; (ii) are neurotoxic oligomers identical to the transmissible agent; (iii) is toxicity disruptive to cells or networks at a biochemical level (such as neurotransmitter transport, cytoskeletal, organelle and synaptic integrity), and to what extent does it cause cell loss and brain atrophy; (iv) what specifically is the cause of oligomer abundance – for example, overproduction, clearance failure (extracellular pathways, such as blood–brain barrier or glymphatic system), or loss of proteostatic integrity within lysosomes, autophagosomes, proteasomes, ubiquitin‐peroxisomal system or any other less understood pathways for misfolded protein degradation and/or excretion out of the cell (Figure 2); (v) do different oligomer sizes (i.e. small oligomers in the range of 2–5 mers (repeat units) and large oligomers (in the range of 6–150 mers) and concentrations have distinct roles in each proteinopathy; (vi) do soluble and structure (such as vesicle) – bound oligomers have distinct roles; (vii) are the current methodological approaches used for oligomer detection and quantification sufficient and what are the challenges for the development of a reproducible and most accurate measurement of oligomer size and concentration in tissues and fluids.
Propagation and transmission
The commonly used term ‘prion‐like property’ describes the ability of aggregated, misfolded proteins, other than prion protein, to act as a template for the formation of seeds from a physiologic form of the same protein, propagate between cells and spread across anatomical pathways. The use of the term ‘prion‐like spread’ has provoked controversy, as it presumes a unifying mechanism of the propagation of misfolded proteins in the nervous system and oversimplifies the complexity of the pathomechanisms underlying the spread of different proteopathic seeds and neurodegeneration development.
An important obvious difference between PrP, Aβ, tau, α‐synuclein and TDP43 is the transmembranous and extracellular location of PrP and Aβ, while tau, α‐synuclein and TDP43 are predominantly intracellular. Therefore, arguably, only the propagation of Aβ peptide (and of ADan and ABri proteins, found in exceptionally rare familial dementias), could be likened to a prion‐like spread, as, similar to prions, these are located in the membrane and extracellularly. Experimental paradigms to test seeding activity include cell models to demonstrate propagation in vitro and animal models to show transmissibility. Another scenario is where transmission occurs between individual organisms within species (such as scrapie and bovine spongiform encephalopathy) or, theoretically, across different species. The following paragraphs will highlight the characteristics of in vivo propagation within an organism (propagation between cells) or scenarios where transmission occurs between individual organisms of the same or different species. Figure 3 summaries the confirmed transmission routes of various proteinopathies.
Figure 3.
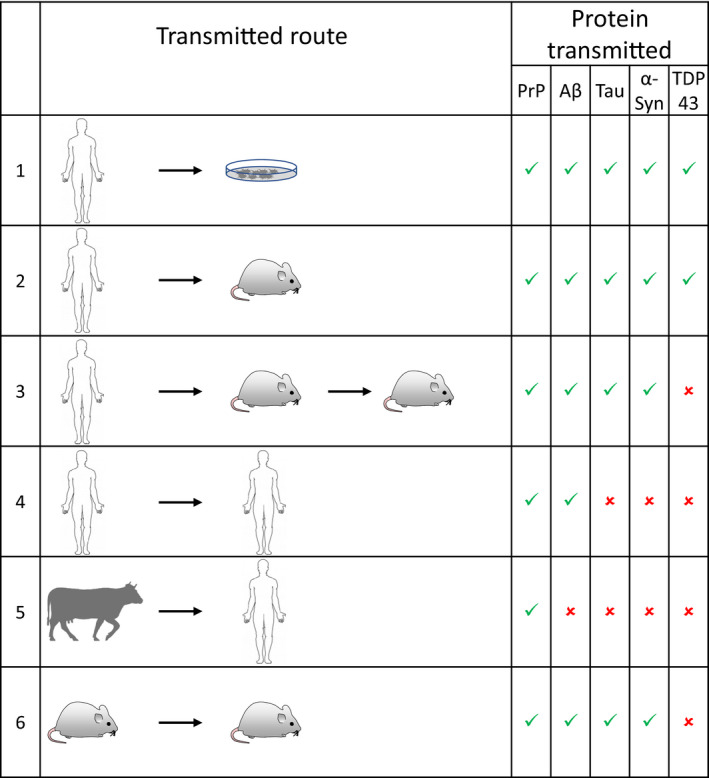
Transmission routes of proteopathic seeds. Rows indicate the demonstrated transmitted route and the transmitted peptides or proteins are shown in the columns on the right. Green ticks indicate evidence of successful transmission and red crosses indicates absence of evidence of successful transmission. Rows 1, 2; transmission into cells and animal models has been accomplished for all 5 proteins. Row 3, serial transmission into animals has been accomplished for PrP, amyloid‐β and α‐synuclein, but so far has not been shown for TDP43; for α‐synuclein and tau conformers this has not been demonstrated for all human diseases. Row 4, transmission between humans is known to occur for PrP and amyloid‐β pathologies, for tau, α‐synuclein or TDP43, this has not been demonstrated and further studies are required. Row 5, transmission from animal species into humans has to date only been reported for prions (bovine spongiform encephalopathy and variant CJD). Row 6, experimental transmission from mice (usually genetically modified) into the same species, has been reported for prions, amyloid‐β, tau, and α‐synuclein.
In vivo propagation between cells
The first observation of cell‐to‐cell transmission in the human brain dates back to 2008 when two groups reported Lewy body pathology development within foetal dopaminergic neurone grafts, which had been implanted in basal ganglia of PD patients many years earlier 107, 108. This ground‐breaking observation was followed by many in vitro and in vivo experiments designed to better understand the mechanism of cell‐to‐cell spread of proteopathic seeds. Many physiological pathways, such as trans‐synaptic and non‐synaptic (interstitial diffusion, micro‐ or macro‐pinocytosis, exosomes, vesicle‐mediated exocytosis–endocytosis or nanotubes 21) are candidates for transcellular spread. While it is beyond the scope of this review to discuss all these pathways in detail, it should be mentioned that there are cell culture models for tau, α‐synuclein, TDP43 22, 109 and more recently also intracellular Aβ seeding and nucleation 38. Also, the role of astrocytes and microglia for efficient trans‐neuronal propagation is increasingly acknowledged. For example, microglial activation in embryonal dopaminergic neurone grafts has recently been shown to be present from early on after graft implantation and well before Lewy body pathology development in these grafts 110. Such observations underpin the complexity of factors that can contribute to the initiation and spread of proteopathic seeds.
Transmission of proteopathic seeds in vivo
The transmission between animals of the same species
The transmission between animals of the same species was first described in prion disease in 1936–1939, when scrapie transmission to sheep, by intraocular injection, was demonstrated by two French veterinary scientists 111, 112, 113. Over subsequent decades many experimental in vivo models have been developed with the overarching aim to investigate the mechanisms of transmission and propagation and seeding potencies of misfolded proteins implicated in neurodegeneration. Aβ transmission in transgenic mice was first reported in 2000 114, mutant P301S tau transmission in transgenic mice in 2009 115 and from human tauopathies into transgenic mice in 2013 59; α‐synuclein transmission in transgenic mice using synthetic fibrils in 2012 116 and human MSA brain extracts in 2013 117, and TDP43 in transgenic mice using human brain‐derived FTLD‐TDP43 extracts in 2018 79. Most common experimental approaches include intracerebral injections of brain extracts derived from transgenic mice or humans into transgenic mice expressing the human form of the protein or into wild‐type mice. For Aβ and tau, intracerebral propagation of respective pathology can also be seen after extended incubation times, following peripheral administration of Aβ 118 and tau aggregates 119 derived from transgenic mouse models either overexpressing APP (APP23) or expressing mutant tau (P301S) respectively. Similarly, neurological impairment and α‐synuclein pathology are demonstrated to develop in the CNS of transgenic mice following intramuscular administration of α‐synuclein fibrils 120. Recently, α‐synuclein transmission in transgenic mice has been demonstrated following oral, intravenous and intraperitoneal administration of recombinant α‐synuclein fibrils, made of human wild‐type α‐synuclein 121.
Experimental transmission between animal species
Experimental transmission between animal species can be useful to determine the species barrier and, thus, the zoonotic potential. Cross‐species transmission of prions has been extensively studied. Scrapie transmission was demonstrated for the first time in goats in 1959 122, and soon after into wild‐type mice 123. The most prominent zoonosis is Bovine Spongiform Encephalopathy (BSE), but several other highly prevalent prion diseases in animals exist with unascertained zoonotic potential. BSE cross‐species transmission was first proven in wild‐type mice (1988) and marmosets (1993) followed by transgenic mice, sheep, macaque and vole. A comprehensive review of transmission studies of prion diseases is in 124. Recently, scrapie transmission after prolonged incubation periods has been demonstrated also in primate, cynomolgus macaque, which show high PrP protein homology to humans 125. Apart from prion disease, there is currently no evidence that other proteins involved in human neurodegeneration can be transmitted between animals. A strong research focus instead is on studying the potential of experimental transmission of diseased human brain tissue into animal models.
Transmission from humans to animals
Transmission from humans to animals is exclusively seen in experimental settings. In transgenic and wild‐type mice, cell‐to‐cell propagation has been demonstrated with intracerebrally injected human brain extracts from AD, various primary tauopathies 59, α‐ synucleinopathy (PD 126 and MSA 117), and more recently also from patients with FTLD‐TDP43 79. The first transmission of a human proteinopathy was prion disease kuru into chimpanzees in 1966 127, 128. More than two decades later, the transmission of Aβ pathology (CAA and parenchymal deposits) 129, 130 and recently, also α‐synuclein Lewy body pathology transmission was accomplished in primates 126. Instead, the transmission of tau and TDP43 pathology into primates has yet to be confirmed.
Transmission of human misfolded proteins into cell models to study in vitro
Transmission of human misfolded proteins into cell models to study in vitro propagation of neurodegenerative disease is an active and extensive research area. These studies are essential to study specific aspects of cell biology and biochemistry of these proteins. As of now, all major neurodegeneration‐related misfolded proteins have been propagated in various cell models (Figure 3) 131, 132, 133, 134, 135.
Transmission between humans
Transmission between humans was first described in prion diseases: kuru was a major epidemic of human prion disease in the people of the Fore linguistic group in the Eastern Highlands of Papua New Guinea. It was transmitted through the practice of engaging in the consumption of dead relatives as a mark of respect and mourning (transumption) 136. Iatrogenic transmission of prion disease was first reported in 1974 in a patient who had received a cadaveric corneal transplant from a donor who, in retrospect, had been identified as having died of CJD 137. Subsequently, the transmission of CJD through cadaver‐derived hormones, most prominently growth hormone (GH), and contaminated dural transplants during neurosurgical (and surgical) procedures have been reported. This resulted in a ban on the use of human cadaver‐sourced hormones in 1985 and cadaver‐derived dural transplants in the 1990s.
For many decades, it was thought that misfolded proteins other than abnormal prion protein do not transmit between humans. This assumption was based on the lack of epidemiological evidence and the absence of any overt clinical neurological manifestations in primate studies 138. Over the last 5 years, this view has fundamentally changed since the publication of several studies providing circumstantial evidence of human transmission of Aβ. In 2015, we observed that patients, who had received pituitary‐derived GH and had died of prion disease, also showed frequent cerebral amyloid angiopathy (CAA) and parenchymal Aβ 41. After this landmark publication, Aβ transmission was also demonstrated in patients who had received contaminated dural grafts 44. Since then, many authors have reproduced these findings in various cohorts of iatrogenic CJD patients 43, 45, 46, 47, 139. In retrospect, Aβ pathology in dural graft‐related iatrogenic CJD patients had already been reported before 140, 141, albeit without association with potential transmissibility. Patients who had not contracted CJD, but died from other causes also show Aβ pathology 43. Furthermore, some have developed complications related to amyloid angiopathy, such as intracerebral haemorrhages 48, 49. The pathogenic role of Aβ seeds has been confirmed by the transmission of pituitary‐derived GH, archived in vials for decades, into transgenic mice that developed accelerated amyloid angiopathy 142. A further mode of Aβ transmission was uncovered when we examined unexplained early‐onset CAA in a small series of patients in the 3rd and 4th decades of life. When exploring the clinical history we found that these patients had neurosurgical interventions during childhood with no evidence of cadaveric dural grafting 42, suggesting Aβ transmission through neurosurgery 42 (Figure 4). To date, CAA‐related haemorrhages have been the main complication of iatrogenically transmitted Aβ 42, 48, 49, prompting public health concerns. Cases reported to date have not shown evidence of advanced AD pathology.
Figure 4.
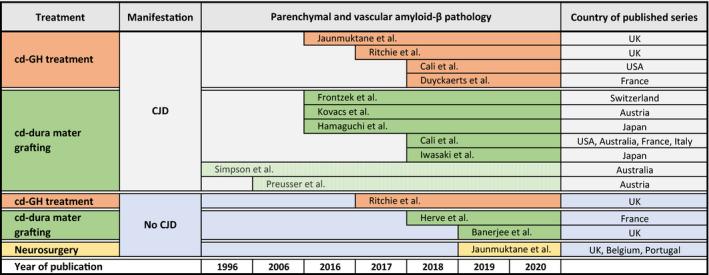
Recent history of published observations on human transmission of amyloid‐β. Initially, the human transmission of amyloid‐β through iatrogenic procedures was reported in the context of transmitted prion disease, with the first landmark study in 2015 documenting widespread parenchymal and vascular amyloid‐β pathology in patients who had received human cadaver derived GH (cd‐GH) treatment several decades earlier. This was followed by a similar observation in patients who had received cadaver derived (cd) dura mater treatment. Later studies described amyloid‐β transmission also independent from prion disease, so far through cadaver derived growth hormone treatment, dura mater grafting and neurosurgical instruments. Two case reports, shown in light green, refer to earlier reports describing amyloid‐β pathology, but not suggesting potential iatrogenic transmissibility. On the far right, the countries of published series are specified.
Human transmission of tau, TDP43 and α‐synuclein pathology has not been proven as yet, but it may just be a matter of time until epidemiological, pathology‐confirmed studies have been conducted. Review of clinical notes from patients, who had died of MSA and PD, showed no evidence of neurosurgical transmission or cadaver‐derived GH treatment 143 and a study of five couples whose spouses had pathologically verified PD, PSP or MSA also did not suggest an increased risk of α‐synucleinopathy development in the other spouse 144. Current absence of evidence, however, is not evidence of the absence of human transmission of misfolded proteins other than prion and Aβ, and further research is necessary before any firm conclusions can be drawn.
A possible explanation of the transmissibility of PrPSc and Aβ, but not tau, α‐synuclein and TDP43, may be related to the differences in cellular localization (extracellular and transmembrane vs. cytoplasmic), rendering prion protein and Aβ more readily available for iatrogenic seed transmission. Another explanation could be related to the incubation periods, which for prion diseases vary from a couple of years to over 40 years 51 and for Aβ, as the current observational studies show (Figure 4), at least 20 years are needed for the pathology to be detectable and manifest clinically. It is plausible that even longer incubation periods, possibly exceeding human lifespan, are required for tau, TDP43 and α‐synuclein pathology development. Further differences could be due to critical mass of initial seeds required, to initiate a self‐amplifying cascade. For example, in vitro studies show that while for tau the minimal propagation unit is as small as tau trimers, larger oligomers comprising up to 100 tau molecules have the greatest seeding efficiently 63. A much higher concentration (at least 10,000 fibrils or oligomers per cell‐like volume) of α‐synuclein, is needed for efficient seeding, with fibrils being more effective at seeding than oligomers 105. In contrast, for tau, using a similar methodological approach (single‐molecular fluorescence resonance energy transfer and kinetic analysis), significantly less oligomers appear to be required for efficient seeding 145. In comparison, in sporadic prion disease in patients with MM genotype at codon 129 of the PRNP gene, the average 50% seeding dose (SD), assessed with RT‐QuIC assay, corresponds to approximately 1010 /g brain, or 1 SD50 unit equivalent to 0.06–0.27 fg of PrPSc 146. As demonstrated in these examples, different methodologies for different neurodegenerative diseases have been applied, with some measuring the oligomer size or number of molecules, and others estimating the concentration or mass.
Of note, research applying kinetic analysis also allow to predict most effective conditions for templated seeding to occur and the suggested relevant factors are: nucleation rate (small number of oligomers or fibrils show most effective seeding at slow nucleation rate) and initial protein concentration and its relationship to critical aggregation concentration (templated seeding is more effective at low concentrations with fewer oligomers needed) 105.
Transmission from animals to humans
Transmission from animals to humans has probably only occurred in the context of BSE. Owing to changes in processing cattle feed, BSE first spread endemically in cattle and was then transmitted, most likely through the food chain, to humans 147. It caused an unusual, early‐onset neurological syndrome, which was later designated as variant CJD (vCJD). Polymorphic variants of the PRNP gene in humans and animals have a strong influence on the transmissibility and development of a clinical phenotype. All but one cases of vCJD occurred in patients with the PRNP codon 129MM genotype. The exception is a patient who presented clinically and radiologically with a sporadic CJD phenotype but showed histologically a prototypical vCJD, and biochemical typing confirmed the BSE/vCJD strain 148. For last several years, no new cases of vCJD have emerged, however, it is debated if the vCJD strain could have adapted into a clinically and histologically classical CJD 149, making it nearly impossible to identify as transmitted form.
Discovery of unusual forms of misfolded protein diseases, such as parkinsonism‐dementia complex of Guam and the recent report of a tauopathy in young East‐African children with nodding syndrome 150, make one consider the possibility of environmental causes, leading to cross‐species transmission of proteins other than prion. As always, such speculations need to be viewed with extreme caution. For tau pathology in particular, its development in patients with long‐standing seizures is well known 151. Thus, tauopathy that has been described in the nodding syndrome, a form of epilepsy with uncontrollable nodding of the head, may well be secondary to the seizures. Recent research, in fact, suggests that epilepsy in patients with nodding syndrome is an immune‐mediated reaction related to parasitic worm onchocerciasis infection 152, further suggesting that tauopathy may, indeed, be secondary.
Conclusion and future perspectives
The concept of a transmissible disease, caused by seeds or template‐directed self‐assembly of proteins has fascinated the scientific community for decades. As highlighted in this review, over the last two decades building on century‐long research in prion diseases, startling revelations, both practical and profound, have been made. The research has furthered the understanding of mechanisms of proteopathic seed development, templated seeding, propagation from cell‐to‐cell and spread from region‐to‐region. While likening misfolded proteins and peptides of common neurodegenerative diseases, such as AD and PD, to prion disease, may have some value in translating the research findings from one field to the other, such comparisons are not always accurate or used attentively. Large gaps remain in understanding the mechanisms of prion and all other neurodegenerative diseases.
The future research focus should be twofold: Firstly, related to public health, to ensure adequate surgical and laboratory instrument decontamination procedures are in place, and prospective epidemiological studies are designed to specifically investigate any potential transmissibility of misfolded proteins and peptides which require long incubation periods to be detectable. There are defined requirements for biosafety procedures to handle cell and animal models, and human tissues in prion disease research and clinical practice. Instead, there are currently no data suggesting that such precautions are warranted for other protein species. Any recommendations to introduce similar requirements for research into other neurodegenerative diseases would add substantial financial and operational barriers and would obstruct advancements in the field. Adherence to good laboratory practice and health and safety guidelines, relevant to human and experimental animal tissue, is currently considered to be appropriate for handling material from neurodegenerative disorders other than prion diseases (see also review: 153). In clinical practice, patients at risk of developing iatrogenic Aβ pathology, notably CAA, should be considered to be followed up with neuroimaging and with appropriate CSF biomarker tests, such as Aβ, tau and other neurodegenerative proteins (e.g. neurofilament light chain). Secondly, further experimental research is needed to unravel the molecular and mechanistic processes leading to initiation of self‐amplification, efficient propagation and spread of proteopathic seeds. The exact role of oligomers in causing neurotoxicity, regional vulnerability, transmissibility and neurodegeneration as discussed in this review needs to be determined. The complexity of genetic and epigenetic alterations, including the role of mosaicism and cell‐ and region‐specific differences in gene and protein expression levels between individuals warrants detailed elucidation. It is likely, that the sum of these research efforts will lead to finding the cure of these devastating diseases.
Conflict of interest
None.
Acknowledgements
ZJ and SB are supported by the Department of Health’s NIHR Biomedical Research Centre’s funding scheme.
Jaunmuktane Z. and Brandner S. (2020) Neuropathology and Applied Neurobiology 46, 522–545 Invited Review: The role of prion‐like mechanisms in neurodegenerative diseases
Contributor Information
Z. Jaunmuktane, Email: z.jaunmuktane@ucl.ac.uk.
S. Brandner, Email: s.brandner@ucl.ac.uk.
References
- 1. Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, et al Normal host prion protein necessary for scrapie‐induced neurotoxicity. Nature 1996; 379: 339–43 [DOI] [PubMed] [Google Scholar]
- 2. Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al Mice devoid of PrP are resistant to scrapie. Cell 1993; 73: 1339–47 [DOI] [PubMed] [Google Scholar]
- 3. Prusiner SB. Some speculations about prions, amyloid, and Alzheimer's disease. N Engl J Med 1984; 310: 661–3 [DOI] [PubMed] [Google Scholar]
- 4. Brandner S, Raeber A, Sailer A, Blattler T, Fischer M, Weissmann C, et al Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci USA 1996; 93: 13148–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136–44 [DOI] [PubMed] [Google Scholar]
- 6. Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 1984; 122: 1131–5 [DOI] [PubMed] [Google Scholar]
- 7. Brion JP, Flament‐Durand J, Dustin P. Alzheimer's disease and tau proteins. Lancet 1986; 2: 1098 [DOI] [PubMed] [Google Scholar]
- 8. Iqbal K, Grundke‐Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al Defective brain microtubule assembly in Alzheimer's disease. The Lancet 1986; 2: 421–6 [DOI] [PubMed] [Google Scholar]
- 9. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA 1998; 95: 6469–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha‐synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 1998; 251: 205–8 [DOI] [PubMed] [Google Scholar]
- 11. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314: 130–3 [DOI] [PubMed] [Google Scholar]
- 12. Feany MB, Dickson DW. Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 1995; 146: 1388–96 [PMC free article] [PubMed] [Google Scholar]
- 13. Probst A, Langui D, Lautenschlager C, Ulrich J, Brion JP, Anderton BH. Progressive supranuclear palsy: extensive neuropil threads in addition to neurofibrillary tangles. Very similar antigenicity of subcortical neuronal pathology in progressive supranuclear palsy and Alzheimer's disease. Acta Neuropathol 1988; 77: 61–8 [DOI] [PubMed] [Google Scholar]
- 14. Joachim CL, Morris JH, Kosik KS, Selkoe DJ. Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann Neurol 1987; 22: 514–20 [DOI] [PubMed] [Google Scholar]
- 15. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009; 132(Pt 11): 2922–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bockman JM, Kingsbury DT, McKinley MP, Bendheim PE, Prusiner SB. Creutzfeldt‐Jakob disease prion proteins in human brains. N Engl J Med 1985; 312: 73–8 [DOI] [PubMed] [Google Scholar]
- 17. Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Ann Rev Neurosci 2015; 38: 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bigio EH, Wu JY, Deng HX, Bit‐Ivan EN, Mao Q, Ganti R, et al Inclusions in frontotemporal lobar degeneration with TDP‐43 proteinopathy (FTLD‐TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD‐FUS), have properties of amyloid. Acta Neuropathol 2013; 125: 463–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie replicate without nucleic acid? Nature 1967; 214: 764–6 [DOI] [PubMed] [Google Scholar]
- 20. Pattison IH, Jones KM. The possible nature of the transmissible agent of scrapie. Vet Rec 1967; 80: 2–9 [DOI] [PubMed] [Google Scholar]
- 21. Guo JL, Lee VM. Cell‐to‐cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014; 20: 130–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smethurst P, Newcombe J, Troakes C, Simone R, Chen YR, Patani R, et al In vitro prion‐like behaviour of TDP‐43 in ALS. Neurobiol Dis 2016; 96: 236–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bennett RE, DeVos SL, Dujardin S, Corjuc B, Gor R, Gonzalez J, et al Enhanced tau aggregation in the presence of amyloid beta. Am J Pathol 2017; 187: 1601–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, et al Amyloid‐beta plaques enhance Alzheimer's brain tau‐seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 2018; 24: 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001; 293: 1491–5 [DOI] [PubMed] [Google Scholar]
- 26. Vergara C, Houben S, Suain V, Yilmaz Z, De Decker R, Vanden Dries V, et al Amyloid‐beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol 2019; 137: 397–412 [DOI] [PubMed] [Google Scholar]
- 27. Rasmussen J, Krasemann S, Altmeppen H, Schwarz P, Schelle J, Aguzzi A, et al Infectious prions do not induce Abeta deposition in an in vivo seeding model. Acta Neuropathol 2018; 135: 965–7 [DOI] [PubMed] [Google Scholar]
- 28. Pattison IH, Millson GC. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J Comp Pathol 1961; 71: 101–9 [DOI] [PubMed] [Google Scholar]
- 29. Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318: 930–6 [DOI] [PubMed] [Google Scholar]
- 30. Scialo C, De Cecco E, Manganotti P, Legname G. Prion and prion‐like protein strains: deciphering the molecular basis of heterogeneity in neurodegeneration. Viruses 2019; 11: 261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giles K, Woerman AL, Berry DB, Prusiner SB. Bioassays and Inactivation of Prions. Cold Spring Harbor Perspect Biol 2017; 9: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Orru CD, Wilham JM, Vascellari S, Hughson AG, Caughey B. New generation QuIC assays for prion seeding activity. Prion 2012; 6: 147–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cohen M, Appleby B, Safar JG. Distinct prion‐like strains of amyloid beta implicated in phenotypic diversity of Alzheimer's disease. Prion 2016; 10: 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watts JC, Condello C, Stohr J, Oehler A, Lee J, DeArmond SJ, et al Serial propagation of distinct strains of Abeta prions from Alzheimer's disease patients. Proc Natl Acad Sci USA 2014; 111: 10323–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qiang W, Yau WM, Lu JX, Collinge J, Tycko R. Structural variation in amyloid‐beta fibrils from Alzheimer's disease clinical subtypes. Nature 2017; 541: 217–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, et al Rapidly progressive Alzheimer's disease features distinct structures of amyloid‐beta. Brain 2015; 138(Pt 4): 1009–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rasmussen J, Mahler J, Beschorner N, Kaeser SA, Hasler LM, Baumann F, et al Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer's disease. Proc Natl Acad Sci USA 2017; 114: 13018–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Olsson TT, Klementieva O, Gouras GK. Prion‐like seeding and nucleation of intracellular amyloid‐beta. Neurobiol Dis 2018; 113: 1–10 [DOI] [PubMed] [Google Scholar]
- 39. Ye L, Rasmussen J, Kaeser SA, Marzesco AM, Obermuller U, Mahler J, et al Abeta seeding potency peaks in the early stages of cerebral beta‐amyloidosis. EMBO Rep 2017; 18: 1536–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C. Detection of misfolded Abeta oligomers for sensitive biochemical diagnosis of Alzheimer's disease. Cell Rep 2014; 7: 261–8 [DOI] [PubMed] [Google Scholar]
- 41. Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, et al Evidence for human transmission of amyloid‐beta pathology and cerebral amyloid angiopathy. Nature 2015; 525: 247–50 [DOI] [PubMed] [Google Scholar]
- 42. Jaunmuktane Z, Quaegebeur A, Taipa R, Viana‐Baptista M, Barbosa R, Koriath C, et al Evidence of amyloid‐beta cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol 2018; 135: 671–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ritchie DL, Adlard P, Peden AH, Lowrie S, Le Grice M, Burns K, et al Amyloid‐beta accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol 2017; 134: 221–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frontzek K, Lutz MI, Aguzzi A, Kovacs GG, Budka H. Amyloid‐beta pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt‐Jakob disease after dural grafting. Swiss Med Weekly 2016; 146: w14287 [DOI] [PubMed] [Google Scholar]
- 45. Cali I, Cohen ML, Hasmall yi US, Parchi P, Giaccone G, Collins SJ, et al Iatrogenic Creutzfeldt‐Jakob disease with Amyloid‐beta pathology: an international study. Acta Neuropathol Commun 2018; 6: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kovacs GG, Lutz MI, Ricken G, Strobel T, Hoftberger R, Preusser M, et al Dura mater is a potential source of Abeta seeds. Acta Neuropathol 2016; 131: 911–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hamaguchi T, Taniguchi Y, Sakai K, Kitamoto T, Takao M, Murayama S, et al Significant association of cadaveric dura mater grafting with subpial Abeta deposition and meningeal amyloid angiopathy. Acta Neuropathol 2016; 132: 313–5 [DOI] [PubMed] [Google Scholar]
- 48. Herve D, Porche M, Cabrejo L, Guidoux C, Tournier‐Lasserve E, Nicolas G, et al Fatal Abeta cerebral amyloid angiopathy 4 decades after a dural graft at the age of 2 years. Acta Neuropathol 2018; 135: 801–3 [DOI] [PubMed] [Google Scholar]
- 49. Banerjee G, Adams ME, Jaunmuktane Z, Alistair Lammie G, Turner B, Wani M, et al Early onset cerebral amyloid angiopathy following childhood exposure to cadaveric dura. Ann Neurol 2019; 85: 284–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kobayashi A, Parchi P, Yamada M, Mohri S, Kitamoto T. Neuropathological and biochemical criteria to identify acquired Creutzfeldt‐Jakob disease among presumed sporadic cases. Neuropathology 2016; 36: 305–10 [DOI] [PubMed] [Google Scholar]
- 51. Rudge P, Jaunmuktane Z, Adlard P, Bjurstrom N, Caine D, Lowe J, et al Iatrogenic CJD due to pituitary‐derived growth hormone with genetically determined incubation times of up to 40 years. Brain 2015; 138(Pt 11): 3386–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, et al Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci USA 2014; 111: E4376–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saijo E, Ghetti B, Zanusso G, Oblak A, Furman JL, Diamond MI, et al Ultrasensitive and selective detection of 3‐repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol 2017; 133: 751–65 [DOI] [PubMed] [Google Scholar]
- 54. Kraus A, Saijo E, Metrick MA 2nd, Newell K, Sigurdson CJ, Zanusso G, et al Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol 2019; 137: 585–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, et al Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci 2017; 37: 11406–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Takeda S, Commins C, DeVos SL, Nobuhara CK, Wegmann S, Roe AD, et al Seed‐competent high‐molecular‐weight tau species accumulates in the cerebrospinal fluid of Alzheimer's disease mouse model and human patients. Ann Neurol 2016; 80: 355–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, et al Inert and seed‐competent tau monomers suggest structural origins of aggregation. Elife 2018; 7: 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014; 82: 1271–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 2013; 110: 9535–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer‐Alicea J, Sharma AM, et al Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 2016; 92: 796–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wegmann S, Maury EA, Kirk MJ, Saqran L, Roe A, DeVos SL, et al Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J 2015; 34: 3028–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jackson SJ, Kerridge C, Cooper J, Cavallini A, Falcon B, Cella CV, et al Short fibrils constitute the major species of seed‐competent tau in the brains of mice transgenic for human P301S tau. J Neurosci 2016; 36: 762–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J Biol Chem 2015; 290: 14893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Furman JL, Vaquer‐Alicea J, White CL 3rd, Cairns NJ, Nelson PT, Diamond MI. Widespread tau seeding activity at early Braak stages. Acta Neuropathol 2017; 133: 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaufman SK, Thomas TL, Del Tredici K, Braak H, Diamond MI. Characterization of tau prion seeding activity and strains from formaldehyde‐fixed tissue. Acta Neuropathol Commun 2017; 5: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, et al Evidence for alpha‐synuclein prions causing multiple system atrophy in humans with Parkinsonism. Proc Natl Acad Sci USA 2015; 112: E5308–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Woerman AL, Oehler A, Kazmi SA, Lee J, Halliday GM, Middleton LT, et al Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol 2019; 137: 437–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song ES, et al Parkinson's disease and multiple system atrophy have distinct alpha‐synuclein seed characteristics. J Biol Chem 2019; 294: 1045–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sano K, Atarashi R, Satoh K, Ishibashi D, Nakagaki T, Iwasaki Y, et al Prion‐like seeding of misfolded alpha‐synuclein in the brains of dementia with lewy body patients in RT‐QUIC. Mol Neurobiol 2018; 55: 3916–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Candelise N, Schmitz M, Llorens F, Villar‐Pique A, Cramm M, Thom T, et al Seeding variability of different alpha synuclein strains in synucleinopathies. Ann Neurol 2019; 85: 691–703 [DOI] [PubMed] [Google Scholar]
- 71. Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, et al Ultrasensitive human prion detection in cerebrospinal fluid by real‐time quaking‐induced conversion. Nat Med 2011; 17: 175–8 [DOI] [PubMed] [Google Scholar]
- 72. Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, et al Alpha‐synuclein RT‐QuIC in the CSF of patients with alpha‐synucleinopathies. Ann Clin Transl Neurol 2016; 3: 812–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Groveman BR, Orru CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al Rapid and ultra‐sensitive quantitation of disease‐associated alpha‐synuclein seeds in brain and cerebrospinal fluid by alphaSyn RT‐QuIC. Acta Neuropathol Commun 2018; 6: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Becker K, Wang X, Vander Stel K, Chu Y, Kordower J, Ma J. Detecting alpha synuclein seeding activity in formaldehyde‐fixed msa patient tissue by PMCA. Mol Neurobiol 2018; 55: 8728–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Laferriere F, Maniecka Z, Perez‐Berlanga M, Hruska‐Plochan M, Gilhespy L, Hock EM, et al TDP‐43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci 2019; 22: 65–77 [DOI] [PubMed] [Google Scholar]
- 76. French RL, Grese ZR, Aligireddy H, Dhavale DD, Reeb AN, Kedia N, et al Detection of TAR DNA‐binding protein 43 (TDP‐43) oligomers as initial intermediate species during aggregate formation. J Biol Chem 2019; 294: 6696–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH, et al Full‐length TDP‐43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia‐TDP patients. Nat Commun 2014; 5: 4824 [DOI] [PubMed] [Google Scholar]
- 78. Kao PF, Chen YR, Liu XB, DeCarli C, Seeley WW, Jin LW. Detection of TDP‐43 oligomers in frontotemporal lobar degeneration‐TDP. Ann Neurol 2015; 78: 211–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, et al Patient‐derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP‐43 pathology in vivo. Nat Commun 2018; 9: 4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Babinchak WM, Haider R, Dumm BK, Sarkar P, Surewicz K, Choi JK, et al The role of liquid‐liquid phase separation in aggregation of the TDP‐43 low‐complexity domain. J Biol Chem 2019; 294: 6306–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S, et al Tau protein liquid‐liquid phase separation can initiate tau aggregation. EMBO J 2018; 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matos CO, Passos YM, do Amaral MJ, Macedo B, Tempone M, Bezerra OCL, et al Liquid‐liquid phase separation and aggregation of the prion protein globular domain modulated by a high‐affinity DNA aptamer. bioRxiv 2019: 659037 10.1101/659037 [DOI] [PubMed] [Google Scholar]
- 83. Li HR, Chiang WC, Chou PC, Wang WJ, Huang JR. TAR DNA‐binding protein 43 (TDP‐43) liquid‐liquid phase separation is mediated by just a few aromatic residues. J Biol Chem 2018; 293: 6090–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mezias C, Raj A. Analysis of amyloid‐beta pathology spread in mouse models suggests spread is driven by spatial proximity, not connectivity. Front Neurol 2017; 8: 653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800 [DOI] [PubMed] [Google Scholar]
- 86. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006; 112: 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003; 24: 197–211 [DOI] [PubMed] [Google Scholar]
- 88. Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al Updated TDP‐43 in Alzheimer's disease staging scheme. Acta Neuropathol 2016; 131: 571–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain 2019; 142: 1503–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Forman MS. Genotype‐phenotype correlations in FTDP‐17: does form follow function? Exp Neurol 2004; 187: 229–34 [DOI] [PubMed] [Google Scholar]
- 91. Wadsworth JD, Joiner S, Linehan JM, Cooper S, Powell C, Mallinson G, et al Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease‐resistant wild‐type and mutant prion protein. Brain 2006; 129(Pt 6): 1557–69 [DOI] [PubMed] [Google Scholar]
- 92. Arneson D, Zhang Y, Yang X, Narayanan M. Shared mechanisms among neurodegenerative diseases: from genetic factors to gene networks. J Genet 2018; 97: 795–806 [PMC free article] [PubMed] [Google Scholar]
- 93. Reynolds RH, Botia J, Nalls MA, Hardy J, Gagliano Taliun SA, Ryten M. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson's disease heritability. NPJ Parkinsons Dis. 2019; 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gordon WS. Advances in veterinary research. Vet Rec 1946; 58: 516–25 [PubMed] [Google Scholar]
- 95. Fritschi SK, Cintron A, Ye L, Mahler J, Buhler A, Baumann F, et al Abeta seeds resist inactivation by formaldehyde. Acta Neuropathol 2014; 128: 477–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Woerman AL, Kazmi SA, Patel S, Freyman Y, Oehler A, Aoyagi A, et al MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol 2018; 135: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schweighauser M, Bacioglu M, Fritschi SK, Shimshek DR, Kahle PJ, Eisele YS, et al Formaldehyde‐fixed brain tissue from spontaneously ill alpha‐synuclein transgenic mice induces fatal alpha‐synucleinopathy in transgenic hosts. Acta Neuropathol 2015; 129: 157–9 [DOI] [PubMed] [Google Scholar]
- 98. Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, et al Induction of cerebral beta‐amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci USA 2009; 106: 12926–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild‐type mice. Mol Neurodegeneration 2011; 6: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yang L, Zhang J, Kamelgarn M, Niu C, Gal J, Gong W, et al Subcellular localization and RNAs determine FUS architecture in different cellular compartments. Hum Mol Genet 2015; 24: 5174–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sangwan S, Zhao A, Adams KL, Jayson CK, Sawaya MR, Guenther EL, et al Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA 2017; 114: 8770–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Simoneau S, Rezaei H, Sales N, Kaiser‐Schulz G, Lefebvre‐Roque M, Vidal C, et al In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3: e125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Li S, Jin M, Liu L, Dang Y, Ostaszewski BL, Selkoe DJ. Decoding the synaptic dysfunction of bioactive human AD brain soluble Abeta to inspire novel therapeutic avenues for Alzheimer's disease. Acta Neuropathol Commun 2018; 6: 121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al In vivo demonstration that alpha‐synuclein oligomers are toxic. Proc Natl Acad Sci USA 2011; 108: 4194–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Iljina M, Garcia GA, Horrocks MH, Tosatto L, Choi ML, Ganzinger KA, et al Kinetic model of the aggregation of alpha‐synuclein provides insights into prion‐like spreading. Proc Natl Acad Sci USA 2016; 113: E1206–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by alpha‐synuclein oligomers provides evidence for spreading of alpha‐synuclein pathology. J Neurochem 2009; 111: 192–203 [DOI] [PubMed] [Google Scholar]
- 107. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host‐to‐graft disease propagation. Nat Med 2008; 14: 501–3 [DOI] [PubMed] [Google Scholar]
- 108. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body‐like pathology in long‐term embryonic nigral transplants in Parkinson's disease. Nat Med 2008; 14: 504–6 [DOI] [PubMed] [Google Scholar]
- 109. Nonaka T, Masuda‐Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, et al Prion‐like properties of pathological TDP‐43 aggregates from diseased brains. Cell Rep 2013; 4: 124–34 [DOI] [PubMed] [Google Scholar]
- 110. Olanow CW, Savolainen M, Chu Y, Halliday GM, Kordower JH. Temporal evolution of microglia and alpha‐synuclein accumulation following foetal grafting in Parkinson's disease. Brain 2019; 142: 1690–700 [DOI] [PubMed] [Google Scholar]
- 111. Cuille J. Experimental transmission of trembling to the goat. C R Seances Acad Sci 1939; 208: 1058–160 [Google Scholar]
- 112. Cuille J, Chelle PL. La maladie dite tremblante du mouton est‐elle inoculable? CR Acad Sci 1936; 203: 1552–4 [Google Scholar]
- 113. Cuille J, Chelle PL. La tremblante du mouton estelle determinee par un virus filterable? CR Acad Sci 1938; 1687–8 [Google Scholar]
- 114. Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, et al Evidence for seeding of beta ‐amyloid by intracerebral infusion of Alzheimer brain extracts in beta ‐amyloid precursor protein‐transgenic mice. J Neurosci 2000; 20: 3606–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11: 909–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, et al Pathological alpha‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science 2012; 338: 949–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, et al Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci USA 2013; 110: 19555–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, et al Peripherally applied Abeta‐containing inoculates induce cerebral beta‐amyloidosis. Science 2010; 330: 980–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, Goedert M, et al Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol 2014; 127: 299–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, et al Intramuscular injection of alpha‐synuclein induces CNS alpha‐synuclein pathology and a rapid‐onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA 2014; 111: 10732–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lohmann S, Bernis ME, Tachu BJ, Ziemski A, Grigoletto J, Tamguney G. Oral and intravenous transmission of alpha‐synuclein fibrils to mice. Acta Neuropathol 2019; 138: 515–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pattison IH, Gordon WS, Millson GC. Experimental production of scrapie in goats. J Comp Pathol 1959; 69: 300–12 [DOI] [PubMed] [Google Scholar]
- 123. Chandler RL. Encephalopathy in mice produced by inoculation with scrapie brain material. The Lancet 1961; 1: 1378–9 [DOI] [PubMed] [Google Scholar]
- 124. Brandner S, Jaunmuktane Z. Prion disease: experimental models and reality. Acta Neuropathol 2017; 133: 197–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Comoy EE, Mikol J, Luccantoni‐Freire S, Correia E, Lescoutra‐Etchegaray N, Durand V, et al Transmission of scrapie prions to primate after an extended silent incubation period. Sci Rep 2015; 5: 11573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Recasens A, Dehay B, Bove J, Carballo‐Carbajal I, Dovero S, Perez‐Villalba A, et al Lewy body extracts from Parkinson disease brains trigger alpha‐synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014; 75: 351–62 [DOI] [PubMed] [Google Scholar]
- 127. Beck E, Daniel PM, Alpers M, Gajdusek DC, Gibbs CJ Jr. Experimental "kuru" in chimpanzees a pathological report. The Lancet 1966; 2: 1056–9 [DOI] [PubMed] [Google Scholar]
- 128. Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a Kuru‐like syndrome to chimpanzees. Nature 1966; 209: 794–6 [DOI] [PubMed] [Google Scholar]
- 129. Ridley RM, Baker HF, Windle CP, Cummings RM. Very long term studies of the seeding of beta‐amyloidosis in primates. J Neural Transm 2006; 113: 1243–51 [DOI] [PubMed] [Google Scholar]
- 130. Baker HF, Ridley RM, Duchen LW, Crow TJ, Bruton CJ. Experimental induction of beta‐amyloid plaques and cerebral angiopathy in primates. Ann N Y Acad Sci 1993; 695: 228–31 [DOI] [PubMed] [Google Scholar]
- 131. Narkiewicz J, Giachin G, Legname G. In vitro aggregation assays for the characterization of alpha‐synuclein prion‐like properties. Prion 2014; 8: 19–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lim S, Haque MM, Kim D, Kim DJ, Kim YK. Cell‐based models to investigate tau aggregation. Comput Struct Biotechnol J 2014; 12: 7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Krauss S, Vorberg I. Prions ex vivo: what cell culture models tell us about infectious proteins. Int J Cell Biol 2013; 2013: 704546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, et al Amyloid beta: structure, biology and structure‐based therapeutic development. Acta Pharmacol Sinica 2017; 38: 1205–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hergesheimer RC, Chami AA, de Assis DR, Vourc'h P, Andres CR, Corcia P, et al The debated toxic role of aggregated TDP‐43 in amyotrophic lateral sclerosis: a resolution in sight? Brain 2019; 142: 1176–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Collinge J, Whitfield J, McKintosh E, Frosh A, Mead S, Hill AF, et al A clinical study of kuru patients with long incubation periods at the end of the epidemic in Papua New Guinea. Philos Trans R Soc Lond B Biol Sci 2008; 363: 3725–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Duffy P, Wolf J, Collins G, DeVoe AG, Streeten B, Cowen D. Letter: possible person‐to‐person transmission of Creutzfeldt‐Jakob disease. N Engl J Med 1974; 290: 692–3 [PubMed] [Google Scholar]
- 138. Walsh DM, Selkoe DJ. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci 2016; 17: 251–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Duyckaerts C, Sazdovitch V, Ando K, Seilhean D, Privat N, Yilmaz Z, et al Neuropathology of iatrogenic Creutzfeldt‐Jakob disease and immunoassay of French cadaver‐sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol 2018; 135: 201–12 [DOI] [PubMed] [Google Scholar]
- 140. Simpson DA, Masters CL, Ohlrich G, Purdie G, Stuart G, Tannenberg AE. Iatrogenic Creutzfeldt‐Jakob disease and its neurosurgical implications. J Clin Neurosci 1996; 3: 118–23 [DOI] [PubMed] [Google Scholar]
- 141. Preusser M, Strobel T, Gelpi E, Eiler M, Broessner G, Schmutzhard E, et al Alzheimer‐type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt‐Jakob disease after dural grafting. J Neurol Neurosurg Psychiatry 2006; 77: 413–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Purro SA, Farrow MA, Linehan J, Nazari T, Thomas DX, Chen Z, et al Transmission of amyloid‐beta protein pathology from cadaveric pituitary growth hormone. Nature 2018; 564: 415–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. De Pablo‐Fernandez E, Cerdan Santacruz D, Warner TT, Holton JL. No evidence of iatrogenic human transmission in autopsy confirmed multiple system atrophy. Move Disord 2018; 33: 1183–4 [DOI] [PubMed] [Google Scholar]
- 144. Rajput AH, Ferguson LW, Robinson CA, Guella I, Farrer MJ, Rajput A. Conjugal parkinsonism ‐ Clinical, pathology and genetic study. No evidence of person‐to‐person transmission. Parkinsonism Relat Disord 2016; 31: 87–90 [DOI] [PubMed] [Google Scholar]
- 145. Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, et al A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun 2015; 6: 7025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Takatsuki H, Satoh K, Sano K, Fuse T, Nakagaki T, Mori T, et al Rapid and quantitative assay of amyloid‐seeding activity in human brains affected with prion diseases. PLoS One 2015; 10: e0126930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Smith PG, Bradley R. Bovine spongiform encephalopathy (BSE) and its epidemiology. Br Med Bull 2003; 66: 185–98 [DOI] [PubMed] [Google Scholar]
- 148. Mok T, Jaunmuktane Z, Joiner S, Campbell T, Morgan C, Wakerley B, et al Variant Creutzfeldt‐Jakob disease in a patient with heterozygosity at PRNP Codon 129. N Engl J Med 2017; 376: 292–4 [DOI] [PubMed] [Google Scholar]
- 149. Asante EA, Linehan JM, Gowland I, Joiner S, Fox K, Cooper S, et al Dissociation of pathological and molecular phenotype of variant Creutzfeldt‐Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc Natl Acad Sci USA 2006; 103: 10759–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Pollanen MS, Onzivua S, Robertson J, McKeever PM, Olawa F, Kitara DL, et al Nodding syndrome in Uganda is a tauopathy. Acta Neuropathol 2018; 136: 691–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Tai XY, Koepp M, Duncan JS, Fox N, Thompson P, Baxendale S, et al Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 2016; 139(Pt 9): 2441–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Johnson TP, Tyagi R, Lee PR, Lee MH, Johnson KR, Kowalak J, et al Nodding syndrome may be an autoimmune reaction to the parasitic worm Onchocerca volvulus. Sci Transl Med 2017; 9: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Jaunmuktane Z, Brandner S. Transmissible human proteopathies: an expanding field. Diagn Histopathol 2019; 25: 16–22 [Google Scholar]
- 154. Blankaart S. The physical dictionary: wherein the terms of anatomy, the names and causes of diseases, chirurgical instruments, and their use, are accurately described. as also the names and virtues of medicinal plants, minerals, stones, gums, salts, earths &c. The Method of Chusing the Best Drugs: the Terms of Chymistry, and of the Apothecary's Art: the Various Forms of Medicines, and the Ways of Compounding Them1726.
- 155. Alzheimer A. Über einen eigenartigen schweren Erkrankungsprozeβ der Hirnrincle. Neurol Central 1906; 25: 146–8. [Google Scholar]
- 156. Hippius H, Neundorfer G. The discovery of Alzheimer's disease. Dialogues Clin Neurosci 2003; 5: 101–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kraepelin E. Psychiatrie. 8 ed, 1910. [Google Scholar]
- 158. Pick A. Ueber die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr 1892; 17: 165–7 [Google Scholar]
- 159. Von Gans A. Betrachtungen fiber Art und Ausbreitung des krankhaften Prozesses in einem Fall yon Pickscher Atrophie des Stirnhirns. 1922.
- 160. Onari K, Spatz H. Anatomische beitrage zur lehre von der pickschen umschriebenen grosshirnrinden‐ atrophie (“Picksche Krankheit”). Z Ges Neurol Psychiatr 1926; 101: 470–511 [Google Scholar]
- 161. Richardson JC, Steele J, Olszewski J. Supranuclear ophthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of "heterogenous system degeneration". Trans Am Neurol Assoc 1963; 88: 25–9 [PubMed] [Google Scholar]
- 162. Rebeiz JJ, Kolodny EH, Richardson EP Jr. Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc 1967; 92: 23–6 [PubMed] [Google Scholar]
- 163. Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol 1982; 11: 592–8 [DOI] [PubMed] [Google Scholar]
- 164. Parkinson J. An essay on the shaking palsy: Whittingham and Rowland, 1817.
- 165. Sanders WR. Case of an unusual form of nervous disease, dystaxia or pseudo‐paralysis agitans, with remarks. Edinb Med J 1865; 987–97 [PMC free article] [PubMed] [Google Scholar]
- 166. Goedert M, Compston A. Parkinson's disease ‐ the story of an eponym. Nat Rev Neurol 2018; 14: 57–62 [DOI] [PubMed] [Google Scholar]
- 167. Dejerine JJ, Thomas A. L’atrophie olivo‐ponto‐cérébelleuse. Paris: Nouvelle iconographie de la Salpêtrière, 1900; 330–70 [Google Scholar]
- 168. Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: a clinical‐pathologic study. Arch Neurol 1960; 2: 511–27 [DOI] [PubMed] [Google Scholar]
- 169. Adams RD, van Bogaert L, van der Eecken H. Dégénérescence nigrostriée ét cerebello‐nigro‐striée (Unicité clinique et variabilité pathologique des dégénérescences préséniles à forme de rigidité extrapyramidale). Psychiat et Neurol 1961; 142: 219–59 [PubMed] [Google Scholar]
- 170. Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry 1969; 32: 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Charcot J‐M. Sclerose des cordons lateraux de la moelle epiniere chez une femme hyste rique atteinte de contracture permanente des quatre membres. Bull Soc Med Paris 1865: 24–35 [Google Scholar]
- 172. Charcot J‐M. Amyotrophies spinales deuteropathiques sclerose laterale amyotrophique. Bureaux du Prog Med 1874; 2: 234–48 [Google Scholar]
- 173. Barrett AM. A case of Alzheimer's disease with unusual neurological disturbances. J Nerv Ment Dis 1913; 40: 361–74 [Google Scholar]
- 174. Masters CL, Gajdusek DC, Gibbs CJ Jr. Creutzfeldt‐Jakob disease virus isolations from the Gerstmann‐Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus‐induced spongiform encephalopathies. Brain 1981; 104: 559–88 [DOI] [PubMed] [Google Scholar]
- 175. Creutzfeldt HG. Uber eine eigenartige herdformige Erkrankung des Zentralnervensystems. Z Ges Neurol Psychiat 1920; 57: 1–18 [Google Scholar]
- 176. Jakob A. Uber eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswerten anatomischen Befunde (Spastische Pseudosklerose‐Encephalomyelopathie mit disseminierten Degenerations‐herden). Z Ges Neurol Psychiat 1921; 64: 147–228 [Google Scholar]
- 177. Spielmeyer W. Histopathologie des Nervensystems. Berlin: J. Springer, 1922. [Google Scholar]
- 178. Spielmeyer W. Die histopathologische Forschung in der Psychiatrie. Klin Wochenschr 1922; 1817–9 [Google Scholar]
- 179. Von Heidenhain A. Klinische und anatomische Untersuchungen fiber eine eigenartige organische Erkrankung des Zentralnervensystems im Praesenium. 1928.
- 180. Gerstmann J, Straussler E, Scheimker I. Über eine eigenartige hereditär‐ familiäre Erkrankung des Zentralnervensystems. Z Gesamte Neurol Psychiatr 1935; 154: 736–62 [DOI] [PubMed] [Google Scholar]
- 181. Gerstmann J. Ober ein noch nicht beschriebenes Reflexphanomen bei einer Erkrankung deszerebellaren Systems. Wein Med Wochenschr 1928; 78: 906–8 [Google Scholar]
- 182. Von Braunmuhl AE. Ober eine eigenartige hereditar‐familiare Erkrankung des Zentralnerven‐systems. Arch Psychiatr vereinigt mil Zeitschr Neurol 1954; 419–49 [DOI] [PubMed] [Google Scholar]
- 183. Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allg Zschr Psychiatr Psych Gerichtl Med 1907; 64: 146–8 [Google Scholar]
- 184. Alzheimer A. Uber eigenartige Krankheitsfalle des spateren Alters. 1911.
- 185. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 10: 333–59 [DOI] [PubMed] [Google Scholar]
- 186. Hauw JJ, Verny M, Delaere P, Cervera P, He Y, Duyckaerts C. Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer's disease and aging. Neurosci Lett 1990; 119: 182–6 [DOI] [PubMed] [Google Scholar]
- 187. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al Association of missense and 5'‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 1998; 393: 702–5 [DOI] [PubMed] [Google Scholar]
- 188. Lewy FH. Paralysis agitans, Handbuch der Neurologie. Berlin: Julius Springer, 1912. [Google Scholar]
- 189. Lafora GR. Contribución á la histopatologia de la parálises agitante. Trab Lab Invest Biol Univers Madrid 1913; 11: 43–54 [Google Scholar]
- 190. Engelhardt E. Lafora and Tretiakoff: the naming of the inclusion bodies discovered by Lewy. Arq Neuropsiquiatr 2017; 75: 751–3 [DOI] [PubMed] [Google Scholar]
- 191. Tretiakoff K. Contribution a l'étude de l'anatomie pathologique du Locus Niger de Soemmering avec quelques déductions relative à la pathogénie des troubles du tonus muscularaire et de la maladie de Parkinson. These de Paris.1919.
- 192. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy‐Drager syndrome). J Neurol Sci 1989; 94: 79–100 [DOI] [PubMed] [Google Scholar]
- 193. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71: 670–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA 1975; 72: 1858–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 1986; 83: 4913–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 11282–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Ou SH, Wu F, Harrich D, Garcia‐Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP‐43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 1995; 69: 3584–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Goate A, Chartier‐Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991; 349: 704–6 [DOI] [PubMed] [Google Scholar]
- 199. Chartier‐Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, et al Early‐onset Alzheimer's disease caused by mutations at codon 717 of the beta‐amyloid precursor protein gene. Nature 1991; 353: 844–6 [DOI] [PubMed] [Google Scholar]
- 200. Naruse S, Igarashi S, Kobayashi H, Aoki K, Inuzuka T, Kaneko K, et al Mis‐sense mutation Val––Ile in exon 17 of amyloid precursor protein gene in Japanese familial Alzheimer's disease. Lancet 1991; 337: 978–9 [DOI] [PubMed] [Google Scholar]
- 201. Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science 1991; 254: 97–9 [DOI] [PubMed] [Google Scholar]
- 202. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature 1995; 375: 754–60 [DOI] [PubMed] [Google Scholar]
- 203. Levy‐Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 1995; 269: 973–7 [DOI] [PubMed] [Google Scholar]
- 204. Strittmatter WJ, Saunders AM, Schmechel D, Pericak‐Vance M, Enghild J, Salvesen GS, et al Apolipoprotein E: high‐avidity binding to beta‐amyloid and increased frequency of type 4 allele in late‐onset familial Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 1977–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Saunders AM, Strittmatter WJ, Schmechel D, George‐Hyslop PH, Pericak‐Vance MA, Joo SH, et al Association of apolipoprotein E allele epsilon 4 with late‐onset familial and sporadic Alzheimer's disease. Neurology 1993; 43: 1467–72 [DOI] [PubMed] [Google Scholar]
- 206. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science 1997; 276: 2045–7 [DOI] [PubMed] [Google Scholar]
- 207. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392: 605–8 [DOI] [PubMed] [Google Scholar]
- 208. Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, et al DJ‐1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol Sci 2003; 24: 159–60 [DOI] [PubMed] [Google Scholar]
- 209. Valente EM, Abou‐Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158–60 [DOI] [PubMed] [Google Scholar]
- 210. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 2004; 44: 601–7 [DOI] [PubMed] [Google Scholar]
- 211. Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, et al Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat Genet 2006; 38: 1184–91 [DOI] [PubMed] [Google Scholar]
- 212. Zimprich A, Benet‐Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al A mutation in VPS35, encoding a subunit of the retromer complex, causes late‐onset Parkinson disease. Am J Hum Genet 2011; 89: 168–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362: 59–62 [DOI] [PubMed] [Google Scholar]
- 214. Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, et al A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 2001; 29: 166–73 [DOI] [PubMed] [Google Scholar]
- 215. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet 2004; 36: 377–81 [DOI] [PubMed] [Google Scholar]
- 216. Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, et al DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004; 74: 1128–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Nishimura AL, Mitne‐Neto M, Silva HC, Richieri‐Costa A, Middleton S, Cascio D, et al A mutation in the vesicle‐trafficking protein VAPB causes late‐onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 2004; 75: 822–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004; 63: 724–6 [DOI] [PubMed] [Google Scholar]
- 219. Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al Mutations in the endosomal ESCRTIII‐complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005; 37: 806–8 [DOI] [PubMed] [Google Scholar]
- 220. Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, et al ANG mutations segregate with familial and 'sporadic' amyotrophic lateral sclerosis. Nat Genet 2006; 38: 411–3 [DOI] [PubMed] [Google Scholar]
- 221. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006; 442: 916–9 [DOI] [PubMed] [Google Scholar]
- 222. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006; 442: 920–4 [DOI] [PubMed] [Google Scholar]
- 223. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319: 1668–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323: 1208–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, et al Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 2009; 84: 85–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Luty AA, Kwok JB, Dobson‐Stone C, Loy CT, Coupland KG, Karlstrom H, et al Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration‐motor neuron disease. Ann Neurol 2010; 68: 639–49 [DOI] [PubMed] [Google Scholar]
- 227. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010; 465: 223–6 [DOI] [PubMed] [Google Scholar]
- 228. Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, et al Familial amyotrophic lateral sclerosis is associated with a mutation in D‐amino acid oxidase. Proc Natl Acad Sci USA 2010; 107: 7556–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Renton AE, Majounie E, Waite A, Simon‐Sanchez J, Rollinson S, Gibbs JR, et al A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 2011; 72: 257–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 2011; 72: 245–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 2011; 477: 211–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, et al SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012; 79: 1556–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, et al Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012; 488: 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Bannwarth S, Ait‐El‐Mkadem S, Chaussenot A, Genin EC, Lacas‐Gervais S, Fragaki K, et al A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014; 137(Pt 8): 2329–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, et al Exome‐wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 2014; 84: 324–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, et al Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 2014; 17: 664–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, et al Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nat Neurosci 2015; 18: 631–6 [DOI] [PubMed] [Google Scholar]
- 238. van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al Genome‐wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 2016; 48: 1043–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, et al NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet 2016; 48: 1037–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, et al CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun 2016; 7: 11253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, et al TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 2017; 95: 808–816.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Smith BN, Topp SD, Fallini C, Shibata H, Chen HJ, Troakes C, et al Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci Transl Med 2017; 9: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Brenner D, Yilmaz R, Muller K, Grehl T, Petri S, Meyer T, et al Hot‐spot KIF5A mutations cause familial ALS. Brain 2018; 141: 688–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, et al Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46: 417–28 [DOI] [PubMed] [Google Scholar]
- 245. Kitamoto T, Tateishi J, Sawa H, Doh‐Ura K. Positive transmission of Creutzfeldt‐Jakob disease verified by murine kuru plaques. Lab Invest 1989; 60: 507–12. [PubMed] [Google Scholar]
- 246. Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N Engl J Med 1957; 257: 974–8 [DOI] [PubMed] [Google Scholar]