

Abstract
Inherited cutis laxa, or inelastic, sagging skin is a genetic condition of premature and generalised connective tissue ageing, affecting various elastic components of the extracellular matrix. Several cutis laxa syndromes are inborn errors of metabolism and lead to severe neurological symptoms. In a patient with cutis laxa, a choreoathetoid movement disorder, dysmorphic features and intellectual disability we performed exome sequencing to elucidate the underlying genetic defect. We identified the amino acid substitution R275W in phosphatidylinositol 4‐kinase type IIα, caused by a homozygous missense mutation in the PI4K2A gene. We used lipidomics, complexome profiling and functional studies to measure phosphatidylinositol 4‐phosphate synthesis in the patient and evaluated PI4K2A deficient mice to define a novel metabolic disorder. The R275W residue, located on the surface of the protein, is involved in forming electrostatic interactions with the membrane. The catalytic activity of PI4K2A in patient fibroblasts was severely reduced and lipid mass spectrometry showed that particular acyl‐chain pools of PI4P and PI(4,5)P2 were decreased. Phosphoinositide lipids play a major role in intracellular signalling and trafficking and regulate the balance between proliferation and apoptosis. Phosphatidylinositol 4‐kinases such as PI4K2A mediate the first step in the main metabolic pathway that generates PI4P, PI(4,5)P2 and PI(3,4,5)P3. Although neurologic involvement is common, cutis laxa has not been reported previously in metabolic defects affecting signalling. Here we describe a patient with a complex neurological phenotype, premature ageing and a mutation in PI4K2A, illustrating the importance of this enzyme in the generation of inositol lipids with particular acylation characteristics.
Keywords: choreoathetosis, cutis laxa, inborn error of metabolism, movement disorder, neurocutaneous disorder, neurometabolism, phosphatidyl inositol, PI4K2A
1. INTRODUCTION
Phosphoinositide lipids coordinate several crucial cellular processes, including vesicular trafficking and intracellular signalling. 1 , 2 Rapid changes in specific cellular pools of phosphoinositide lipids are generated by multiple phosphoinositide kinases, phospholipases and phosphatases. 3
The four isoenzymes that constitute the phosphatidylinositol (PI) 4‐kinases (PI4Ks) catalyse the initial step in the pathways that generates PI(4,5)P2 and PI(3,4,5)P3 by phosphorylating PI at the D4 position to produce PI 4‐phosphate (PI4P). 3 , 4 The four isoenzymes are PI4K2A, PI4K2B, PI4KA and PI4KB (Figure 1). Although the product is always PI4P, the isoforms are targeted to different organelles and show tissue specificity. 4 , 5 This study focuses on the PI 4‐kinase isoform 2‐alpha (PI4K2A), encoded by PI4K2A, which functions in endosomal‐lysosomal trafficking, endosomal exocytosis, Akt activation, packaging of von Willebrand factor in Weibel‐Palade bodies, autophagosome:lysosome fusion and the regulation of sphingomyelin synthesis at the trans‐Golgi network and the trafficking of lysosomal enzymes, including glucocerebrosidase (GBA), to the lysosome. 6 , 7 , 8 , 9 , 10 Despite the fact that cellular PI4P levels are maintained by four different pathways that generate PI 4‐kinase activities, reductions in PI4P generation due to loss of function mutations affecting one of the isozymes can be sufficient to cause neurological disease.
FIGURE 1.
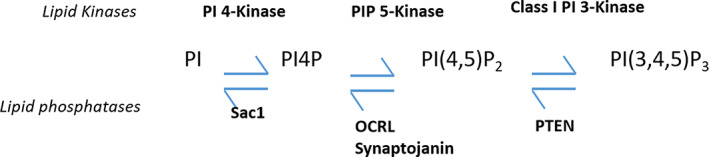
PI 4‐kinases catalyse the ATP‐dependent D4 phosphorylation of PI to generate PI4P. PI4K2A is one of the four mammalian PI 4‐kinase isozymes and it localises mainly to the TGN and endosomes. Both PI4K2A and its PI4P product can bind and recruit proteins required for the formation of trafficking vesicles on intracellular membranes. PI4P can subsequently be phosphorylated by PIP 5‐kinases to form PI(4,5)P2 an important signalling and trafficking molecule. PI(4,5)P2 is in turn a substrate for class I phosphoinositide 3‐kinases that generate PI(3,4,5)P3; however, PI4K2A activity is not thought to be rate limiting for PI(3,4,5)P3 production. Dephosphorylation routes to form PI and PI4P, and the phosphoinositide phosphatases that can potentially catalyse these reactions have also been included
Mutations of the PI4KA activating protein FAM126A (hyccin) result in a 10% to 20% decrease in total cellular PI4P and are associated with a familial leukodystrophy condition featuring hypomyelination and congenital cataract. 11 Interestingly, mutations in PI4KA leading to loss of catalytic activity cause structural brain abnormalities including perisylvian polymicrogyria and cerebellar hypoplasia. 12
These insights suggest that reduced PI4P generation can be deleterious for the structural development of the CNS, but also that diminished PI 4‐kinase activity results in different, mutation‐specific, clinical presentations.
Multiple lines of evidence suggest that alterations in PI4K2A expression can cause disease.
Gene trap Pi4k2a knockout mice exhibit delayed onset neurodegeneration. Mice homozygous for a Pi4k2a gene trap allele develop normally and appear normal as young adults, but exhibit an age‐related neurodegeneration phenotype, consisting of tremor, spastic paraplegia and urinary incontinence. 13
In terms of human disease, changes in PI4K2A expression, possibly due to gene copy number variations, have been reported in breast cancer 14 , 15 and advanced glioblastoma. 16 Recently two siblings, with complete loss of function of PI4K2A, leading to intellectual disability, epilepsy, myoclonus and akathisia, have been described. 17
2. CLINICAL DESCRIPTION
The female patient was born as the fifth and youngest child of non‐consanguineous and healthy Malay parents. Pregnancy and delivery were uncomplicated. Features noted at the age of 12 months included microcephaly, marked hypotonia in an infant with poor muscle volume, wasting of subcutaneous fat and hepatomegaly. Development was globally delayed, with marked intellectual disability. Biochemical evaluations revealed lactic acidemia, 4.2 mmol/L (normal 0‐2.4), ALAT 94 U/L (normal 0‐29), ASAT 260 U/L (normal 0‐36), total bilirubin 38 μmol/L (normal 0‐17), AP 230 U/L (normal 0‐341); yGT 328 U/L (normal 11‐50) and elevation of serum alpha‐fetoprotein. Her serum albumin, uric acid, ammonia, creatine kinase and blood indices were normal. Plasma amino acid analysis showed raised alanine, 606 μmol/L (normal 148‐475) and mildly elevated methionine, 52 μmol/L (normal 5‐34). Urine organic acid analysis showed lactaciduria and presence of phenyl‐derivatives indicating liver impairment.
Dysmorphic features were noted including a mild progeroid appearance with high forehead, large ears, sparse scalp hair, strabismus, hypertelorism, epicanthal folds, blue sclera, flat nasal bridge, bulbous nose, long, prominent philtrum and a high palate (Figure 2). Cutis laxa was displayed with loose and inelastic skin. She had increased laxity of her joints and choreoathetosis with continuous involuntary movements of her limbs and facial musculature. Cerebral MRI revealed delayed cerebral myelination and corpus callosum dysplasia. Her liver function stabilised, and she made slow developmental progress and is now stable at the age of 12 years. Mild lactic acidaemia has persisted, however OXPHOS complex measurements in fibroblasts revealed no decreased enzyme activities or decreased ATP production. mtDNA depletion in fibroblasts was excluded. Transferrin and apolipoprotein CIII isoelectric focusing showed normal N‐ and O‐glycosylation of proteins.
FIGURE 2.

Clinical phenotype of the patient at the age of 6 years old. A, The patient, due to severe hypotonia, is unable to sit up without assistance. B and C, The choreoathetoid movement disorder in combination with hypotonia. D, Several dysmorphic features including a high forehead, hypertelorism, a prominent philtrum, bitemporal narrowing, hypoplastic bridge of the nose, up‐turning nose, long philtrum, thin upper lip, prominent jaw, prominent ears and strabismus
During a physical evaluation at the age of 6 years old the patient demonstrated almost continuous movements of the face and upper extremities. Large amplitude, almost ballistic, movements at the pelvic girdle, and to a lesser degree at the shoulder girdle were noted. In addition, there was frequent facial grimacing with irregular opening of the mouth, partial protrusions of the tongue, opening and closing of the eyes and elevation of the eyebrows. There was posturing of the hands whereby the patient exhibited abduction and adduction of the wrists with a tendency to flex at the wrist, hyperextend at the metacarpophalangeal joints and to flex at the interphalangeal joints in a continuously changing pattern. The feet are intermittently assuming a posture of inversion and flexion of the feet with writhing movements of the toes. The patient demonstrated hyperextension at the elbows and the spine when she sits up and then assumes a partially prone position, with hyperabduction of the hips. There is swaying of the trunk from side to side. There is also intermittent esotropia bilaterally. The findings are consistent with generalised choreoathetosis, associated with joint hypermobility and hypotonia.
Her neurological phenotype remains the same as at the age of 6 years old and she is in stable condition. Mild lactic acidemia has persisted, however OXPHOS complex measurements in fibroblasts revealed no decreased enzyme activities or decreased ATP production. mtDNA depletion in fibroblasts was excluded. Transferrin and apolipoprotein CIII isoelectric focusing showed normal N‐ and O‐glycosylation of proteins.
3. HISTOLOGY
Elastica‐van Gieson (EVG) staining of a skin biopsy revealed almost absent elastin in the papillary dermis and fragmented elastin was visualised in the reticular dermis, confirming the diagnosis of cutis laxa in patient 1 (not shown).
4. METHODS
See Supporting Information files.
5. RESULTS
5.1. Genetic data
Exome sequencing of DNA obtained from the index patient was performed and provided 6.3 Gb of mapped sequencing data, resulting in a median coverage of the exome of 71.5‐fold with at least 20‐fold coverage for 78.3% of the exome. Standard variant filtering resulted in 233 rare, non‐synonymous and canonical splice site variants of high quality. From these 233 variants, we selected the candidate gene variants filtering for gene function, nucleotide conservation and bi‐allelic presence (both homozygous and compound heterozygous). This resulted in two genetic variants, of 39 potential variants that were highly conserved (PhyloP>3.5) in PI4K2A and TWNK (PEO1, TWINKLE, C10orf2 or TWINKLE mtDNA helicase) based on gene function. A homozygous missense mutation was found in PI4K2A, located on chromosome 10. The 823C>T mutation at genomic position 99 416 632 resulted in a R275W amino acid change. For TWNK, also located on chromosome 10, a homozygous missense mutation 1306G>C was found, leading to a G436R amino acid change at position 102 749 463.
The corresponding proteins are known to play a role in metabolic pathways. The variants in these two genes co‐segregated with disease within the family. According to in silico prediction programmes such as SIFT, PolyPhen and MutationTaster, the identified variants were predicted to be ‘deleterious’, ‘probably damaging’ and ‘disease causing’, respectively. Both variants have not been reported in public databases. Both genes have no constraint for missense variants in the GnomAD. Additional assessment of 40 ethnically matched controls showed that both variants were not present.
Additional analysis of PI4K2A and TWNK in 27 patients with cutis laxa and a neurological phenotype revealed no pathogenic variants.
6. FUNCTIONAL STUDIES
6.1. PI4P protein levels
Since decreased PI4P generation could potentially arise due to decreased expression of the PI4K2A enzyme in patient cells, we used Western blotting to compare protein levels in patient vs control cells. We found that PI4K2A expression was not decreased in the patient fibroblasts compared to controls. Instead, the enzyme appeared upregulated when the data were corrected for the loading control, calnexin (Figure 3B).
FIGURE 3.
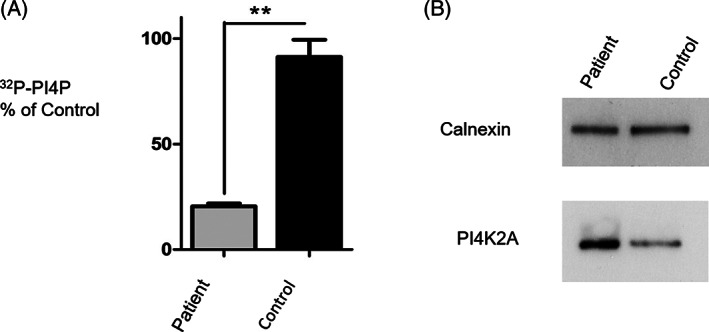
PI 4‐kinase activity measurement in whole cell homogenates prepared from patient fibroblasts. A, Analysis of the catalytic activity of PI4K2A shows that cellular PI4P synthesis is inhibited (**P < .01, n = 3) by 78% in patient cells that express only PI4K2A (R275W). B, Western blots demonstrating that PI4K2A expression is not reduced in patient cells that express the PI4K2A (R275W) variant. Calnexin is used as a protein loading control. The experiment was repeated 3 times with similar results
6.2. Effects of the R275W mutation on PI 4‐kinase activity
To determine whether the R275W mutation impaired PI4K2A function, we measured PI 4‐kinase activity in homogenates prepared from control and patient fibroblasts using our previously established radiometric assay that reports the generation of radiolabelled 32P‐PI4P by this isozyme. 18 PI4P generation was found to be reduced by 77.5 ± 0.5% in patient fibroblasts vs control cells (Figure 3A).
6.3. Effects of the PI4K2A mutation on phosphoinositide levels
The large reduction in PI 4‐kinase activity in patient cells prompted us to investigate whether there were any consequences for cellular phosphoinositide levels, as previous RNAi studies had indicated that PI4K2A activity maintains approximately 10% to 20% of the total cellular PI4P pool. 19 To this end, HPLC‐coupled to mass spectrometry was employed to separate and quantify the different acyl chain variants of PI, PIP, PIP2 and PI(3,4,5)P3 in control and patient cells (Figure 4). This approach revealed that in patient cells the level of the PI4K2A lipid substrate PI was not decreased implying that any reduction in catalytic activity was not due to a generalised depletion of the enzyme's lipid substrate (Figure 4A). Results for PIP, which predominately reports PI4P and, to a lesser degree, the much rarer monophosphorylated PI3P and PI5P lipids, 20 revealed that the 18:0/20:4 and 18:1/18:1 PIP species were reduced in patient fibroblasts by 21% and 17%, respectively and unexpectedly that there was a 32% increase in the very minor 18:0/20:2 PIP pool (Figure 4B,C).
FIGURE 4.
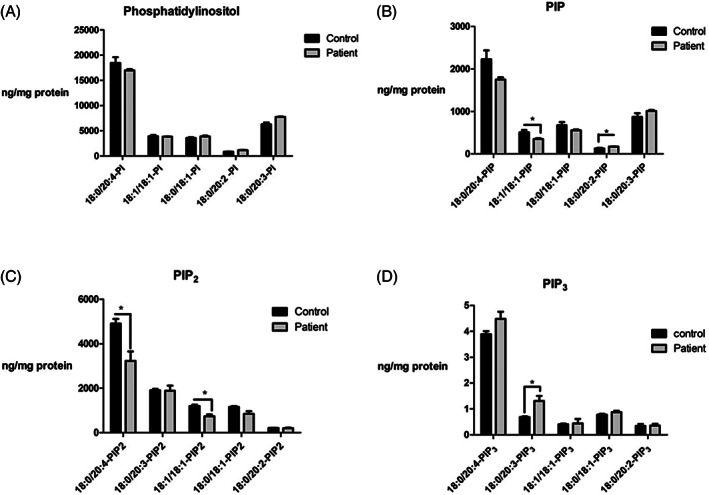
HPLC‐coupled mass spec analysis of different acyl chain inositol phospholipid species from control and patient cells. Mass levels of A, PI; B, PIP; C, PIP2 and D, PIP3 were measured. Data are presented as the mean ± SEM of triplicate determinations, *P ≤ .05
It should be noted though that the mean 21% decrease in 18:0/20:4 PIP was not found to be statistically significant using the unpaired Student's t‐test when the significance threshold was set at P ≤ 0.05. These results indicate that loss of PI4K2A activity only affects particular molecular pools of PI4P (Figure 4B). Extending this approach to the downstream lipid PIP2 which mainly reports PI(4,5)P2, revealed a greater impact of the R275W PI4K2A mutation with a 34% reduction in 18:0/20:4 PIP2 and a 38% decrease in 18:1/18:1 PIP2 (Figure 4C). Finally, mass spectrometric analysis of the quantitatively rare but functionally important phosphoinositide PI(3,4,5)P3 showed little overall change in patient cells except for the 18:0/20:3 structural pool that exhibited a marked 93% increase (Figure 4D).
6.4. Predicted effects of mutations on protein structure
We studied the effect of the R275W mutation using the crystal structure of the catalytic domain of PI4K2A. R275 is located on the surface of the protein, where it directly interacts with the membrane. Supporting Information Figure S1 shows the position of this residue in the octameric complex. Baumlova et al suggested that this arginine is responsible for electrostatic interactions with the lipid headgroups. 21 The R275W mutation, converting arginine into tryptophan, leads to an amino acid with a much larger hydrophobic, but electrostatically neutral amino acid sidechain (Supporting Information Figure S1). Therefore, we predict that mutation R275W will affect membrane anchoring of the PI4K2A protein.
6.5. Complexome profiling
To assess the effect of the mutations in PI4K2A and a possible involvement of TWNK, whole cells and mitochondria enriched fractions from patient and control fibroblasts were analysed by complexome profiling. 22 Complexome profiling enables the investigation of (sub)complex abundance and composition of protein complexes.
To examine whether the mutation in TWNK results in a decreased abundance or an impaired assembly of OXPHOS complexes, mitochondria enriched fractions from patient and control fibroblasts were analysed. Supporting Information Figure S2A shows a heat map representation of the individual migration patterns of all OXPHOS complex subunits identified by complexome profiling. To compare the overall migration pattern of the different complexes, the average abundance profile of all subunits was plotted (Supporting Information Figure S2B). In the experiment shown in Supporting Information Figure S2B, the nuclear encoded subunits SDHC and SDHD gave rise to an additional peak in the complex II profile, we did not observe this subassembly in the complexome profile obtained from whole cells (not shown). In patient mitochondria, the abundance and composition of OXPHOS complexes and supercomplexes comprising mitochondrially encoded subunits was not significantly different from that of the control indicating that the mutation in TWNK had no significant effect on mitochondrial protein biosynthesis, at least in fibroblast.
6.6. TGN‐lysosome trafficking of GBA
Given the potential abnormalities of the lysosomal β‐glucosidase GBA during PI4K2A deficiency, we studied in detail the enzyme in the patient's fibroblasts by a variety of methods.
We examined GBA protein by Western blot analysis using a specific monoclonal antibody. 23 No difference between GBA in patient and control fibroblast were revealed (Supporting Information Figure S3), both showing low (58 kDa) molecular weight species known to coincide with GBA maturation in lysosomes. 24 , 25 Next we measured total GBA activity in wildtype and patient fibroblasts. The levels of activity towards 4‐methylumbelliferyl‐β‐glucoside were comparable: 318 ± 18 nmol/h/mg and 327 ± 16 nmol hydrolysis/h/mg protein in patient and wildtype fibroblasts, respectively.
Finally, we employed activity‐based probes (ABPs) consisting of a cyclophellitol β‐epoxide extended with a BODIPY fluorophore. 26 The ABPs specifically and irreversibly label the nucleophile (glutamate 340) of GBA. They are cell‐permeable and allow the labelling of active GBA molecules in intact cells, which can subsequently be visualised by SDS‐PAGE and fluorescence scanning. 26 Patient and control fibroblasts were incubated with ABP‐BODIPY‐Red to allow labelling of the enzyme in the cells. Next, cells were lysed and the lysate was incubated with ABP‐BODIPY‐Green to label any unreacted GBA. No abnormalities in GBA labelled fibroblast, in cells or in cell lysates, were detected (Supporting Information Figure S3). Finally, we looked for abnormalities in glucosylsphingosine, the sphingoid base formed from glucosylceramide during GBA deficiency in Gaucher disease. No excessive glucosylsphingosine was detected in the patient fibroblasts (not shown), suggesting that the cells contain sufficient functional GBA to degrade glucosylceramide.
7. DISCUSSION AND CONCLUSIONS
The genetic elucidation of novel metabolic cutis laxa syndromes, although recently aided by next generation sequencing, still proves to be a challenge. Analysis of the variants can be complicated and, in most cases, more than one candidate gene was identified. Elaborate functional studies are then needed to assess the pathogenicity of the different variants. In the patient analysed here, homozygous and compound heterozygous missense mutations were found that were predicted to be pathogenic. Study of the cellular function lead to prioritisation of two variants.
The pathogenicity of the R275W mutation in PI4K2A evidently resulted from a 78% reduction in enzyme activity and the decrease of particular acyl species of PI4P and PI(4,5)P2 (Figure 3). Moreover, we demonstrate that this decrease in PI4K2A enzyme activity in patient fibroblasts was not due to PI substrate depletion or reduced enzyme expression (Figure 4). Our results are in concordance with the cellular studies conducted by Zhou et al 27 and Baumlova et al 21 where point substitution mutations of R275 caused an approximately 80% reduction in the kinase activity of PI4K2A. Our results are comparable to those reported from Pi4k2a knockout mice, where loss of the enzyme resulted in >95% decrease in total activity in fibroblasts and >95% decrease in brain membrane associated‐activity. 13 These mice showed a movement disorder, severe wasting of subcutaneous fat and no decrease in elastic fibres in skin. Moreover, the percentage decreases in the steady state cellular levels of PIP and PIP2 that we observed are of a similar magnitude as those previously determined in RNAi studies where PI4K2A expression was ablated in cultured cells. 19 Therefore, our biochemical analyses demonstrate that the R275W mutation inhibits the lipid kinase activity of PI4K2A. Recently published descriptions of the PI4K2A crystal structure revealed that the positively charged R275 residue is situated outside the catalytic substrate binding pocket and on a surface region of the enzyme, which is involved in maintaining tight association with anionic membrane lipids. 21 Substitution of arginine by a bulky and non‐polar tryptophan residue is predicted to inhibit non‐covalent interactions of the enzyme surface with anionic membrane lipids, thus providing a molecular mechanism to explain the marked reduction in PI4K2A activity in the patient cells. Hence, R275 plays a key role in promoting PI4K2A catalytic activity by sustaining electrostatic interactions between the enzyme and anionic lipids at the protein‐membrane interface.
We found that levels of PI(3,4,5)P3, a molecule with potent anti‐apoptotic, pro‐proliferative and pro‐motility properties, were not reduced in the patient cells. This is consistent with previous work established that cellular levels of PI(4,5)P2 do not substrate‐limit the generation of the quantitatively minor lipid PI(3,4,5)P3 (<0.02% of the total phosphoinositide content in this study) by phosphoinositide 3‐kinases. Furthermore, as PI4K2A activity is required for the endosomal trafficking and degradation of activated receptor kinases, 28 it is conceivable that augmented PI(3,4,5)P3 production could arise indirectly through decreased degradative trafficking of activated receptor tyrosine kinases and sustained activation of phosphoinositide 3‐kinases. Indeed we found a small but statistically significant increase in the very low abundance (0.68 ± 0.03 ng/mg protein) PI(3,4,5)P3 species with 18.0/20:3 of acyl chain composition. The functional significance of this alteration to acyl composition is not yet clear, although it is interesting to note that there is some evidence that a switch in phosphoinositide acylation patterns can occur in disease as has been shown for some cancer cell lines with p53 tumour suppressor deletions. 29
Our Western blotting results demonstrated that levels of PI4K2A protein were elevated in patient cells vs controls (Figure 3). This finding indicated that there is a functional link between the catalytic activity of the enzyme and its turnover or expression. Interestingly, work from Mössinger and colleagues revealed that PI4K2A binds the ubiquitin ligase ITCH through a specific N‐terminal PPxY motif resulting in ubiquitination of PI4K2A and concomitant PI4K2A‐mediated ITCH activation. 30 Since the ITCH binding motif of PI4K2A is unaffected by the R275W mutation, it seems plausible that in patient cells, PI4K2A is ubiquitinated but nevertheless subject to delayed degradation due to diminished PI4P‐dependent endo‐lysosomal trafficking. 28 Further work will be required to investigate the possibility that dysfunctional protein degradation results from the R275W mutation and to find out, whether this could affect the patient neurological phenotype.
Another possible disease mechanism to consider is that the cutis laxa and neurological phenotype of the patient may be an indirect consequence of altered PI4K2A activity on the vacuolar ATPase (V‐ATPase). This endosomal proton pump was found to be dysfunctional in related conditions 31 and may be regulated by PI(3,5)P2. 32 , 33 , 34 However, given that the production of PI(3,5)P2 does not require PI 4‐kinase activity, 35 , 36 V‐ATPase involvement seems unlikely. Trans‐Golgi network‐lysosomal trafficking remained unaffected by the R275W mutation (Supporting Information Figure S2). In addition to ruling out a general problem with dysfunctional trafficking, these results make it unlikely that the patient's choreoathetosis is caused by a sphingolipid storage defect similar to the one implicated in Gaucher's–associated Parkinson disease (reviewed by Brockmann and Berg 37 ).
Homozygous TWNK mutations are known to cause a mitochondrial DNA depletion syndrome type 7 (hepatocerebral type) (OMIM 271245). This progressive, neurodegenerative syndrome is characterised by hypotonia, ataxia, opthalmoplegia, hearing loss and seizures leading to early lethality in most cases. 38 We could not exclude that the clinical phenotype of our patient was partly due to the TWNK mutations. However, we found no mtDNA depletion in the fibroblasts of our patient and OXPHOS complex activities were normal. In addition, complexome profiling analysis showed no evidence for a disturbed biogenesis of the OXPHOS complexes comprising subunits encoded by the mitochondrial DNA. Recently the same mutation in TWNK has been described in a patient with ovarian dysgenesis, deafness, neuropathy, ataxia and mild intellectual disability with normal lactate levels, 39 these features are very similar to Perrault syndrome patients with other reported Twinkle mutations (OMIM 616138). 40 No functional evidence for the pathogenicity of these mutations was provided. Our patient shows no signs of ovarian dysgenesis and deafness. However, ovarian dysgenesis typically becomes evident only during puberty while the age of onset for hearing loss is variable. The next few years it should become evident whether or not a Perrault‐like syndrome will also manifest in our patient, who is now 12 years of age. Cutis laxa has never been described in patients with TWNK mutations, making it unlikely that the cutis laxa in our patient relates to mutations in this gene.
The male siblings described by Alkhater et al 17 presented with subtle facial dysmorphism at birth, but showed a normal development till the age of 5 to 6 months. From that age global developmental delay, dystonia, visual inattentiveness and loss of acquired skills were noted. They showed a marked movement disorder characterised by facial dyskinesias and global akathisia and were, and still are, unable to speak or walk. From the age of 9 years old both children had epilepsy. There was no cutis laxa reported in these patients. A homozygous nonsense mutation in PI4K2A was identified in both siblings (99400564C>A, c.65C>A; p.Ser22*). It is to be expected that the biochemical consequence of the homozygous nonsense mutation, a stop codon is introduced after 22 amino acids leading to a complete loss of function of PI4K2A, would be more severe than in our patient. However, the overlap in phenotype is evident.
The knockout mouse model also shows a movement disorder starting from the age of 4 months. 13
In conclusion, our combined observations suggest a disease mechanism involving dyshomeostasis of particular acyl chain phosphoinositide subsets on endosomal membranes.
Based on the presentation in our patient, the patients described by Alkhater et al 17 and the previously characterised mice model we define a novel metabolic defect in the phosphatidylinositol pathway, presenting with a severe neurologic phenotype and expanding the existing phenotype with cutis laxa. Further work is needed to investigate how this might be relevant to the patient's clinical presentation.
CONFLICT OF INTEREST
The authors declare no potential conflict of interests.
AUTHOR CONTRIBUTIONS
Miski Mohamed and Thatjana Gardeitchik participated in the study concept and design, acquisition of data, analysis and interpretation of the data and the drafting of the manuscript. Shanti Balasubramaniam participated in acquisition of data, analysis and interpretation of the data and the critical revision of the manuscript. Sergio Guerrero‐Castillo, Daisy Dalloyaux, Sanne van Kraaij, Hanka Venselaar, Alexander Hoischen, Zsolt Urban, Ulrich Brandt, Michele Frison, Lock‐Hock Ngu, Bert Callewaert, Hans Spelbrink, Wouter W. Kallemeijn and Johannes M. F. G. Aerts participated in the acquisition of data, analysis and interpretation of the data. Raya Al‐Shawi and J. Paul Simons participated in the acquisition of data, analysis and interpretation of the data and the review of study results. Mark G. Waugh, Eva Morava and Ron A. Wevers participated in the study concept and design, acquisition of data, analysis and interpretation of the data, the drafting of the manuscript, critical revision of the manuscript for important intellectual content and supervised the study.
INFORMED CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
ANIMAL RIGHTS
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Supporting information
FIGURE S1 3D‐structure of the catalytic domain of PI4K2A. Panel A: an overview of PI4K2A in which the arrow indicates the position of R275. Panel B shows the position of the R275 residue on the surface of the octameric complex, where interaction with the membrane takes place. In red, the R275W mutant is highlighted and in green the R275.
{kind=link}
FIGURE S2 Complexome profiling analysis of mitochondrial OXPHOS complexes. Panel A: Heat map representation of the migration profiles of all subunits of the five OXPHOS complexes detected in mitochondria enriched fractions from control and patient fibroblasts. Panel B: Migration profiles of the five OXPHOS complexes showing the relative abundance of proteins plotted against their apparent molecular mass. The relative abundance of each complex was calculated as the average of the normalised iBAQ values for all subunits identified. CI, complex I; CII, complex II; CIII, complex III, CIV, complex IV; CV, complex V.
{kind=link}
FIGURE S3 Analysis of GBA. Panel A: Structures of activity‐based probes used to label GBA. Panel B: in vitro ABP 1 labelling of GBA in control and PI4IIα mutant fibroblast homogenate (first row) compared to the labelling of GBA within living cells with ABP 2 (second row), with overlay shown in third row, compared to total amount of GBA protein detected by Western blotting (fourth row). Total protein input was visualised by Coomassie Brilliant Blue staining (fifth row). C, Quantification of Western blotting, and ABP labelling by densitometry of fluorescent bands shown in panel B. D, Enzymatic activity of GBA in cell lysates measured with 4MU‐β‐Glc as substrate.
{kind=link}
DATA S1 Supporting Information: Methods.
ACKNOWLEDGMENTS
This study was financially supported by the Dutch Metakids Foundation (2013, M. M.), the Netherlands Organisation for Scientific Research (NWO) project 017.008.052 (2011, M. M.), the E. C. Noyons Foundation (R. A. W.), the National Institutes of Health (HL090648, Z. U.), the German Research Foundation (DFG) SPP1710; BR1633/3‐1 (U. B.), Research Grant of the EADV (PPRC‐2018‐50, B. C.), Special Research Fund of Ghent University (BOF‐Start 01N04516, B. C.) and the Royal Free Charity (M. G. W.). We thank Dr Qifeng Zhang from Babraham Institute, Cambridge, UK for performing the lipid mass spectrometry analysis. We also thank Prof M. C. Patterson, paediatric neurologist and chair of the Division of Child and Adolescent Neurology at Mayo Clinic (Rochester, Minnesota), for his help with the clinical description of the patient.
Mohamed M, Gardeitchik T, Balasubramaniam S, et al. Novel defect in phosphatidylinositol 4‐kinase type 2‐alpha (PI4K2A) at the membrane‐enzyme interface is associated with metabolic cutis laxa. J Inherit Metab Dis. 2020;43:1382–1391. 10.1002/jimd.12255
Miski Mohamed, Thatjana Gardeitchik, Shanti Balasubramaniam, Sergio Guerrero‐Castillo, Mark G. Waugh, Eva Morava and Ron A. Wevers contributed equally to this study.
Funding information Deutsche Forschungsgemeinschaft, Grant/Award Numbers: SPP1710, BR1633/3‐1; Dutch Metakids Foundation, Grant/Award Number: 2013; EADV, Grant/Award Number: PPRC‐2018‐50; Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NL), Grant/Award Number: 017.008.052; NIH: National Heart, Lung, and Blood Institute (NHLBI), Grant/Award Number: HL090648; Royal Free Charity; Special Research Fund of Ghent University, Grant/Award Number: BOF‐Start 01N04516; Stofwisselkracht
REFERENCES
- 1. Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93:1019‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Waugh MG. PIPs in neurological diseases. Biochim Biophys Acta. 2015;1851:1066‐1082. [DOI] [PubMed] [Google Scholar]
- 3. Clayton EL, Minogue S, Waugh MG. Mammalian phosphatidylinositol 4‐kinases as modulators of membrane trafficking and lipid signaling networks. Prog Lipid Res. 2013;52:294‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clayton EL, Minogue S, Waugh MG. Phosphatidylinositol 4‐kinases and PI4P metabolism in the nervous system: roles in psychiatric and neurological diseases. Mol Neurobiol. 2013;47:361‐372. [DOI] [PubMed] [Google Scholar]
- 5. Balla A, Balla T. Phosphatidylinositol 4‐kinases: old enzymes with emerging functions. Trends Cell Biol. 2006;16:351‐361. [DOI] [PubMed] [Google Scholar]
- 6. Henmi Y, Morikawa Y, Oe N, et al. PtdIns4KIIα generates endosomal PtdIns(4)P and is required for receptor sorting at early endosomes. Mol Biol Cell. 2016;27:990‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banerji S, Ngo M, Lane CF, Robinson CA, Minogue S, Ridgway ND. Oxysterol binding protein‐dependent activation of sphingomyelin synthesis in the golgi apparatus requires phosphatidylinositol 4‐kinase IIα. Mol Biol Cell. 2010;21:4141‐4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ketel K, Krauss M, Nicot AS, et al. A phosphoinositide conversion mechanism for exit from endosomes. Nature. 2016;529:408‐412. [DOI] [PubMed] [Google Scholar]
- 9. Wang H, Sun HQ, Zhu X, et al. GABARAPs regulate PI4P‐dependent autophagosome:lysosome fusion. Proc Natl Acad Sci U S A. 2015;112:7015‐7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hubber A, Arasaki K, Nakatsu F, et al. The machinery at endoplasmic reticulum‐plasma membrane contact sites contributes to spatial regulation of multiple legionella effector proteins. PLoS Pathog. 2014;10:e1004222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baskin JM, Wu X, Christiano R, et al. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat Cell Biol. 2016;18:132‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pagnamenta AT, Howard MF, Wisniewski E, et al. Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum Mol Genet. 2015;24:3732‐3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simons JP, Al‐Shawi R, Minogue S, et al. Loss of phosphatidylinositol 4‐kinase 2alpha activity causes late onset degeneration of spinal cord axons. Proc Natl Acad Sci U S A. 2009;106:11535‐11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Waugh MG. Amplification of chromosome 1q genes encoding the phosphoinositide signalling enzymes PI4KB, AKT3, PIP5K1A and PI3KC2B in breast cancer. J Cancer. 2014;5:790‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J, Lu Y, Zhang J, Kang H, Qin Z, Chen C. PI4KIIalpha is a novel regulator of tumor growth by its action on angiogenesis and HIF‐1alpha regulation. Oncogene. 2010;29:2550‐2559. [DOI] [PubMed] [Google Scholar]
- 16. Waugh MG. Chromosomal instability and phosphoinositide pathway gene signatures in glioblastoma multiforme. Mol Neurobiol. 2016;53:621‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alkhater RA, Scherer SW, Minassian BA, Walker S. PI4K2A deficiency in an intellectual disability, epilepsy, myoclonus, akathisia syndrome. Ann Clin Transl Neurol. 2018;5(12):1617‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clark J, Anderson KE, Juvin V, et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods. 2011;8:267‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Balla A, Kim YJ, Varnai P. Maintenance of hormone‐sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4‐kinase IIIalpha. Mol Biol Cell. 2008;19:711‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sarkes D, Rameh LE. A novel HPLC‐based approach makes possible the spatial characterization of cellular PtdIns5P and other phosphoinositides. Biochem J. 2010;428:375‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baumlova A, Chalupska D, Róźycki B, et al. The crystal structure of the phosphatidylinositol 4‐kinase IIα. EMBO Rep. 2014;15:1085‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heide H, Bleier L, Steger M, et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;16:538‐549. [DOI] [PubMed] [Google Scholar]
- 23. Aerts JM, Donker‐Koopman WE, Murray GJ, Barranger JA, Tager JM, Schram AW. A procedure for the rapid purification in high yield of human glucocerebrosidase using immunoaffinity chromatography with monoclonal antibodies. Anal Biochem. 1986;154:655‐663. [DOI] [PubMed] [Google Scholar]
- 24. Jonsson LM, Murray GJ, Sorrell SH, et al. Biosynthesis and maturation of glucocerebrosidase in Gaucher fibroblasts. Eur J Biochem. 1987;164:171‐179. [DOI] [PubMed] [Google Scholar]
- 25. Van Weely S, Aerts JM, Van Leeuwen MB, et al. Function of oligosaccharide modification in glucocerebrosidase, a membrane‐associated lysosomal hydrolase. Eur J Biochem. 1990;191:669‐677. [DOI] [PubMed] [Google Scholar]
- 26. Witte MD, Kallemeijn WW, Aten J, et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat Chem Biol. 2010;6:907‐913. [DOI] [PubMed] [Google Scholar]
- 27. Zhou Q, Li J, Yu H, et al. Molecular insights into the membrane‐associated phosphatidylinositol 4‐kinase IIα. Nat Commun. 2014;5:3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Minogue S, Waugh MG, De Matteis MA, Stephens DJ, Berditchevski F, Hsuan JJ. Phosphatidylinositol 4‐kinase is required for endosomal trafficking and degradation of the EGF receptor. J Cell Sci. 2006;119:571‐581. [DOI] [PubMed] [Google Scholar]
- 29. Naguib A, Bencze G, Engle DD, et al. p53 mutations change phosphatidylinositol acyl chain composition. Cell Rep. 2015;10:8‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mössinger J, Wieffer M, Krause E, et al. Phosphatidylinositol 4‐kinase IIα function at endosomes is regulated by the ubiquitin ligase Itch. EMBO Rep. 2012;13:1087‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guillard M, Dimopoulou A, Fischer B, et al. Vacuolar H+‐ATPase meets glycosylation in patients with cutis laxa. Biochim Biophys Acta. 2009;1792:903‐914. [DOI] [PubMed] [Google Scholar]
- 32. Deranieh RM, Shi Y, Tarsio M, et al. Perturbation of the vacuolar ATPase: a novel consequence of inositol depletion. J Biol Chem. 2015;290:27460‐27472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ho CY, Choy CH, Wattson CA, Johnson DE, Botelho RJ, et al. The Fab1/PIKfyve phosphoinositide phosphate kinase is not necessary to maintain the pH of lysosomes and of the yeast vacuole. J Biol Chem. 2015;290:9919‐9928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li SC, Diakov TT, Xu T, et al. The signaling lipid PI(3,5)P₂ stabilizes V1‐V(o) sector interactions and activates the V‐ATPase. Mol Biol Cell. 2014;25:1251‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCartney AJ, Zhang Y, Weisman LS, et al. Phosphatidylinositol 3,5‐bisphosphate: low abundance, high significance. Bioessays. 2014;36:52‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shisheva A, Sbrissa D, Ikonomov O. Plentiful PtdIns5P from scanty PtdIns(3,5)P2 or from ample PtdIns? PIKfyve‐dependent models: evidence and speculation (response to: DOI 10.1002/bies.201300012). Bioessays. 2015;37:267‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brockmann K, Berg D. The significance of GBA for Parkinson's disease. J Inherit Metab Dis. 2014;37:643‐648. [DOI] [PubMed] [Google Scholar]
- 38. Nikali K, Lönnqvist T. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. Infantile‐Onset Spinocerebellar Ataxia. Seattle: University of Washington; 1993. ‐2017. [Google Scholar]
- 39. van de Warrenburg BP, Schouten MI, de Bot ST, et al. Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene‐disease associations and unanticipated rare disorders. Eur J Hum Genet. 2016;24:1460‐1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morino H, Pierce SB, Matsuda Y, et al. Mutations in Twinkle primase‐helicase cause Perrault syndrome with neurological features. Neurology. 2014;83:2054‐2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 3D‐structure of the catalytic domain of PI4K2A. Panel A: an overview of PI4K2A in which the arrow indicates the position of R275. Panel B shows the position of the R275 residue on the surface of the octameric complex, where interaction with the membrane takes place. In red, the R275W mutant is highlighted and in green the R275.
FIGURE S2 Complexome profiling analysis of mitochondrial OXPHOS complexes. Panel A: Heat map representation of the migration profiles of all subunits of the five OXPHOS complexes detected in mitochondria enriched fractions from control and patient fibroblasts. Panel B: Migration profiles of the five OXPHOS complexes showing the relative abundance of proteins plotted against their apparent molecular mass. The relative abundance of each complex was calculated as the average of the normalised iBAQ values for all subunits identified. CI, complex I; CII, complex II; CIII, complex III, CIV, complex IV; CV, complex V.
FIGURE S3 Analysis of GBA. Panel A: Structures of activity‐based probes used to label GBA. Panel B: in vitro ABP 1 labelling of GBA in control and PI4IIα mutant fibroblast homogenate (first row) compared to the labelling of GBA within living cells with ABP 2 (second row), with overlay shown in third row, compared to total amount of GBA protein detected by Western blotting (fourth row). Total protein input was visualised by Coomassie Brilliant Blue staining (fifth row). C, Quantification of Western blotting, and ABP labelling by densitometry of fluorescent bands shown in panel B. D, Enzymatic activity of GBA in cell lysates measured with 4MU‐β‐Glc as substrate.
DATA S1 Supporting Information: Methods.